

Abstract
This study determined in primary cultures of human lung cancer cells the cell specificity of chlorin-based photosensitizers. Epithelial cells (EC) preferentially retained 3-[1-hexyloxyethyl]-2-devinylpyropheophorbide-a (HPPH) and related structural variants. Tumor-associated fibroblasts (Fb) differ from EC by a higher efflux rate of HPPH. Immunoblot analyses indicated dimerization of STAT3 as a reliable biomarker of the photoreaction. Compared to mitochondria/ER-localized photoreaction by HPPH, the photoreaction by lysosomally targeted HPPH-lactose showed a trend toward lower STAT3 crosslinking. Lethal consequence of the photoreaction differed between EC and Fb with the latter cells being more resistant. A survey of lung tumor cases indicated a large quantitative range by which EC retain HPPH. The specificity of HPPH retention defined in vitro could be confirmed in vivo in selected cases grown as xenografts. HPPH retention as a function of the tetrapyrrole structure was evaluated by altering side groups on the porphyrin macrocycle. Presence or absence of a carboxylic acid at position 172 proved to be critical. A benzyl group at position 20 enhanced retention in a subset of cancer cells with low HPPH binding. This study indicated experimental tools that are potentially effective in defining the photosensitizer preference and application for individual patient’s cancer lesions.
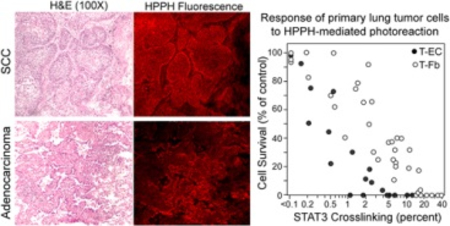
Legend to Graphic Abstract:
Tumor cell-specific binding of HPPH detected in section of human lung squamous cell carcinoma and adenocarcinoma (left) and cell type-specific relationship of HPPH-mediated photoreaction with cell killing (right).
INTRODUCTION
Photodynamic therapy (PDT) of cancer lesions achieves specificity by the enhanced accumulation of the photosensitizer (PS) in tumor tissue, the targeted delivery of therapeutic light, and the lethal action of intracellularly generated reactive oxygen species (1). While these features have further PDT as a treatment of choice for light-accessible tumors, the clinically approved photosensitizers lack cancer cell specificity. Hence, the development of the next generation PSs has the goals: (a) to extend the therapy to light of longer wavelength for deeper tissue penetration, (b) to gain a systemic distribution with enhanced tropism to the tumor lesions, and (c) to achieve preferential retention by cancer cells thereby enhancing tumor cell-specific destruction and reduce skin toxicity (2). Phase I clinical trials of the leading chlorin, 3-[1-hexyloxyethyl]-2-devinylpyropheophorbide-a (HPPH), have confirmed its effectiveness in PDT of Barrett’s esophagus, endobronchial and head and neck lesions (3–5). Despite the encouraging outcome, the data of the head and neck cancer treatments indicated that approximately 20% of the cases did not result in a complete response (6, 7). This finding has been interpreted to mean that an appreciable variation in PS uptake may exist among tumors and the knowledge of the cellular preference and specific action of PSs prior to PDT may allow for the design of optimized treatment parameters for individual cases. The present study presents the experimental approach to identify the cellular specificity of HPPH uptake and action by applying short-term primary cultures of epithelial and stromal cells derived from surgical specimens of human lung cancer tissues. The data indicated pheophorbide derivatives that are specifically retained by lung epithelial tumor cells grown in culture and as a xenograft. Comparative analyses also demonstrated that a large variability in PS preference exists among tumor cell preparations, that subcellular site of PS retention affects cellular response to PDT, and that minor modifications of the porphyrin structure fundamentally alter cellular retention. With the ability to identify the PS preference of the patient’s tumor cells, the selection of the optimal PS and PDT condition for individual cancer cases may become feasible.
MATERIAL AND METHODS
Photosensitizers.
3-[1-Hexyloxyethyl]-2-devinylpyropheophorbide-a (HPPH), 172-methyl-HPPH (HPPH-ME) and 172-lactose-HPPH (HPPH-Lac) (8); 3-(1’-m-iodobenzyloxyethyl)pyropheophorbide-a (PS-531-COOH) and 172-methyl-PS 531 (PS-531-ME) (9); 20-benzoic acid-HPPH (PS-821) and 20-benzoic acid-(172-methyl)-HPPH (PS-820) (R. K. Pandey, P. Joshy et al, unpublished; manuscript in preparation) were generated in the laboratory of Dr. R. K. Pandey. Structures of these PSs are shown in Fig. 6. Stock PS solutions in 5% glucose in distilled water containing 2% ethanol and 0.1% Tween 80 were serially diluted in culture medium immediately prior addition to cells. Photofrin-II (Profimer sodium) (10) and Foscan® (Temoporfin) (11, 12) were provided by the PDT Center at RPCCC.
Figure 6.

Specificity of pheophorbide retention by human lung tumor cells. Primary cultures of lung T-ECs were grown to confluence in KSFM. A standard PS treatment was applied for every cell preparation: Uptake of the PSs indicated at the right was carried by incubating the cultures for 24 h in RPMI containing10%FBS and 1600 nM PSs followed by 24 h chase in PS-free medium. The cell-associated PS fluorescence was imaged by microscopy at 100X magnification using 2 sec exposures. All images were identically processed. Examples of PS retention patterns are shown. For comparison, the PS retention pattern of N-Fb is included at the left and the two most distinct patterns detected in cancer cells derived from PDX tissue (TEC-1–2 and TEC-21, still including murine stromal cells) are shown on the right.
Cells.
Under the terms of an IRB-approved protocol, Tissue Procurement Service at RPCCC provided surgically resected non-small cell lung cancer (NSCLC) tissues from 119 cases (laboratory designations L201 to L384) for preparing primary cell cultures. The cancers were diagnosed as stage 1 (N=35), stage 2 (N=73), stage 3 (N=10), or stage 4 (N=1) and represented 75 adenocarcinomas, 42 squamous carcinomas and 2 large cell carcinomas. Primary cultures of tumor epithelial cells (T-EC) and stromal fibroblasts (T-Fb) were generated as described previously (13, 14). Since no significant differences in HPPH uptake and action could be associated with the specific cancer type of the originating tissue (adenocarcinoma or squamous cell carcinoma), the results obtained with each EC preparation were treated as single category termed T-EC. Whenever possible, non-tumor lung tissue was used to prepare the “normal” epithelial cells (N-EC). EC were selectively grown on collagen-1 matrix (PureCol®, Advanced BioMatrix, San Diego, CA) in serum-free keratinocyte medium (KSFM; Invitrogen). Recovery of proliferating T-EC cultures was successful in 49% of the cases, whereas the recovery of N-EC was 91%. Tumor cells derived from 24 different xenografts of human NSCLC procured at RPCCC, which had been successfully passaged at least 3 times in SCID mice (15, 16), were propagated in tissue culture using medium containing 10% FBS and termed TEC-1 to TEC-24. Cells derived from xenografts subsequently generated from in vitro TEC cultures were labeled with hyphenated numbers (e.g., TEC-1–1, etc). Co-cultures were assembled by plating EC (5 ×103 cells per cm2) onto a collagen-1 matrix followed 24 h later by the addition of T-Fb (1 ×105 cells per cm2) in RPMI containing 10% FBS. To facilitate detection of Fb within co-cultures, Fb were stained with carboxyfluorescein succinimidyl ester (CFSE; Invitrogen) prior to their addition to EC (13). In reconstituted co-cultures containing CSFE-labeled Fb, the precise location of the non-labeled T-EC is recognizable by comparing the corresponding phase-contrast and green fluorescent images included in Figs. 1 and 5.
