

Abstract
CRISPR-Cas systems are RNA-guided nucleases that provide adaptive immune protection for bacteria and archaea against intruding genomic materials. The programmable nature of CRISPR targeting mechanisms has enabled their adaptation as powerful genome engineering tools. Cas9, a type II CRISPR effector protein, has been widely used for gene editing applications owing to the fact that a single guide RNA can direct Cas9 to cleave desired genomic targets. An understanding of the role of different domains of the protein and guide RNA-induced conformational changes of Cas9 in selecting target DNA has been and continues to enable development of Cas9 variants with reduced off-targeting effects. It has been previously established that an arginine-rich bridge helix (BH) present in Cas9 is critical for its activity. In the present study, we show that two proline substitutions within a loop region of the BH of Streptococcus pyogenes Cas9 impair the DNA cleavage activity by accumulating nicked products and reducing target DNA linearization. This in turn imparts higher selectivity in DNA targeting. We discuss the probable mechanisms by which the BH-loop contributes to target DNA recognition.
Keywords: CRISPR, Cas9 endonucleases, SpyCas9, Bridge helix, target specificity, off-target DNA cleavage
graphical abstract
INTRODUCTION
CRISPR-Cas (Clustered Regularly Interspaced Short Palindromic Repeats-CRISPR associated) systems are RNA-protein based adaptive immune systems present in bacteria and archaea.1, 2 Using an RNA molecule as a guide, the CRISPR-Cas complexes cleave DNA and/or RNA of the invading genetic elements that carry a complementary region corresponding to the guide RNA.3–8 In the most current classification, CRISPR-Cas systems are organized into two classes and further into six types (I through VI) and several sub-types based on the locus organization and the Cas endonuclease that cleaves the intruding genetic element.8–11
Cas9, the signature protein for the type II CRISPR systems, requires two native RNA components, CRISPR RNA (crRNA) and a trans-activating CRISPR RNA (tracrRNA), for its DNA targeting activity.12 The crRNA contains the “guide” region that is used for locating complementarity in the target DNA. These two RNA molecules can be fused to produce a single-guide-RNA (sgRNA) without affecting the functionality.12 The ease of using a single Cas9 protein and an sgRNA for DNA targeting has been monumental for genome editing13–21 and other applications such as site-specific DNA repression and activation, proteomic analyses, and is being investigated for use in gene therapy applications.22–24
Cas9 is a multi-domain protein. Crystal structures of Cas9 orthologs from different subtypes of type II CRISPR reveal a common architecture, where the protein folds into a bi-lobed architecture consisting of a nuclease (NUC) lobe and a recognition (REC) lobe.25–33 The NUC and REC lobes are connected to each other by a long arginine-rich bridge helix (BH). The NUC lobe consists of two endonuclease domains, HNH and RuvC, and a domain responsible for recognizing the DNA protospacer-adjacent-motif (PAM), a 2–8 nucleotides (nt) long region that is essential to discriminate between self and foreign DNA.4, 7, 12, 34, 35 The REC lobe of Cas9 and BH are involved in subtype-specific tracrRNA-crRNA recognition.30, 31, 33
The apo-Cas9 protein undergoes large conformational re-arrangement upon sgRNA binding to form the binary complex, including a 65Å rigid body movement of the REC-III domain of REC lobe.28, 29 The core region of the sgRNA makes extensive interactions with REC domains and the BH. Interestingly, majority of the interactions of Cas9 with the crRNA-guide involves the RNA sugar-phosphate backbone, resulting in a solvent exposed pre-ordered “seed” region that is poised to search and locate a target DNA with an approximately 20 nt complementary segment called “protospacer”.28, 31 The first step in DNA targeting by Cas9 is locating the PAM region in the target and the longevity of the ternary complex (Cas9-sgRNA-DNA) is enhanced by the presence of a cognate PAM flanking the protospacer.36 Following PAM recognition, the crRNA-guide region searches for complementarity in the flanking DNA by unwinding the DNA duplex subsequently forming an R-loop between the crRNA-guide and the protospacer. Once the complementarity between the target DNA and the RNA guide is established, the target DNA cleavage is brought about by two independent cleavage reactions performed by HNH on the strand complementary to the crRNA-guide and RuvC on the non-complementary DNA strand.12, 27
The binary complex undergoes a smaller degree of conformational change upon target DNA binding to form a ternary complex, mostly involving the HNH domain. Once the R-loop complementarity reaches 14–17 nt long, the HNH movement occurs, after which it is positioned ideally to cleave the complementary strand of DNA.32, 37–39 The movement of HNH to the active position acts as an allosteric switch that activates the RuvC domain such that the coordinated activities of both endonuclease sites bring about a concerted DNA cleavage.27, 40 The positioning of, not cleavage by, HNH is essential for RuvC activity when both endonuclease domains are present in the protein.40 Interestingly, it was shown that Campylobacter jejuni Cas9 nicks DNA using the RuvC domain when HNH domain is absent, indicating the complexities of the interplay between different domains of the protein.33 The coordinated activity also implements specificity in DNA cleavage. It was recently reported that REC-II domain has to move to facilitate the positioning of the HNH domain.38 Thus, the conformational changes in response to RNA and DNA binding not only enable ideal binding environments, but also impart fidelity in the cleavage process.
Even though relatively simple to use compared to other gene-editing techniques, Cas9’s primary drawback is off-target DNA cleavage, which arises due to the tolerance of Cas9 to mismatches between the sgRNA-guide and the target DNA. The stringency of the interdependence between RNA-DNA complementarity and DNA cleavage efficiency varies along different regions of the protospacer.12, 37 While PAM proximal mismatches greatly reduce DNA cleavage, PAM-distal mismatches are tolerated to varying degrees. Within the PAM proximal region, mismatches at different positions have been observed to differentially affect activity, with nt 3 to 6 having the most detrimental effects on target cleavage as compared to nt 1 and 2 and others beyond the 6th nucleotide.37 Interestingly, in Streptococcus pyogenes (Spy) Cas9, the presence of PAM and at least 9 nt of perfect match in the seed region (PAM proximal region) is sufficient to produce a protein-RNA-DNA complex that has similar stability as that of a complex with fully matched (20 nt) target DNA,41 indicating that mismatches beyond the 9 nt seed region affect steps in the mechanism that are subsequent to stable ternary complex formation.
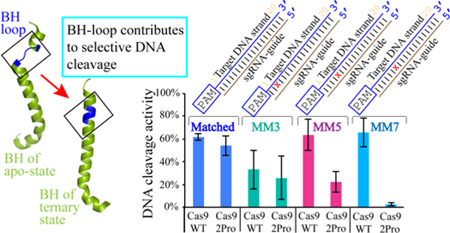
In this work we focused on investigating the role of BH in target DNA cleavage. The BH is an Arginine Rich Motif (ARM) and it is a universal feature of Cas9. BH plays a central role in function as it bridges the NUC and REC lobes and makes direct and indirect interactions with crRNA, tracrRNA and target DNA (Figure 1A and 1B).27, 29–31, 33 It was shown in several Cas9 orthologs that mutating the arginine residues in the BH significantly reduced its activity.31, 37, 43 A comparison of apo-, Cas9-sgRNA, and Cas9-sgRNA-DNA structures of SpyCas9 shows that a short loop in the BH in the apo-protein (residues Leu64-Thr67, called BH-loop hereafter) is transformed into a helix in the nucleic acid bound forms (Figure 1C). To gain insights into the role of loop-to-helix conversion of the BH-loop in SpyCas9 function, we introduced two proline substitutions at positions L64 and K65 to generate a variant called SpyCas92Pro. The prolines are expected to interfere with the transition to the contiguous helix upon interacting with the sgRNA. Our results reveal that compared to the wild type protein (SpyCas9WT), DNA cleavage activity of SpyCas92Pro decreases substantially against those with PAM-proximal mismatches. We propose that, in the wild type SpyCas9, the full helical conformation of the BH when bound to sgRNA and the interaction of K65 in the BH-loop with the phosphate lock loop region promote Cas9-DNA interactions that result in tolerance to RNA-DNA mismatches. The mechanistic insights on BH will aid further development of Cas9 variants with reduced off-target cleavage.
Figure 1. Interactions involving BH in SpyCas9.
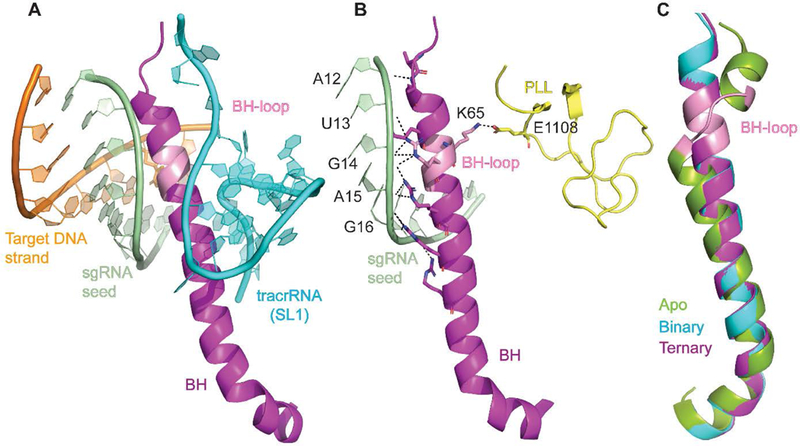
A) BH inserted into the nucleic acid interface B) Interactions of BH with the sgRNA seed region and the phosphate lock loop (PLL). Dashed lines represent interactions that are within 3.5 Å. C) Superposition of SpyCas9 BH from different crystal structures (apo-: PDB ID: 4CMP,29 binary: PDB ID: 4ZT0,28 ternary: PDB ID: 5F9R27). SL1- Stem loop 1. Figures were made using Pymol.42
MATERIALS AND METHODS
Protein mutagenesis, overexpression and purification.
