

Abstract
The consequences of immune dysfunction in B-Chronic Lymphocytic Leukemia (CLL) likely relate to the incidence of serious recurrent infections and second malignancies that plague CLL patients. The well-described immune abnormalities are not able to consistently explain these complications. Here, we report bone marrow (BM) hematopoietic dysfunction in early and late stage untreated CLL patients. Numbers of CD34+ BM hematopoietic progenitors responsive in standard CFU assays, including CFU-GM/GEMM and CFU-E, were significantly reduced. Flow cytometry revealed corresponding reductions in frequencies of all hematopoietic stem and progenitor cell (HSPC) subsets assessed in CLL patient marrow. Consistent with the reduction in HSPCs, BM resident monocytes and natural killer (NK) cells were reduced, a deficiency recapitulated in blood. Finally, we report increases in protein levels of the transcriptional regulators HIF-1α GATA-1, PU.1, and GATA-2 in CLL patient BM, providing molecular insight into the basis of HSPC dysfunction. Importantly, PU.1 and GATA-2 were rapidly increased when healthy HSPCs were exposed in vitro to TNFα, a cytokine constitutively produced by CLL B cells. Together, these findings reveal BM hematopoietic dysfunction in untreated CLL patients that provides new insight into the etiology of the complex immunodeficiency state in CLL.
Introduction
Individuals with CLL are highly susceptible to infections and secondary cancers, owing to diminished innate and adaptive immune repertoires and function1. Peripheral immune suppression, either as a consequence of progressive disease or secondary to cytotoxic therapy, could account for much of the immune incompetence, but changes in bone marrow (BM)-derived hematopoietic cells that replenish them has not been sufficiently studied. This is especially the case for the hematopoietic status of untreated CLL (herein referred to as “CLL”) patients. One study reported no change in frequencies of CD34+ cells among nucleated BM cells between CLL patients and controls, but numbers of colony forming progenitors (CFU) were reduced2. However, this study did not directly evaluate the clonogenic potential of isolated CD34+ BM progenitors or take into account leukemic burden levels in the BM when assaying CFU-responding cells. Thus, the reduction in CFU could reflect dilution of marrow by infiltrating CLL B cells. In contrast, another study reported significant reductions in frequencies of BM CD34+ cells in untreated CLL patients3. Importantly, in that study, anemic CLL patients had reduced proportions of phenotypically-defined BM erythroid progenitors, a deficiency suggested to be due to increased levels of TNFα, and independent of the degree of BM leukemic B cell infiltration. These conflicting results support additional studies to determine the status of BM hematopoietic function and its contribution to the complex immune deficiency well documented in CLL patients.
Numerous factors have been described to modulate normal hematopoiesis in the setting of malignancies, but few have been discussed in the context of hematopoietic stem and progenitor cells (HSPCs). Accumulating evidence suggests that key factors of hematopoietic regulation may play a role in modulating hematopoiesis in CLL. Levels of the transcription factor hypoxia-inducible factor 1-alpha (HIF-1α) are tightly regulated in hematopoietic stem cells (HSCs), control cell division4, 5, and play a key role in regulation of hematopoietic differentiation6, 7. HIF-1α is implicated in the biology of many malignancies8, including CLL9–11. Importantly, HIF-1α contributes to CLL B cell homing and survival by modulating VEGF and CXCR4 production in CLL B cells11 and VEGF in BM mesenchymal stromal cells9. A prominent HIF-1α target is GATA-1, a master erythroid lineage determinant6. Importantly, dysregulated, sustained expression of GATA-1 can inhibit erythroid differentiation12, 13. BM infiltration of CLL cells, which constitutively produce TNFα14, 15, may further inhibit erythropoiesis by introducing TNFα to the hematopoietic niche and altering the balance between transcription factors expressed in HSPCs, particularly PU.1 and GATA-2. Cumulatively, based on these published findings and with the goal of better understanding the etiology of immune dysfunction in CLL, we assessed BM HSPC functional capacity and performed an in-depth characterization of hematopoietic progenitor subsets in untreated CLL patients. We further set out to determine if the innate immune repertoire in blood reflected BM hematopoietic dysfunction and elucidate contributing molecular mechanism(s) for impaired myeloid and erythroid differentiation.
Methods
Patients
Aged-matched healthy control (HC) patients undergoing hip replacement (Mayo Clinic IRB 1062–00), healthy apheresis blood donors (IRB exempt), and CLL patients (Mayo Clinic IRB 1827–00) (Table 1, Table S1) signed informed consent per the Declaration of Helsinki and Mayo Clinic guidelines and provided BM and/or blood samples. The BM and blood were provided as either freshly obtained samples or previously cryopreserved by the Mayo Clinic CLL Tissue Bank. Freshly obtained HC and CLL marrow was subject to ACK lysis to remove mature erythrocytes and utilized immediately for the described studies. All cryopreserved HC marrow was previously ACK-lysed, whereas cryopreserved CLL blood and marrow was previously subject to ficoll separation. Healthy control cryopreserved blood samples underwent ficoll separation before freezing. Table 1 identifies fresh and frozen samples. All comparisons of cell population frequencies were made between samples that were handled identically. We recognize that cryopreserved samples are exposed to high levels of oxygen during thawing that may alter transcription factor expression. To minimize these effects, cells were used immediately after thawing, kept on ice, and equivalent numbers of frozen and fresh samples were analyzed in both the HC and CLL cohorts.
Table 1.
CLL patient clinical and prognostic features.
| Untreated CLL Patients | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Patient Number |
Age | Sex | Rai Stage |
Cytogenetics | ZAP-70 | CD38 | CD49d | IGHV | Leukemic marrow involvement (%)- hematopathology |
Leukemic marrow involvement(%)- Flow Cytometry |
| 1 | 70 | M | I | 11 q- | + | - | - | UM | 90 | 86.17%# |
| 2 | 62 | M | 0 | 17p- | - | + | + | M | 90 | 83.28%# |
| 3 | 42 | F | II | Trisomy 12 | + | - | + | UM | 30 | 61.99%# |
| 4 | 86 | M | 0 | 11 q- | + | + | - | UM | 90 | 85.72%# |
| 5 | 78 | F | III | 13q- | - | - | - | N/A | 30 | 30.85%# |
| 6 | 82 | M | I | Trisomy 12 | + | + | + | UM | 80 | 66.05%# |
| 7 | 78 | M | I | 11 q- | - | - | + | UM | 60 | 65.01%# |
| 8 | 72 | F | IV | 13q- | - | - | - | M | 80 | 47.62%# |
| 9 | 72 | M | I | 13q- | - | - | + | M | 90 | 94.95%# |
| 10 | 68 | M | IV | 13q- | - | - | + | M | 95 | 59.72%# |
| 11 | 56 | M | IV | 13q- | + | + | - | UM | 70 | 77.93%# |
| 12 | 75 | M | I | 13q- | - | - | M | 2 | 31.17%# | |
| 13 | 68 | M | II | 13q- | - | - | UM | 80 | 24.15%# | |
| 14 | 73 | F | IV | 13q- | - | - | M | 80 | 90.93%# | |
| 15 | 59 | M | 0 | Trisomy 12 | - | + | M | 75 | 48.64%# | |
| 16 | 80 | M | IV | 13q- | + | - | UM | 75 | 69.36%# | |
| 17 | 69 | M | I | Normal | - | + | UM | 80 | 62.49%# | |
| 18 | 68 | M | I | Normal | + | - | UM | 80 | 92.00%# | |
| 19 | 75 | M | 0 | 13q- | - | + | M | 80 | 94.40%# | |
| 20 | 55 | M | IV | 13q- | - | - | M | 50 | 71.69%# | |
| 21 | 59 | M | III | Trisomy 12 | + | + | + | UM | 95 | 94.00%# |
| 22 | 60 | F | II | N/A | + | + | + | UM | 80 | 27.46%# |
| 23 | 47 | M | I | 13q- | + | + | - | UM | 80 | 80.16%# |
| 24 | 38 | M | I | Normal | + | - | + | M | 20 | 45.72%# |
| 25 | 72 | M | I | 13q- | - | - | + | M | 90 | 77.05%* |
| 26 | 67 | F | II | Normal | - | - | + | M | 90 | 75.14%* |
| 27 | 64 | F | IV | 17p- | + | - | - | UM | 100 | 82.14%* |
| 28 | 68 | M | I | Other | + | + | - | UM | 30 | 87.62%* |
| 29 | 59 | M | II | 17p- | + | + | + | UM | 95 | 94.58%* |
| 30 | 75 | M | 0 | Normal | - | + | + | M | 60 | 67.77%* |
| 31 | 61 | M | I | 11 q- | - | - | UM | 60 | 91.54%* | |
| 32 | 52 | F | I | Other | - | - | UM | 60 | 86.56%* | |
| 33 | 66 | M | IV | Trisomy 12 | + | + | UM | >90 | 88.73%# | |
| 34 | 60 | M | I | 11 q- | + | - | UM | 20 | 68.66% * | |
| 35 | 64 | M | 0 | 11 q- | N/A | - | UM | 20 | 60.88%* | |
| 36 | 53 | M | I | 13q- | - | M | 20 | 47.11%* | ||
| 37 | 86 | M | I | 17p- | - | UM | 60 | 93.58% * | ||
| 38 | 73 | M | I | Normal | + | - | UM | 90 | 86.22% * | |
| 39 | 58 | F | III | 13q- | - | - | M | >90 | 91.36%* | |
| 40 | 60 | M | IV | Normal | - | + | M | >90 | 72.09% * | |
| 41 | 52 | F | I | Normal | + | - | - | UM | 95 | 92.66% * |
| 42 | 75 | F | IV | 11 q- | - | - | - | UM | 90 | 93.05% * |
| 43 | 68 | F | I | 13q- | - | - | - | M | 10 | 13.68%# |
| 44 | 80 | M | I | Trisomy 12 | + | - | + | UM | 90 | 86.29%# |
| 45 | 68 | M | I | 11 q- | - | - | - | UM | 90 | 80.34%# |
| 46 | 58 | M | I | N/A | + | - | - | UM | 90 | 73.12%# |
Each CLL patient’s information is indicated as well as clinically relevant prognostic features including ZAP-70, CD38, and CD49d expression where available, mutational status of the immunoglobulin heavy chain variable region (IGHV), and CLL marrow involvement. 11q deletion (11q-), 17p deletion (17p-), 13q deletion (13q-), prognostic factor positive (+), prognostic factor negative (−), unmutated IGHV (UM), mutated IGHV (M), and not available (N/A). Notation of (#) indicates that the sample was used fresh following ACK lysis whereas (*) designates use of a previously ficolled and cryopreserved sample. Additional patient information can be found in Table S1.