Figure 1.

EC-specific retention of HPPH determined in reconstituted co-cultures of TEC-1 and CSFE-labeled lung (L364) tumor stromal cells. (A and B), Co-cultures were incubated in serum-free RPMI containing 3.2 μM HPPH (A) or HPPH-Lac (B) at 37°C for 30 min and then followed by a chase in fresh RPMI-10%FBS for 4 and 24h. The culture morphology was assessed by phase microscopy followed by imaging CSFE and PS fluorescence. (C) Comparison of cellular retention of HPPH, Foscan and Photofrin II. Co-cultures of TEC-1 and CSFE-stained stromal cells were first incubated for 4 h at 37°C in RPMI-10% FBS containing either 3.2 μM HPPH, 3.2 μM Foscan or 10 μg/ml Photofrin-II followed by a 24 h chase in PS-free medium. The culture and cell-associated PS were imaged by phase and fluorescent microscopy.
Figure 5.

HPPH dose-dependent and cell type-specific PDT response. A co-culture of TEC-1–2 (passage 37) and CSFE-labeled T-Fb (L323) was incubated for 24 h with 800nM HPPH in DMEM-10% FBS and then chased for 24 h in HPPH-free medium. Cellular level of retained HPPH was determined by fluorescent microscopy prior to treatment with therapeutic light (Day 0). The culture was imaged daily for 3 days post PDT to record the reorganization of CSFE-labeled stromal cells and fate of TEC-1–2 cell clusters. HPPH is stably retained by photo-damaged cells and is detectable by fluorescence present in aggregates of killed TEC-1–2 cells. Surviving TEC-1–2 clusters are recognized by the low to non-detectable HPPH fluorescence and physical exclusion of stromal cells from the collagen-support matrix.
PS uptake and photoreaction.
Cells were incubated with PS-containing medium for time periods ranging from 30 min on ice (for surface binding) to 24h at 37° (maximal uptake). To achieve complete internalization of PS, cells were incubated for additional 4h (Fb) or 24 h (EC) in PS-free culture medium containing 10% FBS (chase). Cell-associated PS was visualized at 100X or 400X magnification on an inverted fluorescence microscope (Zeiss Axiovert 200; filter setting λex 410/440 nm and λem 675/750 nm). Images were captured with a Q-Imaging camera in a 16 bits/channel format by 0.1 to 3 sec exposure to ensure recording emitted fluorescent light within the linear range of detection. Fluorescence was quantified by integrating pixel values within defined areas using ImageQuant TL program (G.E. Health Sciences, Malborough, MA) and corrected for background (fluorescence recorded for culture wells incubated with PS-containing medium but without cells). Net fluorescence intensity values were calculated for an equivalent of a 1-sec image exposure (termed fluorescence units, FU). In co-cultures containing CSFE-stained Fb, 510–530 nm fluorescence was recorded before that of the PSs.
Photoreaction was performed by illuminating PS-treated cell cultures within a cell culture incubator at 37° with 665 or 675 nm light of an argon-pumped dye laser for 9 min at a dose of 5.6 mW/cm2 to a total fluence of 3 J/cm2. Depending upon the experimental design, cells were either extracted immediately after light treatment or incubated at 37° in serum-containing medium for up to 3 days.
Cell analyses.
Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer. Aliquots of extracts were subjected to Western blotting as described (9, 14). STAT3 crosslinking was expressed by the percent conversion of monomeric STAT3 into the homodimeric form I of the STAT3-crosslinked complexes. PDT-mediated cell killing was determined 24 h after exposure to therapeutic light by counting viable cells in a hematocytometer using trypan blue dye exclusion.
PS uptake in vivo.
Groups of age-matched (8–10 week; both gender) SCID mice (C.B-lgh-lb/lcrTac-PrMscid/Ros; Department of Laboratory Animal Resources, RPCCC) received single s.c. implantation of human lung tumor tissue or cells (2×106 in 50 μl PBS) on the dorsal sides of the left hind leg. When palpable tumor mass reached a diameter of 8–10 mm, the animals were injected intravenously (10 μl/g body weight) with 5% glucose in water and containing PS (3 nmole/g). Groups of 4 animals were euthanized 6, 24 and 48 h later. Tumor, liver (after perfusion with phosphate buffered saline), lung and kidney were excised and 3–5 mm cubes were immediately embedded in OCT and frozen on dry ice. Five-μm cryosections were examined for cellular distribution and level of PS fluorescence by microscopy as described above for tissue cultures. Cryosections of organs from tumor-bearing SCID mice, but not injected with PS, were used to define background fluorescence values. The animal studies were approved by IACUC.
Statistical evaluation.
Mean and standard deviation of experiments carried out at least in triplicate were calculated and used for the graphic presentations. Significance between control and experimental groups was examined using a Student t-test. A value of P <0.05 was regarded as significant.
RESULTS
Analysis of cell-specific uptake and retention of HPPH
Previous studies have suggested that lung EC and Fb differ in their ability to retain pheophorbides, i.e., HPPH (13, 17). What has yet to be determined is: (a) whether the level of PS accumulation is subject to change as a consequence of oncogenic transformation of ECs, (b) whether specific retention of PS is consistent among the same cell types derived from separate tumor cases, (c) whether the distinct subcellular localization of the PS influences the outcome of PDT, and (d) whether the differential retention is dictated by the molecular structure of the porphyrin.
To answer these questions, the assay of primary cells derived from human lung tumor tissue was applied. One caveat of cultured cells in place of tissue in situ is that the culture system primarily involves cells that are capable of proliferating in vitro and, thus, do not fully represent in vivo differentiated tissue. Moreover, a limitation of primary EC cultures is that proliferation is seldom maintained beyond few passages. Our compiled information on successful primary EC cultures indicated that only in 14 of 58 cases of T- ECs could be cultured for >5 passages and only 3 cases exhibited an immortalized phenotype with crisis-free growth for >50 passages from the time point of initial tumor tissue dissociation. We concluded that the definition of the basic profile for PS retention by lung tumor cells had to rely on assaying primary cultures within the first 3 passages after establishment.