Proline substitutions were introduced at the 64th and 65th amino acid positions of SpyCas9WT plasmid (Addgene-PMJ806, UniProt protein ID- CAS9 Q99ZW2) using polymerase chain reaction (PCR) (Table S1). The correctness of the sequence was confirmed by DNA sequencing covering the whole reading frame of the gene. Sequence-confirmed clones were transformed into Escherichia coli Rosetta strain 2 (DE3) for protein expression. Protein purification followed published protocols12 and is detailed in the supplementary methods.
RNA transcription.
This work used two sgRNAs, a full-length (122 nt, sgRNAFL) and a variant with deletions in the repeat-antirepeat region (98 nt, sgRNAdel) [Figure S1A and Table S2A]. These sgRNAs are similar to previous reports12, 31 except for the spacer region. The guide region of both the sgRNAs is 20 nt long. The sequences as shown in Table S2A were ordered as gBlock gene fragments from Integrated DNA Technologies (IDT), and cloned into pUC19 vector in between KpnI and EcoRI sites and transformed into DH5α cells [New England Biolabs (catalog number C2987H), for sgRNAFL) and E. cloni cells [Lucigen (catalog number 60106–1), for sgRNAdel]. E. cloni cells facilitated production of sgRNAdel without mutations in the gene sequence. To facilitate in vitro transcription, a T7 promoter sequence was introduced ahead of the sgRNA sequence, and a BbsI restriction site was placed to linearize the plasmid at the end of the sgRNA sequence. The BbsI-linearized plasmids were used as template for in vitro transcription. The transcription reaction followed established protocols and is detailed in the supplementary methods.
In-vitro DNA cleavage assays.
Protospacer strands for the MM5 DNA (mismatched substrate) were ordered as oligos from IDT, annealed and ligated into pUC19 vector (Table S1). The oligos contained a 30 nt long protospacer with a 20 nt match to the guide region towards the 3’ end and a PAM (GGG). The oligo was inserted between BamHI and EcoRI sites of pUC19. Wild-type substrate and other mismatched (MM) substrates (MM3, MM7, MM16, MM18, MM19–20, MM17–20) were generated with mutagenic primers using MM5 plasmid following either site-directed mutagenesis,44 Sequence and Ligation Independent Cloning (SLIC),45 or Single-Primer Reactions IN Parallel (SPRINP) method46 and transformed into DH5α or E. cloni cells.
For cleavage assay, protein was diluted to 1 µM in 20 mM HEPES pH 7.5, 150 mM KCl, 2 mM TCEP, and 2 mM EDTA. The sgRNA was annealed using the following steps: heat at 95°C for 2 minutes, cool at room temperature for 2 minutes, add annealing buffer (20 mM TRIS-HCl pH 7.5, 100 mM KCl, 1 mM MgCl2), and transfer it back to the heat block that has been turned off for slow cooling. The cleavage assays were carried out in a final volume of 10 µL and typically contained the following: 20 mM HEPES pH 7.5, 150 mM KCl, 2 mM TCEP, 100 ng plasmid (substrate DNA). MgCl2 was at 1 mM, 5 mM, or 10 mM concentration. The protein-RNA was at equimolar ratio and the concentration varied for the different experiments. There was no pre-incubation of protein and RNA; protein was added as the last component of the cleavage reaction. The reaction was carried out at 37°C for 15 minutes. The reaction was stopped using 50 mM EDTA and 1% SDS and products were resolved on a 1% agarose gel. The gel was post-stained with ethidium bromide and imaged using a BioRad ChemiDoc MP apparatus.
To quantify the cleavage activities, each gel image was analyzed using the Image J software47 to record intensities corresponding to nicked (N), linear (L), and supercoiled (SC) bands, which are designated respectively as IN, IL and ISC. Background-corrected total activity (TA) was calculated as following:
| (1) |
with the values with the “0” subscript [e.g.,] representing those calculated with the respective signals observed at the no enzyme control lane of each gel.
To compare the total activities, , the ratio of the total activity between SpyCas92Pro and SpyCas9WT, was computed following equations 1a through 1c.
First, at each enzyme complex concentration, the value was computed as:
| (1a) |
Since all measurements showed saturation behaviors at enzyme complex concentrations above 50 nM (see Results), values at 100 nM, 150 nM, and 200 nM protein-RNA complex concentration were averaged:
| (1b) |
To account for experimental errors, values from different replications were averaged, and designated as, which was used to evaluate differences between SpyCas92Pro and SpyCas9WT:
| (1c) |
with n representing the number of replications (n ≥ 3).
To analyze the effect of BH-loop mutation on the type of products produced, background corrected nicked and linear products were calculated as:
| (2) |
| (3) |
with the values with the “0” subscript representing those calculated with the respective signals observed at the no enzyme control lane of each gel. In addition, RL/N, the ratio of Linear vs. Nicked DNAs, was calculated from the background-corrected Linear and Nicked products as following:
| (4) |
For each reported data point, average values were obtained from a minimum of three replications. Standard deviation (SD) and standard error of mean (SEM) were calculated based on the number of replications using the following equations:
| (5) |
where R is a data value from each replication, RAV is average of data values of all the replications, and n is the number replications.
| (6) |
where n is the number replications.
Electrophoretic Mobility Shift Assay (EMSA).
sgRNAdel was dephosphorylated using Alkaline phosphatase (New England Biolabs) and 5’ end labelled with 32P (γ−32P ATP purchased from PerkinElmer) using T4 polynucleotide kinase (New England Biolabs). The labeled sgRNAdel was purified using BioSpin column P-30 (BioRad) and a 100% recovery was assumed for calculations. The binding reaction was setup with increasing concentrations of protein (10 nM to 70 nM) at a constant RNA concentration of ~50 nM in a buffer containing 20 mM HEPES pH 7.5, 150 mM KCl, 2 mM TCEP, 1mM MgCl2. The exact amount of sgRNA may be lower since the concentration was not measured after the labeling procedure. After incubation at room temperature for 15 minutes, the components were resolved on a 6% native acrylamide gel. The gel and the running buffer composition included 0.25X Tris-Borate (TB) buffer pH 8.6 and 1mM MgCl2. The bands were visualized by phosphor imaging with Typhoon FLA 7000 system (GE life sciences). Three independent replications of the assay were performed. Graph was generated by plotting the average of three replications of bound complex over different protein concentrations and SEM is shown.
Limited Proteolysis.
SpyCas9 (6 µg) with or without bound sgRNA was digested with 0.0125 µg of trypsin (480:1 mass ratio) in a buffer containing 50 mM Tris-HCl pH 8.0 and 20 mM CaCl2. For the sgRNA-bound reactions, there was a pre-incubation of protein and sgRNAdel or sgRNAFL (protein to RNA ratio, 1:1.2) for 10 minutes at room temperature before the addition of trypsin. The digestion was stopped at 15 minutes with SDS-PAGE dye and the samples were resolved on a 10% SDS-PAGE gel. The protein bands were visualized by coomassie brilliant blue G-250 staining.
Cell-based activity assay.
SpyCas92Pro construct used for genome editing study was made from wild-type gene backbone, pCSDest2-SpyCas9-NLS-3XHA-NLS (Addgene#69220),48 following the same method that was used to generate the bacterial SpyCas92Pro variants (Table S1). The sgRNAdel backbone (pLKO.1-puro-U6) was obtained from Addgene (50920)49 and the guide region was replaced for the different target sites that were tested (Tables S2B and S3). Full-length sgRNA for the cell-based study was constructed by Gibson assembly method using the pLKO.1-puro-U6 backbone (Table S2B).50
We used separate pCSDest2-SpyCas9-NLS-3XHA-NLS (driven by the CMV IE94 promoter) and pLKO.1-puro-U6sgRNA (driven by the U6 promoter) plasmids for the expression of SpyCas9 and its sgRNA (Table S4). Cell-based assays followed previously published protocols.51 The culturing medium for HEK293T cells contained DMEM with 10% FBS and 1% Penicillin/Streptomycin (Gibco) and the cells were grown in a 37°C incubator supplemented with 5% CO2. 200 ng Cas9-expressing plasmid, 200 ng sgRNA-expressing plasmid and 10 ng mCherry plasmid were transfected into ~1.5 × 105 cells using Polyfect transfection reagent (Qiagen) in a 24-well plate, following manufacturer’s protocol. The mCherry plasmid was used to analyze the quality of transfection. The genomic DNA was extracted using DNeasy Blood and Tissue kit (Qiagen) after 72 hours of transfection. PCR-amplification was carried out using 50 ng of genomic DNA and primers specific for each genomic site (Table S5) with High Fidelity 2X PCR Master Mix (New England Biolabs). Indel analysis was performed by TIDE (Tracking of Indels by Decomposition)52 using 20 ng of purified PCR product (Zymo Research). The trace files were analyzed using the TIDE web tool (https://tide.deskgen.com). For T7E1 analysis, 0.5 μl T7 Endonuclease I (10 U/μl, New England Biolabs) was added to 10 μl of pre-annealed PCR product in 1X NEB Buffer 2 for 1 hour. The bands were resolved on a 2.5% agarose gel and visualized using SYBR-safe stain (ThermoFisher Scientific).
Off-target analysis by targeted DNA deep-sequencing.
