

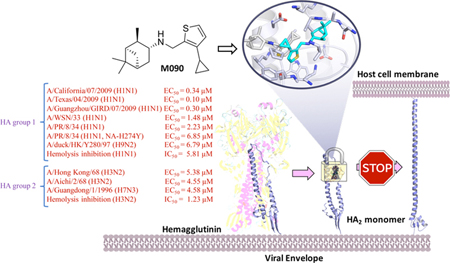
ABSTRACT
Influenza pandemic is a constant major threat to public health caused by influenza A viruses (IAV). IAVs are sub-categorized by the surface proteins: hemagglutinin (HA) and neuraminidase (NA), in which they are both essential targets for drug discovery. While it is of great concern that NA inhibitor oseltamivir resistant strains are frequently identified from human or avian influenza virus, structural and functional characterization of influenza HA has raised hopes for new antiviral therapies. In this study, we explored a structure-activity relationship (SAR) of pinanamine-based antivirals and discovered a potent inhibitor M090 against amantadine-resistant viruses, including the 2009 H1N1 pandemic strains, and oseltamivir- resistant viruses. Mechanism of action studies, particularly hemolysis inhibition, indicated that M090 targets influenza HA and it occupied a highly conserved pocket of the HA2 domain and inhibited virus-mediated membrane fusion by “locking” the bending state of HA2 during the conformational rearrangement process. This work provides new binding sites within the HA protein and indicates that this pocket may be a promising target for broad-spectrum anti-influenza A drug design and development.
SYNOPSIS
INTRODUCTION
Influenza is a severe respiratory disease caused by influenza viruses. The virus belongs to the Orthomyxoviridae family and is an enveloped negative-sense RNA virus.1 There are three genera of influenza viruses, A, B and C, but only influenza A virus (IAV) causes pandemics.2 In the last two decades, we have witnessed the outbreak of influenza epidemics and pandemics, such as highly pathogenic avian influenza (HPAI), which occurred in 2005 and 2013 with the subtypes H5N1 and H7N9, respectively.3,4 Furthermore, in the 2009–2010 influenza season, the H1N1 influenza (swine flu) quickly spread worldwide and caused substantial morbidity and mortality globally.5 In recent years, another HPAI, H5N6, was identified in the south of China and cases of human infection were reported.6 Although anti-influenza vaccines are available, the efficacy is limited by antigenic drift or shift of virus.7,8 Two classes of antiviral drugs are available on the market, the neuraminidase (NA) inhibitors (oseltamivir, zanamivir, and peramivir) and Matrix 2 (M2) ion channel inhibitors (amantadine and rimantadine) (Chart S1). An issue facing both classes of drugs is emerging drug resistance. Nearly all current influenza virus strains are resistant to amantadine and its derivatives.9–13 The only available orally administered anti-influenza drug, oseltamivir, has been documented to have lost its effect in some strains.14–16 Therefore, discovery of a novel generation of anti-influenza agents and an understanding of their molecular mechanisms are urgently needed.
Among the list of drug targets that are currently being pursued in preclinical and clinical developments, M2 proton channel remains a hot topic. More than 95% of current circulating influenza A viruses carry the amantadine-resistant AM2-S31N mutation,17 rendering it a high-profile drug target.18–28 During the course of developing AM2-S31N inhibitors, it was found that although some of the compounds inhibit viral replication through AM2-S31N channel blockage, there are several analogs that inhibit viral replication through AM2-S31N-independent mechanisms. Following drug resistance testing selection experiments reveal several mutations in hemagglutinin (HA),29–31 indicating HA might be the drug target instead of M2. Nevertheless, the exact binding site and detailed mechanisms of action of these inhibitors have not yet been fully explored.
Small molecules can be used to probe key features of the mechanism of a specific binding site with a functional protein. Forward chemical genetics operates by using an effective compound to identify the genetic underpinnings of drug targets.32 Using this approach, in this study, we report our discovery of a potent antiviral, M090, and its mechanism of action (MOA). Compound M090 was discovered through a structure-activity relationship (SAR) study (Table 1) of a series of pinanamine-based antivirals.33–35 Structural similarity between M090 and reported AM2-S31N inhibitors led us to hypothesize that M090 inhibits viral replication through AM2-S31N blockage. However, electrophysiology experiments revealed that M090 did not inhibit the AM2- S31N channel. Given the potent antiviral activity and the high selectivity index, we therefore are interested in applying M090 as a chemical probe to dissect its MOA. Through resistance selection experiment, we identified a novel binding pocket that is located at HA2. The proposed MOA was further supported by molecular dynamics simulations and hemolytic fusion assays. Overall this work suggests a new binding site and the mechanism underlying the interaction of small molecules with the HA protein of IAV and indicates that this more conserved pocket may be an advanced target for antiviral drug design and development.
Table 1.
Inhibitory effect of the synthesized compounds on influenza virus-infected MDCK cellsa.
 | |||
|---|---|---|---|
| Compd. | Structure | Activity (EC50, μM)b |
MCC (μM)c |
| 1 | 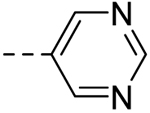 |
−d | >100 |
| 2 | 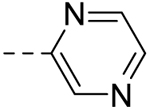 |
− | >100 |
| 3 | 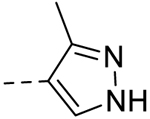 |
− | >100 |
| 4 | 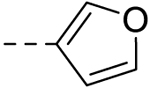 |
60.46 ± 1.75 | >100 |
| 5 | 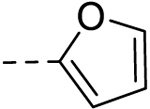 |
− | 50 |
| 6 | 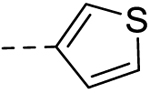 |
28.60 ±0.24 | 50 |
| 7 | 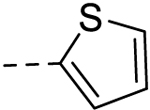 |
6.42 ± 0.18 | 25 |
| 8 | 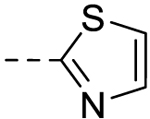 |
− | >100 |
| 9 | 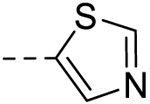 |
− | >100 |
| 10 | 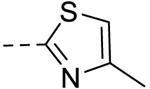 |
75.00 ± 0.08 | >100 |
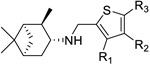 | |||||
|---|---|---|---|---|---|
| R1 | R2 | R3 | |||
| 11 | Me | H | H | − | 10 |
| 12 | Et | H | H | − | 25 |
| 13 | Br | H | H | − | 50 |
| 14 | n-Pr | H | H | − | 50 |
| l5 | n-Bu | H | H | 10.33 ± 2.17 | 25 |
| 16 | OEt | H | H | − | 50 |
| 17 | vinyl | H | H | 1.82 ± 1.04 | 50 |
|
18 (M090) |
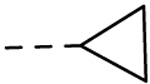 |
H | H | 0.30 ± 0.04 | 25 |
| l9 | 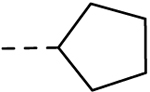 |
H | H | 0.69 ± 0.12 | 12.5 |
| 20 | 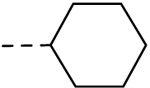 |
H | H | − | 1.56 |
| 21 | 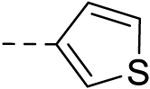 |
H | H | − | 12.5 |
| 22 | 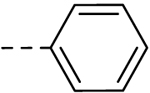 |
H | H | − | 12.5 |
| 23 | H | Br | H | 5.16 ± 1.08 | 6.25 |
| 24 | H | Me | H | 0.20 ± 0.004 | 12.5 |
| 25 | H | Et | H | − | 25 |
| 26 | H | n-Pr | H | − | 12.5 |
| 27 | H | n-Bu | H | 0.46 ± 0.07 | 12.5 |
| 28 | H | vinyl | H | − | 6.25 |
| 29 | H | 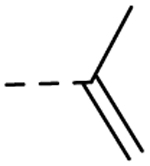 |
H | − | 25 |
| 30 | H | 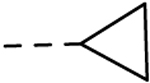 |
H | 6.68 ± 0.42 | 25 |
| 31 | H | 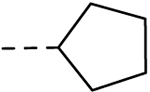 |
H | − | 1.56 |
| 32 | H | 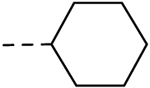 |
H | − | 0.78 |
| 33 | H | 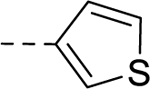 |
H | − | 25 |
| 34 | H | 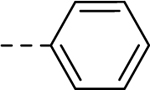 |
H | − | 5 |
| 35 | H | H | Cl | − | 25 |
| 36 | H | H | Me | − | 0.78 |
| 37 | H | H | Br | − | 43 |
| 38 | H | H | Et | − | 1.56 |
| 39 | H | H | t-Bu | − | 0.78 |
| 40 | H | H | 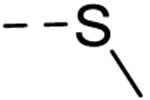 |
− | 25 |
| 41 | H | H | 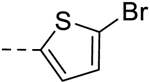 |
− | 0.78 |
| Amantadine | >100 | >100 | |||
Virus strain, A/Guangzhou/GIRD/07/2009 (H1N1).
All the compounds were also tested for the inhibitory effect against A/WSN/33 and A/HK/68 strains based on a CCK-8 reagent measurement method44. Compounds 6, 7, 10, 11–18, 23, 24, 26, 28, 30, 36 and 40 exhibited range of micromolar to sub-micromolar activity against both strains (the data were not shown), consistent with this experiment with the Guangzhou strain, compound 18 (M090) was the most potent inhibitor of the WSN and HK strains too.
MCC: minimum cytotoxic concentration, or concentration which led to minimal change in cell morphology after 48 h incubation with compound.
“−” represent that no viral inhibition occurs when the concentrations of compounds were lower than MCC.
RESULTS
Chemistry and SAR study.