Depending upon their structural and physicochemical properties, pheophorbides exhibit two basic modes of cellular uptake: 1) by transmembrane diffusion into intracellular compartment with preferential accumulation in the ER and the outer membrane of mitochondria as noted for HPPH, or 2) by binding to the cell surface followed by endocytosis and deposition in lysosomes as found with HPPH-lactose (HPPH-Lac) (8, 17). We confirmed that the two distinct uptake mechanisms applied to cultures of lung tumor ECs (Supporting Information, Fig. S1A). After 24h incubation, the characteristically distinct subcellular distribution of HPPH and HPPH-Lac was evident.
Cellular uptake and retention of porphyrin-based PS is strongly influenced by the presence of plasma proteins known to competitively bind PSs (18, 19). Since in vitro propagation of primary lung EC cultures demanded the use of hormonally defined medium, the retention of PS by ECs maintained in KSFM was noted to be lasting (Fig. S1B, upper panels). Although the presence of 10% FBS promoted differentiation of lung EC in vitro (Fig. S1B, lower panels), the comparative assessment of PS retention by cultured tumor and stromal cells was carried out using biologically relevant assay conditions involving serum-containing RPMI medium. Beside plasma proteins, the expression of ATP-dependent transporter proteins, in particular ABCG2, has been found to reduce retention of HPPH by certain cell types (20, 21). However, western blot analyses of lung EC and Fb cultures indicated only three cases of T-EC with a detectable level of ABCG2 protein. Hence, the contribution of ABCG2 to the PS uptake phenotype of the primary cultures was not further pursued as part of this study.
The cell type specificity of PS uptake was evaluated in reconstituted co-cultures of primary human lung T-EC and T-Fb. This assay permits the comparative visualization of PS binding to the cell surface, the kinetics of internalization, and preferential retention (Fig. 1). The time course of cellular distribution indicated the differential PS retention between the two cell types. While the initial uptake of HPPH by EC and Fb appeared comparable, the retention of HPPH by EC during the subsequent chase period exceeded several-fold that by Fb, which generally showed an efflux of HPPH with t1/2 ~ 1–2h (Fig. 1A). In contrast, HPPH-Lac, a potential ligand for galactoside-specific lectins, demonstrated a strong cell-surface binding followed by endocytosis and predominant lysosomal deposition (Fig. 1B). With a single exception (L340 T-EC, Fig. S2), none of the lung EC and Fb combination indicated an appreciably enhanced cell type-specific binding and uptake of HPPH-Lac. Lysosomal retention was often more pronounced in Fb than EC (13) with a t1/2 of egress ranging from 12 to 48h. The fluorescence detectable in cells after 72 h still represented intact HPPH-Lac without evidence of lysosomal hydrolysis, as determined by thin-layer chromatographic analysis of cell extracts (data not shown).
The co-culture assay with EC and Fb was applied to Photofrin and Foscan, two PSs already approved for clinical use. The results indicated that neither PS exhibited a preferential accumulation in tumor EC over Fb, and thus, strikingly differed from HPPH (Fig. 1C).
Photoreaction mediated by mitochondrial and lysosomal HPPH
The precise relationship of the subcellular site of HPPH-mediated photoreaction (mitochondria or lysosomes) with the immediate cellular signaling reactions and cell death was evaluated in monotypic cell cultures (Fig. 2). PS level within the cells was quantitated by fluorescence imaging (Fig. 2A) and related to the immediate light-dependent reaction resulting in covalent STAT3 crosslinking, degradation of EGFR, activation of p38 MAPK pathway (Fig. 2B), and by cell death manifested 24 h later (Fig. 2C). The results demonstrated that maximal uptake of HPPH-Lac by non-tumor and tumor EC reached approximately half the level of HPPH. Despite this difference, when normalized to the cellular concentration, both PSs appeared to be similarly effective in mediating cellular responses to photoreactions. A consistent difference was detected in the higher activation of the p38 MAPK pathway and cell death by the mitochondrial HPPH photoreaction.
Figure 2.

PS-dose dependent photoreactions in primary lung ECs. Confluent monolayers of N-EC and T-EC (L374, passage 3) were treated with increasing concentrations of HPPH or HPPH-Lac for 24 h in RPMI containing 10% FBS followed by a chase for 24 h in PS-free medium. (A) The cell-associated PS fluorescence was determined by microscopy. Only the fluorescent images of T-EC cells at 400X treated with the highest PS concentration are shown to demonstrate the distinct subcellular distribution. (B) The levels of fluorescence in each culture were quantitated and expressed in arbitrary fluorescence units (FU). After 9 min light treatment (3 J/cm2) the products of the PS-mediated photoreaction were determined by immunoblot analyses including the covalent crosslinking of STAT3, the loss of EGFR and the activation of p38 kinase. (C) In parallel cultures, the percentage of PDT-surviving ECs were determined after additional 24 h-incubation of the light-treated cells in RPMI containing10% FBS.
Quantitative relationship of HPPH concentration and PDT response
Since assays of PS uptake and action in individual lung EC preparations were technically restricted to a few cultures per case, the broader definition of PS action had to rely on the compiled information gathered from multiple patients’ samples analyzed over the course of this study. The analyses compared HPPH and HPPH-Lac uptake, determined by the fluorescence measurement before light treatment, with the level of crosslinked STAT3 immediately after light treatment (Fig. 3) and, in replicated cultures, to cell survival 24 h later (Fig. 4). Although there was considerable variation in the PS uptake among cell cultures derived from separate tumor specimens, a close correlation of PS level with photoreaction could nevertheless be detected and appeared to be similar in non-tumor and tumor EC cultures (Fig. 3A and B) and was not appreciably different from the reference cell line, TEC-1–2 (Fig. 3C). The compilation also revealed a trend of the lysosomal PDT with HPPH-Lac being somewhat less effective in eliciting a photoreaction leading to STAT3 crosslinking than mitochondrial HPPH.
Figure 3.

Compilation of data derived from individual cultures of N-EC (A), T-EC (B) and TEC-1–2 (C) correlating percentage of STAT3 crosslinking with cellular level of HPPH or HPPH-Lac (FU). The treatments of the cultures were the same as applied in Fig. 2. Each circle represents the result of individual cultures derived from a total of 54 separate lung samples.
Figure 4.

Compilation of data derived from individual cultures of N-EC (A), T-EC (B), TEC-1–2 (C), and T-Fb (D) correlating cellular survival with the percentage of STAT3 crosslinking. Duplicate cell cultures were subjected to incubation with HPPH and HPPH-Lac and subsequent light treatment as applied in Fig. 3, but in each set, one of the duplicate cultures was incubated for additional 24 h in RPMI containing 10% FBS for determining the percentage of surviving cells. Each circle represents the result of individual duplicate cultures.