For off-target DNA cleavage analysis, we used sites that were identified as off-targets for DTS7 editing through GUIDE-seq analysis.51 The genomic DNA following transfection was used for deep-sequencing. We used a two-step PCR amplification to produce DNA fragments for on-target and off-target sites following previous protocols.48 The first step used locus-specific primers containing universal overhangs with complementary ends to the TruSeq adaptor sequences (Table S6), while the second step used a universal forward primer and an indexed reverse primer to introduce the TruSeq adaptors (Table S7). The PCR program is as per published protocols.51 Equal amounts of the products from each treatment group were mixed and purified using DNA Clean & Concentrator kit (Zymo Research). The library was deep sequenced using a paired-end 150 bp MiSeq run. The sequencing results and statistical analysis were done using R as described before.48, 53
RESULTS
Proline substitutions in the BH-loop affect total activity on DNA targets.
To investigate the role of the BH-loop in Cas9 activity, we substituted two amino acids in this loop of SpyCas9 (L64 and K65) to prolines (SpyCas92Pro). DNA cleavage activity assays were performed at different Mg2+ concentrations using varying concentrations of an enzyme complex containing equimolar Cas9 and sgRNA. Figure 2 shows data obtained with an sgRNA having deletions in the repeat and tracrRNA regions (designated as sgRNAdel in the current study, Figure S1A). At a total reaction time of 15 minutes, for each concentration of the enzyme-RNA complex tested, SpyCas9WT and SpyCas92Pro gave similar total activity (sum of linear and nicked products, equation 1) with a DNA substrate containing a 20 nt target sequence complementary to the guide region of the sgRNAdel (matched DNA, Figure S1B) at 5 mM Mg2+ (Figure 2A). Very similar data were obtained at 10 mM Mg2+ (Figure S2A). The total activity of both SpyCas92Pro (43%) and SpyCas9WT (59%) was diminished at 1 mM Mg2+ compared to that at 5 mM and 10 mM Mg2+, and the reduction was more pronounced for SpyCas92Pro (Figure S2B). In addition, experiments with a full-length sgRNA (sgRNAFL) that contains the full repeat-antirepeat regions showed similar activity for both SpyCas92Pro and SpyCas9WT at 10 mM (~80% for both) and a lower activity at 1 mM Mg2+ (~54% and ~67% respectively) (Figure S3). This indicates that the extra regions present in sgRNAFL slightly enhance the cleavage activity under low Mg2+ concentrations, but do not provide significant favorable interactions that may impact functional studies of the BH-loop substitutions.
Figure 2. Comparison of SpyCas9WT and SpyCas92Pro activities using different DNA substrates.
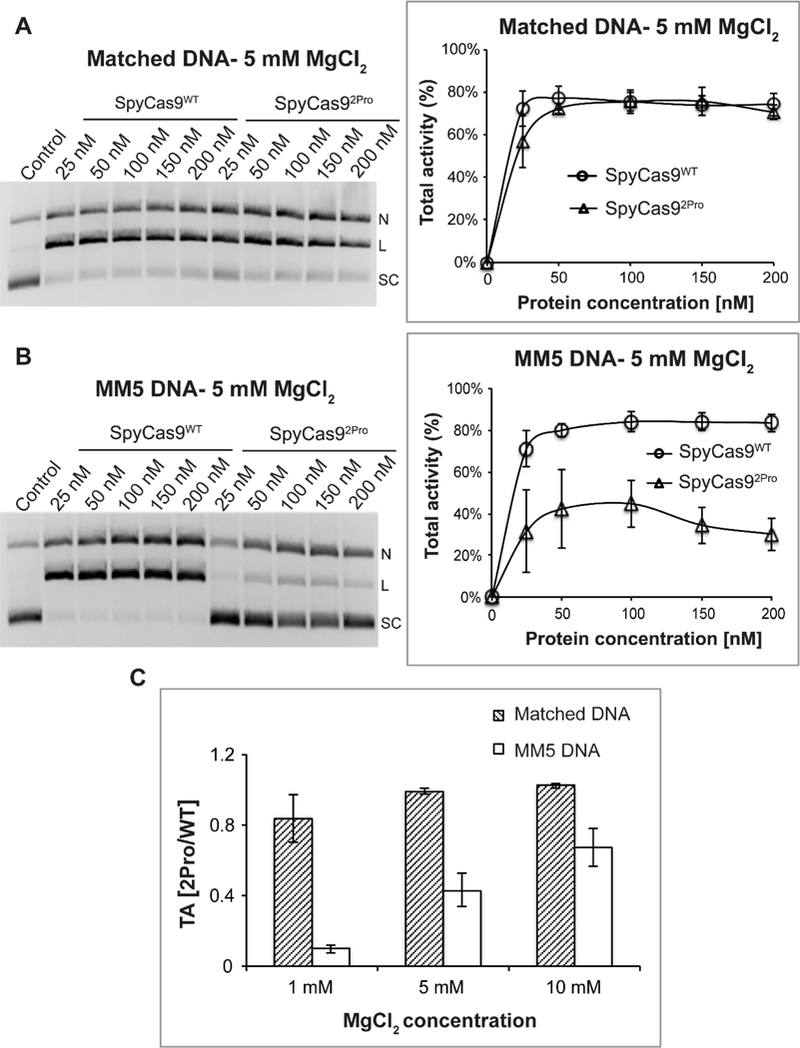
A) Total activity with a fully matched DNA substrate at 5 mM Mg2+. Shown on the left is a representative gel presenting the DNA cleavage with varying amounts of protein: sgRNA complex. Supercoiled (SC), linear (L), and nicked (N) DNA bands are indicated. Shown on the right is a plot of the total activity vs. the enzyme complex concentration. Average values from three replications were plotted against protein concentrations to produce a line graph. B) Total activity with a mismatched DNA (MM5) substrate at 5 mM Mg2+. Organization of the panel is the same as that in panel A. C) The averaged ratio of total DNA cleavage activities between SpyCas92Pro and SpyCas9WT, , at different Mg2+ concentrations. For all the panels, data shown were obtained with a reaction time of 15 minutes, and error bars represent standard error mean (SEM). Each experiment was typically conducted in replicates of three, using proteins from two different batches of purification.
We further tested the effect of BH-loop mutation on a DNA target containing mismatches (MM) to the sgRNA guide (Figure 3A). At 15-minutes reaction time, with a substrate containing a mismatch at the 5th nt from the PAM proximal side (MM5) and at 1mM Mg2+ concentration, SpyCas92Pro exhibited very minimal total activity (~5%), while SpyCas9WT showed ~50% DNA cleavage (Figure S4A). At 5 mM Mg2+, SpyCas92Pro regained ~40% total cleavage with MM5, while total activity of SpyCas9WT increased to ~80% (Figure 2B). The total activity at 10 mM Mg2+ increased to ~60% for SpyCas92Pro and to ~85% for SpyCas9WT (Figure S4B), indicating that higher Mg2+ concentration can only partially compensate the effect caused by the BH-loop mutation.
Figure 3. Comparison of SpyCas9WT and SpyCas92Pro activities using sgRNAdel on different DNA substrates at 5 mM Mg2+ ions.
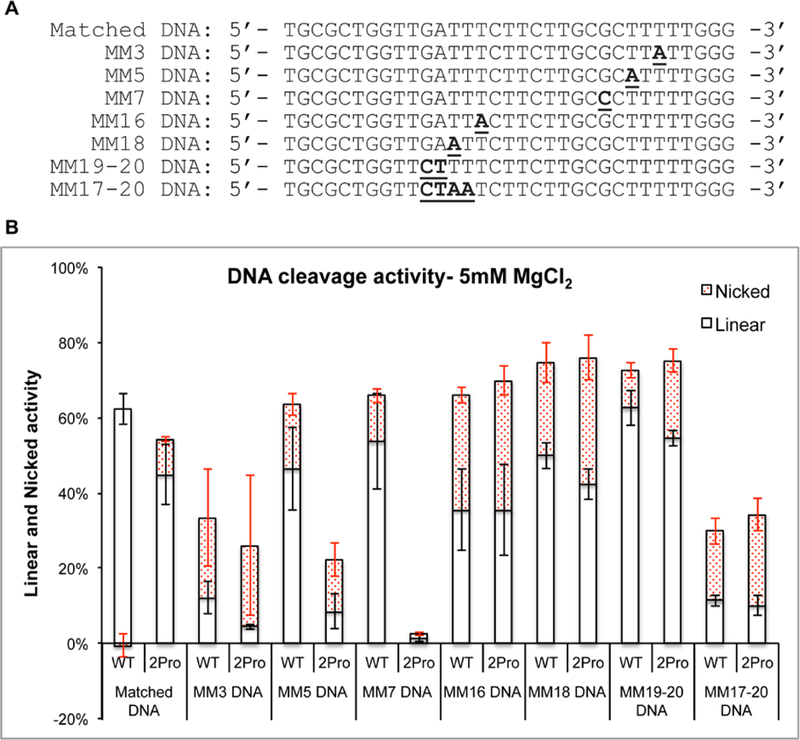
A) Sequences of DNA substrates (the sequence of non-complementary DNA strand is shown) used in this study. Bold and underlined sequences are mismatches in the protospacer while annealing to sgRNA. B) Graph shows the total activity with separate regions indicating the percentage of nicked (red shaded region) and linear products. The enzyme concentration was at 50 nM. For matched DNA and MM5 DNA, there are nine and six replications respectively, while for the rest there are three replications. Error bars represent SEM.