Flow cytometry
The erythrocyte-depleted BM and blood cells were stained with monoclonal antibodies against surface and/or intranuclear proteins (Table S2). The fixable viability dye and surface markers were stained in 1X PBS at 4oC whereas the intranuclear proteins were stained in each respective fixation and permeabilization buffer at 4oC or room temperature, per manufacturer’s instructions. After staining, all samples were fixed in 1% paraformaldehyde (Electron Microscopy Sciences) for analysis. Flow cytometry data was collected on a FACSCanto (Beckton Dickinson) that was standardized to allow direct mean fluorescence intensity (MFI) comparisons across experiments using a modified protocol outlined by Perfetto et al.16, 17. Analysis of cellular populations (defined in Table S3) was performed with FlowJo 10.3 (Tree Star).
Colony forming unit (CFU) assays
Total CD34+ cells were isolated from freshly obtained BM samples (Miltenyi Biotec). Following isolation, 500 CD34+ cells were plated in triplicate in semisolid Methocult H4435 (StemCell Technologies) media that allows enumeration of CFU-GM, CFU-GEMM, and CFU-E based on morphology. The assays were incubated for 11–12 days at 37oC 5%CO2. Colonies were manually counted, scored, and averaged across triplicates.
TNFα culture assays
Isolated CD34+ cells from freshly obtained HC BM were incubated in serum-free media (CellGenix) plus the human recombinant proteins stem cell factor (10ng/mL), IL-6 (10ng/mL), erythropoietin (1ng/mL), and IL-3 (1ng/mL). Cells were further supplemented with 25ng/mL TNFα (Peprotech), a concentration within the experimental range of previous reports known to modify hematopoietic cells18, 19, or media alone for 12 or 24 hours, followed by flow cytometry.
Statistics
Statistical analysis was performed with GraphPad Prism 6. Mann-Whitney U-tests were applied to all comparisons between HC and CLL cohorts. The TNFα experiments utilized a paired t-test, and all correlations analyses were determined with a two-tailed Spearman correlation. For all tests; *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, and no asterisk or “N.S.” indicates non-statistical significance. Sample numbers were of limited availability, so a sample size power calculation was not performed. Summary statistics of bar graphs represent the mean ± the standard error of the mean (SEM). Given that, the variance between groups compared are similar. The sample and experiment replicate number for each figure is reported in each appropriate figure legend.
Results
BM infiltration by CLL B cells and equalization of the BM cellular compartment by CD19 exclusion
Our analysis focused on 46 untreated CLL patients with no clinically detectable autoimmune disease (Table 1, Table S1) that represent the clinical spectrum from early to late Rai stages (0-I: 61%, II: 11%, III-IV: 28%), a relatively even distribution of relevant prognostic indicators, and that reproduce the sex bias of males in CLL disease (34/46, 73.9%). To evaluate BM hematopoiesis, we first used flow cytometry to characterize the frequency of BM progenitor subsets in CLL patients and age-matched healthy controls (HCs). All CLL patients, regardless of Rai stage, had varying BM infiltration of CD19+CD5+ B cells as determined by both clinical pathology scores and flow cytometry (Figure 1A, Table 1, representative flow cytometry plots in Figure 1B). Importantly, and in relation to analysis, we recognized that the infiltration introduces a proportional calculation bias by the diluting CLL cells when assessing cell frequency as a function of total BM cells. To assess the infiltration bias, we evaluated the frequency of CD19- BM cells, which contain the hematopoietic cell populations of interest. As expected, CLL patient BM had significantly less CD19- cells compared to HCs (P<0.0001, HC mean ± SEM: 74.2 ± 2.8, CLL mean ± SEM: 25.8 ± 3.9). This outcome prohibits comparative analysis of BM non-B/leukemic cell populations to be assessed between HCs and CLL patients. To address this, we sought to evaluate the BM cellular content between the CLL and HC cohorts before and after CD19 exclusion (Figure 1C, left and right columns). Interestingly, the frequency of the remaining, CD19-excluded BM cells (our base comparator for all BM cell frequencies analyzed) was comparable between the two cohorts, as exemplified by the equalization of polymorphonuclear leukocyte (PMN) frequencies before and after CD19 exclusion (Figure 1D, before: P<0.0001, after: P=0.0537). Next, we compared the remaining cells in the BM of HC and CLL patients after CD19-exclusion (Figure S1). We found the majority of remaining BM populations were PMNs and T cells, the latter of which is expanded in CLL BM compared to HCs. Thus, CD19 exclusion allows for comparisons of BM cell frequencies between the two cohorts. Importantly, all BM progenitor populations were evaluated as either a percentage of lineage (Lin) marker negative BM cells, which included an antibody to CD19, or total CD19-excluded BM cells (Table S3).
Figure 1. Equalization of CLL patient and HC BM cellular compartments by CD19-exclusion.
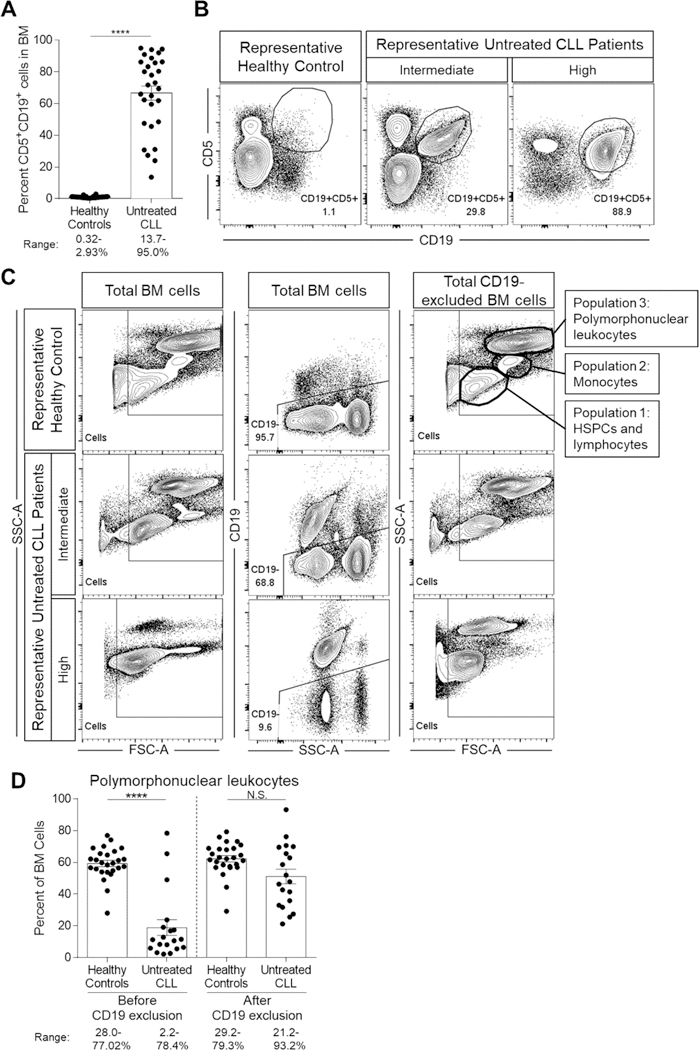
Freshly isolated ACK-lysed BM cells from CLL patients and HCs were evaluated by flow cytometry for total cellular composition and frequencies of CD19+CD5+ and CD19- cells. (A) Frequencies of CD19+CD5+ cells in CLL patient BM samples compared to HCs. HC n=34 and CLL n=29 (60 individual experiments). (B) Representative flow cytometry plots exhibiting variable CLL B cell infiltration in BM compared to a representative HC example. (C) Representative flow cytometry plots demonstrating BM cellular profiles before (left column) and after (right column) CD19-exclusion (middle column). Exclusion of all CD19+ cells, including the infiltrating CLL B cells, allows for equalization of the remaining (CD19-) BM cells. The post-CD19 exclusion HC flow cytometry plot (top right) illustrates three key cellular populations that can be identified by distinct FSC-A and SSC-A characteristics: 1) lymphocytes and HSPCs, 2) monocytes, and 3) polymorphonuclear leukocytes and granulocytes. (D) Evaluation of polymorphonuclear leukocyte frequencies in BM before and after CD19 exclusion. HC n=26 and CLL n=19 (40 individual experiments). Data in (A) and (D) is represented as the mean and SEM with each point indicating an individual BM sample. Numbers under each figure represent the range for each parameter evaluated for HCs and CLL patients. N.S. = not significant and ****P<0.0001 by Mann-Whitney U-test.