A more striking difference emerged when STAT3 crosslinking was correlated with cell killing. HPPH PDT was essentially 100% lethal for all EC cultures in which ~2% STAT3 crosslinking was achieved (Fig. 4A–C). That amounted to a photoreaction mediated by ~ 50 fluorescence units of HPPH (Fig. 3). In contrast, lysosomal HPPH-Lac PDT required a greater photoreaction for a ~100% lethal outcome that resulted in ~5–10% STAT3 crosslinking. Even more resistant proved to be stromal cells. In agreement with previous findings (13), T-Fb differed from EC by requiring a photoreaction yielding ~10–20% STAT3 crosslinking of STAT3 to achieve 100% lethality (Fig. 4D). These results suggest that successful lung tumor PDT by HPPH is dependent on a minimal photoreaction that can be predicted to result in ~2% STAT3 crosslinking, a level of photoreaction that will kill the tumor epithelial cells but should spare co-resident stromal cells.
The relevance of the differential retention of HPPH for PDT of lung tumors is demonstrated by the treatment of reconstituted co-cultures of T-EC and T-Fb (Fig. 5). T-EC, which proliferate in the presence of FBS, were combined with stromal cells to form segregated cell clusters as observed in original cancer tissue or its xenograft grown in SCID mice (see below). These co-cultures were treated with HPPH and subjected to a 24-h chase period. While all T-EC clusters retained HPPH at a level above that of Fb, there was a notable range of HPPH detectable among clonally expanded T-EC. This variation may reflect, in part, the heterogeneity of cells isolated from the tumor tissue. Exposure of the cultures to therapeutic light, followed by continued maintenance in growth medium, revealed the differential killing of the tumor cells. PDT-mediated killing was dictated by the level of HPPH at the time of light treatment and was in agreement with the data shown in Fig. 4C. In the same co-cultures, essentially all T-Fb, which, by the time of light treatment had already lost >90% of HPPH initially taken up, survived as predicted by the data in Fig. 4D. Within few days of post-PDT culture, Fb had infiltrated the sites of PDT-killed EC clusters. These sites are recognizable in Fig. 5 by the HPPH fluorescence stably retained by remnants of dead EC clusters. Also evident was that surviving Fb accumulated and proliferated at the sites of dead ECs. The same co-culture also indicated that those T-EC cell clusters, which escaped lethal PDT due to a low level of HPPH, did resume proliferation under physical exclusion of Fb from the growth site (Fig. 5, right panel).
Variability in the specificity of pheophorbide retention by T-EC
The initial characterization of T-EC was restricted to HPPH and HPPH-Lac because these compounds directed fundamentally distinct subcellular deposition, allowing assessment of the effects of photoreactions triggered at these subcellular sites. To address the question of the impact of cell transformation on the specificity of HPPH uptake, we devised a categorization of patient-derived cell preparations based on the comparative retention of 6 reference HPPH derivatives. The HPPH variants represented combinations of three specific structural modifications added to peripheral positions of the porphyrin macrocycle (Fig. 6, right side). The modifications included: (a) a methyl group at 172 position to neutralize the acidic charge of HPPH (HPPH and HPPH-ME); (b) iodophenyl group on position 3 (PS-531-COOH and PS-531-ME); and (c) a benzoic acid group on position 20 (PS-820 and PS-821). Positions 3, 172 and 20 were considered potential attachment points for targeting molecules to be evaluated in future studies. The structural modifications were also intended to assess the role of these side groups to affect the porphyrin’s interaction with components at the different subcellular sites.
Based on the experience gained with the initial study (see above Figs 1–5), a standard assay was applied to primary T-EC (passages 1–3, N=15). T-ECs were grown on collagen matrix in KSFM and, upon confluence, the medium was changed to RPMI containing 10% FBS and 1600nM of each of the 6 PSs. After 24 h incubation, the cells were subjected to a 24 h chase in the same medium, but without PS. The PSs retained by the cells were visualized by microscopy using identical imaging conditions. This comparative assay was also applied to N-EC (N=6) and T-Fb (N=6), as well as to cells recovered from patient-derived xenograft (PDX) (N=7; denoted TEC-1, −2, −9, −10, −17, −21, and −24).
Representative PS retention patterns are shown in Fig. 6. The key findings are as follows. Although the two groups consisting of T-Fb and N-EC showed diagnostic quantitative differences in their cellular retention of the 6 PSs, each group displayed a strikingly constant profile among individual cell preparations. All Fb cultures demonstrated a short retention time for each PS, with a particularly efficient egress of the carboxylated compounds HPPH, PS-531-COOH and PS-821. In contrast, all N-EC preparations exhibited a strong retention of the carboxylated compounds. Major variations in PS retention profiles were detected among T-EC cultures. Only 5/15 cases showed a PS preference comparable to that of N-EC, manifested by a strong retention of HPPH and PS-531-COOH. Among these 5 cases, only two strongly retained PS-821. In contrasts, the other 10/15 T-EC cases demonstrated a drastically reduced retention of HPPH, PS-531-COOH and PS-821, but sustained retention of the neutral derivatives HPPH-ME and PS-531-ME. The cross-comparison of all T-EC cultures indicated a rather uniform retention of PS-820, which in few cases resulted in this PS form becoming the most prominent compound retained after a 24 h chase period. However, Fb cultures also showed an elevated retention of PS-820. A particularly altered PS retention pattern was detected in one T-EC preparation (L237) (Fig. 6). Within the 24-h chase period, these cells lost each of the 6 PSs with a retention level below that of stromal cells.
Although the survey of primary T-EC cultures was restricted to early passages, in 4 cases the cells could be maintained beyond 5 passages and the impact of prolonged in vitro culture on PS uptake was re-assessed in later passages. In each case, essentially the same PS retention profile was identified as determined for the early passages (data not shown), suggesting a phenotypic stability of the tumor-evolved PS retention mechanisms.
The analyses of cells from 7 PDX cases indicated only two, TEC-21 (Fig. 6) and TEC-24, with a strong retention of the carboxylated PS. The cells derived from the other 5 PDX cases (including TEC-1 and TEC-1–2) showed a highly diminished retention of the carboxylated PS, but, as already described for several primary T-EC cases, the retention of the neutralized PS, HPPH-ME and PS-531-ME was substantially retained.
The patterns of PS retention (Fig. 6) tightly correlated with the photoreactions (representative example of TEC-1–2 in Fig. 7A) and cell killing (Fig. 7B) mediated by the individual PSs. The relationship of PS fluorescence value with PDT reactions mediated by the 6 HPPH-derivatives were in the quantitative range as predicted by the data in Figs 3 and 4.
Figure 7.

PS-specific photoreaction in lung T-EC. Confluent cultures of TEC-1–2 cells (passage 12) were treated for 24 h in RPMI containing 10% FBS and 1600 nM of the PSs indicated at the top, followed by 24 h chase in PS-free medium. (A) The cellular level of PS was determined by imaging and expressed as AU. The cultures were subjected to PDT and immediately analyzed for the level of crosslinked STAT3, EGFR and phosphorylated p38 MAPK. (B) Duplicate cultures were identically treated and PDT-surviving cells determined after 24 h culture.