We note that repetitions for each of the DNA cleavage experiments gave characteristically very similar behaviors on the dependence of enzyme concentrations, although the absolute values of the activities show some variations, presumably reflecting variability in the amount of active enzyme complex in the different preparations. In addition, all measurements showed saturation behaviors at enzyme complex concentrations above 50 nM (Figures 2A, 2B, S2 and S4). Therefore, to quantitatively evaluate differences between SpyCas92Pro and SpyCas9WT, the ratio of the total activity between SpyCas92Pro and SpyCas9WT, was calculated at saturating enzyme concentrations from multiple repetitions (see equations 1a -1c). The analyses show that with the matched DNA substrate, is close to 1 at all three Mg2+ concentrations tested (Figure 2C). For the mismatched substrate MM5, values are all significantly less than 1, increasing from 0.1 at 1 mM Mg2+ to 0.7 at 10 mM Mg2+ (Figure 2C). Together with the results from varying protein-RNA concentrations (Figure 2B), the data indicate that total activity of SpyCas92Pro is compromised against the MM5 substrate, although the activity can be partially restored at higher Mg2+ concentrations.
Effects of BH-loop proline substitution on total activity vary depending on the mismatch positions.
Expanding on the finding that total activity of SpyCas92Pro is compromised against the MM5 mismatched substrate, we investigated how the positioning of the mismatch affects SpyCas92Pro activity. Studies on the matched and MM5 substrates have shown that the activity levels plateau at a protein-RNA concentration of 50 nM and above, and that the activity levels vary depending on Mg2+ concentrations (Figures 2A, 2B, S2 and S4). Based on these results, we chose an enzyme complex concentration of 50 nM and Mg2+ concentrations of 1 mM and 5 mM to conduct detailed analysis of the effect of mismatch positions on DNA cleavage with the BH-loop substitution.
It was recently established that positions 3–6 at the PAM proximal side are more crucial than positions 1–2 for target DNA cleavage by SpyCas9.37 We tested the effect of mismatches at the 3rd and 7th nt positions (MM3 and MM7, Figure 3A) on target DNA cleavage and compared it with that of MM5. Even though the total activity of both SpyCas92Pro and SpyCas9WT were reduced on MM3 (26% and 33% respectively) and MM5 (13% and 50% respectively), SpyCas92Pro has a greater reduction compared to SpyCas9WT (Figures 3B and S5). The most significant difference was found for the MM7 substrate, where SpyCas9WT showed a cleavage of 66% while SpyCas92Pro possessed only 3% activity at 5 mM Mg2+ (Figures 3B and S5A). Similar results were observed at 1 mM Mg2+ concentration, where SpyCas9WT possessed 43% cleavage and SpyCas92Pro showed no significant activity (5%) on MM7 (Figures S5B and S7). These results show that SpyCas92Pro is more effective in discriminating PAM-proximal mismatches than SpyCas9WT and the level of enhanced discrimination depends on the mismatch position.
We then tested whether the BH-loop mutation will affect the cleavage of DNA substrates with mismatches at the PAM-distal side (Figures 3, S6 and S7). Both single and multiple mutations were created at the PAM distal segment of the substrate (MM16, MM18, MM19–20 and MM17–20, Figure 3A). The cleavage activity on substrates with single mutations at 16th (SpyCas9WT at 66% vs. SpyCas92Pro at 70%) and 18th (SpyCas9WT at 74% vs. SpyCas92Pro at 76%) nt positions at 5mM Mg2+ were slightly higher for SpyCas92Pro compared to SpyCas9WT (Figures 3B and S6A). An analysis of the same reaction at 1 mM Mg2+ shows 18% for SpyCas9WT and 33% for SpyCas92Pro for MM16 and 44% for SpyCas9WT and 28% for SpyCas92Pro for MM18 (Figures S6B and S7). A double mutant at 19th and 20th nt positions (MM19–20) has similar activities with both SpyCas9WT and SpyCas92Pro (~70% at 5 mM for both proteins, and ~32% for SpyCas9WT and ~24% for SpyCas92Pro at 1 mM Mg2+, Figures 3B, S6 and S7). A quadruple mutant from positions 17th to 20th (MM17–20) has negligible cleavage at 1 mM Mg2+ and the cleavage increased to ~30% for SpyCas9WT and ~34% SpyCas92Pro in the presence of 5 mM Mg2+ (Figures 3B, S6 and S7). Overall, the data indicate that the difference in activity between SpyCas92Pro and SpyCas9WT are much smaller on the PAM-distal mismatched substrates as compared to the PAM-proximal ones.
To further characterize the activity of SpyCas92Pro, the reaction rates for precursor cleavage (kobs) were measured for the matched, MM5, and MM18 DNA targets [SI methods, SM 3]. At 50 nM protein-RNA concentration, SpyCas92Pro cleaves the MM5 DNA 5.8 times slower compared to SpyCas9WT, while a reduction of 2.2 times is observed for the matched DNA (Figure S8). This is consistent with the reduced total activity observed (Figure 2) and supports the conclusion that SpyCas92Pro is compromised against the PAM-proximal mismatched MM5 substrate. Since SpyCas92Pro can eventually attain a similar total activity on matched DNA (Figure 2), these data suggest that there are differences in the DNA cleavage mechanisms of SpyCas9WT and SpyCas92Pro. Interestingly, SpyCas92Pro cleaves MM18, a PAM-distal mismatch, at a slightly higher rate (1.9 times) compared to SpyCas9WT (Figure S8). This is consistent with the slightly higher total activities observed for PAM-distal mismatches (Figure 3) and suggest that the BH-loop variations induce differences in target DNA engagement with respect to PAM-proximal and PAM-distal mismatches. Further studies are required to completely characterize these differences.
Proline substitution in the BH-loop reduces linearization of mismatched substrates.
During analyses of DNA cleavage by SpyCas92Pro and SpyCas9WT, we observed that the two proteins gave different amounts of nicked and linear products (Figure 4). As shown in Figure 4, at 5 mM Mg2+ and matched DNA, while the total activity at saturation was comparable between SpyCas9WT and SpyCas92Pro (~70%), SpyCas9WT produced slightly more linear product (~65%), compared to that of SpyCas92Pro (~54%), (Figure 4A). With the mismatched MM5 substrate, SpyCas92Pro (~20%) showed a clear reduction in the percentage of linear product as compared to SpyCas9WT (~60%), which accounted for the majority of the reduction in the total activity (Figure 4B). Similar differences between SpyCas92Pro and SpyCas9WT in the pattern of nicked and linear products were observed at 10 mM Mg2+ for both matched and MM5 substrates (Figures S9A and S10A). The pattern stayed the same with sgRNAFL on both matched and MM5 substrates (Figures S11A and S12A), indicating that the reduction in linearization of mismatched DNA by SpyCas92Pro is prevalent under the different conditions tested and does not change even in the presence of a full-length sgRNA. Interestingly, both SpyCas92Pro and SpyCas9WT produce more nicked products with either matched or MM5 substrates at 1 mM Mg2+, even though the absolute values are lower for SpyCas92Pro in all the conditions that were tested (Figures S9B, S10B, S11B and S12B).
Figure 4. Comparison of the linearization and nicking activities of SpyCas9WT and SpyCas92Pro.
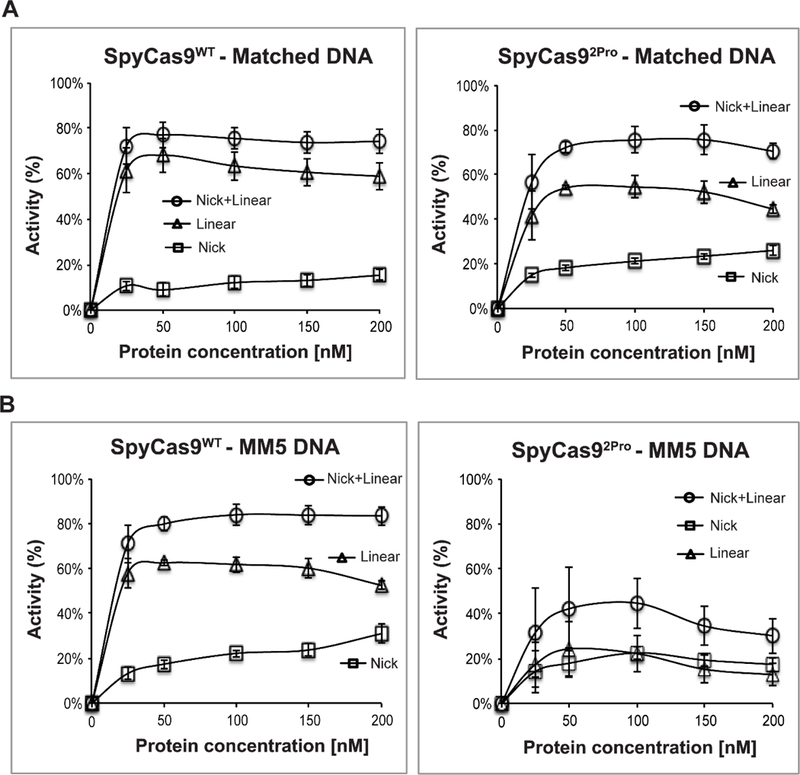
A) Analysis of cleavage pattern of SpyCas9WT (left) and SpyCas92Pro (right) with a fully matched DNA substrate at 5 mM Mg2+ ions. B) Analysis of cleavage pattern of a mismatched (MM5) DNA substrate at 5 mM Mg2+ ions using SpyCas9WT (left) and SpyCas92Pro (right). The average values for nicked (%), linear (%), and nicked + linear (%) (see Materials and Methods) are plotted against protein concentration. Data were obtained from three replications with a reaction time of 15 minutes and error bars represent SEM.
