

SUMMARY
Gap-junction-mediated cell-cell communication enables tumor cells to synchronize complex processes. We previously found that glioblastoma cancer stem cells (CSCs) express higher levels of the gap junction protein Cx46 compared to non-stem tumor cells (non-CSCs) and that this was necessary and sufficient for CSC maintenance. To understand the mechanism underlying this requirement, we use point mutants to disrupt specific functions of Cx46 and find that Cx46-mediated gap-junction coupling is critical for CSCs. To develop a Cx46 targeting strategy, we screen a clinically relevant small molecule library and identify clofazimine as an inhibitor of Cx46-specific cell-cell communication. Clofazimine attenuates proliferation, self-renewal, and tumor growth and synergizes with temozolomide to induce apoptosis. Although clofazimine does not cross the blood-brain barrier, the combination of clofazimine derivatives optimized for brain penetrance with standard-of-care therapies may target glioblastoma CSCs. Furthermore, these results demonstrate the importance of targeting cell-cell communication as an anti-cancer therapy.
Graphical Abstract
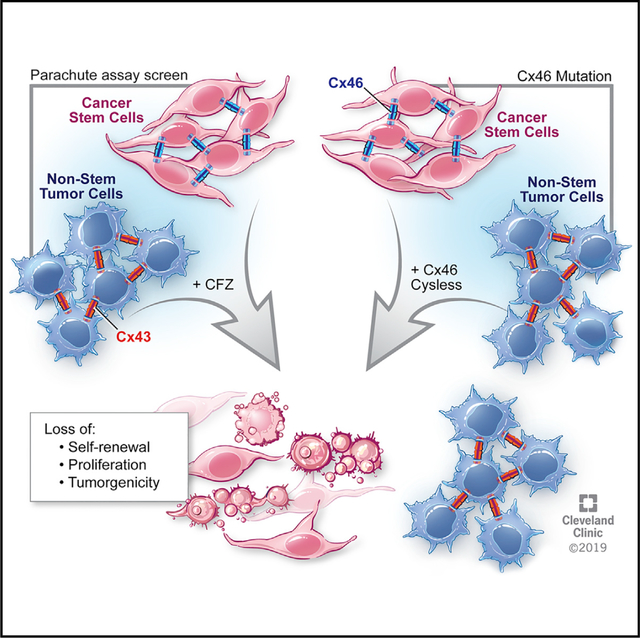
In Brief
Cx46 was previously shown to be essential for glioblastoma cancer stem cell maintenance. Here, Mulkearns-Hubert et al. show that cancer stem cells depend on Cx46-mediated cell-cell communication and identify a Cx46 inhibitor, clofazimine. Clofazimine preferentially inhibits Cx46-mediated communication and targets cancer stem cells to decrease tumor growth.
INTRODUCTION
Glioblastoma (GBM; grade IV astrocytoma), the most commonly occurring primary malignant brain tumor, remains uniformly fatal despite aggressive therapy that includes surgery, radiation, and chemotherapy. Increased understanding of the molecular alterations underlying tumorigenesis has not translated to clinical success; patient prognosis remains poor, with a median survival of only 14–16 months and 5-year survival rates of less than 3% (McGirt et al., 2009; Stupp et al., 2009, 2015). One factor underlying the difficulty in treating GBM is the cellular diversity present within these tumors. Heterogeneous populations of cancer stem cells (CSCs) exhibit essential characteristics of sustained self-renewal, persistent proliferation, and ability to initiate tumors when transplanted into mice (Lathia et al., 2015), and they display resistance to the GBM standard-of-care therapies: radiation and temozolomide (Bao et al., 2006; Chen et al., 2012; Liu et al., 2006). Efforts to treat GBM are focused on the ability to target CSCs, because this may lead to the development of more effective therapies for GBM with increased clinical success.