Lung tumor cell-specific retention of HPPH in vivo
The characterization of PS retention by tumor cells in vitro has the caveat that the results may not adequately represent the same occurring in vivo. Therefore, we determined the impact of in vivo growth of TEC-1 on HPPH uptake in xenografts subcutaneously implanted into SCID mice. The initial experiments analyzed the uptake and cellular localization of HPPH in tumor tissue. The liver, kidney and lung served as reference for systemic deposition of HPPH. The localization and quantification of HPPH involved fluorescent microscopic analyses of 5-μm cryosections of mouse organs (Fig. 8A). A high level HPPH accumulation in the liver, kidney and lung was noted 6 h after PS injection that exceeded that for the tumor by about ~2-fold (Fig. 8B). However, by 24 h the levels of HPPH in the normal organs had substantially declined whereas that in the tumor had further increased resulting in a level that equaled that of the liver. By 48 h, tumor cells still retained HPPH at a level above the other organs. The microscopic inspection (Fig. 8A) revealed that HPPH is primarily confined to the regions of tumor cell clusters within the TEC-1 tissue, equivalent to what has been observed in in vitro (Fig. 1A). The quantitation of HPPH in cryosections agreed with the temporal biodistribution of HPPH previously defined in mice using 14C-HPPH as tracer (8) and also fell within the pharmacokinetic profile of HPPH in patients (22).
Figure 8.

Uptake and retention of HPPH by organs in TEC-1 xenograft-bearing SCID mice. Aliquots of TEC-1 xenograft tissue were implanted into two separate groups of 12 SCID mice and grown for 4 weeks to a size ranging from 8 to 10 mm diameter. HPPH (3 μmole/kg) was injected into the tail vein. After 6, 24 and 48 h, sets of four mice were euthanized and the indicated organs collected, embedded into OCT and 5-μm cryosections prepared. The sections were imaged by fluorescent microscope at 100X magnification with capture of the images by 3 sec exposure. (A) The fluorescent images of a representative set of the organ sections are reproduced. (B) The net HPPH fluorescence densities were determined for each organ section (using 4 to 6 separate areas) and expressed in arbitrary units (AU). The average AU values for HPPH fluorescence were used as measure of HPPH concentration in each organ of individual mice. Mean and S.D. of the AU values for both sets of animals (N=8) are shown.
The TEC-1 xenograft model also offered the opportunity to evaluate the in vivo biology of HPPH-Lac that, based on in vitro assay (Fig. 1B), did not suggest a suitable tropism for the tumor. Injection of HPPH-Lac into TEC-1-bearing SCID mice demonstrated an even greater complication, one that could not have been determined in the reconstituted tissue culture system (Fig. S3). Six hours following intravenous injection, most of HPPH-Lac was localized to the liver with a minor amount detectable in the TEC-1 tumor. By 24 h, the level of HPPH-Lac in the liver and kidney had substantially declined. No further increase of HPPH-Lac was measurable in the tumor where it remained substantially below that determined for HPPH in similarly treated animals (Fig. 8A). The difference in retention of HPPH-Lac and HPPH in tumor tissue in vivo mirrors qualitatively the difference observed in tissue culture (Figs. 1 and 2).
The experimental tools of in vitro and in vivo analysis of PSs also allowed addressment of the final question, as to whether the information about the specificity of lung tumor cells to retain HPPH in vitro (Fig. 6) will apply to in vivo. To do so, xenograft-derived TEC-1 and TEC-21 cells, which significantly differ in HPPH binding, were tested under identical conditions for the level of HPPH retained by these cells in Fb co-culture in vitro (Fig. 9A) and as subcutaneous tumor in SCID mice (Fig. 9B). The results confirmed that the ~4-fold higher HPPH retention by TEC-21 cells in culture was similarly manifested in the TEC-21 xenograft. The preferential HPPH retention of TEC-21 cells was particularly evident by the substantially elevated HPPH level in TEC-21 tumor tissue compared to the liver. A histological examination indicated that most of HPPH was localized to the tumor cell clusters (Fig. 9C). Taken together these findings support the conclusion that information gained from a cell culture assay are representative for the biological properties of the same tumor cells in vivo. The data also suggest that the mechanisms responsible for the lung EC-specific retention of HPPH is not subject to an appreciable change as a function of culture conditions. This study has provided the tumor models and test systems for approaching the future goal to identify the underlying biochemistry of cell type-specific retention of HPPH.
Figure 9.

Comparison of HPPH retention by lung tumor cells grown in vitro and in vivo. Duplicate sets of SCID mice bearing either a TEC-1 or TEC-21 xenograft were generated. (A) From one set of animals, the tumor was collected and processed for tissue culture of the cancer cells along with residual murine stromal cells using DMEM-10% FBS as growth medium (passage 0). After 3 weeks, the cultures contained clusters of tumor cells within a background monolayer of stroma cells. These cultures were treated for 24 h with medium containing 3 μM HPPH followed by 24 h chase period. The cellular retention of HPPH was determined by fluorescent microscopy using identical imaging setting. (B) A second set of animals was injected with 3 umole/kg HPPH and after 24 h, the tumor and liver were collected. The relative level of HPPH in these organs was determined by fluorescent microscopy of 5-μm cryosections. The images of HPPH fluorescence for each section were processed using identical conditions. (C) A cryosection of TEC-21 tumor was stained with hematoxylin and eosin (H&E) and compared to the HPPH fluorescent image (red colorized) of the same area on an adjacent section. The white bar shown at the top of the right panel indicates 250 μm.
DISCUSSION
Tissue cultures and xenografts were used as tools to determine the specificity and action of HPPH as these apply to PDT of carcinomas of the lung. The experimental approach was designed to gain diagnostic information about tumor cells that potentially could be subjected to PDT. Specifically, the assay aimed at evaluating individual tumor cases in respect to uptake and retention of HPPH. Since this information could not be gained by in situ treatment of patients, the best possible alternative assessment was by expanding the patient’s tumor cells from tumor samples ex vivo and determining in these the preference for PS. Despite the modifying effects anticipated to occur due to in vitro propagation of the cells, the study supported the usefulness of the techniques and indicated biomarkers that could assist in designing and monitoring PDT of tumor lesions.