Expanding on the analyses of matched and MM5 substrates, we analyzed the amount of linear and nicked products produced by SpyCas9WT and SpyCas92Pro with substrates containing mismatch(es) at various protospacer positions (Figure S13). Using data obtained at 50 nM enzyme complex, we computed RL/N, the ratio of Linear vs. Nicked DNAs [equation (4)], for each replication, then averaged RL/N over three or more replications. At 5 mM Mg2+, SpyCas9WT gave higher average RL/N values than SpyCas92Pro for all 8 substrates tested (Figure S13A). This shows that SpyCas92Pro produces a lower relative fraction of linearized product compared to SpyCas9WT, and therefore, is acting more like a “nickase”. Reduction in linearizing activity of SpyCas92Pro varies depending on the position of the mismatch (Figure S13A).
For MM5 and MM7, two of the PAM-proximal single mismatch substrates that cause the most reduction in total cleavage by SpyCas92Pro when compared to SpyCas9WT (Figures 2–4), the average RL/N of SpyCas92Pro was reduced by ~8 times for MM5 and ~30 times for MM7 as compared to that of SpyCas9WT (Figure S13A). Further analyses showed that at 1 mM Mg2+, SpyCas92Pro had lower RL/N values for the matched and PAM-proximal mismatched substrates when compared to SpyCas9WT, while the ratios are comparable for PAM-distal mismatches, except for MM18 that produced more linearization by SpyCas92Pro (Figure S13B). The observations support the notion that BH-loop mutations cause a reduction in linearizing activity and that the effects are more pronounced at the PAM-proximal region.
Overall all, the pronounced nicking activity of SpyCas92Pro, especially on the mismatched DNA substrates, implies that the cleavage ability of one of the endonucleases is compromised in SpyCas92Pro and that the impairment is more pronounced on target DNA with PAM-proximal mismatches.
Structural flexibility of SpyCas9WT and SpyCas92Pro binary complexes varies.
As the BH-loop undergoes a loop-to-helix transition upon binding sgRNA and makes direct RNA contacts (Figures 1A and 1B), the substitutions in the BH-loop likely affect the binary Cas9-sgRNA complex. EMSA measurements showed that at approximately 50 nM sgRNAdel, a 1:1 molar ratio of sgRNA and protein gave ~70% complex for SpyCas92Pro and ~85% for SpyCas9WT (Figures 5A and S14). As such, under experimental conditions used to assess DNA cleavage (i.e., 50 nM equimolar protein and RNA, see Figures 2 –4), the functional differences observed is not due to a significant reduction of sgRNA binding in SpyCas92Pro, but rather due to the structural and/or dynamic differences in the binary complex. This is also consistent with the observation that for matched DNA, SpyCas92Pro and SpyCas9WT can cleave the precursor DNA to a comparable degree, albeit at a slower rate by SpyCas92Pro (Figures 2C and S8).
Figure 5. RNA binding and limited proteolysis of SpyCas9WT and SpyCas92Pro.
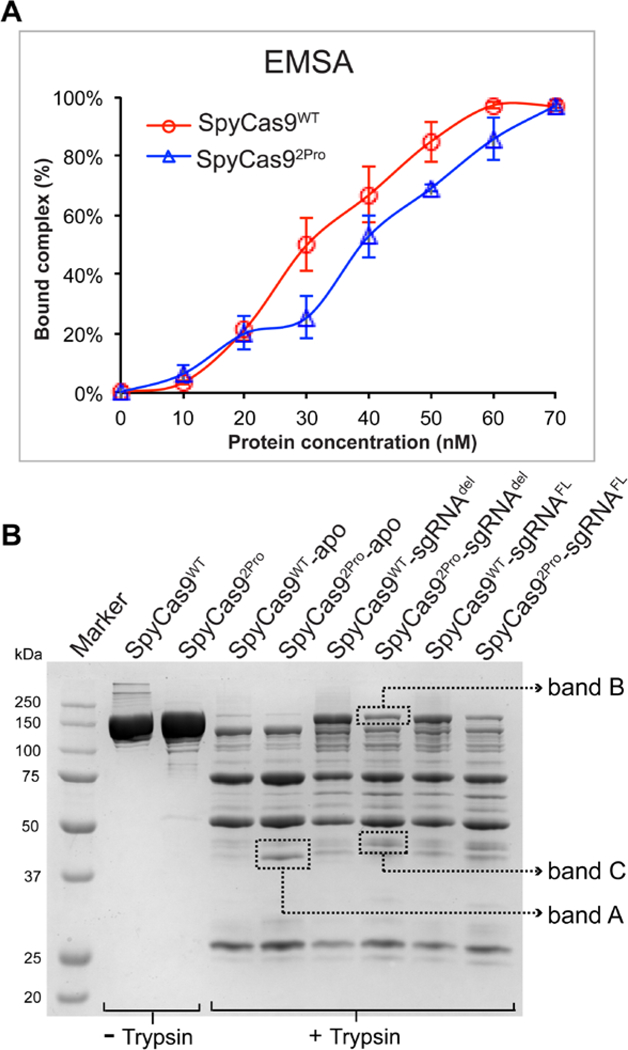
A) Graph showing quantification of binary complex formed by SpyCas92Pro and SpyCas9WT. EMSA was conducted using 5’−32P labelled sgRNAdel. The protein concentration was increased from 10 nM to 70 nM relatively to sgRNA concentration (~50 nM). Graph shows the average of bound complex from three independent replications over different protein concentrations. The data indicate that the RNA binding property of SpyCas92Pro is not significantly reduced compared to SpyCas9WT. B) Trypsin digestion of SpyCas9WT and SpyCas92Pro with or without sgRNA. In the apo-form, the digestion profiles for both proteins are similar except for increased intensity of Band A in SpyCas92Pro. The sgRNA bound form of SpyCas92Pro is not protected to the same extent as SpyCas9WT-sgRNA complex (see the difference in intensity of Band B). In addition, Band C is more prominent in SpyCas92Pro-sgRNA complex, indicating conformational differences between the two binary complexes.
To further support the notion that differences exist between SpyCas9WT and SpyCas92Pro in the binary protein-RNA complexes, we performed limited trypsin proteolysis. Comparing the apo- forms of SpyCas9WT and SpyCas92Pro, the banding pattern was similar for both proteins, except for an increase in the amount of a band in between 37 kDa and 50 kDa in SpyCas92Pro (Figure 5B, Band A). The binary complexes show different digestion patterns as compared to the apo-proteins, with more pronounced variations between SpyCas92Pro and SpyCas9WT (Figure 5B). SpyCas92Pro protein bound to sgRNA (both deleted and full-length versions) is more easily degraded by trypsin compared to SpyCas9WT bound to sgRNA, as indicated by the reduction of the full-length SpyCas92Pro compared to SpyCas9WT (Figure 5B, Band B). In addition, another band in between 37 kDa and 50 kDa (Figure 5B, Band C) is more intense in sgRNA bound SpyCas92Pro as compared to that of SpyCas9WT-sgRNA complex. These data indicate differences in the flexibility of the sgRNA-bound complexes of SpyCas92Pro and SpyCas9WT, which may lead to increased accessibility of trypsin to internal regions of SpyCas92Pro and therefore loss of full-length protein. This implicates that the loop-to-helix transition of the BH and its interactions with sgRNA as observed in the crystal structures may be essential in organizing an efficient binary complex, although further work is required to reveal the details.
SpyCas92Pro shows moderate activity in cell-based assays and exhibit a reduced off-target DNA cleavage compared to SpyCas9WT.
We tested the ability of SpyCas92Pro to produce lesions at seven different genomic sites of HEK293T cells using a TIDE assay (Table S3). SpyCas92Pro showed varying efficiencies in producing lesions on the seven target sites examined (Figure 6A). One of the sites (DTS7) has comparable efficiencies for both proteins (68% lesion for SpyCas9WT and 42% for SpyCas92Pro) and another site (DTS55) has moderate cleavage efficiency in the case of SpyCas92Pro (18%) compared to SpyCas9WT (65%) (Figure 6A and Table S8A). At the rest of the five sites, the amount of lesions produced by SpyCas92Pro is lower (varied between 1–3%) compared to SpyCas9WT (varied between 2–76%) (Table S8A). There was no difference in the cleavage efficiency using a full length or a shorter version of sgRNA, similar to results observed in in vitro activity assays (Figures 6B and S15). Furthermore, while SpyCas9WT is not affected by a 20-nt or 21-nt guide region in the sgRNA construct, SpyCas92Pro worked slightly more efficiently with a 20-nt guide region (Figure 6B). The reduced efficiency of 21-nt gRNA to induce lesions has been previously observed for Cas9 variants developed for reduced off-targeting effect (High-fidelity Cas9, enhanced Cas9).54, 55 The reduced targeting and cleavage efficiencies of SpyCas92Pro indicates that the BH-loop is critical in a cellular environment compared to an in vitro setting where the reduction in total activity is not so pronounced especially while targeting a completely complementary DNA. It is possible that the BH-loop substitution is promoting more nicking under the cellular conditions, similar to in vitro assays (Figure S13). Since nicks can be efficiently repaired in a cellular environment56, this can be translated into a reduction in the on-target DNA cleavage efficiency. Further experiments are required to confirm this.
Figure 6. Activity analysis of SpyCas9WT and SpyCas92Pro in HEK293T cells.