Reductions in phenotypic BM hematopoietic progenitor subsets and functional CFU-responding cells
Flow cytometry analysis revealed significantly decreased proportions of total CD34+ HSPCs among Lin-CD19- BM cells in CLL patients compared to HCs (Figure 2A, Figure S2, P<0.0001). The CD34+ fraction of BM encompasses progenitors that give rise to colonies in standard CFU assays. We found significantly reduced total CFU-responding cells from CLL patient-derived CD34+ cells compared to HCs (Figure 2B, P=0.0034). We further conducted an enumeration of CFU-GM/GEMM and CFU-E colonies within the total CFU responding cells and found both significantly decreased in CLL patients (Figure 2C–D, P=0.0084 and P=0.0434, respectively). Importantly, the number of total CFU responding cells in CLL patient BM did not correlate with the level of BM leukemic burden (r=−0.4636, P=0.0699), and included CLL patients with low to high marrow involvement.
Figure 2. Reductions in BM HSPC frequency and functional clonogenic progenitors in CLL patients.
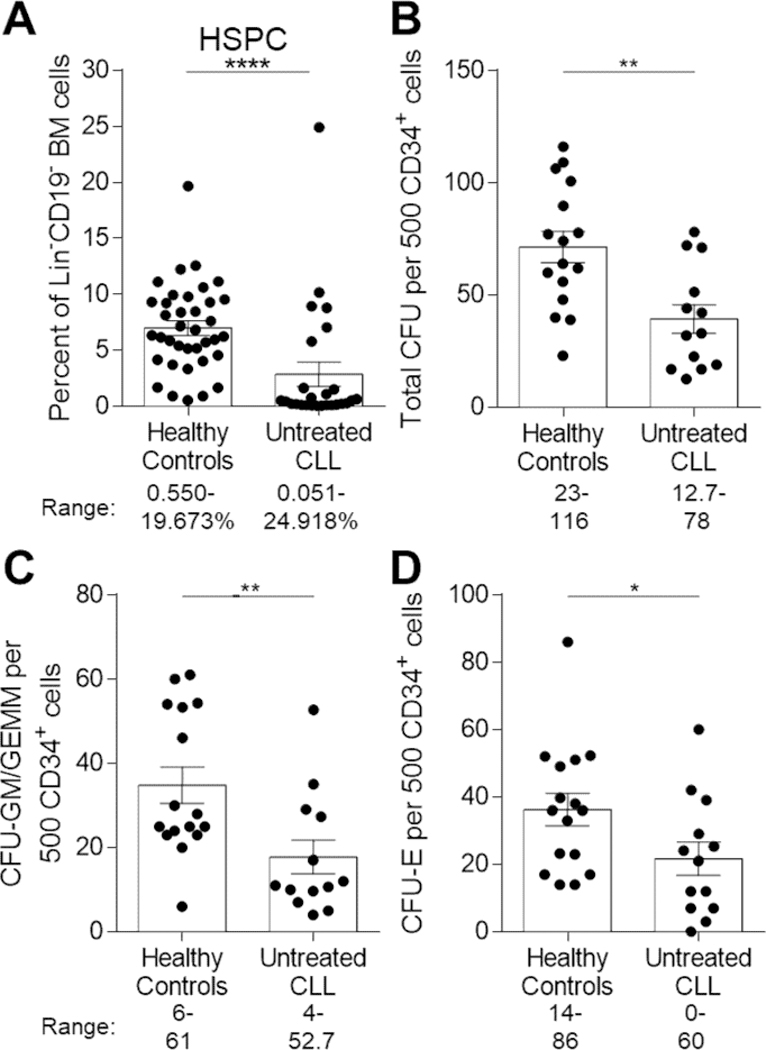
(A) Freshly isolated ACK-lysed BM cells were stained with combinations of antibodies to distinguish frequencies of Lin-CD19-CD34+ HSPCs between HCs (n=37) and CLL patients (n=26) (57 individual experiments). Data is represented as the mean and SEM with each point indicating an individual BM sample. Numbers under the figure represent the range for each cohort evaluated. (B-D) Numbers of (B) total CFU, (C) CFU-granulocyte, monocyte/granulocyte, erythrocyte, monocyte, megakaryocyte (CFU-GM/GEMM), or (D) CFU-erythroid (CFU-E) colonies per 500 input CD34+ cells. HC n=16 and CLL n=13 (24 individual experiments). Data is represented as the mean and SEM of triplicate platings with each point indicating an individual subject. Numbers under each figure represent the range for each colony evaluated for HCs and CLL patients *P<0.05, **P<0.01, and ****P<0.0001 were obtained by Mann-Whitney U-test.
To determine if reductions in CFUs were reflected in decreased myeloid and erythroid lineage progeny in vivo, we evaluated frequencies of CD33+CD14+ monocyte and CD33+CD71+ myelo-erythroid progenitors by flow cytometry (Figure 3A–B, Figure S3). Frequencies of monocyte (P=0.0089) and myelo-erythroid progenitors (P<0.0001) were significantly reduced in CLL BM compared to HCs. Interestingly, frequencies of PMNs (granulocytes, Figure 1D), a subset of differentiated progeny of CMPs, were not significantly different between HCs and CLL patients (P=0.0537). However, we recognize the variability within CLL patients is quite high and may reflect a subset of individuals lacking robust granulocyte generation. To determine if the reductions in monocyte/erythroid lineage cells was due to deficiencies in select HSPCs, we evaluated frequencies of HSCs, MPPs, and CMPs and found significant reductions in CLL BM (Figure 3C–E, HSC and MPP P<0.0001, CMP P=0.0019). The reduction in HSPCs was also reflected in frequencies of CLPs (Figure 3F, P<0.0001). Accompanying the reduction in CLPs, natural killer (NK) cell progenitors were reduced (Figure 3G, Figure S3, P=0.0023). These data suggest that while HSPCs are present at low levels, they are functionally compromised given reduced CFU numbers. The reduction in CFU-responding cells, even after removal from the tumor microenvironment, suggests a cell intrinsic alteration in a subset of CD34+ HSPCs.
Figure 3. BM hematopoietic progenitor frequencies are diminished in CLL patients.