Cell-cell communication is mediated through the connexin family of proteins and the gap junction (GJ) channels that these proteins comprise. Six connexin proteins assemble into a channel through the plasma membrane that can exchange small molecules between the cytoplasm and the extracellular space as hemichannels. When these channels dock with a compatible hexamer on a neighboring cell, a GJ is formed. GJ intercellular communication (GJIC) exchanges ions, microRNAs (miRNAs), small metabolites such as glucose, antioxidants, and peptides between cells, allowing them to coordinate their phenotypes and respond to environmental conditions (Goodenough and Paul, 2009). Connexin proteins serve three main cellular functions: exchange of small molecules between cells as GJs, exchange of small molecules between a cell and the extracellular space as hemichannels, and binding to intracellular proteins (Goodenough and Paul, 2003, 2009; Leithe et al., 2018; Stout et al., 2004).
Previous work based mainly on connexin 43 (Cx43) suggested that connexins act as tumor suppressors (Aasen et al., 2016). However, we have identified pro-tumorigenic connexins in prostate cancer (Zhang et al., 2015), breast cancer (Thiagarajan et al., 2018), leukemia (Sinyuk et al., 2015), and GBM (Hitomi et al., 2015). GBM CSCs express higher levels of Cx46 compared to non-CSCs, and Cx46 is required for CSC proliferation, survival, self-renewal, and tumor formation (Hitomi et al., 2015). Pan-GJ inhibitors slowed tumor growth in mice with intracranial tumors, but these compounds inhibit connexins as an off-target effect. Therefore, these compounds would likely cause side effects in patients based on their broad effects targeting multiple connexins that play essential roles in many normal organs.
Here, we used mutational analysis and identified the dominant function of Cx46 in GBM CSCs to be cell-cell communication through GJs (GJIC) rather than hemichannel activity. We thus hypothesized that targeting of CSCs through specific inhibition of Cx46 would slow tumor growth and lead to the development of new therapies for patients with GBM. A screen of U.S. Food and Drug Administration (FDA)-approved small molecules identified the anti-leprosy drug clofazimine as a preferential inhibitor of Cx46-mediated cell-cell communication and GBM CSC maintenance. Because clofazimine was unable to penetrate the blood-brain barrier at physiological dosages, we propose that Cx46 future derivatization of the compound is required to permeate the blood-brain barrier and may produce an optimal targeting drug for GBM CSCs. Altogether, our data suggest that repurposing and derivatization of this and similar compounds may benefit patients with GBM.
RESULTS
Cx46-Mediated Cell-Cell Communication Is Essential to Maintain GBM CSCs
Our previous studies identified Cx46 as a potential anti-CSC target (Hitomi et al., 2015). To develop a strategy to specifically inhibit Cx46, we first sought to determine the function of Cx46 required to maintain GBM CSC properties. To achieve this, we identified a panel of Cx46 mutations that would allow us to deduce the individual importance of GJIC and hemichannel activity. Two Cx46 point mutations have been reported in human patients with cataracts (Hansen et al., 2006; Santhiya et al., 2010). These mutations, L11S and T19M, are both located in the N-terminal tail of the Cx46 protein (Figure 1A) and have been functionally investigated in the context of the rat protein in Xenopus oocytes (Tong et al., 2013, 2015). When co-expressed with wild-type Cx46, the presence of the L11S mutation was shown to dramatically reduce both GJIC and hemichannel activity (Tong et al., 2013). In contrast, while expression of Cx46 T19M alone was not sufficient for GJIC, co-expression of the Cx46 T19M mutant with wild-type Cx46 increased hemi-channel activity but did not affect GJIC (Tong et al., 2015). We also used a cysless mutant previously engineered in Cx43 that disrupts the three disulfide bonds necessary to maintain the structure of connexins required for GJ docking. This mutant was reported to block GJIC without affecting hemichannel activity of Cx43 in both Xenopus oocytes and ovarian granulosa cells (Bao et al., 2004; Tong et al., 2007), and we confirmed its inhibition of GJIC in HeLa cells (data not shown).
Figure 1. Mutational Analysis Indicates that Cell-Cell Communication Is Essential to Maintain Glioblastoma Cancer Stem Cells.

(A) Schematic showing the location of introduced Cx46 point mutants in the protein.