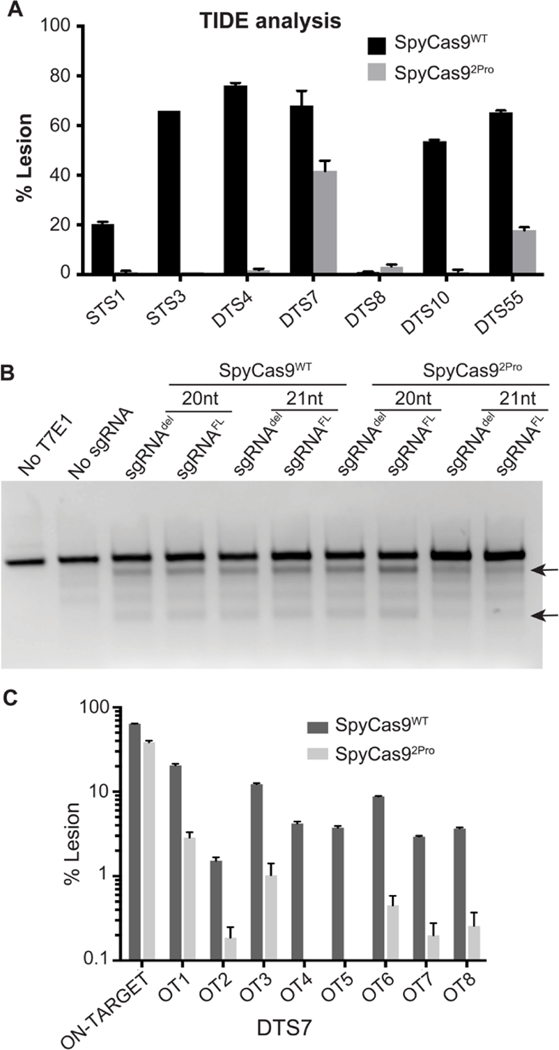
A) TIDE analysis of cleavage by SpyCas9WT and SpyCas92Pro at different genomic loci. B) T7 endonuclease assay for DTS7 spacer (with 20 nt or 21 nt of length) and using shortened (del) or full length (FL) repeat-tracrRNA region. Black arrows indicate cleavage products produced by T7E1 on mismatches created as a result of Cas9 editing. C) Off-target activity of SpyCas9WT and SpyCas92Pro as measured by targeted deep sequencing, the unmodified controls show no editing.
We proceeded to analyze the off-target effects of SpyCas92Pro. We compared the off-target editing profile following targeting of DTS7 genomic site of HEK293T cells by SpyCas9WT and SpyCas92Pro. We analyzed this by targeted deep sequencing of sites that have been previously shown as off-target sites for SpyCas9WT (Table S6)51 by GUIDE-seq.57 The results show an average on-target activity of 64% for SpyCas9WT and 39% for SpyCas92Pro (Table S8B). Interestingly, the off-target activity of SpyCas92Pro was much lower compared to SpyCas9WT (Figure 6C). SpyCas9WT produced significant levels of cleavage at two of the eight off-target areas that were tested (an average of 20% on site 1 and 12% on site 3). The amount of lesion produced by SpyCas92Pro on site 1 is 3% and site 3 is 1%, and the rest of the sites averaged to 0% (Table S8B). Thus, the specificity of DNA cleavage by SpyCas92Pro that was manifested under in vitro conditions is translatable to cellular assays. An analysis of the mismatches present in the off-target regions is shown in Figure S16.
DISCUSSION
SpyCas92Pro shows a higher degree of selectivity in DNA targeting.
The combined in vitro and cell-based analyses show that introducing two prolines in the BH-loop affects the DNA cleavage function of SpyCas9, with the effects being more pronounced in a cellular environment. In in vitro studies, SpyCas92Pro shows significantly reduced total cleavage activities against targets with PAM-proximal mismatch(es) as compared to SpyCas9WT (Figures 2B, 2C, and 3B). The ability of SpyCas92Pro to better discriminate against mismatched DNA is maintained in cellular assays as they demonstrate smaller degrees of off-target cleavage (Figure 6C). Interestingly, in vitro analyses show that there is more nicked product formation by SpyCas92Pro (Figures 4 and S13), suggesting that the activity of one of the endonuclease sites, RuvC or HNH, is impacted in SpyCas92Pro compared to SpyCas9WT. In the cell-based assays, SpyCas92Pro produces indels efficiently at only two out of the seven on-target sites tested (Figure 6A). This is likely linked to the impairment of one of the endonuclease sites of SpyCas92Pro that prevents double-stranded DNA breaks. Since nicked DNA can be repaired by the cellular machinery56, deficiency of one of the nucleases’ activity can lead to reduction in the number of indels produced. Previous work has shown that HypaCas9 variant acted on 19 out of the 24 endogenous sites tested, compared to 18 out of 24 in SpyCas9-HF1 and 23 out of 24 in eSpyCas9(1.1).38, 54, 55 This shows that substitutions in SpyCas9 affect the ability of the protein to act on different genomic sites perhaps due to weakened protein-nucleic acid interactions that in turn can potentially reduce off-target DNA cleavage. The reduction in on-target cleavage may be compounded in SpyCas92Pro due to reduction in the linearization activity at the target sites. Overall, the data indicate that SpyCas92Pro exhibits a higher degree of specificity in DNA targeting.
BH-loop substitution potentially affects protein-RNA-DNA interactions and impacts multiple aspects of Cas9 activity.
Our results show that the disruption of BH-loop affects more than one step in the catalytic cycle of Cas9. The BH-loop makes direct interactions with sgRNA and phosphate lock loop (PLL) (Figure 1B), yet SpyCas92Pro and SpyCas9WT form a similar amount of binary protein-sgRNA complex (Figure 5A). Interestingly, while SpyCas92Pro can cleave matched DNA to a similar extent as compared to SpyCas9WT (Figure 2), the rate of DNA cleavage is reduced in SpyCas92Pro (Figure S8). These results indicate that BH-loop disruption is not confined to a simple effect of RNA binding, but rather affects processes downstream of binary complex formation. Based on the available crystal structures and results reported here, we propose that proline substitutions in the BH-loop affect the conformational flexibility of Cas9-sgRNA binary complex, unwinding of DNA and stabilization of the nascent R-loop, and cross-talk between the two endonuclease sites. The reasonings are as follows.
The search of complementarity between a DNA target and the RNA guide is facilitated by a pre-organized seed region of the RNA guide in Cas9 and several protein-sgRNA interactions favor positioning of the seed region. For example, in both binary and ternary complexes of SpyCas9, the residues R63, R66, R70, R74, and R78 from the BH makes phosphate backbone interactions with the seed region (C18, G16, A15, G14 (PAM-proximal)) of the sgRNA (Figure 1).27, 28, 31 Substituting R66, R70, or R74 markedly reduces the activity of SpyCas9 and it was demonstrated that the interactions between the BH and the seed region of sgRNA are essential for R-loop initiation.37 The residue R66 lies in the BH-loop region and interacts with 14th and 15th nt in the seed region of the sgRNA through a direct H-bond. Water-mediated H-bonds are observed between R66 and the 62nd and 63rd nt of the tracrRNA in one of the SpyCas9 crystal structures (PDB ID: 4OO8).31 Even though R66 is not being directly substituted in the present study, the introduction of two consecutive prolines likely impacts helix formation in this region. This may affect positioning of R66 for interacting with sgRNA. It was previously shown that sgRNA without the seed sequence cannot induce conformational changes similar to that of sgRNA with the seed region,28 implicating that defects in organizing the seed region in SpyCas92Pro could be translated to downstream conformational changes. We note that trypsin digestions indicate that the BH-loop substitutions alter the structure and dynamics of Cas9-sgRNA complex (Figure 5B). However, further work is required to reveal the detailed changes in protein-RNA-DNA interactions in SpyCas92Pro binary and ternary complexes.
In addition to the direct interaction of BH-loop with the sgRNA, BH-loop is indirectly involved in DNA unwinding. The PLL, which contacts the phosphate backbone of the DNA at +1 position to initiate strand switching of DNA for R-loop formation,25 interacts with the BH-loop. This interaction is through a H-bond between K65 of BH-loop and E1108 of PLL and this H-bond is maintained even in the binary complex (PDB-ID: 4ZT0), ready and poised for strand switching.28 In our experiments, K65 has been substituted with a proline. The absence of this pre-organization can potentially affect DNA-unwinding in SpyCas92Pro.
Our experiments show that SpyCas92Pro has reduced activity with DNA substrates having PAM-proximal mismatches. We propose that the defects due to the absence of BH-loop conformational transition is compensated at least partially by the strength of DNA-RNA base pairing along the initial regions of the guide region in a matched DNA target. It is reasonable to envisage that in the case of SpyCas92Pro and target DNA with PAM-proximal mismatches, the compensatory RNA-DNA interactions are compromised. This may affect a productive R-loop formation, causing reduced activity with such DNA targets. For the PAM-distal mismatches, SpyCas92Pro shows similar or slightly higher total activity as that of SpyCas9WT. It has been reported that the pairing between 1 to ~14 nt in the RNA-DNA hybrid stabilizes the ternary complex and initiates HNH movement,37–39 with the HNH movement being modulated by mismatches at the PAM-distal end.38 Our data suggest that the BH-loop residues may also play a subtle role in modulating the HNH movement, although further investigations are needed.