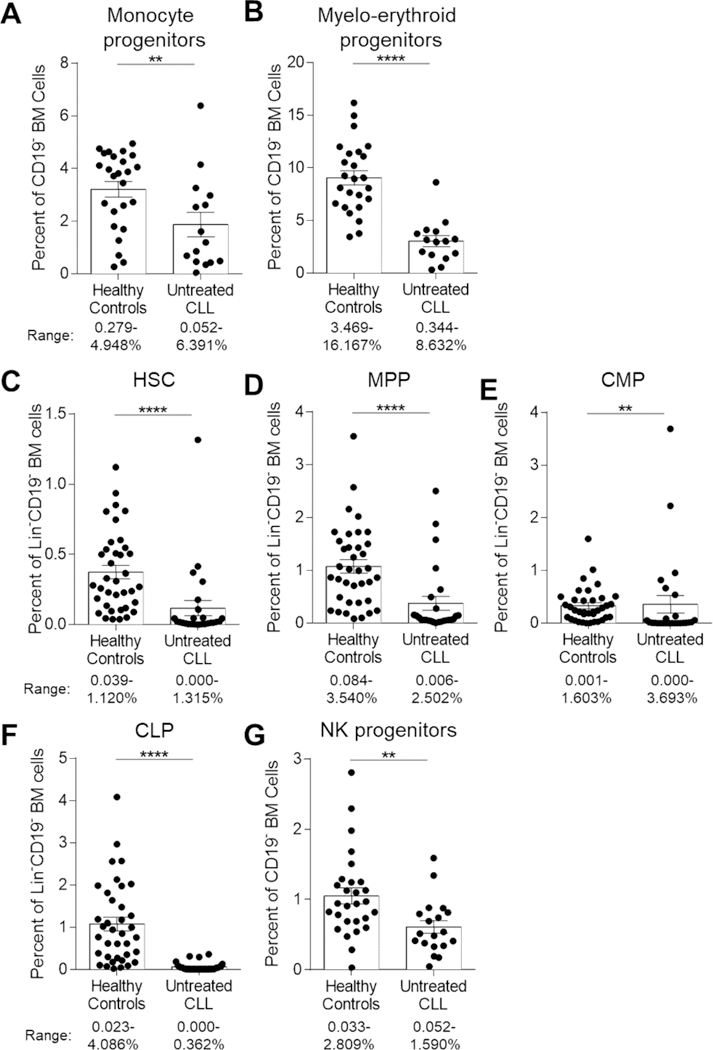
(A-B) Freshly isolated ACK-lysed BM cells were stained with combinations of antibodies to distinguish specific progenitor frequencies of lineage-biased BM progenitors as a function of total CD19-excluded cells. (A) Monocyte and (B) Myelo-erythroid cell progenitors. HC n=25 and CLL n=15 (38 individual experiments). (C-F) Frequencies of specific progenitor frequencies were evaluated as a function of Lin-CD19- cells. (C) HSC, hematopoietic stem cell (D) MPP, multipotent progenitor, (E) CMP, common myeloid progenitor, and (F) CLP, common lymphocyte progenitor. HC n=37, CLL n=26 (57 individual experiments). (G) NK cell progenitors were assessed as a function of total CD19-excluded cells. HC n=28 and CLL n=19 (44 individual experiments). See Table S3 for phenotypic definitions for each population. Bars and error represents mean and SEM of each group whereas each point indicates individual subjects. Numbers under each figure represent the range for each parameter evaluated for HCs and CLL patients. **P<0.01, and ****P<0.0001 were obtained by Mann-Whitney U-test.
Association of frequencies of innate subsets in BM and blood in CLL patients
Steady-state production of innate immune effector cells is required for immune surveillance and eradication of infectious agents. The paucity of myeloid progenitors and monocytes in CLL patient BM could significantly impair immune homeostasis, resulting in increased propensity to serious life-threatening infections. To determine if the blood innate immune repertoire reflected BM hematopoietic dysfunction, we performed association analyses between BM and blood. Paired CLL patient BM and blood samples were selected as either intermediate marrow cellularity (20–60% leukemic marrow involvement by pathology) or high cellularity (greater than 90% leukemic marrow involvement by pathology) cohorts. T cells, NK cells, and monocytes were evaluated by flow cytometry after CD19+CD5+ exclusion to remove the proportional calculation bias of the diluting CLL cells (Figure 4, Figure S4). Additionally, HC blood and BM samples (unmatched and unpaired) were analyzed in the same manner and averaged to obtain a control reference value for each tissue and cellular subset. All HC and CLL samples were previously cryopreserved, eliminating any freeze/thaw bias. We focused on NK cells and monocytes because sample processing of cryopreserved samples utilized ficoll separation, which eliminate granulocytes. As an internal control, we compared T cell frequencies in BM and blood (Figure 4A). We observed no appreciable difference in frequencies of T cells between intermediate and high cellularity patients, and the frequency of T cells correlated between BM and blood (Figure S5, r=0.8810, P=0.0072). We next evaluated total NK cells and CD56brightand CD56dim NK cell subsets (Figure 4B–D). We found that NK cells were significantly more diminished in the high cellularity patients compared to those with intermediate cellularity, and that the frequency of NK cells in the BM mirrored those in the blood (r=0.9286, P=0.0022), including CD56bright (r=0.8095, P=0.0218) and CD56dim (r=0.8810, P=0.0072) subsets. Finally, we evaluated classical (CD14+CD16-) and non-classical (CD14-CD16+) monocytes (Figure 4E–F) and found that like NK cells, they were more diminished in CLL patients with higher marrow involvement. However, while classical monocytes in the blood mirrored the BM population (r=0.9048, P=0.0046), non-classical monocytes were discordant (r=0.6429, P=0.0962). Overall, these data show that the reduced output of innate immune cells in BM is associated with diminished, phenotypically identical innate immune progeny in blood.
Figure 4. Comparative immune subset analysis of CLL patient blood and BM.
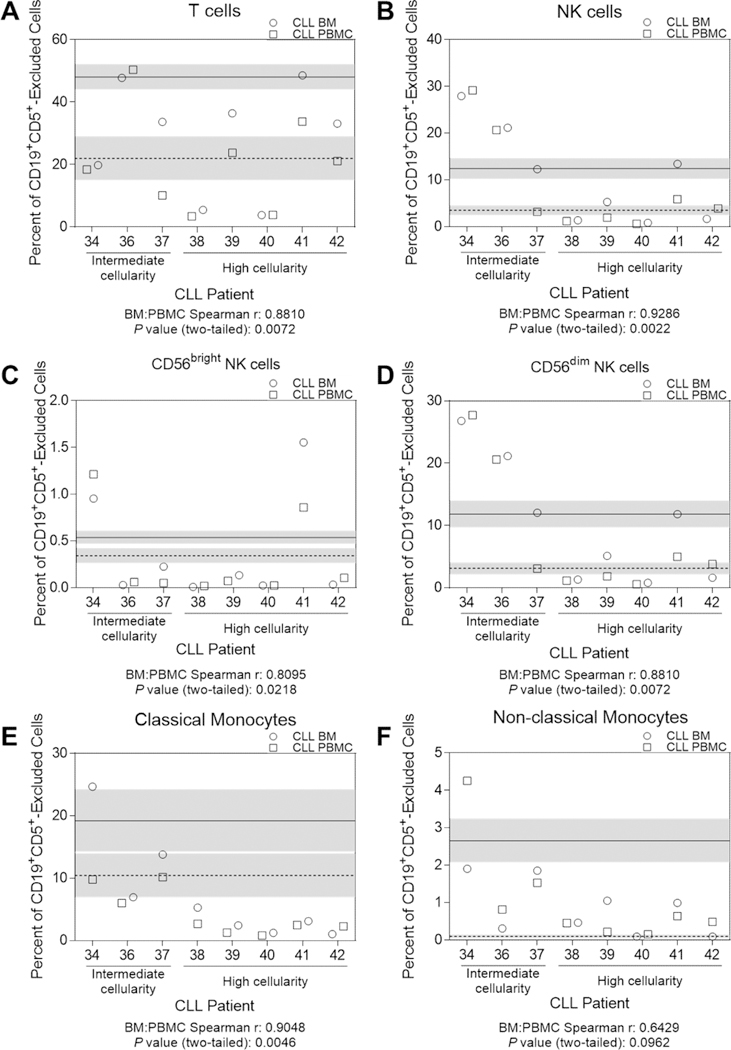
Previously cryopreserved paired BM and blood samples from CLL patients (total n=8, intermediate cellularity n=3 and high cellularity n=5, visit dates less than four weeks apart between BM and blood collections) and previously cryopreserved HC BM (n=7) and blood samples (n=6) (unmatched and unpaired) were evaluated by flow cytometry for frequencies of (A) T cells, (B-D) NK cells and subsets, and (E-F) monocyte subsets. All populations were gated after exclusion of infiltrating CD19+CD5+ cells. The HC BM and blood data were each averaged together for a reference value and indicated by horizontal solid (blood) or dashed (BM) lines. Shaded areas represent the SEM of the reference values. Each individual CLL patient is represented by a patient number that matches Tables 1 and S1. Cell frequencies from the BM are represented by an open circle and those from blood by an open square. See Table S3 for phenotypic definitions for each population. Correlation r and P values between BM and blood cells were determined by a Spearmen two-tailed correlation (see Figure S6). All data from one individual experiment.