(B) CSCs from the patient-derived xenograft specimen T4121 were transfected with wild-type or mutant Cx46, and the number of cells was measured at the indicated times after plating using CellTiter-Glo. The values shown are relative to day 0. n = 4 experiments performed in triplicate. *p < 0.05, **p <0.01, ***p < 0.001 by two-way ANOVA compared to vector to test for significant differences between the curves.(C and D) Transfected CSCs from the PDX specimens T4121 (C) and T387 (D) were assessed for active caspase-3/7 on day 1 using Caspase-Glo. The values shown are normalized to the CellTiter-Glo signal at day 1 and are given relative to vector. n = 4 experiments for T4121 and n = 3 for T387, all performed in triplicate. *p < 0.05, **p < 0.01, ***p <0.001 by Student’s unpaired t test with Welch’s correction compared to vector.(E and F) Transfected CSCs from the patient-derived xenograft specimens T4121 (E) and T387 (F) were plated in a limiting-dilution format (between 1 and 20 cells/well of a 96-well plate), and the number of spheres per well was counted between days 10 and 14. The stem cell frequency was calculated using the online algorithm described in the STAR Methods section. The values shown are relative to the stem cell frequency of the vector-transfected cells. n = 3 experiments, with 24 technical replicates per cell number per experiment. *p < 0.05, **p < 0.01, ***p <0.001 by c2 test compared to the vector control. Data are represented as mean ± SEM for (B)–(D) and mean ± range for (E) and (F).
See also Figure S1.
We introduced these mutations into human Cx46 cDNA and transfected the DNA into GBM CSCs isolated from two patient-derived xenografts (PDXs; T4121 and T387). Using qPCR, we were able to detect the expression of each Cx46 mutant in CSCs at the mRNA level (Figures S1A and S1B). Expression of Cx46 T19M or overexpression of wild-type Cx46 in the presence of endogenous Cx46 had little effect on CSC proliferation (Figure 1B; Figure S1C), apoptosis (Figures 1C and 1D), or self-renewal (Figures 1E and 1F), a hallmark of the CSC state, which was assessed by limiting dilution sphere-formation analysis, while we observed small but signifi-cant decreases in proliferation and self-renewal with expression of Cx46 L11S. However, expression of Cx46 cysless dramatically decreased CSC proliferation, increased apoptosis, and decreased self-renewal in both patient-derived specimens. Expression of these mutants in non-CSCs, which express low levels of Cx46 (Hitomi et al., 2015), had little effect beyond that of expressing wild-type Cx46 (Figures S1D–S1G). Altogether, these observations demonstrate that when expressed with endogenous Cx46, the cysless mutant, which has been shown to have the greatest effect on cell-cell communication, also had the greatest effect on CSC maintenance compared to the other mutants (Table S1) and led us to conclude that GJIC mediated by Cx46 is essential to maintain GBM CSC proliferation, survival, and self-renewal.
A Screen of FDA-Approved Small Molecules Identifies Clofazimine as an Inhibitor of Cx46-Mediated GJIC
Based on our observation that GBM CSCs require Cx46-mediated GJIC for survival, we designed an assay system to screen for inhibitors of this process. We assessed GJIC using a quantitative calcein transfer assay (Figure 2A) (Hitomi et al., 2015), a modification of the parachute dye-uptake assay (Ziambaras et al., 1998). In this assay, cells labeled with both a GJ-permeable dye (calcein red-orange acetoxymethyl [AM], shown in black) and a non-spreading membrane dye (1,1’-dioctadecyl-3,3,3’,3’- tetramethylindodicarbocyanine, 4-chlorobenzenesulfonate salt [DiD], shown in magenta) were added to a subconfluent mono-layer of unlabeled cells. The formation of GJs is indicated by membrane dye-negative cells that become calcein positive with time. HeLa cells express low levels of endogenous connexins (Elf-gang et al., 1995) and display minimal dye coupling (Figure 2B; Figure S2). However, stable expression of Cx46 or transient expression of Cx43 in HeLa cells established functional GJs and coupling between cells, as evidenced by the spread of calcein dye (shown in black) between cells (Figure 2B; Figure S2). Using stable Cx46-expressing HeLa cells, we then screened the 727 compounds of the NIH Clinical Collection of FDA-approved small molecules for their ability to inhibit Cx46-mediated GJIC at a concentration of 10 mM over a treatment time of 3 h (Figure 2C). The spread of calcein between treated cells was imaged and compared to both vehicle (DMSO) treatment and treatment with the pan-GJ inhibitor carbenoxolone (CBX; 200 nM). We identified several compounds that blocked Cx46-mediated GJIC compared to CBX as a positive control (Figure 2D). Several of the top hits were screened at concentrations between 0.1 and 10 mM, and we found that the FDA-approved anti-mycobacterial drug clofazimine inhibited Cx46 GJIC at the lowest concentrations compared to the other hits (Figure 2E), with little effect on Cx46 hemichannel activity, as determined by the amount of calcein lost from sparsely plated cells (Figure 2F). Altogether, these results demonstrate that clofazi-mine is a candidate to inhibit Cx46 GJIC without affecting hemi-channel activity.