To study the SAR of pinanamine derivatives using a cytopathic effect (CPE) assay, we conducted a phenotypic screen for synthetic compounds against the swine-origin human influenza A/Guangzhou/GIRD/07/2009 (H1N1)36 virus. As shown in Scheme 1 and Table 1. The synthesis of pinanamine-heterocyle derivatives (compounds 1 to 10, Table 1), 5-position substituted thiophene linked derivatives (compounds 35 to 41, Table 1) and compounds 11, 13, 16, 21, 22, 23, 24, 33 and 34 were obtained from commercially available heterocyclic formaldehyde materials reacting with 3-pinanamine through an easily accessible reductive amination reaction (Method E).37 For the synthesis of 3- and 4- substituted thiophene derivatives, the routes started from 3-bromothiophene to 3-alkyl substituted thiophene through a Kumada-Corriu coupling reactions (for compounds 19, 20, 31 and 32, Method A),38 subsequently, a Vilsmeier–Haack reaction was conducted to afford corresponding 3- (Method B) or 4- (Method C) substituted thiophenecarboxaldehyde (for compounds 12, 14, 15, 25, 26 and 27),39 or 3- and 4- substituted thiophenecarboxaldehyde reacted with potassium alkyltrifluoroborate directly (for compounds 17, 18, 28, 29 and 30, Method D) through a Suzuki-Miyaura cross-coupling reactions (Method A),40,41 then the same reductive amination reaction gave to the desired compounds (details were described in the Experimental Section).
Scheme 1. Synthetic methods of pinanamine derivatives.
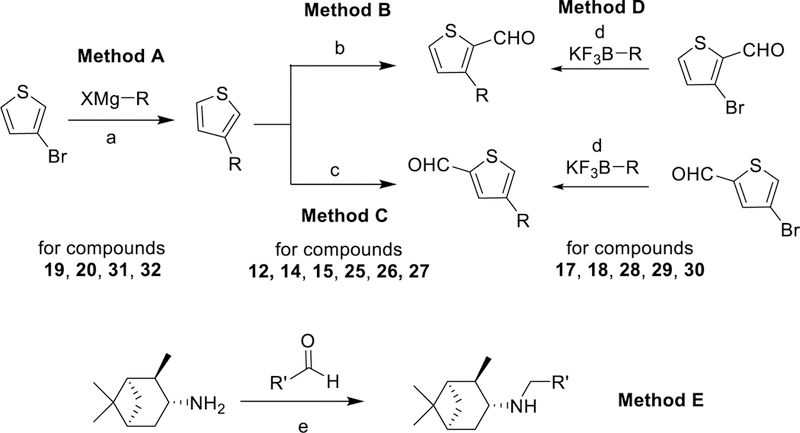
Reagents and conditions: (a) Ni(dppp)Cl2, ether, reflux; (b) POCl3, DMF, r.t; (c) n-BuLi, −78°C, THF, then DMF; (d) Pd(dppf)Cl2, K3PO4, H2O/toluene=1:3, 100 °C; (e) NaBH(OAc)3, CH2Cl2, AcOH, r. t.
Firstly, we explored the SAR of pinanamine-heterocyle derivatives (compounds 1 to 10) and identified compound 7 as the most potent compound in primary CPE assay. These results indicated that thiophene group may be a prior pharmacophore. We then further obtained several active compounds through the SAR studies of thiophene based pinanamine derivatives. SAR exhibited that aliphatic substitutions with middle size in the 3- or 4-position (R1 or R2 in Table 1) of thiophene revealed potent activities (Table 1). The most potent compound with the highest selectivity index, M090, was selected for follow-up inhibition studies on multiple strains (including amantadine-resistant and oseltamivir-resistant strains) of IAV using CPE (Table 2) and a plaque reduction assay (PRA) (Figure 1 and S1). Gratifyingly, M090 showed potent antiviral activities against A/California/07/2009, H1N1 (amantadine-resistant, oseltamivir- sensitive) and A/Texas/04/2009, H1N1 (amantadine-resistant, oseltamivir-resistant) strains,42 with an PRA EC50 of 0.34 ± 0.016 μM and 0.1 ± 0.009 μM respectively, (Figure 1B and 1C). As the structure of M090 fits the pharmacophore model of AM2-S31N inhibitors,26–28 we hypothesize that the broad-spectrum antiviral activity of M090 might result from its inhibition of the AM2-S31N proton channel. Therefore, we tested the channel blockage of AM2-S31N by M090 series in two- electrode voltage clamp electrophysiological assay (Table S1).43 Surprisingly, M090 showed negligible channel blockage, which suggests its antiviral activity is independent of AM2-S31N channel blockage. Given the broad-spectrum antiviral activity of M090, we were intrigued to explore its novel MOA.
Table 2.
Effect against a broader panel of influenza A viruses.
| Compound | M090 | Amantadine | Oseltamivir | ||
|---|---|---|---|---|---|
|
Antiviral activity (EC50, μM)a |
H1N1 | A/WSN/33 | 1.48 ± 0.22 | >100 | 0.40 ± 0.08 |
| A/PR/8/34 | 2.23 ± 0.60 | >100 | 0.13 ± 0.03 | ||
| A/PR/8/34 (NA-H274Y) |
6.85 ± 1.85 | >100 | >10 | ||
| H3N2 | A/HK/68 | 5.38 ± 0.32 | 1.00 ± 0.50 | 2.30 ± 0.90 | |
| A/Aichi/2/68 | 4.55 ± 0.80 | NDb | 0.99 ± 0.05 | ||
| H7N3 | A/duck/Guangd ong/1/1996 |
4.58 ± 0.66 | ND | 2.67 ± 0.56 | |
| H9N2 | A/duck/HK/ Y280/97 | 6.79 ± 0.44 | ND | >10 | |
The anti-influenza activity of M090 was evaluated by CPE assay and validated by PRA assay (Figure S1).
ND, not detected.
Figure 1.
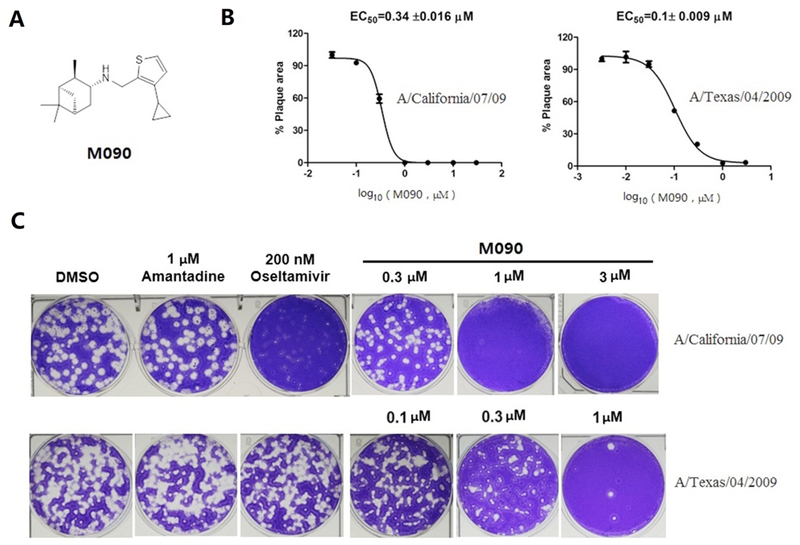
Identification of M090 as a potent antiviral against A/California/07/09 and A/Texas/04/2009. (A) Chemical structure of M090. (B, C) Inhibitory curves and plaques for M090; The EC50 was analyzed by measurement of percent area of plaques. Each point corresponds to the mean ± s.d.; data were obtained from two independent experiments.
Mechanism of Action Study.
The replication cycle of the influenza virus begins at absorption to and ends at the production of its progeny viruses; the whole cycle requires approximately 8 to 10 h. There are three steps, relative to virus entry at time 0, in this process: the early stage of attachment and entry (−1 to 2 h), the middle stage of endosomal release, genome replication and viral protein translation (2 to 8 h), and the late stage of virion assemble and release of progeny viruses (8 to 10 h).45 Defining −1 h as the time point of absorption, an unbiased drug time-of- addition (TOA) experiment46 was performed.
The whole life cycle of virus was divided into eight treatment intervals of 1 to 2 h each (Figure 2A); M090 was administered for treatment, and the yield was measured using TCID50 (virus infectious dose that can produce pathological change in 50% of cell cultures inoculated.) testing. The intervals of −1 to 2 h and 0 to 2 h post-virus absorption showed inhibition due to the presence of M090 (Figure 2B). This finding indicates that M090 blocked the early steps of the virus life cycle, as well as the entry stage, which includes attachment, endocytosis and fusion.47
Figure 2. Time-of-addition approach to identify the targeted stage of M090.