SpyCas92Pro demonstrated consistently more nicking with the different DNA substrates, especially with mismatches, compared to SpyCas9WT. The coordination between HNH and RuvC by means of conformational changes to bring about double-strand DNA cleavage is well documented.40 The BH is directly linked to RuvC motif-II in the primary protein sequence. In addition, it was suggested based on molecular dynamics simulations that N844 and K848 of HNH can form interactions with E60 and T58 of BH.58 These interactions suggest that BH-loop substitution can possibly affect the positioning of the endonuclease sites and the allosteric communication between these sites, though further studies are needed to clarify this. Cas9 substitutions affecting the positioning of endonuclease sites were previously observed in eSpyCas9(1.1) and SpyCas9-HF1.38. In eSpyCas9(1.1) and SpyCas9-HF1, HNH is trapped in an intermediate, inactive state when they bind to mismatched DNA targets, and since the positioning of HNH is important for RuvC activity, the off-target DNA cleavage is reduced in these Cas9 variants.38, 39 Interestingly, it was shown that mismatches between crRNA and protospacer can promote formation of nonproductive protein-RNA complex that causes accumulation of DNA nicks.59 These previous studies and our data support our hypothesis that the communication between the endonuclease sites is impaired in SpyCas92Pro and that the effect is more pronounced when SpyCas92Pro binds to mismatched targets, thus reducing DNA linearization.
Gene-editing capabilities of SpyCas92Pro.
The cell-based analysis shows that SpyCas92Pro is not comparable to SpyCas9WT in its gene-editing capabilities. The impairment of the cross-talk between the endonuclease domains may be the strong contributor for this, since nicks are efficiently repaired in a cellular environment.56 Interestingly, Cas9 “nickase” has been shown as a strategy60, 61 to reduce off-target effects. It might be possible to improve the on-target activity of SpyCas92Pro using two sgRNAs to nick individual strands within a target genomic site. Similarly, the BH-loop substitutions can be tested along with other high-efficient Cas9 variants to analyze the presence of synergistic effects. Further elaborate studies are required to develop SpyCas92Pro as an efficient gene-editing tool.
Cas9 utilizes structuring of ARM region in response to RNA-binding as found in other RNA binding proteins.
ARM is an RNA-binding motif that consists of around 8–10 amino acids, usually enriched in basic amino acids, especially arginine. The ARM motif is able to recognize and bind specific RNA structural elements such as stem loop or bulge regions.62 The ARM regions in several RNA-binding proteins have been shown to be disordered or with lower helical content in the apo-form, with an increase in the helical content after binding to specific RNA.63 ARM can adopt different protein structural elements such as beta hairpins, alpha helix, and random coils after binding specific RNA targets.64
In SpyCas9, the BH adopts a helix-loop-helix conformation in the apo- structure but converts to a contiguous long helix in the binary and ternary complexes.27–29 In the case of Actinomyces naeslundii (Ana) Cas9 (type II-C), the BH and certain regions of REC domain are disordered in one of the two available apo- crystal forms (PDB ID: 4OGC), while they are ordered in another crystal form (PDBID: 4OGE).29 These facts imply that further studies are essential to conclusively show that loop-to-helix conversion occurs in Cas9 with response to sgRNA binding and whether Cas9 subtype-specific differences exist in this mechanism.
The structure of sgRNA before binding to Cas9 is not known. Most interactions of sgRNA with the BH-loop region are through the phosphate backbone and the specific structure at this region is highly essential for interactions with the BH.28 It is possible that the BH that is in a helix-loop-helix state in the apo- form inserts into a folded sgRNA molecule to convert the BH into a contiguous helix. However, this may cause significant topological challenges as the BH is an interior helix in a multi domain protein. Another possibility is the sgRNA folding into its specific structure after interacting with the BH. The positioning of BH in the binary complex supports the second possibility. In the binary and ternary complex structures, BH is inserted between the crRNA and tracrRNA regions of sgRNA.27, 28, 31 It can be envisioned that sgRNA undergoes certain folding transition upon interacting with BH, with a concurrent BH loop-to-helix conversion. Further studies are required to determine the structural changes in sgRNA with respect to Cas9 binding.
Crystal structures show that BH-RNA interactions are present in other Cas proteins such as Cas12a (formerly Cpf1, type V-A) (PDB-ID: 5NFV)65 and Cas12b (formerly C2C1, type V-B) (PDB-ID: 5U31)66, even though the exact positioning and length of BH is different. As such, it is possible that fine-tuning BH-RNA interactions can modulate substrate specificity in other families of Cas proteins as well.
Supplementary Material
ACKNOWLEDGMENTS
We thank E. Sontheimer and his laboratory for performing cell-based assays and E. Sontheimer for helpful discussions during the development of this manuscript. We thank P. Liu and X.D. Gao for helping with data analysis of deep sequencing experiments. We thank H.P. Parameshwaran for help with radioactive experiments and checking biochemical data for accuracy, and all Rajan lab members for critical discussions during preparation of the manuscript. We thank the OU Protein Production Core (PPC) facility for protein purification services and instrument support. The OU PPC is supported by an IDeA grant from NIGMS [grant number P20GM103640].
FUNDING
Work reported here was supported in part by grants from the National Science Foundation [grant number MCB-1716423, RR; grant number CHE-1213673, PZQ; grant number MCB-1716744, PZQ] and an Institutional Development Award (IDeA) grant from the National Institute of General Medical Sciences of the National Institutes of Health [grant number P20GM103640 to RR laboratory] and in part by a grant from the Research Council of the University of Oklahoma Norman Campus to RR.
Footnotes
AVAILABILITY OF DATA AND MATERIALS
There is a U.S. Patent pending on the subject matter of the manuscript, filed by the University of Oklahoma. The deep sequencing data from this study have been submitted to the NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) under accession number SRP186584.
ACCESSION CODES
UniProt protein ID: CAS9 Q99ZW2
NCBI protein ID: NP_269215.1
SUPPLEMENTARY DATA: Supplementary data is provided.
CONFLICTS OF INTEREST
The authors declare no competing financial interests.
REFERENCES
- [1].Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, and Horvath P (2007) CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712. [DOI] [PubMed] [Google Scholar]
- [2].Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, Dickman MJ, Makarova KS, Koonin EV, and van der Oost J (2008) Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321, 960–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Marraffini LA, and Sontheimer EJ (2010) CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat Rev Genet 11, 181–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Marraffini LA, and Sontheimer EJ (2010) Self versus non-self discrimination during CRISPR RNA-directed immunity. Nature 463, 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Marraffini LA, and Sontheimer EJ (2009) Invasive DNA, chopped and in the CRISPR. Structure 17, 786–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hale CR, Zhao P, Olson S, Duff MO, Graveley BR, Wells L, Terns RM, and Terns MP (2009) RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell 139, 945–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Garneau JE, Dupuis ME, Villion M, Romero DA, Barrangou R, Boyaval P, Fremaux C, Horvath P, Magadan AH, and Moineau S (2010) The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468, 67–71. [DOI] [PubMed] [Google Scholar]
- [8].Abudayyeh OO, Gootenberg JS, Konermann S, Joung J, Slaymaker IM, Cox DB, Shmakov S, Makarova KS, Semenova E, Minakhin L, Severinov K, Regev A, Lander ES, Koonin EV, and Zhang F (2016) C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 353, aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Mohanraju P, Makarova KS, Zetsche B, Zhang F, Koonin EV, and van der Oost J (2016) Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems. Science 353, aad5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Shmakov S, Smargon A, Scott D, Cox D, Pyzocha N, Yan W, Abudayyeh OO, Gootenberg JS, Makarova KS, Wolf YI, Severinov K, Zhang F, and Koonin EV (2017) Diversity and evolution of class 2 CRISPR-Cas systems. Nat. Rev. Microbiol. 15, 169–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Koonin EV, Makarova KS, and Zhang F (2017) Diversity, classification and evolution of CRISPR-Cas systems. Curr. Opin. Microbiol. 37, 67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, and Charpentier E (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Pennisi E (2013) The CRISPR craze. Science 341, 833–836. [DOI] [PubMed] [Google Scholar]
- [14].Doudna JA, and Sontheimer EJ (2014) Methods in Enzymology. The use of CRISPR/Cas9, ZFNs, and TALENs in generating site-specific genome alterations. Preface. Methods Enzymol. 546, xix-xx. [DOI] [PubMed] [Google Scholar]
- [15].Ma H, Marti-Gutierrez N, Park SW, Wu J, Lee Y, Suzuki K, Koski A, Ji D, Hayama T, Ahmed R, Darby H, Van Dyken C, Li Y, Kang E, Park AR, Kim D, Kim ST, Gong J, Gu Y, Xu X, Battaglia D, Krieg SA, Lee DM, Wu DH, Wolf DP, Heitner SB, Belmonte JCI, Amato P, Kim JS, Kaul S, and Mitalipov S (2017) Correction of a pathogenic gene mutation in human embryos. Nature 548, 413–419. [DOI] [PubMed] [Google Scholar]
- [16].Zhou Y, Zhu S, Cai C, Yuan P, Li C, Huang Y, and Wei W (2014) High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature 509, 487–491. [DOI] [PubMed] [Google Scholar]