Figure 2. A Screen of FDA-Approved Small Molecules Identifies Clofazimine as an Inhibitor of Cx46-Mediated Cell-Cell Communication.

(A) Schematic of calcein dye transfer between HeLa cells expressing no exogenous connexin proteins and HeLa cells expressing Cx43 or Cx46. Cells are labeled with Vybrant DiD (pseudocolored magenta), which cannot pass between cells, and calcein red-orange AM (pseudocolored black), which spreads between cells through gap junctions.
(B) Parachute dye transfer assay of parental HeLa cells, stable Cx46-expressing HeLa cells, and transiently transfected Cx43-expressing HeLa cells. Unlabeled recipient cells were plated in a subconfluent monolayer, and dual-labeled cells were added. If gap junctions formed between labeled and unlabeled cells, the calcein dye (shown in black) diffused into cells that were not labeled with DiD (magenta). Scale bar, 50 μm.
(C) Schematic of the parachute dye transfer assay with timing used to test the NIH Clinical Collection compounds for inhibition of Cx46-mediated cell-cell communication in stable HeLa-Cx46 cells. A subconfluent monolayer of HeLa-Cx46 cells was plated and incubated with drugs at 10 μM for 3 h. A separate population was labeled with calcein AM and Vybrant DiD and added to the recipients, and dye transfer was imaged for 5 h.
(D) Summary graph of the degree to which the drugs from the NIH Clinical Collection inhibited Cx46-mediated GJIC. Percent inhibition is relative to DMSO vehicle control treatment (0%) and the pan-gap-junction inhibitor CBX (100%).
(E and F) Validation of the screen results. Cells were treated with increasing concentrations (0.1, 1, and 10 mM) of the top four hits from the screen and one hit that did not show inhibition (purple). Those cells were then either plated and incubated with a labeled population of donor cells (E) to measure GJIC or plated sparsely(F) to assay dye leakage through hemichannels. 0 on the plot in (E) indicates no inhibition was observed. Data are normalized to DMSO (0% inhibition) and CBX (100%), and these experiments were performed in triplicate.
See also Figure S2.
Cx46 Is More Sensitive Than Other Connexins Expressed in GBM to Inhibition by Clofazimine
To specifically target Cx46 in CSCs, the lead compound should have limited efficacy against the 20 other human connexins. To test the specificity of clofazimine for Cx46, we first screened for the additional connexins expressed in GBM using bioinformatics. Using both RNA sequencing and microarray data from the GlioVis database (http://gliovis.bioinfo.cnio.es/) (Bowman et al., 2017), we identified the connexins most highly expressed in GBM compared to normal brain (Figure 3A). In addition to Cx46, which was the most highly expressed relative to normal brain tissue, Cx45 and Cx37 were detected at higher levels in GBM. We also screened clofazimine against Cx43, the most ubiquitously expressed connexin throughout the body (Oyamada et al., 2005). HeLa cells expressing any of these four connexins displayed GJ coupling, as evidenced by the spread of calcein dye (black) from DiD (magenta)-labeled donor cells to unlabeled recipient cells (Figure 3B). As expected, the pan-GJ inhibitor CBX inhibited calcein spread mediated by each connexin. However, while coupling of HeLa cells expressing Cx46 was blocked by clofazimine, cells expressing Cx43, Cx37, and Cx45 continued to exhibit GJIC even in the presence of clofazimine (Figure 3C). These data indicate that of the connexins tested, Cx46-mediated GJIC was specifically inhibited by clofazimine.