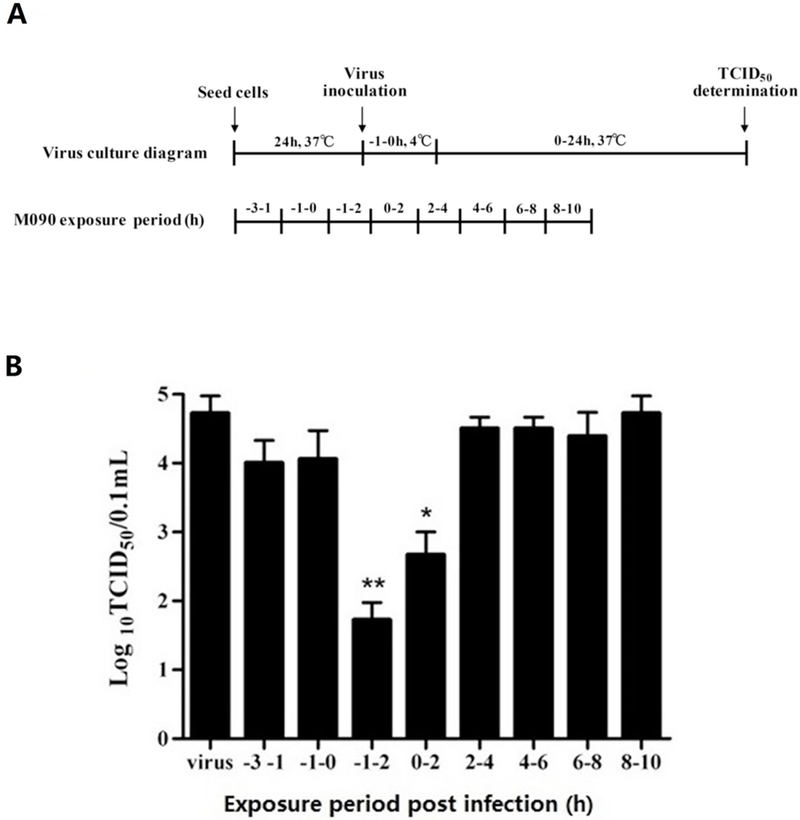
(A) Time course design. MDCK cells were infected with H1N1 (A/PR/8/34) at 100 TCID50. The virus absorption was indicated as −1 h and M090 was added at −3 to −1, −1 to 0, −1 to 2, 0 to 2, 2 to 4, 4 to 6, 6 to 8, 8 to 10 h post-infection. After each treatment period, M090 was removed and cells were incubated with culture medium till the end point. (B) M090 was added at each interval, and the virus yield was determined by measuring the TCID50. *p<0.05, ** p<0.01, compared to the condition without drug treatment.
The attachment, endocytosis and fusion process is related to two viral proteins of IAV, M2 and HA.8,48 As M090 did not inhibit the AM2-S31N proton channel as shown by the TEVC assay results, we hypothesize that the HA protein may be the target of this series of antiviral compounds such as M090. HA is encoded by the fourth viral RNA segment, and its precursor, HA0, undergoes extra- or intracellular cleavage into two subunits, HA1 and HA2.49,50 In the virus entry progress, HA1 regulates virus attachment to the sialic acid (SA) receptor of the host cell, which appears as hemagglutination.51,52 As shown in hemagglutination inhibition (HI) assays, M090 did not inhibit the virus-induced aggregation of chicken erythrocytes (Figure S2), revealing that there was no interaction between M090 and the HA1 domain.
Identification of Binding Sites by Drug Resistance Selection Experiments.
To identify the binding site of M090, resistance-induction experiments were performed against the A/Guangzhou/GIRD/07/2009 (H1N1) virus in MDCK cells (Table 3). At each passage, serial 2-fold dilutions of M090 were selected (43 ~ 0.042 μM) and virus that developed 50% CPE was picked for the subsequent passage. At the first two passages, the virus progeny maintained full drug sensitivity. Drug resistance developed from passage 3, and the EC50 increased three-fold. Resistance gradually increased in passages 4 to 6 and became significant after passage 6. After passage 7, the virus was fully resistant to M090.
Table 3.
Resistance Development under Pressure of M090a.
| Passage no. | EC50 (μM)b |
|---|---|
| 0 | 0.30 |
| 1 | 0.34 |
| 2 | 0.32 |
| 3 | 1.70 |
| 4 | 8.70 |
| 5 | 13.8 |
| 6 | 25.6 |
| 7 | >43.0 |
| 8 | >43.0 |
| 9 | >43.0 |
Resistance to M090 developed from passage 3 and became significant after passage 6.
The parent strain is A/Guangzhou/GIRD/07/2009.
EC50 values tested after each passage.
We sequenced the M and HA RNA segments of the resistant strain of passage 8 and compared the sequences with those of the parent A/Guangzhou/GIRD/07/2009 virus, which was reported in the Influenza Research Database.53 Two substitutions were found (changes in basic groups are shown in Figure S3). Both substitutions occurred in the HA protein, one is A15T, which located at the signal peptide of HA50 and does not exist in the mature virus protein. Logically, this substitution could not emerge from drug pressure. The other site, HA-E418D, known as HA2- E74D, is located on the top of the a-helix of the HA2 trimer (Figure 3). This mutant is the sole reasonable active site of M090.
Figure 3. Crystal structure (PDB entry: 3AL4) and M090-induced substitution of HA.
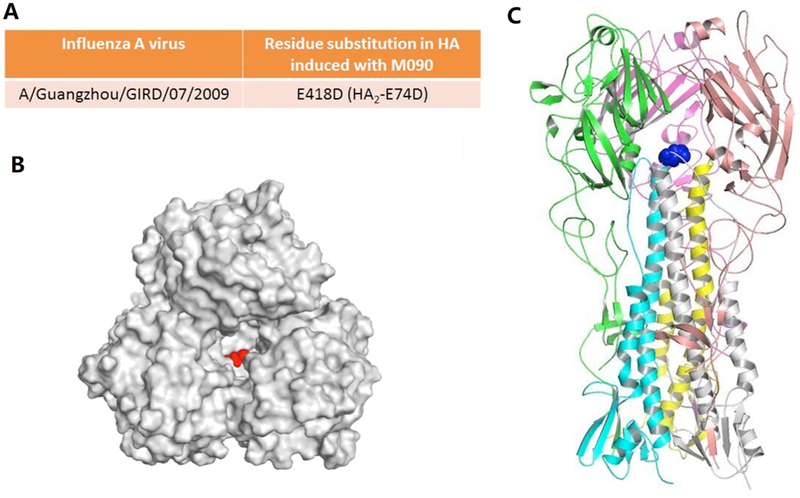
HA structure from the swine-origin A (H1N1)-2009 influenza A virus A/California/04/2009.54 The RNA sequences of this strain are identical to those of A/California/07/2009 and A/Guangzhou/GIRD/07/2009 that we have used. (A) Amino-acid substitution found in HA. (B, C) Top and side view of the HA model. The M090-resistant site HA2-E74 is highlighted by spherical models in red and blue, respectively.
Molecular Dynamics (MD) Simulations.
The potential binding sites of M090 (Figure 4) were characterized using SiteMap,55 and M090 was docked into these sites using Glide.56 The top-ranked binding poses were clustered. The most favorable docking score in each cluster (Figure S4) was chosen and further refined by MD simulations.
Figure 4. Potential binding sites of M090 in wild type HA protein (PDB entry: 3AL4).
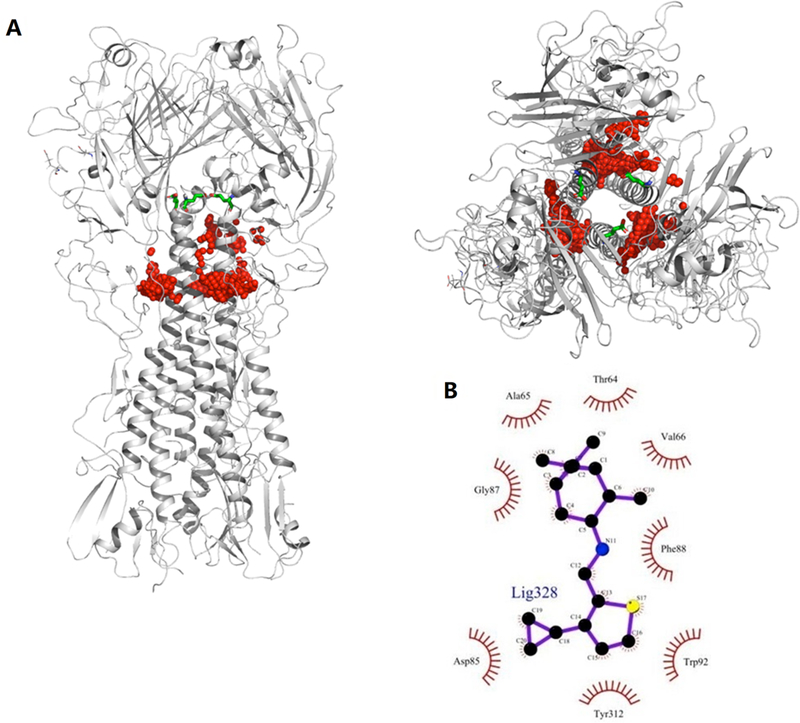
(A) Front and top views of potential binding sites (highlighted in red), HA2-E74 is highlighted by stick model in green. (B) 2D model of the HA binding pocket suggested by docking.
The stability of the predicted conformations of the M090-HA2 complex were evaluated by the average root-mean-square deviation (RMSD) value of HA protein and M090. As shown in Figure 5, in the HA2 monomer, a short helix and a long helix are linked by a loop (Figure 5A). The predicted binding pose of M090 in the binding pocket is located at the interface of the long helix (residues 82 to 93) and loop (residues 57 to 69) (numbering started from the HA2 segment). M090 fits well in the pocket. Its pinane scaffold forms hydrophobic interactions with HA2-Ala65 and HA2- Val66, the thiophene ring lays in the hydrophobic region formed by HA2-Ile89, Tyr312, and HA2-Trp92, the cyclopropyl group forms hydrophobic interactions with HA2-Phe88 and Pro303, and the hydrogen bond between N-H and Tyr305 enhances the binding of the compound with the protein (Figure 5B).
Figure 5. Predicted M090-HA2 structure.