The data demonstrate (a) the difference between stromal and epithelial cells in HPPH retention accounting for the observed enrichment in tumor tissue, (b) the impact malignant transformation on cellular retention of specific prophyrin structures, (c) the relevance of peripheral modification of the tetrapyrrole for uptake and subcellular deposition, and (d) cell-type specific difference in lethal action of PDT. The key information is the description of markers that permits linking the level of cellular HPPH with the photoreaction and cellular response. These markers are meant for evaluating patient’s tumors considered for PDT and for adjusting treatment conditions in those cases indicating a phenotype of low HPPH retention. Such adjustments can potentially include: (a) the application of a higher dose of HPPH, (b) a light treatment at an earlier time point after HPPH injection when HPPH in the tumor has not yet fallen below a level necessary for an effective PDT, or (c) the choice of an HPPH derivative recognized to have a higher retention than HPPH by the particular cancer case.
The experimental work highlighted several features related to HPPH retention and photoreaction: The first finding is the impact of the subcellular site on HPPH deposition and photoreaction. Oxidative crosslinking of cytoplasmic STAT3 suggests that the effective ROS generated by HPPH (mitochondria/ER) and HPPH-Lac (lysosomes) are able to reach latent STAT3 (23). One tentatively attributes the somewhat lower STAT3 action by HPPH-Lac (Fig. 4) to the distinct subcellular sites of PS (Fig. 3A) coupled with the limited reach of singlet oxygen and other ROS (24, 25). The issue of regional position of PS and ROS action appears to be even more relevant in explaining the differences triggering sufficient cellular damage resulting in lethal outcome (Fig. 5). While the leakage of enzymes from photo-damaged lysosomes may exert damaging action (26, 27), it seems to be not sufficiently prominent to match the lethal effects of mitochondrial/ER-originating photoreaction. A wealth of studies (reviewed in (28, 29) has abundantly demonstrated that mitochondrial stress reactions, including caspase-mediated destruction and apoptosis, are key effectors of PDT.
HPPH-Lac has been used in this study as an example of a lysosomal PS. Its action is considered to be representative for other porphyrin-derivatives that have been modified to provide potential target-specific functions. Such derivatives include PS with covalently attached ligands for cell surface receptors, adhesion proteins, lectins receptor, or PS as a part of immunoglobulins or nanostructures (30–32). All these compounds are predicted to bind to the external face of the plasma membrane and to be subject to endocytosis and lysosomal deposition comparable to what has been observed for HPPH-Lac. Thus, one expects these PS conjugates to mediate a similar progression of photoreaction. The lack of tumor cell specificity of HPPH-Lac (Fig. 2B) suggests that lung T-EC do not express sufficient galactoside-specific cell surface lectins to serve as cancer specific target. The substantial binding to the cell surface of both tumor and stromal cells is tentatively attributed to the porphyrin moiety of HPPH-Lac. The carbohydrate moiety is considered to prevent transmembrane diffusion. Based on binding studies not presented here, lung T-EC and T-Fb indicated a saturable binding of HPPH-Lac at 0°C with a Kd value of ~800 nM. This binding was not appreciably altered by the addition of 100-fold molar excess of lactose, supporting the interpretation that the observed HPPH-Lac interaction primarily involved non-lectin type functions.
The characterization of primary cultures of lung EC as well as established lines of lung cancer cells have indicated that the cell type-specific retention of HPPH and related PS is primarily associated with the interaction of these PSs with mitochondria/ER (Fig. 6). The studies revealed a preference of lung T-EC to retain HPPH over stromal cells. Since HPPH is accumulated in the same mitochondrial/ER compartments in EC and Fb, we have to interpret the cell-specific difference is probably due to different membrane compositions of these organelles. The identity of the relevant membrane molecules remains to be determined.
The second major finding of this study is that cell cultures are suitable to identify PS structures, which are preferentially retained by cancer cells with weak retention of HPPH. A particularly strong manifestation of altered PS preference of lung tumor cells is the high level retention of neutral forms (methyl ester derivatives) for pheophorbides by approximately 2/3 of the tested cancer cases. Moreover, the screen (e.g., Fig. 6) indicated that pheophorbide with a benzoic acid at position 20 (PS-820) converted the porphyrin into a PS that is retained by a broader spectrum of EC than observed with HPPH. The two carboxylic groups present in PS-821 altered the porphyrin into a compound that was preferentially retained by a sub-group of EC. That subgroup was not predictable based on the results with HPPH or PS-531-COOH (Fig. 6). We interpret the altered PS retention patterns to be caused by changes of membrane components as a function of the cellular transformation. Considering that the positions of the side groups affect the conformation of the porphyrin and consequently the interaction of the PS form with the lipid bilayer (33, 34) and its constituents of the subcellular organelles, the comparative analysis as shown in Fig. 6 emerges as suitable tool to interrogate the cellular phenotype optimally suited for a specific HPPH subtype.
Xenografts allow for the evaluation of HPPH specificity in vivo. Although xenografts do not perfectly mirror the cancer tissue as it exists in the originating tumor of a patient (16), it permits the in vivo assessment of HPPH uptake as a function of porphyrin structure and systemic distribution (Figs. 8 and 9). In agreement with the findings of past studies that used radiolabeled HPPH as tracers for systemically administered compound (8), the liver has been noted to be a substantial site of PS binding and clearance. This property was magnified in the example of HPPH-Lac and was probably caused by the highly effective hepatic clearance mechanism for compounds with terminal galactose residues (35–37). In contrast, HPPH with substantially lower sequestration in the liver and other organs than HPPH-Lac is subject to prolonged temporal accumulation in lung cancer cells accounting for the elevated concentration after 24h. Hence, knowing this property for tumor cases considered for PDT, will allow a design for more optimal treatment conditions. The current work has provided the tools for assessing this property for newly diagnosed cancer cases.
Supplementary Material
Figure S1. Cellular uptake of HPPH and HPPH-Lac by lung cells. (A) Time course of internalization was monitored by the cellular localization of PS fluorescence in TEC-1 cells. Binding, under suppression of endocytosis, was determined in cells incubated for 30 min in serum-free RPMI containing 3.2 μM HPPH on ice. Uptake and accumulation in intracellular compartments was evaluated after return of the cultures to fresh, PS-free medium and incubation at 37°C for 30 min, 4 h and 24 h. Following raising the culture temperature, plasma membrane-associated HPPH was internalized within 30min with subcellular retention in mitochondria and ER. HPPH-Lac, more stably interacted with cell surface where it was still detectable after 30 min incubation at 37°C. Subsequent endocytosis and lysosomal accumulation required several hours of incubation. Only the fluorescent images of the cells, taken under identical conditions at 400X magnification, are shown. (B) Effect of serum on cellular morphology and retention of HPPH by primary N-ECs (L364, passage 3). ECs were grown to confluence in KSFM. The cells were then incubated for 30 min at 37°C in either KSFM or RPMI containing 3.2 μM HPPH. After recording the cell-associated PS-fluorescence, the incubation of the cells with fresh, PS-free medium was continued for 4 and 24 h. The cellular morphology and PS level was imaged by phase and fluorescent microscopy at 100X and 400X magnification. Serum-induced differentiation of EC is evident by gaining a squamous morphology. All images at 100X magnification were made by using identical camera settings. Detection of HPPH fluorescence at 400X magnification in cells after 24 h chase in serum-containing RPMI required a 6-times longer exposure.