- [17].Wang Q, Chen S, Xiao Q, Liu Z, Liu S, Hou P, Zhou L, Hou W, Ho W, Li C, Wu L, and Guo D (2017) Genome modification of CXCR4 by Staphylococcus aureus Cas9 renders cells resistance to HIV-1 infection. Retrovirology 14, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Fogarty NME, McCarthy A, Snijders KE, Powell BE, Kubikova N, Blakeley P, Lea R, Elder K, Wamaitha SE, Kim D, Maciulyte V, Kleinjung J, Kim JS, Wells D, Vallier L, Bertero A, Turner JMA, and Niakan KK (2017) Genome editing reveals a role for OCT4 in human embryogenesis. Nature 550, 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hou Z, Zhang Y, Propson NE, Howden SE, Chu LF, Sontheimer EJ, and Thomson JA (2013) Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc. Natl. Acad. Sci. U S A 110, 15644–15649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, and Liu DR (2017) Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 551, 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hess GT, Tycko J, Yao D, and Bassik MC (2017) Methods and Applications of CRISPR-Mediated Base Editing in Eukaryotic Genomes. Mol. Cell 68, 26–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Doudna JA (2015) Genomic engineering and the future of medicine. JAMA 313, 791–792. [DOI] [PubMed] [Google Scholar]
- [23].Gao XD, Tu LC, Mir A, Rodriguez T, Ding Y, Leszyk J, Dekker J, Shaffer SA, Zhu LJ, Wolfe SA, and Sontheimer EJ (2018) C-BERST: defining subnuclear proteomic landscapes at genomic elements with dCas9-APEX2. Nat. Methods 15, 433–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Xiao-Jie L, Hui-Ying X, Zun-Ping K, Jin-Lian C, and Li-Juan J (2015) CRISPR-Cas9: a new and promising player in gene therapy. J. Med. Genet. 52, 289–296. [DOI] [PubMed] [Google Scholar]
- [25].Anders C, Niewoehner O, Duerst A, and Jinek M (2014) Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 513, 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hirano H, Gootenberg JS, Horii T, Abudayyeh OO, Kimura M, Hsu PD, Nakane T, Ishitani R, Hatada I, Zhang F, Nishimasu H, and Nureki O (2016) Structure and Engineering of Francisella novicida Cas9. Cell 164, 950–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Jiang F, Taylor DW, Chen JS, Kornfeld JE, Zhou K, Thompson AJ, Nogales E, and Doudna JA (2016) Structures of a CRISPR-Cas9 R-loop complex primed for DNA cleavage. Science 351, 867–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Jiang F, Zhou K, Ma L, Gressel S, and Doudna JA (2015) STRUCTURAL BIOLOGY. A Cas9-guide RNA complex preorganized for target DNA recognition. Science 348, 1477–1481. [DOI] [PubMed] [Google Scholar]
- [29].Jinek M, Jiang F, Taylor DW, Sternberg SH, Kaya E, Ma E, Anders C, Hauer M, Zhou K, Lin S, Kaplan M, Iavarone AT, Charpentier E, Nogales E, and Doudna JA (2014) Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 343, 1247997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Nishimasu H, Cong L, Yan WX, Ran FA, Zetsche B, Li Y, Kurabayashi A, Ishitani R, Zhang F, and Nureki O (2015) Crystal Structure of Staphylococcus aureus Cas9. Cell 162, 1113–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Nishimasu H, Ran FA, Hsu PD, Konermann S, Shehata SI, Dohmae N, Ishitani R, Zhang F, and Nureki O (2014) Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 156, 935–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Huai C, Li G, Yao R, Zhang Y, Cao M, Kong L, Jia C, Yuan H, Chen H, Lu D, and Huang Q (2017) Structural insights into DNA cleavage activation of CRISPR-Cas9 system. Nat Commun 8, 1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Yamada M, Watanabe Y, Gootenberg JS, Hirano H, Ran FA, Nakane T, Ishitani R, Zhang F, Nishimasu H, and Nureki O (2017) Crystal Structure of the Minimal Cas9 from Campylobacter jejuni Reveals the Molecular Diversity in the CRISPR-Cas9 Systems. Mol. Cell 65, 1109–1121 e1103. [DOI] [PubMed] [Google Scholar]
- [34].Deveau H, Barrangou R, Garneau JE, Labonte J, Fremaux C, Boyaval P, Romero DA, Horvath P, and Moineau S (2008) Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J Bacteriol 190, 1390–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Gasiunas G, Barrangou R, Horvath P, and Siksnys V (2012) Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. U S A 109, E2579–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Sternberg SH, Redding S, Jinek M, Greene EC, and Doudna JA (2014) DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507, 62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zeng Y, Cui Y, Zhang Y, Zhang Y, Liang M, Chen H, Lan J, Song G, and Lou J (2018) The initiation, propagation and dynamics of CRISPR-SpyCas9 R-loop complex. Nucleic Acids Res. 46, 350–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Chen JS, Dagdas YS, Kleinstiver BP, Welch MM, Sousa AA, Harrington LB, Sternberg SH, Joung JK, Yildiz A, and Doudna JA (2017) Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature 550, 407–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Yang M, Peng S, Sun R, Lin J, Wang N, and Chen C (2018) The Conformational Dynamics of Cas9 Governing DNA Cleavage Are Revealed by Single-Molecule FRET. Cell Rep. 22, 372–382. [DOI] [PubMed] [Google Scholar]
- [40].Sternberg SH, LaFrance B, Kaplan M, and Doudna JA (2015) Conformational control of DNA target cleavage by CRISPR-Cas9. Nature 527, 110–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Singh D, Sternberg SH, Fei J, Doudna JA, and Ha T (2016) Real-time observation of DNA recognition and rejection by the RNA-guided endonuclease Cas9. Nat Commun 7, 12778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].DeLano WL (2002) Pymol: An open-source molecular graphics tool. CCP4 Newsletter On Protein Crystallography, 82–92. [Google Scholar]
- [43].Sampson TR, Saroj SD, Llewellyn AC, Tzeng Y-L, and Weiss DS (2013) Erratum: Corrigendum: A CRISPR/Cas system mediates bacterial innate immune evasion and virulence. Nature 501, 262–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Bachman J (2013) Site-directed mutagenesis. Methods Enzymol. 529, 241–248. [DOI] [PubMed] [Google Scholar]
- [45].Scholz J, Besir H, Strasser C, and Suppmann S (2013) A new method to customize protein expression vectors for fast, efficient and background free parallel cloning. BMC Biotechnol. 13, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Edelheit O, Hanukoglu A, and Hanukoglu I (2009) Simple and efficient site-directed mutagenesis using two single-primer reactions in parallel to generate mutants for protein structure-function studies. BMC Biotechnol. 9, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Schneider CA, Rasband WS, and Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Bolukbasi MF, Gupta A, Oikemus S, Derr AG, Garber M, Brodsky MH, Zhu LJ, and Wolfe SA (2015) DNA-binding-domain fusions enhance the targeting range and precision of Cas9. Nat. Methods 12, 1150–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kearns NA, Genga RM, Enuameh MS, Garber M, Wolfe SA, and Maehr R (2014) Cas9 effector-mediated regulation of transcription and differentiation in human pluripotent stem cells. Development 141, 219–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA 3rd, and Smith HO (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345. [DOI] [PubMed] [Google Scholar]
- [51].Amrani N, Gao XD, Liu PP, Edraki A, Mir A, Ibraheim R, Gupta A, Sasaki KE, Wu T, Donohoue PD, Settle AH, Lied AM, McGovern K, Fuller CK, Cameron P, Fazzio TG, Zhu LJ, Wolfe SA, and Sontheimer EJ (2018) NmeCas9 is an intrinsically high-fidelity genome-editing platform. Genome Biol 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Brinkman EK, Chen T, Amendola M, and van Steensel B (2014) Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 42, e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Ihaka R, and Gentleman R (1996) R: A Language for Data Analysis and Graphics. J. Comput. Graph. Statist. 5, 299–314. [Google Scholar]
- [54].Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, and Joung JK (2016) High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529, 490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, and Zhang F (2016) Rationally engineered Cas9 nucleases with improved specificity. Science 351, 84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Vriend LE, and Krawczyk PM (2017) Nick-initiated homologous recombination: Protecting the genome, one strand at a time. DNA Repair (Amst) 50, 1–13. [DOI] [PubMed] [Google Scholar]
- [57].Tsai SQ, Zheng Z, Nguyen NT, Liebers M, Topkar VV, Thapar V, Wyvekens N, Khayter C, Iafrate AJ, Le LP, Aryee MJ, and Joung JK (2015) GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 33, 187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Zuo Z, and Liu J (2017) Structure and Dynamics of Cas9 HNH Domain Catalytic State. Sci. Rep. 7, 17271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Szczelkun MD, Tikhomirova MS, Sinkunas T, Gasiunas G, Karvelis T, Pschera P, Siksnys V, and Seidel R (2014) Direct observation of R-loop formation by single RNA-guided Cas9 and Cascade effector complexes. Proc. Natl. Acad. Sci. U S A 111, 9798–9803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Mali P, Aach J, Stranges PB, Esvelt KM, Moosburner M, Kosuri S, Yang L, and Church GM (2013) CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 31, 833–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Tsai SQ, Wyvekens N, Khayter C, Foden JA, Thapar V, Reyon D, Goodwin MJ, Aryee MJ, and Joung JK (2014) Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat. Biotechnol. 32, 569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Weiss MA, and Narayana N (1998) RNA recognition by arginine-rich peptide motifs. Biopolymers 48, 167–180. [DOI] [PubMed] [Google Scholar]
- [63].Calnan BJ, Biancalana S, Hudson D, and Frankel AD (1991) Analysis of arginine-rich peptides from the HIV Tat protein reveals unusual features of RNA-protein recognition. Genes Dev. 5, 201–210. [DOI] [PubMed] [Google Scholar]
- [64].Casu F, Duggan BM, and Hennig M (2013) The arginine-rich RNA-binding motif of HIV-1 Rev is intrinsically disordered and folds upon RRE binding. Biophys. J. 105, 1004–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Swarts DC, van der Oost J, and Jinek M (2017) Structural Basis for Guide RNA Processing and Seed-Dependent DNA Targeting by CRISPR-Cas12a. Mol. Cell 66, 221–233 e224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Yang H, Gao P, Rajashankar KR, and Patel DJ (2016) PAM-Dependent Target DNA Recognition and Cleavage by C2c1 CRISPR-Cas Endonuclease. Cell 167, 1814–1828 e1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.