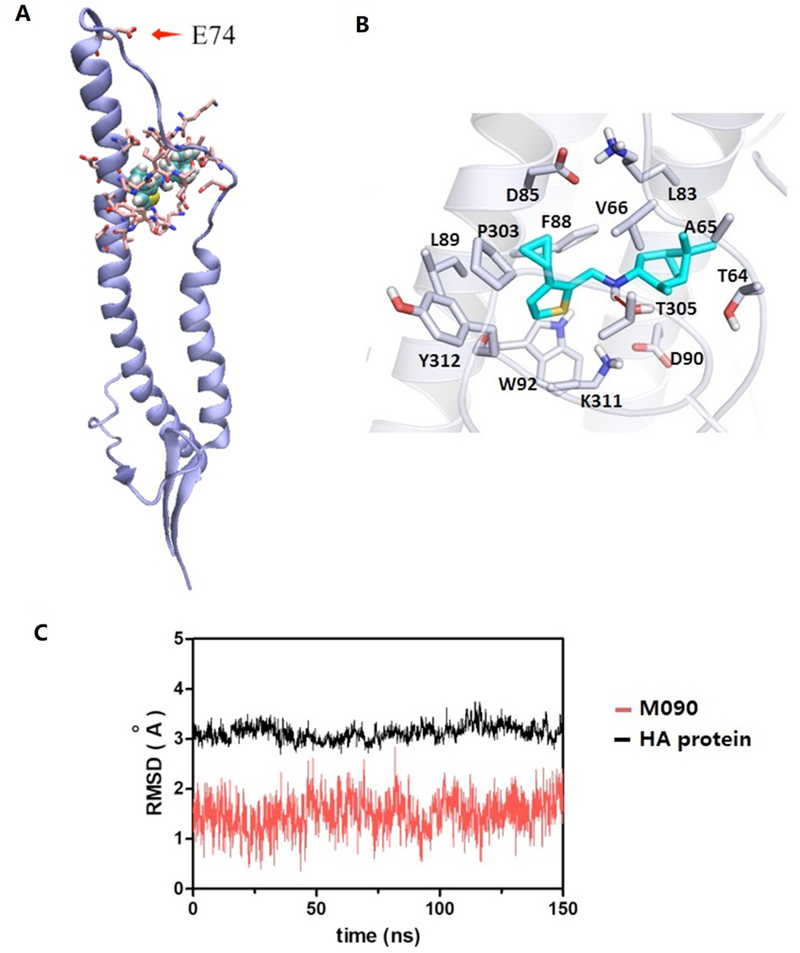
(A) Adjacent to HA2-74, M090 fits well in the pocket between a long α-helical segment and a loop of the HA2 monomer. (B) Binding mode of M090 with protein. (C) Time evolution of the RMSD values of M090 and HA protein during the MD simulation.
During low pH-triggered membrane-fusion, the conformation of the HA2 monomer changes from a folding shape to a needle shape, and HA2 attaches to the host cell membrane (Figure S5)57. Around the experimentally observed M090-induced mutant site HA2-E74, Glu74 directly interacts with the adjacent Arg76 residues to form three hydrogen bonds to stabilize the bent conformation of HA2 (Figure 6A). MD simulations indicated that in the presence of M090, site 1, which is close to the binding pocket, has higher H-bond occupation than the other two binding sites (Figure 6B). Stabilized by M090, the initial HA2 bending state is “locked”, and the HA2 monomer is accordingly blocked to from forming a needle-shaped conformation, which helps the protein attach to the host cell. The in silico results are consistent with the experimental drug- resistant mutation E74D, which indicates that the Glu74 mutant is a “shorter form”, namely, Asp, and the hydrogen bond interaction is weakened and breaks down easily despite drug binding.
Figure 6. Mode of M090 action studied by MD simulation.
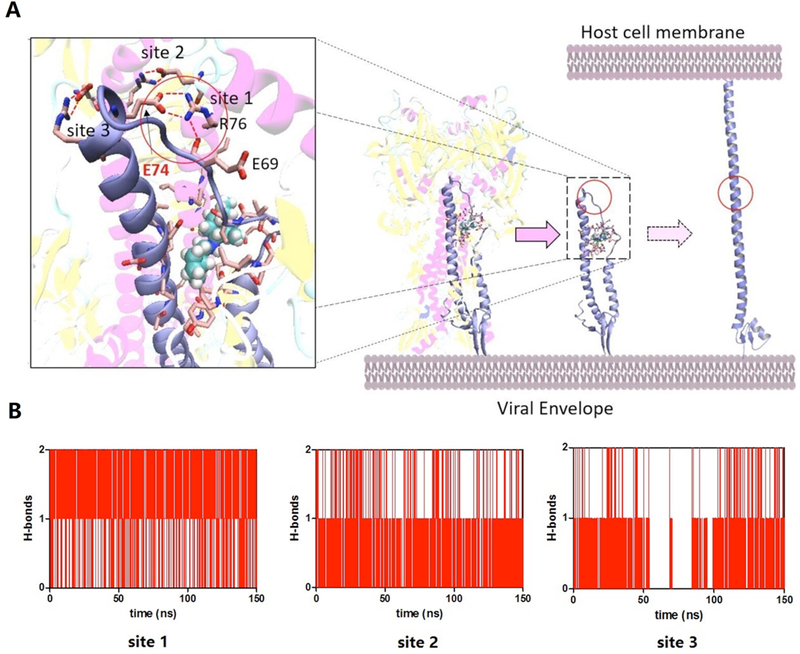
(A) M090 blocks the conformational change of the HA2 monomer by enhancing the H-bond interaction between the long helix and the loop. (B) Time evolution of the number of H-bonds of each site during the MD simulation.
Hemolytic Fusion Assays.
MD simulations demonstrated that M090 inhibits the membranefusion process by blocking the conformational changes of HA2; thus, the HA-mediated hemolysis inhibitory effect of M090 was tested for validation.
The optical density (OD540) value was measured after the chicken red blood cells (CRBC) were mixed with the virus together with different concentrations of M090. Two different viral strains (H1N1, A/PR/8/34 and H3N2, A/Aichi/2/68, which represent the two HA phylogenetic groups respectively) were used to induce hemolysis. A known hemolysis inhibitor, Arbidol58, was used as our control. As shown in Figure 7, the hemolysis inhibition was occurred in dose-dependent manner using the two virus strains.
Figure 7. Hemolysis inhibition measurements.
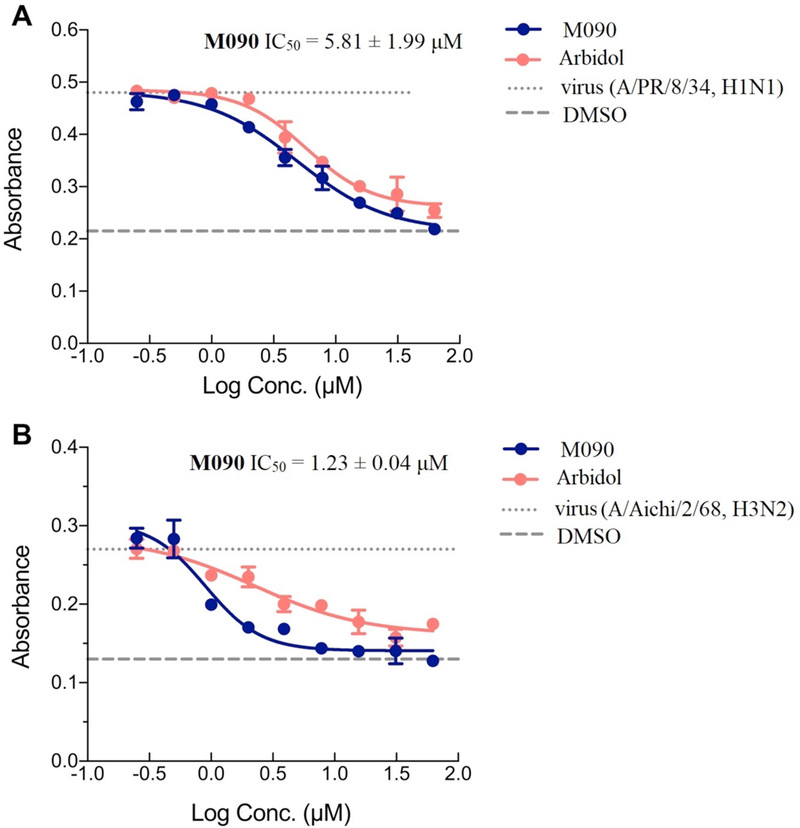
(A) H1N1, A/PR/8/34. (B) H3N2, A/Aichi/2/68. The level of hemolysis inhibition was measured at different concentration of M090. At each concentration, the inhibition effect of M090 (blue curves) was more potent than the positive control, Arbidol (red curves). Each point corresponds to the mean ± s.d.; The results were repeated in two independent experiments.
DISCUSSION
Based on the differences in nucleotide sequences, the HA subtypes are divided into two phylogenetic groups, group 1, including H1, H5 and H9, and group 2, including H3 and H7.59 Many fusion inhibitors were reported in literature, lots of them were HA group 1 specific, such as BMS-19994560, RO5464466 series,61,62 CL-385319 series63–65 and so on.66–71 Recently, a potent peptidic inhibitor of HA was identified, it was also group 1 specific.72 In the case of group 2, THBQ and its analogues73–75 and a series of azaspiro derivatives76 were identified to target H3 subtype. Another broad-spectrum antiviral drug, Arbidol, was characterized as a HA2 inhibitor.58 The co-crystallization of it with HA was analyzed and revealed that its binding site was similar with THBQ.77 We aligned the sequences which cover the potential binding pocket of M090 from two sets of HA subtype strains, set 1 was comprised of five H1, one H5, one H9 strains, and set 2 was comprised of two H3, two H7 strains (Figure S6). It was found that the sequence similarity of the strains from group 1 was more than 90 % while the strains from group 2 has more than 50% similarity. Remarkably, the escape mutation site, E418, is highly conserved among the two sets. Comparing to the antiviral effect of M090 on different virus strains including H1 and H9 (belong to group 1) as well as, H3 and H7 (belong to group 2) (Table 2), we found that this region is not absolutely group specific and may be used to design advanced inhibitors that target both phylogenetic groups of HAs and potentially explore broad-spectrum antiviral drugs. At the same time of preparing this paper, a series of aniline-based HA inhibitors were published and reported their antiviral activities against H1 and H5 HAs, the MD simulation study demonstrate that the binding site of these inhibitors overlaps with the binding cavity of THBQ,78 it may also inhibit the group 2 HA viruses. That work partially support our research strategy and demonstrated the rationality of the new binding site we found. These results can serve as a starting point for our work in coming future including the further optimization of M090 and its co-crystal complex with HA aiming for the analysis of binding site confirmation.