Figure S2. Enhanced binding of HPPH-Lac detected in one case of T-EC. Co-cultures of T-EC and stromal cells (L370 and L372) were treated for 30 min on ice with serum-free medium containing 3 μM HPPH-Lac. Cell-associated PS fluorescence was visualized by microscopy at 100X magnification. In each panel, the segregated cluster of T-EC is shown on the left and the surrounding Fb on the right half of the images.
Figure S3. Hepatic sequestration of HPPH-Lac. TEC-1 xenograft-bearing SCID mice were tested for organ distribution of HPPH-Lac by using identical procedures as described for HPPH in Fig. 8. Mice were injected i.v. with HPPH-Lac (3 μmole/kg) and organs were collected after 6 and 24 h. The PS fluorescence in 5-μm cryosections of tumor, liver and kidney were imaged by microscopy.
ACKNOWLEDGEMENTS
This work was supported by NCI grants P01CA055791 and RPCCC Support Grant P30CA16056. The authors thank Joseph Missert for preparations of photosensitizers, Barbara Owczarczak and Kimberly D. Ramsey for assistance in treatment of xenograft-bearing mice, the members of the Department of Pathology and Tissue Procurement Services at RPCCC for surgical lung tissue, Dr. Elizabeth A. Repasky, Dr. Bonnie L. Hylander and Rosemarie F. Pitoniak of the Department of Immunology for human lung tumor xenografts grown in SCID mice, Dr. Sandra Sexton and the staff of the animal facility at RPCCC for animal care, the laser core of the PDT Center for assistance in laser application, and Dr. Barbara Henderson for manuscript review.
Footnotes
SUPPORTING INFORMATION
Additional Supporting Information is available in the online version of this article:
REFERENCES
- 1.Agostinis P, Berg K, Cengel KA, Foster TH, Girotti AW, Gollnick SO, et al. Photodynamic therapy of cancer: an update. CA Cancer J Clin. 2011;61(4):250–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abrahamse H, Hamblin MR. New photosensitizers for photodynamic therapy. Biochem J. 2016;473(4):347–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nava HR, Allamaneni SS, Dougherty TJ, Cooper MT, Tan W, Wilding G, et al. Photodynamic therapy (PDT) using HPPH for the treatment of precancerous lesions associated with Barrett’s esophagus. Lasers Surg Med. 2011;43(7):705–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Loewen GM, Pandey R, Bellnier D, Henderson B, Dougherty T. Endobronchial photodynamic therapy for lung cancer. Lasers Surg Med. 2006;38(5):364–70. [DOI] [PubMed] [Google Scholar]
- 5.Simone CB 2nd, Cengel KA. Photodynamic therapy for lung cancer and malignant pleural mesothelioma. Semin Oncol. 2014;41(6):820–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rigual N, Shafirstein G, Cooper MT, Baumann H, Bellnier DA, Sunar U, et al. Photodynamic therapy with 3-(1’-hexyloxyethyl) pyropheophorbide a for cancer of the oral cavity. Clin Cancer Res. 2013;19(23):6605–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shafirstein G, Rigual NR, Arshad H, Cooper MT, Bellnier DA, Wilding G, et al. Photodynamic therapy with 3-(1’-hexyloxyethyl) pyropheophorbide-a for early-stage cancer of the larynx: Phase Ib study. Head Neck. 2016;38 Suppl 1:E377–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zheng X, Morgan J, Pandey SK, Chen Y, Tracy E, Baumann H, et al. Conjugation of 2-(1’-hexyloxyethyl)-2-devinylpyropheophorbide-a (HPPH) to carbohydrates changes its subcellular distribution and enhances photodynamic activity in vivo. J Med Chem. 2009;52(14):4306–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Srivatsan A, Wang Y, Joshi P, Sajjad M, Chen Y, Liu C, et al. In vitro cellular uptake and dimerization of signal transducer and activator of transcription-3 (STAT3) identify the photosensitizing and imaging-potential of isomeric photosensitizers derived from chlorophyll-a and bacteriochlorophyll-a. J Med Chem. 2011;54(19):6859–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dougherty TJ. An update on photodynamic therapy applications. J Clin Laser Med Surg. 2002;20(1):3–7. [DOI] [PubMed] [Google Scholar]
- 11.Senge MO, Brandt JC. Temoporfin (Foscan(R), 5,10,15,20-tetra(m-hydroxyphenyl)chlorin)--a second-generation photosensitizer. Photochem Photobiol. 2011;87(6):1240–96. [DOI] [PubMed] [Google Scholar]
- 12.Lorenz KJ, Maier H. Photodynamic therapy with meta-tetrahydroxyphenylchlorin (Foscan) in the management of squamous cell carcinoma of the head and neck: experience with 35 patients. Eur Arch Otorhinolaryngol. 2009;266(12):1937–44. [DOI] [PubMed] [Google Scholar]
- 13.Tracy EC, Bowman MJ, Pandey RK, Henderson BW, Baumann H. Cell-type selective phototoxicity achieved with chlorophyll-a derived photosensitizers in a co-culture system of primary human tumor and normal lung cells. Photochem Photobiol. 2011;87(6):1405–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tracy EC, Bowman MJ, Henderson BW, Baumann H. Interleukin-1alpha is the major alarmin of lung epithelial cells released during photodynamic therapy to induce inflammatory mediators in fibroblasts. Br J Cancer. 2012;107(9):1534–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bogner PN, Patnaik SK, Pitoniak R, Kannisto E, Repasky E, Hylander B, et al. Lung cancer xenografting alters microRNA profile but not immunophenotype. Biochem Biophys Res Commun. 2009;386(2):305–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hylander BL, Punt N, Tang H, Hillman J, Vaughan M, Bshara W, et al. Origin of the vasculature supporting growth of primary patient tumor xenografts. J Transl Med. 2013;11:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Srivatsan A, Pera P, Joshi P, Wang Y, Missert JR, Tracy EC, et al. Effect of chirality on cellular uptake, imaging and photodynamic therapy of photosensitizers derived from chlorophyll-a. Bioorg Med Chem. 2015;23(13):3603–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moan J, Rimington C, Evensen JF, Western A. Binding of porphyrins to serum proteins. Adv Exp Med Biol. 1985;193:193–205. [DOI] [PubMed] [Google Scholar]
- 19.Gyulkhandanyan A, Gyulkhandanyan L, Ghazaryan R, Fleury F, Angelini M, Gyulkhandanyan G, et al. Assessment of new cationic porphyrin binding to plasma proteins by planar microarray and spectroscopic methods. J Biomol Struct Dyn. 2013;31(4):363–75. [DOI] [PubMed] [Google Scholar]