Increased expression levels of select lineage-determining transcription factors in CLL patient BM HSPCs
Blood cell genesis from HSCs is complex and dictated by temporal activation, repression, and cross-antagonism among lineage-determining regulatory proteins20–22. The hematopoietic progenitor reductions observed may be due to altered differentiation programs and transcription factor expression patterns. We therefore sought to evaluate the levels of four key HSPC transcription factors in BM progenitor cells. HIF-1α is a master regulator of hematopoiesis5, is stably expressed at low levels in normal HSCs and early progenitors23, and is upregulated in CLL patient BM MSCs9. Importantly, elevated expression of HIF-1α impairs HSC function4, 5 and, to our knowledge, has not been evaluated in CLL patient HSPCs. We therefore assessed levels of HIF-1α in HSC/MPP, lineage-biased, and myelo-erythroid progenitors (Figure 5A–C, Figure S6, Table S3). There was a significant increase in levels of HIF-1α protein in lineage-biased progenitors (P=0.0235), but not HSC/MPP or myelo-erythroid progenitors in CLL patients compared to HCs. HIF-1α is a key molecular regulator of the erythroid-lineage transcription factor GATA-16. Examination of the lineage-biased and myelo-erythroid progenitor compartments revealed significant increases in levels of GATA-1 protein between HCs and CLL patient BM cells in these BM progenitor compartments (Figure 5E–F, P=0.0009, and P=0.0009 respectively). Similar to HIF-1α, levels of GATA-1 in the HSC/MPP compartment remained largely unperturbed (Figure 5D, P=0.0738).
Figure 5. Alterations in transcription factor expression levels in BM HSPC subsets in CLL patients.
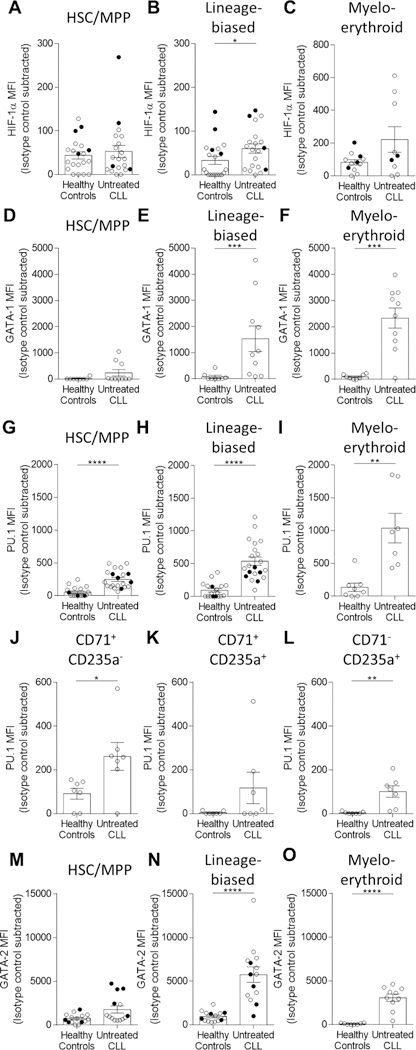
Fresh or frozen, thawed BM cells were analyzed for intranuclear expression of transcription factors by flow cytometry. (A-C) HIF-1α (A-B: HC n=20, CLL n=20, 10 individual experiments, C: HC n=12, CLL n=9, 7 individual experiments), (D-F) GATA-1 (HC n=8, CLL n=10, 1 individual experiment), (G-L) PU.1 (G-H: HC n=18, CLL n=21, 9 individual experiments, I: HC n=8, CLL n=7, 3 individual experiments and G-I: HC n=8, CLL n=8, 3 individual experiments), and (M-O) GATA-2 (M-N: HC n=14, CLL n=14, 4 individual experiments, O: HC n=8, CLL n=10, 1 individual experiment). Cellular populations defined as: HSC/MPP (Lin-CD19-CD34+CD38-CD45RA-), lineage-biased (Lin-CD19-CD34+CD38+), myelo-erythroid (CD19-CD33+CD71+) progenitor, and erythroid development (delineated by CD71 and CD235a) (see Table S3). MFI values for each transcription factor were subtracted against the isotype control and presented as the mean and SEM where each point indicates individual subjects. Freshly isolated samples are designated as closed symbols and previously cryopreserved samples as open symbols to allow for sample distinction. *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001 were obtained by Mann-Whitney U-test.
We next evaluated expression levels of the transcription factor PU.1, a critical regulator of HSC function and myelopoiesis24, 25 and direct antagonist of GATA factors and erythropoiesis24–26. Strikingly, PU.1 protein was significantly increased in HSC/MPP (P<0.0001), lineage-biased (P<0.0001), and myelo-erythroid (P=0.0012) progenitors in CLL patients versus HCs (Figure 5G–I). PU.1 levels were also significantly increased in CD71+CD235a- pro-erythroblasts (P=0.0175) and CD71-CD235a+ erythrocytes (P=0.0012), but not the intermediate CD71+CD235a+ erythroblasts (P=0.1352) (Figure 5J–L)27.
We also evaluated expression levels of GATA-2, a direct PU.1 antagonist28 whose mRNA transcripts are elevated CLL HSCs29. GATA-2 is also a critical repressor of the GATA-2/GATA-1 switch for erythropoiesis30, 31. We documented increased levels of GATA-2 protein in CLL patient lineage-biased and myelo-erythroid progenitors (Figure 5N–O, P<0.0001 and P<0.0001, respectively). Interestingly, similar to HIF-1α and GATA-1, GATA-2 protein is not elevated in CLL patient HSC/MPPs (Figure 5M, P=0.0608), suggesting that specific progenitor population susceptibility, timing of GATA-2 expression, and/or mRNA stability may be relevant in CLL. Taken together, these experimental findings show dysregulated expression of four key transcriptional regulators of HSPC biology in CLL patient BM.
TNFα selectively modulates transcription factor levels in BM HSPCs
Cytokines are known mediators of hematopoiesis, in part by influencing transcription factor expression levels. To this point, CLL B cells constitutively secrete TNFα14, 15, an inflammatory mediator, well-known modulator of HSPC activity32, 33, and inhibitor of erythropoiesis. The erythroid inhibition has been attributed to increased PU.118, which is upregulated in normal HSPCs after exposure to TNFα18. Importantly, in CLL patients, TNFα levels are higher in BM plasma than in blood34. To investigate if TNFα could contribute to the transcription factor imbalance we documented in ex vivo BM HSPCs, CD34+ cells were isolated from fresh HC BM and incubated with media or TNFα18, 19 for 12 or 24 hours then evaluated by flow cytometry. We found minimal or no changes in levels of the proliferation antigen Ki-67 or HIF-1α (Figure S7, Figure S8). Thus, short-term exposure to TNFα does not inhibit the proliferation of early progenitors, consistent with our finding that Ki-67 levels in ex vivo hematopoietic progenitor subsets were comparable between HCs and CLL patients (Figure S8I–J).
However, we found a significant increase in PU.1 levels 12 and 24 hours post-TNFα exposure in HSC/MPP (P<0.0001 and P<0.0001) and lineage-biased progenitors (P=0.0012 and P=0.0049, Figure 6A–B, Figure S6). In addition, GATA-2 levels were significantly increased in HSC/MPP (P=0.0008 and P=0.0005) and lineage-biased (P=0.0002 and P<0.0001) progenitors with TNFα exposure (Figure 6C–D). Together, these data suggest that the increase in PU.1 and GATA-2 proteins documented in CLL patient HSPCs may be partially modulated by CLL-derived TNFα.
Figure 6. TNFα modulates transcription factor expression levels in BM HSPCs.
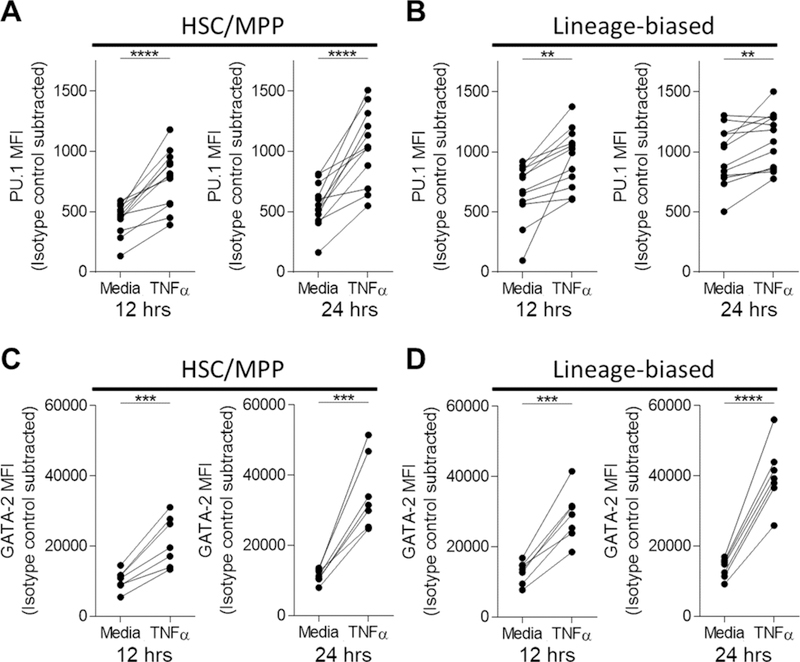
(A-D) CD34+ cells from fresh, ACK-lysed HC BM were isolated and incubated in serum-free media supplemented with HSPC-supportive cytokines (see Methods). Cultures were additionally supplemented with TNFα (25ng/mL) or media only for 12 or 24 hours followed by intranuclear staining for (A-B) PU.1 (n=12, 9 individual experiments) or (C-D) GATA-2 (n=7, 6 individual experiments) among the HSC/MPP (CD34+CD38-CD45RA-) and lineage-biased (CD34+CD38+) compartments. All MFI values were subtracted against the isotype control and presented such that each point and connecting line indicates a paired sample. **P<0.01, ***P<0.001, and ****P<0.0001 were obtained by a paired t-test.