CONCLUSION
Influenza always threatens causes potential new pandemics and annual epidemics; thus, new therapeutics, especially with new targets and mechanisms of action, constitute a crucial strategy to combat these outbreaks. The process of HA growth and the function of HA offers targets for therapeutic intervention.79 Here, using a forward chemical genetics approach, we identified a potent antiviral agent, M090, and its binding site in the HA2 subunit. Extensive MOA studies of M090 indicated that small molecules can bind to a highly conserved segment of HA2, a new binding site located in the space of the long helix and the loop of the HA2 monomer. In the presence of inhibitor, low pH-induced membrane fusion was blocked via conformational rearrangements that led to inhibition of HA2.
This work identifies highly potent inhibitors against amantadine- and oseltamivir-resistant influenza A viruses and provides new insights into the specific binding site of small molecules with HA protein and implications regarding structure-based rational drug design. M090 and its further-modified compounds potentially could be components in combination therapy with approved anti-influenza drugs to overcome highly pathogenic virus strains. Indeed, this study proposed a promising binding site for the design and development of broad-spectrum antiviral drugs, and these small molecules may have a more resistant genetic barrier under drug pressure due to the conservation of the binding site. In addition, HA contributes to the early stage of the viral cycle, and newly designed HA inhibitors based on this binding site can be used for their prophylactic and therapeutic efficacy.
EXPERIMENTAL SECTION
Chemistry.
All commercially available compounds and solvents were reagent grade and were used without further treatment unless otherwise noted. Reactions were monitored by TLC using Qing Dao Hai Yang GF254 silica gel plates (5 × 10 cm); zones were detected visually under ultraviolet irradiation (254 nm) by either spraying with an ethanol solution of ninhydrin or by treatment with iodine gas. Compounds were purified by silica gel column chromatography performed on silica gel (200 to 300 mesh) from Qing Dao Hai Yang and characterized by 1H NMR, 13C NMR and ESI-MS. The NMR spectra were recorded on a Bruker NMR AVANCE 400 (400 MHz) or a Bruker NMR AVANCE 500 (500 MHz), and the NMR reagents CDCl3 and DMSO-d6 were used as internal standards. Chemical shifts (δ) were recorded in parts per million and coupling constants (J) in hertz (Hz). MS data were measured on an Agilent MSD-1200 ESI- MS system. The purity of compounds was analyzed by HPLC performed on an Agilent Sunfire C18 (150×4.6mm, 3.5μm) column, solvent, H2O (0.1 % HCOOH), 0.8 mL/min flow rate or 16 min gradient, 10 % to 100 % TFA in H2O (10 % to 80% KH2PO4 in H2O for compounds 25 and 37), 1.0 mL/min flow rate, the peak was detected at 254 nm. All compounds submitted for testing in biological assays were confirmed to be >95% purity by HPLC. Detailed regarding inhibitor characterization are provided in the Supporting Information. For use in assays, compounds were prepared as hydrochlorides or phosphates and dissolved in DMSO.
Detailed synthesis procedures.
Method A.
To a solution of Ni(dppp)Cl2 (1 mol%) in anhydrous ether (20 mL), in the N2 atmosphere, Cyclopentylmagnesium chloride or Cyclohexylmagnesium chloride (2M in ether, 30 mmol) was added dropwise at 0 °C, then the 3-Bromothiophene (25 mmol) in ether was added dropwise. The resulting solution was refluxed for 15h, then cooled to r. t. and quenched by 2M HCl. The mixture was extracted using ethyl acetate (100 mL×3). The organic layer was washed with brine, dried with Na2SO4 and the solvents were removed. The residue was purified by silica gel column chromatography to afford the corresponding alkylthiophene.
Method B.
To a solution of DMF (5 mL), POCl3 (15 mmol) was added, the mixture was stirred for 1 h at r. t. Then the alkylthiophene (10 mmol) in DMF (5 mL) was added dropwise and stirred for 1 h at 100 °C. The solution was then cooled to 0 °C and quenched by saturated NaHCO3 solution. The mixture was extracted by ethyl acetate (3 × 20 mL), the organic layer was washed with brine, dried with Na2SO4 and the solvents were removed. The residue was purified by silica gel column chromatography to afford the corresponding aldehyde.
Method C.
To a solution of alkylthiophene (10 mmol) in anhydrous ether (10 mL), n-BuLi (2M in ether, 15 mmol) was added dropwise at 0 °C in the N2 atmosphere. The mixture was refluxed for 1 h. The solution was then cooled to r. t. and DMF (1 mL) was added. The resulting solution was stirred for 2 hs and quenched by saturated NH4Cl solution. The mixture was extracted by ethyl acetate (3 × 20 mL), the organic layer was washed with brine, dried with Na2SO4 and the solvents were removed. The residue was purified by silica gel column chromatography to afford the corresponding aldehyde.
Method D.
To a solution of 3- or 4- substituted thiophenecarboxaldehyde (35 mmol), K3PO4 (105 mmol), Pd(dppf)Cl2.CH2Cl2 (0.7 mmol) and in toluene/H2O = 3/1 (140 mL), Potassium Alkyltrifluoroborate (42mmol) was added in the N2 atmosphere. The resulting solution was refluxed for 10 h. Then 50 mL water was added. Subsequently, the mixture was extracted using ethyl acetate (100 mL×3). The organic layer was washed with brine, dried with Na2SO4 and the solvents were removed. The residue was purified by silica gel column chromatography to afford the corresponding alkylthiophene.
Method E.
To a solution of (1R,2R,3R,5S)-(–)-isopinocampheylamine (6.5 mmol) in CH2Cl2 (20 mL), aldehyde (1.5 equiv., 9.8 mmol) was added. The reaction mixture was stirred at r.t. for 1 h, followed by the addition of sodium triacetoxyborohydride (26 mmol). The reaction was then stirred for 10 h and quenched by the addition of H2O and extracted with CH2Cl2 (3×20 mL). The combined organic layers were washed with brine, dried with Na2SO4 and evaporated to dryness. The residue was purified by silica gel column chromatography, treated with saturated HCl/CH3OH (20 mL), then evaporated to dryness. The insoluble material was washed with diethyl ether (3×20 mL) to afford the secondary amines as the corresponding hydrochloride salts.
Cytopathic Effect (CPE) Assay.
In the CPE assay, MDCK cells were grown to a confluent monolayer in a 96-well culture plate at a concentration of 5×104/well for 24 h. The medium was removed, and the cells were rinsed twice. An infectious virus at 100 TCID50 (the influenza virus infectious dose that can produce pathological change in 50% of cells during 48 hours inoculated time period) was inoculated into the MDCK cells, which were then incubated for 2 h at 37 °C in 5% CO2. The virus supernatant was removed, which was followed by the addition of serial two-fold dilutions of antiviral compounds in DMEM containing 1.5 μg/mL trypsin. After being incubated at 34 °C in 5% CO2 for 48 h, the infected cells displayed 100% CPE under the microscope, and the CPE percentages in the antiviral compound-treated groups were recorded. The EC50 values were calculated using a non-linear regression model in GraphPad Prism.
Cytotoxicity Assay.
The cytotoxicity assay was carried out using similar method as the CPE assay. MDCK cells were grown to a monolayer in a 96-well culture plate at a concentration of 5× 104/well. After 24 hours, the medium was removed, and the cells were rinsed with PBS twice. Compounds with two-fold serial dilution of compounds in DMEM were added into the cells. After incubation for 48 hours, the cell survival rate of each well was observed under the microscope and the concentration which led to minimal change for cell morphology was counted as the minimum cytotoxic concentration.
Plaque Reduction Assay.
A monolayer of A549 cells was infected with 0.01 MOI influenza A viruses for 1 h at 37 °C. The inocula were removed, and the cells were washed twice with phosphate-buffered saline (PBS). The cells were overlaid with 1% agar DMEM with amantadine or one of the synthesized compounds in the presence of 2 μg/mL trypsin and 0.3% BSA. Two to three days after infection, the monolayers were fixed and stained with 0.1% crystal violet solution. The viruses used for this assay were A/California/07/2009 (H1N1) and A/Texas/04/2009 (H1N1). To use the virus strains A/WSN/33 (H1N1), A/HK/68 (H3N2) and A/PR/8/34 (H1N1), MDCK cells were used instead of A549 cells.
Time-of-addition approach.
MDCK cells were grown to a confluent monolayer in a 24-well culture plate and washed twice with PBS. 100 TCID50 of influenza A virus (A/PR/8/34) at was inoculated into the MDCK cells (0.5 mL per well) and allowed to absorb at 4 °C for 1 h. The wells were then washed with PBS. The plates received medium containing 1.5 μg/mL TPCK- treated trypsin (0.5 mL per well) and were incubated in a CO2 incubator at 37 °C for 10 h. Compound (10 μM) was added at −3 to −1, −1 to 0, −1 to 2, 0 to 2, 2 to 4, 4 to 6, 6 to 8, 8 to 10 h post-infection. After each treatment period, the monolayer was washed of drug with PBS, and medium containing 1.5 μg/mL TPCK (0.5 mL per well) was added. After 24 h, the plate was frozen and thawed, the supernatant was collected, and TCID50 was measured to determine the virus yield.