- 20.Robey RW, Steadman K, Polgar O, Bates SE. ABCG2-mediated transport of photosensitizers: potential impact on photodynamic therapy. Cancer Biol Ther. 2005;4(2):187–94. [PubMed] [Google Scholar]
- 21.Liu W, Baer MR, Bowman MJ, Pera P, Zheng X, Morgan J, et al. The tyrosine kinase inhibitor imatinib mesylate enhances the efficacy of photodynamic therapy by inhibiting ABCG2. Clin Cancer Res. 2007;13(8):2463–70. [DOI] [PubMed] [Google Scholar]
- 22.Bellnier DA, Greco WR, Loewen GM, Nava H, Oseroff AR, Pandey RK, et al. Population pharmacokinetics of the photodynamic therapy agent 2-[1-hexyloxyethyl]-2-devinyl pyropheophorbide-a in cancer patients. Cancer Res. 2003;63(8):1806–13. [PubMed] [Google Scholar]
- 23.Sehgal PB. Paradigm shifts in the cell biology of STAT signaling. Semin Cell Dev Biol. 2008;19(4):329–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuimova MK, Yahioglu G, Ogilby PR. Singlet oxygen in a cell: spatially dependent lifetimes and quenching rate constants. J Am Chem Soc. 2009;131(1):332–40. [DOI] [PubMed] [Google Scholar]
- 25.Hatz S, Lambert JD, Ogilby PR. Measuring the lifetime of singlet oxygen in a single cell: addressing the issue of cell viability. Photochem Photobiol Sci. 2007;6(10):1106–16. [DOI] [PubMed] [Google Scholar]
- 26.Oleinick NL, Morris RL, Belichenko I. The role of apoptosis in response to photodynamic therapy: what, where, why, and how. Photochem Photobiol Sci. 2002;1(1):1–21. [DOI] [PubMed] [Google Scholar]
- 27.Tsubone TM, Martins WK, Pavani C, Junqueira HC, Itri R, Baptista MS. Enhanced efficiency of cell death by lysosome-specific photodamage. Sci Rep. 2017;7(1):6734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kessel D, Oleinick NL. Cell Death Pathways Associated with Photodynamic Therapy: An Update. Photochem Photobiol. 2018;94(2):213–8. [DOI] [PubMed] [Google Scholar]
- 29.Apoptosis Kessel D., Paraptosis and Autophagy: Death and Survival Pathways Associated with Photodynamic Therapy. Photochem Photobiol. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laville I, Figueiredo T, Loock B, Pigaglio S, Maillard P, Grierson DS, et al. Synthesis, cellular internalization and photodynamic activity of glucoconjugated derivatives of tri and tetra(meta-hydroxyphenyl)chlorins. Bioorg Med Chem. 2003;11(8):1643–52. [DOI] [PubMed] [Google Scholar]
- 31.Pellosi DS, De Jesus P, Tedesco AC. Spotlight on the delivery of photosensitizers: different approaches for photodynamic-based therapies. Expert Opin Drug Deliv. 2017;14(12):1395–406. [DOI] [PubMed] [Google Scholar]
- 32.Hodgkinson N, Kruger CA, Abrahamse H. Targeted photodynamic therapy as potential treatment modality for the eradication of colon cancer and colon cancer stem cells. Tumour Biol. 2017;39(10):1010428317734691. [DOI] [PubMed] [Google Scholar]
- 33.Bronshtein I, Afri M, Weitman H, Frimer AA, Smith KM, Ehrenberg B. Porphyrin depth in lipid bilayers as determined by iodide and parallax fluorescence quenching methods and its effect on photosensitizing efficiency. Biophys J. 2004;87(2):1155–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dror SB, Bronshtein I, Garini Y, O’Neal WG, Jacobi PA, Ehrenberg B. The localization and photosensitization of modified chlorin photosensitizers in artificial membranes. Photochem Photobiol Sci. 2009;8(3):354–61. [DOI] [PubMed] [Google Scholar]
- 35.Ashwell G, Harford J. Carbohydrate-specific receptors of the liver. Annu Rev Biochem. 1982;51:531–54. [DOI] [PubMed] [Google Scholar]
- 36.Stockert RJ, Morell AG. Hepatic binding protein: the galactose-specific receptor of mammalian hepatocytes. Hepatology. 1983;3(5):750–7. [DOI] [PubMed] [Google Scholar]
- 37.Breitfeld PP, Simmons CF Jr., Strous GJ, Geuze HJ, Schwartz AL. Cell biology of the asialoglycoprotein receptor system: a model of receptor-mediated endocytosis. Int Rev Cytol. 1985;97:47–95. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Cellular uptake of HPPH and HPPH-Lac by lung cells. (A) Time course of internalization was monitored by the cellular localization of PS fluorescence in TEC-1 cells. Binding, under suppression of endocytosis, was determined in cells incubated for 30 min in serum-free RPMI containing 3.2 μM HPPH on ice. Uptake and accumulation in intracellular compartments was evaluated after return of the cultures to fresh, PS-free medium and incubation at 37°C for 30 min, 4 h and 24 h. Following raising the culture temperature, plasma membrane-associated HPPH was internalized within 30min with subcellular retention in mitochondria and ER. HPPH-Lac, more stably interacted with cell surface where it was still detectable after 30 min incubation at 37°C. Subsequent endocytosis and lysosomal accumulation required several hours of incubation. Only the fluorescent images of the cells, taken under identical conditions at 400X magnification, are shown. (B) Effect of serum on cellular morphology and retention of HPPH by primary N-ECs (L364, passage 3). ECs were grown to confluence in KSFM. The cells were then incubated for 30 min at 37°C in either KSFM or RPMI containing 3.2 μM HPPH. After recording the cell-associated PS-fluorescence, the incubation of the cells with fresh, PS-free medium was continued for 4 and 24 h. The cellular morphology and PS level was imaged by phase and fluorescent microscopy at 100X and 400X magnification. Serum-induced differentiation of EC is evident by gaining a squamous morphology. All images at 100X magnification were made by using identical camera settings. Detection of HPPH fluorescence at 400X magnification in cells after 24 h chase in serum-containing RPMI required a 6-times longer exposure.
Figure S2. Enhanced binding of HPPH-Lac detected in one case of T-EC. Co-cultures of T-EC and stromal cells (L370 and L372) were treated for 30 min on ice with serum-free medium containing 3 μM HPPH-Lac. Cell-associated PS fluorescence was visualized by microscopy at 100X magnification. In each panel, the segregated cluster of T-EC is shown on the left and the surrounding Fb on the right half of the images.
Figure S3. Hepatic sequestration of HPPH-Lac. TEC-1 xenograft-bearing SCID mice were tested for organ distribution of HPPH-Lac by using identical procedures as described for HPPH in Fig. 8. Mice were injected i.v. with HPPH-Lac (3 μmole/kg) and organs were collected after 6 and 24 h. The PS fluorescence in 5-μm cryosections of tumor, liver and kidney were imaged by microscopy.