Discussion
Here, we report hematopoietic dysfunction in untreated CLL patients as reflected by reduced frequencies and function of BM HSPCs and their progeny. This deficiency is particularly striking as it is evident across all progenitor populations, except granulocytes, and in early and late stage disease patients. The reduced frequencies of hematopoietic progenitors was similarly reflected in diminished CFU-responding progenitors. These deficiencies represent a novel and previously undescribed mechanism for the impaired ability of CLL patients to robustly contain infections and surveil for secondary malignancies. This is particularly noted in our finding of the mirroring of BM and blood NK and classical monocyte populations and represents a potential innate immune signature of BM deficiency.
Importantly, and in contrast to other reports2, 3, 29, we evaluated the frequency of all BM progenitor populations in both CLL patients and HCs as a function of CD19-excluded BM cells, equalizing both cohorts and allowing for a more accurate assessment of BM cellular composition. The equalization of CD19- BM cells is critical as CLL patients have variable leukemic cell infiltration in BM that proportionally dilutes the frequency of other cellular populations, an important consideration for other marrow-infiltrative diseases.
We pursued insight into the mechanism of impaired hematopoiesis in CLL BM. Infiltration by CLL B cells into BM could result in direct or indirect tissue-site hypoxia, resulting in increased HIF-1α expression in HSPCs. The role of hypoxia and HIF-1α in the BM is well studied and widely regarded as a major maintenance and control factor of HSCs and early progenitors5. Our finding that HIF-1α is overexpressed in CLL BM lineage-biased progenitors provide important clues to the reduced hematopoietic output observed. Overexpression of HIF-1α can inhibit HSPC differentiation4, 5 and drive aberrant GATA-1 expression6, potentially contributing to global decreases in progenitor frequency and function we report. We also found elevated expression levels of PU.1, an additional regulator of HSPC differentiation and direct antagonist of GATA factors.
The observation that GATA-2 is increased in lineage-biased and myelo-erythroid progenitors, but not HSC/MPPs, as opposed to PU.1 which is overexpressed in each of these progenitor compartments, provides a possible explanation for HSPC dysfunction. GATA-2 is a critical regulator of HSPC maintenance, proliferation, and differentiation35, and downregulation of GATA-2 is required for expression of GATA-1 and progression of erythroid development. The significant increase in levels of GATA-2 in CLL patient lineage-biased progenitors (which contain erythroid progenitors) could contribute to CLL-induced hematopoietic dysfunction by preventing the GATA-2/GATA-1 switch that is required for normal erythropoiesis30, 31 in addition to directly antagonizing PU.1. Overexpression of GATA-2 and HIF-1α has been shown to induce quiescence in human HSCs by directly affecting HSC cell cycle and reducing colony formation5, 36. However, we found no differences in the levels of the proliferation marker Ki-67 between HC and CLL patient HSPCs. As Ki-67 expression only provides a snapshot of cell cycle status, additional experiments are required to assess the proliferative status of HSPCs in CLL patient BM. Together, overexpression of these key transcription factors may contribute to the inability of HSPCs to successfully undergo lineage commitment, resulting in the global decrease of HSPC populations reported here.
The immune-modulating cytokines secreted by CLL B cells could be implicated in dysregulated expression of transcription factors that orchestrate hematopoietic differentiation programs. We show that TNFα has rapid direct effects on Lin-CD34+ HSPCs. The selective and significant modulation of PU.1 and GATA-2 by TNFα indicate that CLL B cell-derived factors are capable of influencing HSPCs directly. We note that the TNFα exposure results cannot be directly compared to the HC and CLL ex vivo data due to differences in experimental protocol including the supportive cytokines added for CD34+ survival (see Methods). In addition to TNFα-mediated inhibition by upregulation of PU.118, TNFα also inhibits formation of CD235a+ (glycophorin A) erythroid progenitors as well as limiting the proliferation of mature erythrocytes19. This mechanism has previously been proposed to be a contributing factor to CLL-related anemia3. Neutralizing antibodies to TNFα restored peripheral blood CFU-responding cell numbers from CLL patients15, suggesting that elevation in systemic (including BM34) levels of TNFα may be a major contributor to hematopoietic dysfunction. Of related interest, increased TNFα levels in CLL patient plasma has been correlated with CLL disease progression37.
Dysfunctional hematopoiesis in CLL is likely to have complex etiologies. There are other secreted factors produced by CLL B cells that may contribute to dysfunctional hematopoiesis. The chemokine CCL3 is produced by CLL B cells after BCR stimulation in supportive environments, such as BM38, 39, and is known to inhibit erythropoiesis in acute myeloid leukemia40. TGF-β and VEGF, additional cytokines constitutively secreted by CLL B cells41, may also contribute to hematopoietic dysfunction by inducing HSC quiescence42, 43 and regulating HSC survival44 and hematopoiesis45, respectively. Interestingly, TGF-β has been known to have a role in inducing and modulating erythropoiesis46–48 and high levels of GATA-2 negatively regulate this process49. Exosomes and microvesicles, two classes of extracellular vesicles (EVs) secreted by CLL B cells, are capable of modulating the BM microenvironment, as we and others have shown9, 50–52, and have defined roles in modulating normal and malignant hematopoiesis53–55. EVs represent another potential mechanism of CLL-induced hematopoietic dysfunction. Future studies are planned to address the role(s) of CCL3, TGF-β, VEGF, and EVs in BM hematopoietic dysfunction in CLL patients.
The BM is a complex organ consisting of many cell lineages in different stages of differentiation and a multifaceted microenvironment consisting of stromal cells, osteoblasts, osteoclasts, and accessory cells56. It is possible that CLL BM infiltration and interaction with elements of the microenvironment contributes to dysfunctional hematopoiesis. Indeed, we and others have shown that CLL B cells are capable of directly and indirectly modulating mesenchymal stromal cells that reside in the BM9, 57. These alterations in BM stromal cells, and possibly other components of the microenvironment, such as protective nurse-like cells58, likely contribute to decreased hematopoietic capacity in CLL patients59.
There is currently much interest in dissecting the presence of any genetic abnormalities in CLL patient HSPCs as it has been hypothesized that genetic lesions found in the CLL clone may arise in BM progenitors29, 60, 61. These genetic alterations, when present, may contribute to hematopoietic dysfunction, but have yet to be investigated. Further, the suspected genetic lesions are not mutually exclusive with intrinsic or extrinsic modulation of HSPCs in CLL patient BM and will be of interest for future studies.
Finally, given the chronic nature of CLL, decreased BM hematopoietic output would be predicted to be reflected in the appropriate cellular constituents in blood. Indeed, we show severe reductions in NK cells and monocytes in CLL patients with higher BM leukemic burden that is reflected in reduced phenotypically comparable blood cell frequencies. Importantly, regardless of the level of leukemic BM infiltration, the frequency of NK and classical monocytes in BM mirrored the blood. Future studies will need to build on this preliminary observation and seek to relate it to clinically-relevant complications. This study provides, for the first time, a novel consideration for marrow infiltration of CLL B cells that limits the developmental capacity of hematopoietic progenitors. Importantly, these findings challenge the current paradigm that the immune deficiency in CLL is solely the result of a peripheral global immunodeficiency that has excluded investigation of the effect of CLL infiltration into the marrow on steady-state hematopoiesis, particularly in early stage patients. The advance in our knowledge of BM hematopoiesis and how it relates to immune status in CLL will provide a new platform upon which to subsequently devise therapeutic strategies and interventions to enhance CLL patient immune status and hematopoietic output.
Supplementary Material
Key Points.
Cell-intrinsic defects in BM hematopoietic stem and progenitor cells (HSPC) in untreated CLL patients
Altered levels of specific nuclear factors regulating HSPC differentiation and function in untreated CLL patients
Acknowledgements
This work was supported by funding provided by the Mayo Clinic Center for Biomedical Discovery to K.L.M., W.D., and N.E.K. B.A.M. is supported by a NIH T32 Training Grant in Basic Immunology (NIH AI07425).
Research funding has been provided to the institution from Pharmacyclics, Morphosys, and AbbVie for clinical studies in which Dr. Sameer Parikh is a principal investigator.
Dr. Sameer Parikh has also participated in Advisory Board meetings of Pharmacyclics, AstraZeneca, and AbbVie (he was not personally compensated for his participation).