Two-Electrode Voltage Clamp (TEVC) Assay.80
Selected compounds were tested in a TEVC assay using Xenopus laevis frog oocytes microinjected with RNA expressing the wild type or S31N mutant of the A/M2 protein. The potency of the inhibitors was expressed as percentage inhibition of A/M2 current, as observed after 2 min of incubation with 100 Μm compounds at pH 5.5. All measurements were repeated two times with different cells.
Hemagglutination inhibition (HI) assay.
Compound from a serial two-fold dilution in PBS was mixed with an equal volume of influenza virus (A/PR/8/34). After a 1-h incubation in a V- bottom 96-well plate, 50 μL of compound-virus preparation was added to an equal volume of 0.5% freshly prepared chicken red blood cells in Alsever’s solution. The mixture was incubated for 40 min at room temperature before observing erythrocyte aggregation on the plate.
Serial passage experiments and resistant mutation identification.
MDCK cells were infected with influenza H1N1 virus (A/Guangzhou/GIRD07/2009) at 100 TCID50 for 2 h; the inoculum was removed, and cells were incubated with M090 at the concentrations of two-fold serial dilutions beginning at MCC or in the absence of M090 for 2 days to evaluate drug pressure-selected mutations in vitro. At the endpoint of each passage, the viruses that developed a significant 50% CPE were harvested and subjected to the following passages. The titer sample of harvested virus at the 8th passage was subjected to sequencing. Influenza viral RNA was isolated using a QIAamp viral RNA Mini Kit (Qiagen, Hilden, Germany). The eight segment cDNAs were generated with reverse-transcription polymerase chain reaction (PCR) using specific primers (Table S2). Reverse-transcription PCR products were identified by agarose gel electrophoresis. PCR products were sequenced by BGI, Inc. (Shenzhen, CHN).
Docking and molecular dynamics (MD) simulations.
The crystal structure of swine-origin A (H1N1)-2009 influenza A virus HA (PDB: 3AL4) was obtained from the Protein Data Bank (http://www.rcsb.org/pdb/) and prepared using the Protein Preparation Wizard.81 The missing sidechains were added, and the protonation states were determined using PROPKA. The structure thus obtained further underwent a restrained energy minimization. The compound was prepared using LigPrep (Schrödinger, LLC), and 30 conformations were generated for docking. The binding sites were characterized using SiteMap,55 and all conformations were docked into the active site using Glide.56
MD simulations were performed using the GROMACS code.82 The Amber99SB-ildn force field83 was used for the protein, and the GAFF force field84 was used for the compound. Each HA/compound complex was solvated in a 15*15*15 nm3 cubic box with 144538 TIP3P85 water molecules. To neutralize the system and to prepare a final NaCl concentration of 0.15 M, 304 Na+ and 296 Cl- ions were added. The system was energy minimized with the steepest descent algorithm for 1000 steps. Thereafter, 200-ps isothermal-isochoric (NVT, with T=300 K) and 200-ps isothermal-isobaric (NPT, with T=300 K and P=1 atm) MD simulations were performed to equilibrate the system. The harmonic restraint with a force constant of 1000 kJ mol−1 nm−2 was used during the simulations. The equilibrated system was then subjected a simulation of 150 ns without any restraint. During the equilibration and production runs, the time step was 2 fs, and the LINCS algorithm86 was applied to constrain the bonds involving hydrogens. The van der Waals and short-range electrostatic interactions cutoff was set to 10 Å, and the particle-mesh Ewald (PME) method87 was used to treat long-range electrostatics.
Hemolysis inhibition assay.
A 100-μL influenza virus (A/PR/8/34) or (A/Aichi/2/68) sample was incubated with serial two-fold dilution of compounds in PBS for 30 minutes at 37 °C, and an equal volume of chicken red blood cells with citric acid-sodium citrate (pH 5.0) was added. After incubation at 37 °C for 30 minutes, the mixture was centrifuged (400×g, 8 min, 4 °C). Then, 300 μL of the supernatant was transferred to a 96-well plate, and the absorbance was measured at 540 nm with a Thermo Multiskan MK3 Spectrum spectrophotometer. The value of the mock-infected samples served as the background. IC50 was expressed as the compound concentration that caused 50% inhibition of hemolysis.
Supplementary Material
ACKNOWLEDGMENT
We are grateful for the support of this work by National Natural Science Foundation of China (81302648 to X.Z.; 81761128014 to Zifeng Y.), the State Scholarship Fund of China Scholarship Council (201604910441 to X.Z.), the Youth Innovation Promotion Association CAS (2016319 to X.Z.), the Pearl River S&T Nova Program of Guangzhou (201806010115 to X.Z.), the Ministry of Science and Technology of China (2015DFM30010 to Zifeng Y.), Science research project of the Guangdong Province (2016A050503047 to Zifeng Y.), the Science and Technology Development Fund in Macao Special Administrative Region (084/2015/A to Zifeng Y.), Start-up grant from Guangzhou Medical University (to W.H.), the grant of State Key Laboratory of Respiratory Disease (SKLRD2016ZJ0002 to W.H.). Computer time for this work was awarded by a grant from the Swedish Infrastructure Committee (SNIC) for the project “Modeling of protein-ligand binding” (to Y.T.) and the State Scholarship Fund of China Scholarship Council (201600160013 to Y.Z.). CKP. M. thanks support from Health and Medical Research Fund of Hong Kong (Grant no.17160792). J. W. thanks support from the NIH AI119187.
ABBREVIATIONS
- IAV
influenza A viruses
- HA
hemagglutinin
- NA
neuraminidase
- HPAI
highly pathogenic avian influenza
- MOA
mechanism of action
- SAR
structure-activity relationship
- CPE
cytopathic effect
- PRA
plaque reduction assay
- TEVC
two-electrode voltage clamp
- TOA
time-of-addition
- SA
sialic acid
- HI
hemagglutination inhibition
- MD
molecular dynamics
- PME
particle-mesh Ewald
- RMSD
root-mean-square deviation
- RBC
red blood cells
Footnotes
ASSOCIATED CONTENT
Supporting Information.
The Supporting Information is available free of charge on the ACS Publications website
Details regarding inhibitor synthesis, characterization and 1H NMR and 13CNMR spectra of new compounds (PDF)
Molecular formula strings (CSV)
The authors declare no competing financial interest.
REFERENCES
- (1).Bouvier N; Palese P The biology of influenza viruses. Vaccine, 2008, 26, D49–D53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Salomon R; Webster RG The influenza virus enigma. Cell 2009, 136, 402–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Munster VJ; Wallensten A; Baas C; Rimmelzwaan GF; Schutten M; Olsen B; Osterhaus AD; Fouchier RA Mallards and highly pathogenic avian influenza ancestral viruses, northern Europe. Emerg. Infect. Dis. 2005, 11, 1545–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Mao C; Wu XY; Fu XH; Di MY; Yu YY; Yuan JQ; Yang ZY; Tang JL An internet-based epidemiological investigation of the outbreak of H7N9 avian influenza A in China since early 2013. J. Med. Internet Res. 2014, 16, e221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).https://www.cdc.gov/h1n1flu. Aug 11, 2010.