Abbreviations used in text:
- BM
bone marrow
- HSPC
hematopoietic stem/progenitor cell
- Lin
lineage marker flow cytometry antibody cocktail
- HSC
hematopoietic stem cell
- MPP
multipotent progenitor
- CMP
common myeloid progenitor
- CLL
untreated chronic lymphocytic leukemia
- HC
healthy control
- CFU
colony forming unit
- PMN
polymorphonuclear leukocyte
- HIF-1α
natural killer cell (NK), hypoxia-inducible factor 1 alpha
- TNFα
tumor necrosis factor alpha
Footnotes
Disclosure of conflicts of interest
References
- 1.Forconi F, Moss P. Perturbation of the normal immune system in patients with CLL. Blood 2015; 126(5): 573–581. [DOI] [PubMed] [Google Scholar]
- 2.Sala R, Mauro FR, Bellucci R, De Propris MS, Cordone I, Lisci A, et al. Evaluation of marrow and blood haemopoietic progenitors in chronic lymphocytic leukaemia before and after chemotherapy. Eur J Haematol 1998. July; 61(1): 14–20. [DOI] [PubMed] [Google Scholar]
- 3.Tsopra OA, Ziros PG, Lagadinou ED, Symeonidis A, Kouraklis-Symeonidis A, Thanopoulou E, et al. Disease-related anemia in chronic lymphocytic leukemia is not due to intrinsic defects of erythroid precursors: a possible pathogenetic role for tumor necrosis factor-alpha. Acta Haematol 2009; 121(4): 187–195. [DOI] [PubMed] [Google Scholar]
- 4.Eliasson P, Rehn M, Hammar P, Larsson P, Sirenko O, Flippin LA, et al. Hypoxia mediates low cell-cycle activity and increases the proportion of long-term–reconstituting hematopoietic stem cells during in vitro culture. Experimental Hematology 2010 2010/April/01/; 38(4): 301–310.e302. [DOI] [PubMed] [Google Scholar]
- 5.Takubo K, Goda N, Yamada W, Iriuchishima H, Ikeda E, Kubota Y, et al. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell 2010. September 03; 7(3): 391–402. [DOI] [PubMed] [Google Scholar]
- 6.Zhang F-L, Shen G- M, Liu X- L, Wang F, Zhao Y- Z, Zhang J- W. Hypoxia-inducible factor 1–mediated human GATA1 induction promotes erythroid differentiation under hypoxic conditions. Journal of Cellular and Molecular Medicine 2012; 16(8): 1889–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoon D, Ponka P, Prchal JT. Hypoxia. 5. Hypoxia and hematopoiesis. American Journal of Physiology - Cell Physiology 2011; 300(6): C1215. [DOI] [PubMed] [Google Scholar]
- 8.Schito L, Semenza GL. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends in Cancer 2016. 12//; 2(12): 758–770. [DOI] [PubMed] [Google Scholar]
- 9.Ghosh AK, Secreto CR, Knox TR, Ding W, Mukhopadhyay D, Kay NE. Circulating microvesicles in B-cell chronic lymphocytic leukemia can stimulate marrow stromal cells: implications for disease progression. Blood 2010. March 04; 115(9): 1755–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koczula KM, Ludwig C, Hayden R, Cronin L, Pratt G, Parry H, et al. Metabolic plasticity in CLL: adaptation to the hypoxic niche. Leukemia 2016. 01//print; 30(1): 65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Valsecchi R, Coltella N, Belloni D, Ponente M, ten Hacken E, Scielzo C, et al. HIF-1α regulates the interaction of chronic lymphocytic leukemia cells with the tumor microenvironment. Blood 2016; 127(16): 1987–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Whyatt D, Karis A, Harkes I, Verkerk A, Gillemans N, Elefanty A, et al. The level of the tissue-specific factor GATA-1 affects the cell-cycle machinery. Genes and Function 1997; 1(1): 11–24. [DOI] [PubMed] [Google Scholar]
- 13.Whyatt D, Lindeboom F, Karis A, Ferreira R, Milot E, Hendriks R, et al. An intrinsic but cell-nonautonomous defect in GATA-1-overexpressing mouse erythroid cells. Nature 2000. August 03; 406(6795): 519–524. [DOI] [PubMed] [Google Scholar]
- 14.Foa R, Massaia M, Cardona S, Tos AG, Bianchi A, Attisano C, et al. Production of tumor necrosis factor-alpha by B-cell chronic lymphocytic leukemia cells: a possible regulatory role of TNF in the progression of the disease. Blood 1990. July 15; 76(2): 393–400. [PubMed] [Google Scholar]
- 15.Michalevicz R, Porat R, Vechoropoulos M, Baron S, Yanoov M, Cycowitz Z, et al. Restoration of in vitro hematopoiesis in B-chronic lymphocytic leukemia by antibodies to tumor necrosis factor. Leuk Res 1991; 15(2–3): 111–120. [DOI] [PubMed] [Google Scholar]
- 16.Perfetto SP, Ambrozak D, Nguyen R, Chattopadhyay P, Roederer M. Quality assurance for polychromatic flow cytometry. Nat Protoc 2006; 1(3): 1522–1530. [DOI] [PubMed] [Google Scholar]
- 17.Perfetto SP, Ambrozak D, Nguyen R, Chattopadhyay PK, Roederer M. Quality assurance for polychromatic flow cytometry using a suite of calibration beads. Nat Protoc 2012. December; 7(12): 2067–2079. [DOI] [PubMed] [Google Scholar]
- 18.Grigorakaki C, Morceau F, Chateauvieux S, Dicato M, Diederich M. Tumor necrosis factor alpha-mediated inhibition of erythropoiesis involves GATA-1/GATA-2 balance impairment and PU.1 over-expression. Biochem Pharmacol 2011. July 15; 82(2): 156–166. [DOI] [PubMed] [Google Scholar]
- 19.Xiao W, Koizumi K, Nishio M, Endo T, Osawa M, Fujimoto K, et al. Tumor necrosis factor-alpha inhibits generation of glycophorin A+ cells by CD34+ cells. Exp Hematol 2002. November; 30(11): 1238–1247. [DOI] [PubMed] [Google Scholar]
- 20.Cvejic A Mechanisms of fate decision and lineage commitment during haematopoiesis. Immunol Cell Biol 2016. March; 94(3): 230–235. [DOI] [PubMed] [Google Scholar]
- 21.Lunger I, Fawaz M, Rieger MA. Single-cell analyses to reveal hematopoietic stem cell fate decisions. FEBS Lett 2017. August; 591(15): 2195–2212. [DOI] [PubMed] [Google Scholar]
- 22.Nakajima H Role of transcription factors in differentiation and reprogramming of hematopoietic cells. Keio J Med 2011; 60(2): 47–55. [DOI] [PubMed] [Google Scholar]
- 23.Nombela-Arrieta C, Pivarnik G, Winkel B, Canty KJ, Harley B, Mahoney JE, et al. Quantitative imaging of haematopoietic stem and progenitor cell localization and hypoxic status in the bone marrow microenvironment. Nat Cell Biol 2013. May; 15(5): 533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arinobu Y, Mizuno S-i, Chong Y, Shigematsu H, Iino T, Iwasaki H, et al. Reciprocal Activation of GATA-1 and PU.1 Marks Initial Specification of Hematopoietic Stem Cells into Myeloerythroid and Myelolymphoid Lineages. Cell Stem Cell 2007. October/11/; 1(4): 416–427. [DOI] [PubMed] [Google Scholar]
- 25.Fukuchi Y, Ito M, Shibata F, Kitamura T, Nakajima H. Activation of CCAAT/Enhancer-Binding Protein α or PU.1 in Hematopoietic Stem Cells Leads to Their Reduced Self-Renewal and Proliferation. STEM CELLS 2008; 26(12): 3172–3181. [DOI] [PubMed] [Google Scholar]
- 26.Burda P, Laslo P, Stopka T. The role of PU.1 and GATA-1 transcription factors during normal and leukemogenic hematopoiesis. Leukemia 2010. July; 24(7): 1249–1257. [DOI] [PubMed] [Google Scholar]
- 27.van Lochem EG, van der Velden VHJ, Wind HK, te Marvelde JG, Westerdaal NAC, van Dongen JJM. Immunophenotypic differentiation patterns of normal hematopoiesis in human bone marrow: Reference patterns for age-related changes and disease-induced shifts. Cytometry Part B: Clinical Cytometry 2004; 60B(1): 1–13. [DOI] [PubMed] [Google Scholar]
- 28.Walsh JC, DeKoter RP, Lee H-J, Smith ED, Lancki DW, Gurish MF, et al. Cooperative and Antagonistic Interplay between PU.1 and GATA-2 in the Specification of Myeloid Cell Fates. Immunity 2002 2002/November/01/; 17(5): 665–676. [DOI] [PubMed] [Google Scholar]
- 29.Kikushige Y, Ishikawa F, Miyamoto T, Shima T, Urata S, Yoshimoto G, et al. Self-renewing hematopoietic stem cell is the primary target in pathogenesis of human chronic lymphocytic leukemia. Cancer Cell 2011. August 16; 20(2): 246–259. [DOI] [PubMed] [Google Scholar]
- 30.Bresnick EH, Katsumura KR, Lee HY, Johnson KD, Perkins AS. Master regulatory GATA transcription factors: mechanistic principles and emerging links to hematologic malignancies. Nucleic Acids Res 2012. July; 40(13): 5819–5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moriguchi T, Yamamoto M. A regulatory network governing Gata1 and Gata2 gene transcription orchestrates erythroid lineage differentiation. Int J Hematol 2014. November; 100(5): 417–424. [DOI] [PubMed] [Google Scholar]
- 32.Mizrahi K, Askenasy N. Physiological functions of TNF family receptor/ligand interactions in hematopoiesis and transplantation. Blood 2014; 124(2): 176. [DOI] [PubMed] [Google Scholar]
- 33.Rusten LS, Jacobsen SE. Tumor necrosis factor (TNF)-alpha directly inhibits human erythropoiesis in vitro: role of p55 and p75 TNF receptors. Blood 1995; 85(4): 989. [PubMed] [Google Scholar]
- 34.Bojarska-Junak A, Hus I, Szczepanek EW, Dmoszyńska A, Roliński J. Peripheral blood and bone marrow TNF and TNF receptors in early and advanced stages of B-CLL in correlation with ZAP-70 protein and CD38 antigen. Leukemia Research 2008 2008/February/01/; 32(2): 225–233. [DOI] [PubMed] [Google Scholar]
- 35.Vicente C, Conchillo A, Garcia-Sanchez MA, Odero MD. The role of the GATA2 transcription factor in normal and malignant hematopoiesis. Crit Rev Oncol Hematol 2012. April; 82(1): 1–17. [DOI] [PubMed] [Google Scholar]
- 36.Tipping AJ, Pina C, Castor A, Hong D, Rodrigues NP, Lazzari L, et al. High GATA-2 expression inhibits human hematopoietic stem and progenitor cell function by effects on cell cycle. Blood 2009. March 19; 113(12): 2661–2672. [DOI] [PubMed] [Google Scholar]
- 37.Ferrajoli A, Keating MJ, Manshouri T, Giles FJ, Dey A, Estrov Z, et al. The clinical significance of tumor necrosis factor-alpha plasma level in patients having chronic lymphocytic leukemia. Blood 2002. August 15; 100(4): 1215–1219. [PubMed] [Google Scholar]
- 38.Hartmann EM, Rudelius M, Burger JA, Rosenwald A. CCL3 chemokine expression by chronic lymphocytic leukemia cells orchestrates the composition of the microenvironment in lymph node infiltrates. Leuk Lymphoma 2016; 57(3): 563–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sivina M, Hartmann E, Kipps TJ, Rassenti L, Krupnik D, Lerner S, et al. CCL3 (MIP-1alpha) plasma levels and the risk for disease progression in chronic lymphocytic leukemia. Blood 2011. February 03; 117(5): 1662–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Y, Gao A, Zhao H, Lu P, Cheng H, Dong F, et al. Leukemia cell infiltration causes defective erythropoiesis partially through MIP-1[alpha]/CCL3. Leukemia 2016. 09//print; 30(9): 1897–1908. [DOI] [PubMed] [Google Scholar]
- 41.Lotz M, Ranheim E, Kipps TJ. Transforming growth factor beta as endogenous growth inhibitor of chronic lymphocytic leukemia B cells. The Journal of Experimental Medicine 1994; 179(3): 999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blank U, Karlsson S. TGF-beta signaling in the control of hematopoietic stem cells. Blood 2015. June 04; 125(23): 3542–3550. [DOI] [PubMed] [Google Scholar]
- 43.Fan X, Valdimarsdottir G, Larsson J, Brun A, Magnusson M, Jacobsen SE, et al. Transient disruption of autocrine TGF-beta signaling leads to enhanced survival and proliferation potential in single primitive human hemopoietic progenitor cells. J Immunol 2002. January 15; 168(2): 755–762. [DOI] [PubMed] [Google Scholar]
- 44.Gerber HP, Malik AK, Solar GP, Sherman D, Liang XH, Meng G, et al. VEGF regulates haematopoietic stem cell survival by an internal autocrine loop mechanism. Nature 2002. June 27; 417(6892): 954–958. [DOI] [PubMed] [Google Scholar]
- 45.Xue Y, Chen F, Zhang D, Lim S, Cao Y. Tumor-derived VEGF modulates hematopoiesis. J Angiogenes Res 2009. December 23; 1: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fortunel NO, Hatzfeld A, Hatzfeld JA. Transforming growth factor-beta: pleiotropic role in the regulation of hematopoiesis. Blood 2000. September 15; 96(6): 2022–2036. [PubMed] [Google Scholar]
- 47.Zermati Y, Fichelson S, Valensi F, Freyssinier JM, Rouyer-Fessard P, Cramer E, et al. Transforming growth factor inhibits erythropoiesis by blocking proliferation and accelerating differentiation of erythroid progenitors. Experimental Hematology 2000 2000/August/01/; 28(8): 885–894. [DOI] [PubMed] [Google Scholar]
- 48.Zermati Y, Varet B, Hermine O. TGF-β1 drives and accelerates erythroid differentiation in the Epo-dependent UT-7 cell line even in the absence of erythropoietin. Experimental Hematology 2000 2000/March/01/; 28(3): 256–266. [DOI] [PubMed] [Google Scholar]
- 49.Dong XM, Yin RH, Yang Y, Feng ZW, Ning HM, Dong L, et al. GATA-2 inhibits transforming growth factor-beta signaling pathway through interaction with Smad4. Cell Signal 2014. May; 26(5): 1089–1097. [DOI] [PubMed] [Google Scholar]
- 50.Paggetti J, Haderk F, Seiffert M, Janji B, Distler U, Ammerlaan W, et al. Exosomes released by chronic lymphocytic leukemia cells induce the transition of stromal cells into cancer-associated fibroblasts. Blood 2015. August 27; 126(9): 1106–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic Mark M, et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015. November 19; 527(7578): 329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lin LY, Du LM, Cao K, Huang Y, Yu PF, Zhang LY, et al. Tumour cell-derived exosomes endow mesenchymal stromal cells with tumour-promotion capabilities. Oncogene 2016. November 17; 35(46): 6038–6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kumar B, Garcia M, Weng L, Jung X, Murakami JL, Hu X, et al. Acute myeloid leukemia transforms the bone marrow niche into a leukemia-permissive microenvironment through exosome secretion. Leukemia 2018. March; 32(3): 575–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pando A, Reagan JL, Quesenberry P, Fast LD. Extracellular vesicles in leukemia. Leuk Res 2018. January; 64: 52–60. [DOI] [PubMed] [Google Scholar]
- 55.Quesenberry PJ, Goldberg L, Aliotta J, Dooner M. Marrow Hematopoietic Stem Cells Revisited: They Exist in a Continuum and are Not Defined by Standard Purification Approaches; Then There are the Microvesicles. Front Oncol 2014; 4: 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Asada N, Takeishi S, Frenette PS. Complexity of bone marrow hematopoietic stem cell niche. International Journal of Hematology 2017 2017/July/01; 106(1): 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Janel A, Dubois-Galopin F, Bourgne C, Berger J, Tarte K, Boiret-Dupre N, et al. The chronic lymphocytic leukemia clone disrupts the bone marrow microenvironment. Stem Cells Dev 2014. December 15; 23(24): 2972–2982. [DOI] [PubMed] [Google Scholar]
- 58.Boissard F, Fournie JJ, Quillet-Mary A, Ysebaert L, Poupot M. Nurse-like cells mediate ibrutinib resistance in chronic lymphocytic leukemia patients. Blood Cancer J 2015. October 02; 5: e355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lagneaux L, Delforge A, Dorval C, Bron D, Stryckmans P. Excessive production of transforming growth factor-beta by bone marrow stromal cells in B-cell chronic lymphocytic leukemia inhibits growth of hematopoietic precursors and interleukin-6 production. Blood 1993; 82(8): 2379. [PubMed] [Google Scholar]
- 60.Damm F, Mylonas E, Cosson A, Yoshida K, Della Valle V, Mouly E, et al. Acquired Initiating Mutations in Early Hematopoietic Cells of CLL Patients. Cancer Discovery 2014; 4(9): 1088. [DOI] [PubMed] [Google Scholar]
- 61.Marsilio S, Khiabanian H, Fabbri G, Vergani S, Scuoppo C, Montserrat E, et al. Somatic CLL mutations occur at multiple distinct hematopoietic maturation stages: documentation and cautionary note regarding cell fraction purity. Leukemia 2017. 12/05/online; 32: 1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.