- (6).Yang ZF; Mok CK; Peiris JS; Zhong NS Human infection with a novel avian influenza A(H5N6) virus. N. Engl. J. Med. 2015, 373, 487–489. [DOI] [PubMed] [Google Scholar]
- (7).Krammer F; Palese P Advances in the development of influenza virus vaccines. Nat. Rev. Drug Discov. 2015, 14, 167–182. [DOI] [PubMed] [Google Scholar]
- (8).De Clercq E Antiviral agents active against influenza A viruses. Nat. Rev. Drug Discov. 2006, 5, 1015–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Bright RA; Shay DK; Shu B; Cox NJ; Klimov AI Adamantane resistance among influenza A viruses isolated early during the 2005–2006 influenza season in the United States. J. Am Med. Assoc. 2006, 295, 891–894. [DOI] [PubMed] [Google Scholar]
- (10).Wan XF; Carrel M; Long LP; Alker AP; Emch M Perspective on emergence and re-emergence of amantadine resistant influenza A viruses in domestic animals in China. Infect. Genet. Evol. 2013, 20, 298–303. [DOI] [PubMed] [Google Scholar]
- (11).Bashashati M; Vasfi Marandi M.; Sabouri F Genetic diversity of early (1998) and recent (2010) avian influenza H9N2 virus strains isolated from poultry in Iran. Arch. Virol. 2013, 158, 2089–2100. [DOI] [PubMed] [Google Scholar]
- (12).Govorkova EA; Baranovich T; Seiler P; Armstrong J; Burnham A; Guan Y; Peiris M; Webby RJ; Webster RG Antiviral resistance among highly pathogenic influenza A (H5N1) viruses isolated worldwide in 2002–2012 shows need for continued monitoring. Antiviral Res. 2013, 98, 297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Salter A; Ni Laoi B.; Crowley B Emergence and phylogenetic analysis of amantadine- resistant influenza a subtype H3N2 viruses in Dublin, Ireland, over six seasons from 2003/2004 to 2008/2009. Intervirology 2011, 54, 305–315. [DOI] [PubMed] [Google Scholar]
- (14).Baz M; Abed Y; Papenburg J; Bouhy X; Hamelin ME; Boivin G Emergence of oseltamivir-resistant pandemic H1N1 virus during prophylaxis. N. Engl. J. Med. 2009, 361, 2296–2297. [DOI] [PubMed] [Google Scholar]
- (15).Marjuki H; Mishin VP; Chesnokov AP; Jones J; De La Cruz. JA; Sleeman K; Tamura D; Nguyen HT; Wu HS; Chang FY; Liu MT; Fry AM; Cox NJ; Villanueva JM; Davis CT; Gubareva LV Characterization of drug-resistant influenza A(H7N9) variants isolated from an oseltamivir-treated patient in Taiwan. J. Infect. Dis. 2015, 211, 249–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Pizzorno A; Abed Y; Plante PL; Carbonneau J; Baz M; Hamelin ME; Corbeil J; Boivin G Evolution of oseltamivir resistance mutations in influenza A(H1N1) and A(H3N2) viruses during selection in experimentally-infected mice. Antimicrob. Agents Chemother. 2014, 58, 6398–6405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Furuse Y; Suzuki A; Oshitani H Large-scale sequence analysis of M gene of influenza A viruses from different species: mechanisms for emergence and spread of amantadine resistance. Antimicrob. Agents Chemother. 2009, 53, 4457–4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Wang J; Ma C; Fiorin G; Carnevale V; Wang T Hu F; Lamb RA; Pinto LH; Hong M; Klein ML; DeGrado WF Molecular dynamics simulation directed rational design of inhibitors targeting drug-resistant mutants of influenza A virus M2. J. Am. Chem. Soc. 2011, 133, 12834–12841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Zhao X; Jie Y; Rosenberg MR; Wan J; Zeng S; Cui W; Xiao Y; Li Z; Tu Z; Casarotto MG; Hu W Design and synthesis of pinanamine derivatives as anti-influenza A M2 ion channel inhibitors. Antiviral Res, 2012, 96, 91–99. [DOI] [PubMed] [Google Scholar]
- (20).Rey-Carrizo M; Torres E; Ma C; Barniol-Xicota M; Wang J; Wu Y; Naesens L; DeGrado WF; Lamb RA; Pinto LH; Vázquez S 3-Azatetracyclo [5.2.1.15,8.01,5] undecane derivatives: from wild-type inhibitors of the M2 ion channel of influenza A virus to derivatives with potent activity against the V27A mutant. J. Med. Chem. 2013, 56, 9265–9274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Wang J; Ma C; Wang J; Jo H; Canturk B; Fiorin G; Pinto LH; Lamb RA; Klein ML; DeGrado WF Discovery of novel dual inhibitors of the wild-type and the most prevalent drug-resistant mutant, S31N, of the M2 proton channel from influenza A virus. J. Med. Chem. 2013, 56, 2804–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Wang J; Wu Y; Ma C; Fiorin G; Wang J; Pinto LH; Lamb RA; Klein ML; DeGrado WF Structure and inhibition of the drug-resistant S31N mutant of the M2 ion channel of influenza A virus. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 1315–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Rey-Carrizo M; Barniol-Xicota M; Ma C; Frigolé-Vivas M; Torres E; Naesens L; Llabrés S; Juárez-Jiménez J; Luque FJ; DeGrado WF; Lamb RA; Pinto LH; Vázquez S Easily accessible polycyclic amines that inhibit the wild-type and amantadine- resistant mutants of the M2 channel of influenza A virus. J. Med. Chem. 2014, 57, 5738–5747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Barniol-Xicota M; Gazzarrini S; Torres E; Hu Y; Wang J; Naesens L; Moroni A; Vázquez S Slow but steady wins the race: dissimilarities among new dual inhibitors of the wild-type and the V27A mutatn M2 channels of influenza A virus. J. Med. Chem. 2017, 60, 3727–3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Hu Y; Wang Y; Fang L; Ma C, Wang J Design and expeditious synthesis of organosilanes as potent antivirals targeting multidrug-resistant influenza A viruses. Eur. J. Med. Chem. 2017, 135, 70–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Li F; Hu Y; Wang Y; Ma C; Wang J Expeditious lead optimization of isoxazole- containing influenza A virus M2-S31N inhibitors using the Suzuki-Miyaura cross-coupling reaction. J. Med. Chem. 2017, 60, 1580–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Li F; Ma C; Hu Y; Wang Y; Wang J Discovery of potent antivirals against amantadine-resistant influenza A viruses by targeting the M2-S31N proton channel. ACS Infect. Dis. 2016, 2, 726–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Li F; Ma C; DeGrado WF; Wang J Discovery of highly potent inhibitors targeting the predominant drug-resistant S31N mutant of the influenza A virus M2 proton channel. J. Med. Chem. 2016, 59, 1207–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Torres E; Duque MD; Vanderlinden E; Ma C; Pinto LH; Camps P; Froeyen M; Vázquez S; Naesens L Role of the viral hemagglutinin in the anti-influenza virus activity of newly synthesized polycyclic amine compounds. Antiviral Res, 2013, 99, 281–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Torres E; Leiva R; Gazzarrini S; Rey-Carrizo M; Frigolé-Vivas M; Moroni A; Naesens L; Vázquez S Azapropellanes with anti-influenza A virus activity. ACS Med. Chem. Lett, 2014, 5, 831–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kolocouris A; Tzitzoglaki C; Johnson FB; Zell R; Wright AK; Cross TA; Tietjen I; Fedida D; Busath DD Aminoadamantanes with persistent in vitro efficacy against H1N1 (2009) influenza A. J. Med. Chem, 2014, 57, 4629–4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Walsh DP; Chang YT Chemical genetics. Chem. Rev. 2006, 106, 2476–2530. [DOI] [PubMed] [Google Scholar]
- (33).Hu W; Zeng S; Li C; Jie Y; Li Z; Chen L Identification of hits as matrix-2 protein inhibitors through the focused screening of a small primary amine library. J. Med. Chem. 2010, 53, 3831–3834. [DOI] [PubMed] [Google Scholar]
- (34).Zhao X; Li C; Zeng S; Hu W Discovery of highly potent agents against influenza A virus. Eur. J. Med. Chem. 2011, 46, 52–57. [DOI] [PubMed] [Google Scholar]
- (35).Dong J; Chen S; Li R; Cui W; Jiang H; Ling Y; Yang Z Hu, W. Imidazole-based pinanamine derivatives: discovery of dual inhibitors of the wild-type and drug-resistant mutant of the influenza A virus. Eur. J. Med. Chem. 2016, 108, 605–615. [DOI] [PubMed] [Google Scholar]
- (36).Yang ZF; Zhao J; Zhu YT; Wang YT; Liu R; Zhao SS; Li RF; Yang CG; Li JQ; Zhong NS The tree shrew provides a useful alternative model for the study of influenza H1N1 virus. Virol. J 2013, 10, 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Li S; Chiu G; Pulito VL; Liu J; Connolly P, J.; Middleton, S. A. 1- Arylpiperazinyl-4-cyclohexylamine derived isoindole-1,3-diones as potent and selective a- 1a/1d adrenergic receptor ligands. Bioorg. Med. Chem. Lett. 2007, 17, 1646–1650. [DOI] [PubMed] [Google Scholar]
- (38).Hölzer B; Hoffmann RW Kumada-Corriu coupling of Grignard reagents, probed with a chiral Grignard reagent. Chem. Commun. 2003, 0, 732–733. [DOI] [PubMed] [Google Scholar]
- (39).Hays DS; Detty MR Studies towards alkylthiophene-2-carboxaldehydes. reduction of 3-alkenylthiophenes with triethylsilane/trifluoroacetic acid. regioselectivity in formylation reactions of 3-alkylthiophenes. Heterocycles 1995, 40, 925–937. [Google Scholar]
- (40).Fang GH; Yan ZJ; Deng MZ Palladium-catalyzed cross-coupling of stereospecific potassium cyclopropyl trifluoroborates with aryl bromides. Org. Lett, 2004, 6, 357–360. [DOI] [PubMed] [Google Scholar]
- (41).Molander GA; Brown AR Suzuki-Miyaura cross-coupling reactions of potassium vinyltrifluoroborate with aryl and heteroaryl electrophiles. J. Org. Chem. 2016, 71, 9681–9686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Ma C; Zhang J; Wang J Pharmacological characterization of the spectrum of antiviral activity and genetic barrier to drug resistance of M2-S31N channel blockers. Mol. Pharmacol. 2016, 90, 188–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Wang J; Cady SD; Balannik V; Pinto LH; DeGrado WF; Hong M Discovery of spiro-piperidine inhibitors and their modulation of the dynamics of the M2 proton channel from influenza A virus. J. Am. Chem. Soc. 2009, 131, 8066–8076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Zhao X; Zhang ZW; Cui W; Chen S; Zhou Y; Dong J; Jie Y; Wan J; Xu Y; Hu W Identification of camphor derivatives as novel M2 ion channel inhibitors of influenza A virus. Med. Chem. Commun. 2015, 6, 727–731. [Google Scholar]
- (45).Shapiro GI; Gurney TJ; Krug RM Influenza virus gene expression: control mechanisms at early and late times of infection and nuclear-cytoplasmic transport of virus- specific RNAs. J. Virol. 1987, 61, 764–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Daelemans D; Pauwels R; De Clercq E; Pannecouque C A time-of-drug addition approach to target identification of antiviral compounds. Nat. Protoc, 2011, 6, 925–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Dimitrov DS Virus entry: molecular mechanisms and biomedical applications. Nature Rev. Microbiol. 2004, 2, 109–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Du J; Cross TA; Zhou HX Recent progress in structure-based anti-influenza drug design. Drug Discov. Today. 2012, 17, 1111–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Yang J; Li M; Shen X; Liu S Influenza A virus entry inhibitors targeting the hemagglutinin. Viruses 2013, 5, 352–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Sriwilaijaroen N; Suzuki Y Molecular basis of the structure and function of H1 hemagglutinin of influenza virus. Proc. Jpn. Acad. Ser. B. 2012, 88, 226–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Pedersen JC Hemagglutination-inhibition test for avian influenza virus subtype identification and the detection and quantitation of serum antibodies to the avian influenza virus. Methods Mol. Biol. 2008, 436, 53–66. [DOI] [PubMed] [Google Scholar]
- (52).Yu M; Si L; Wang Y; Wu Y; Yu F; Jiao P; Shi Y; Wang H; Xiao S; Fu G; Tian K; Wang Y; Guo Z; Ye X; Zhang L; Zhou D Discovery of pentacyclic triterpenoids as potential entry inhibitors of influenza viruses. J. Med. Chem. 2014, 57, 10058–10071. [DOI] [PubMed] [Google Scholar]
- (53).https://www.fludb.org/brc/fluSegmentDetails.spg?ncbiProteinId=ADD92535&decorator=influenza&context=1527208477058. Apr 16, 2014. [Google Scholar]
- (54).Zhang W; Qi J; Shi Y; Li Q; Gao F; Sun Y; Lu X; Lu Q; Vavricka CJ; Liu D; Yan J; Gao GF Crystal structure of the swine-origin A (H1N1)-2009 influenza A virus hemagglutinin (HA) reveals similar antigenicity to that of the 1918 pandemic virus. Protein Cell 2010, 1, 459–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Halgren TA Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [DOI] [PubMed] [Google Scholar]
- (56).Friesner RA; Murphy RB; Repasky MP; Frye LL; Greenwood JR; Halgren TA; Sanschagrin PC; Mainz DT Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [DOI] [PubMed] [Google Scholar]
- (57).Kalani MR, Moradi A, Moradi M & Tajkhorshid E Characterizing a histidine switch controlling pH-dependent conformational changes of the influenza virus hemagglutinin. Biophys. J. 2013, 105, 993–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Leneva IA; Russell RJ; Boriskin YS; Hay AJ Characteristics of Arbidol- resistant mutants of influenza virus: implications for the mechanism of anti-influenza action of Arbidol. Antiviral Res. 2009, 81, 132–140. [DOI] [PubMed] [Google Scholar]
- (59).Tong S; Li Y; Rivailler P; Conrardy C; Castillo DA; Chen LM; Recuenco S; Ellison JA; Davis CT; York IA; Turmelle AS; Moran D; Rogers S; Shi M; Tao Y; Weil MR; Tang K; Rowe LA; Sammons S; Xu X; Frace M; Lindblade KA; Cox NJ; Anderson LJ; Rupprecht CE; Donis RO A distinct lineage of influenza A virus from bats. Proc. Natl. Acad. Sci. U S A, 2012, 109, 4269–4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Cianci C; Yu K-L; Dischino DD; Harte W; Deshpande M; Luo G; Colonno RJ; Meanwell NA; Krystal MJ pH-Dependent changes in photoaffinity labelling patterns of the H1 influenza virus hemagglutinin by using an inhibitor of viral fusion. Virology 1999, 73, 1785–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Zhu L; Li Y; Li S; Li H; Qiu Z; Lee C; Lu H; Lin X; Zhao R; Chen L; Wu J; Tang G; Yang W Inhibition of influenza A virus (H1N1) fusion by benzenesulfonamide derivatives targeting viral hemagglutinin. PLoS One 2011, 6, e29120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Tang G; Lin X; Qiu Z; Li W; Zhu L; Wang L; Li S; Li H; Lin W; Yang M; Guo T; Chen L; Lee D; Wu J; Yang W Design and synthesis of benzenesulfonamide derivatives as potent anti-influenza hemagglutinin inhibitors. ACS Med. Chem. Lett. 2011,2, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Plotch SJ; O’Hara B; Morin J; Palant O; LaRocque J; Bloom JD; Lang SA Jr.; DiGrandi MJ; Bradley M; Nilakantan R; Gluzman Y Inhibition of influenza A virus replication by compounds interfering with the fusogenic function of the viral hemagglutinin. J. Virology 1999, 73, 140–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Liu S; Li R; Zhang R; Chan CCS; Xi B; Zhu Z; Yang J; Poon VKM; Zhou J; Münch J; Kirchhoff F; Pleschka S; Haarmann T; Dietrich U; Pan C; Du L; Jiang S; Zheng B CL-385319 inhibits H5N1 avian influenza A virus infection by blocking viral entry. Eur. J. Pharmacol. 2011, 660, 460–467. [DOI] [PubMed] [Google Scholar]
- (65).Li R; Song D; Zhu Z; Xu H; Liu S An induced pocket for the binding of potent fusion inhibitor CL-385319 with H5N1 influenza virus hemagglutinin. PLoS One 2012, 7, e41956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Luo G; Colonno R; Krystal M Characterization of a hemagglutinin-specific inhibitor of influenza A virus. Virology 1996, 226, 66–76. [DOI] [PubMed] [Google Scholar]
- (67).Luo G; Torri A; Harte WE; Danetz S; Cianci C; Tiley L; Say S; Mullaney D; Yu K-L; Ouellet C; Dextraze P; Meanwell N; Colonno R; Kyrstal M Molecular mechanism underlying the action of a novel fusion inhibitor of influenza A virus. J. Virol. 1997, 71, 4062–4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Deshpande MS; Wei J; Luo G; Cianci C; Danetz S; Torri A; Tiley L; Krystal M; Yu K-L; Huang S; Gao Q; Meanwell NA An approach to the identification of potent inhibitors of influenza virus fusion using parallel synthesis methodology. Bioorg. Med. Chem. Lett. 2001, 11, 2393–2396. [DOI] [PubMed] [Google Scholar]
- (69).White KM; De Jesus P; Chen Z; Abreu P Jr.; Barile E; Mak PA; Anderson P; Nguyen QT; Inoue A; Stertz S; Koenig R; Pellecchia M; Palese P; Kuhen K; García-Sastre A; Chanda SK; Shaw ML Potent anti-influenza compound blocks fusion through stabilization of the prefusion conformation of the hemagglutinin protein. ACS Infect. Dis. 2015, 1, 98–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Basu A; Antanasijevic A; Wang M; Li B; Mills DM; Ames JA; Nash PJ; Williams JD; Peet NP; Moir DT; Prichard MN; Keith KA; Barnard DL; Caffrey M; Rong L; Bowlin TL New small molecule entry inhibitors targeting hemagglutinin-mediated influenza A virus fusion. J. Virol. 2014, 88, 1447–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Basu A; Komazin-Meredith G; McCarthy C; Antanasijevic A; Cardinale SC; Mishra RK; Barnard DL; Caffrey M; Rong L; Bowlin TL Molecular mechanism underlying the action of the influenza A virus fusion inhibitor MBX2546. ACS Infect. Dis. 2017, 3, 330–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Kadam RU; Juraszek J; Brandenburg B; Buyck C; Schepens WBG; Kesteleyn B; Stoops B; Vreeken RJ; Vermond J Goutier W; Tang C Vogels R; Friesen RHE; Goudsmit J; van Dongen MJP; Wilson IA Potent peptidic fusion inhibitors of influenza virus. Science 2017, 358, 496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Bodian D; Yamasaki R; Buswell R; Stearns J; White J; Kuntz I Inhibition of the fusion-inducing conformational change of influenza hemagglutinin by benzoquinones and hydroquinones. Biochemistry 1993, 32, 2967–2978. [DOI] [PubMed] [Google Scholar]
- (74).Russell RJ; Kerry PS; Stevens DJ; Steinhauer DA; Martin SR; Gamblin SJ; Skehel JJ Structure of influenza hemagglutinin in complex with an inhibitor of membrane fusion. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 17736–17741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Antanasijevic A; Hafeman NJ; Tundup S; Kingsley C; Mishra RK; Rong L; Manicassamy B; Wardrop D; Caffrey M Stabilization and improvement of a promising influenza antiviral: making a PAIN PAINless. ACS Infect. Dis. 2016, 2, 608–615. [DOI] [PubMed] [Google Scholar]
- (76).Vanderlinden E; Goktas F; Cesur Z; Froeyen M; Reed ML; Russell CJ; Cesur N; Naesens L Novel inhibitors of influenza virus fusion: structure-activity relationship and interaction with the viral hemagglutinin. J. Virol. 2010, 84, 4277–4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Kadam RU; Wilson IA Structural basis of influenza virus fusion inhibition by the antiviral drug Arbidol. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Leiva R; Barniol-Xicota M; Codony S; Ginex T Vanderlinden, E. Montes, M.; Caffrey, M.; Javier Luque, F.; Naesens, L. Vázquez, S. Aniline-based inhibitors of influenza H1N1 virus acting on hemagglutinin-mediated fusion. J. Med. Chem. 2018, 61, 98–118. [DOI] [PubMed] [Google Scholar]
- (79).Li F; Ma C; Wang J Inhibitors targeting the influenza virus hemagglutinin. Curr. Med. Chem. 2015, 22, 1361–1382. [DOI] [PubMed] [Google Scholar]
- (80).Balannik V; Lamb RA; Pinto LH The oligomeric state of the active BM2 ion channel protein of influenza B virus. J. Biol. Chem. 2008, 283, 4895–4904. [DOI] [PubMed] [Google Scholar]
- (81).Sastry GM; Adzhigirey M; Day T; Annabhimoju R; Sherman W Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided. Mol. Des. 2013, 27, 221–234. [DOI] [PubMed] [Google Scholar]
- (82).Abraham MJ; Murtola T; Schulz R; Pall S; Smith JC Hess B Lindahl E Gromacs: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar]
- (83).Wickstrom L; Okur A; Simmerling C Evaluating the performance of the FF99SB force field based on NMR scalar coupling data. Biophys. J. 2009, 97, 853–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Wang J; Wang W; Kollman PA; Case DA Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [DOI] [PubMed] [Google Scholar]
- (85).Price DJ; Brooks CL A modified TIP3P water potential for simulation with Ewald summation. J. Chem. Phys. 2004, 121, 10096–10103. [DOI] [PubMed] [Google Scholar]
- (86).Hess B; Bekker H; Berendsen HJC; Fraaije JGEM LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar]
- (87).Essmann U; Perera L; Berkowitz ML Darden, T.; Lee, H.; Pedersen, L. G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.