Figure 3. Cx46 Is More Sensitive Than Other Connexins Expressed in GBM to Inhibition by Clofazimine.

(A) Heatmap of connexin mRNA expression in GBM compared to normal brain tissue by both RNA sequencing and microarray. Data are from The Cancer Genome Atlas (TCGA) and were obtained from GlioVis. Red indicates higher expression compared to normal brain, while blue indicates lower expression than normal brain tissue.
(B) Parachute dye transfer assay of HeLa cells expressing different connexin proteins. HeLa cells were transfected with connexin proteins, unlabeled cells were plated in a subconfluent monolayer, and cells dual labeled with Vybrant DiD (shown in magenta) and calcein red-orange AM (shown in black) were treated with DMSO, 1 mM clofazimine (CFZ), or 200 mM carbenoxolone (CBX) for 3 h and added to the unlabeled cells. The presence of calcein dye (black) in cells that are not magenta indicates GJIC. Scale bar, 50 μm.
(C) Quantification of (B). The percent inhibition of GJIC with clofazimine is shown compared to that of vehicle and the pan-gap-junction inhibitor CBX. ***p < 0.001 by unpaired Student’s t test with Welch’s correction compared to the DMSO-treated control. Data are represented as mean ± SEM. n = 3.
Clofazimine Preferentially Targets GBM CSCs Compared to Non-CSCs
Our previous studies identified Cx46 as an essential connexin expressed by GBM CSCs, and our preceding results indicated that clofazimine preferentially inhibits coupling of cells expressing Cx46. Based on these results, we hypothesized that clofazimine would specifically target GBM CSCs compared to non-CSCs. Treatment of CSCs and non-CSCs with increasing concentrations of clofazimine from 0.05 to 5 μ M allowed us to calculate half maximal inhibitory concentration (IC50)values of approximately 2 μ M for the CSC population of four PDX specimens (Figure 4A). In contrast, the non-CSC populations did not reach 50% growth inhibition within the same concentration range of clofazimine (Figure 4A; Figure S3A). For comparison, the IC50 value of the immortalized, non-transformed fibroblast cell line NIH 3T3 was calculated to be approximately 86 μ M, indicating that CSCs were dramatically more sensitive than other cell types to clofazimine. Limiting dilution analysis showed a significant and striking inhibitory effect of clofazimine on CSC self-renewal, even at a concentration at which proliferation was only minimally affected (0.5 μM (Figure 4B; Figure S3A). This inhibition of CSC growth and self-renewal was accompanied by a concentration-dependent increase in apoptosis in the CSC population, with minimal induction of apoptosis in the non-CSCs (Figure 4C).
Figure 4. Clofazimine Preferentially Targets GBM CSCs Compared to Non-CSCs.

(A) Summary of IC50 values for clofazimine (CFZ) in four patient-derived xenograft matched CSCs and non-CSCs and the NIH 3T3 untransformed fibro-blast cell line. Cells were treated with increasing concentrations of clofazimine for 3 days, and cell number was measured using CellTiter-Glo. n = at least 3 experiments with cells plated in triplicate. Data are represented as the mean.
(B) CSCs were plated into drug-containing medium at increasing cell densities (1, 5, 10, and 20 cells/well of a 96-well plate), and the number of wells containing spheres was counted after 10–14 days. The online algorithm described in the STAR Methods section was used to calculate stem cell frequency. ***p < 0.001 by c2 test compared to the DMSO-treated control. Data are represented as mean ± range. n = 3 experiments, with 24 technical replicates per cell number per experiment.
(C) CSCs and non-CSCs were treated with clofazi-mine for 3 days, and active caspase-3/7 was measured using Caspase-Glo. The values shown are normalized to the number of total cells at the same time point and are relative to the DMSO control for each cell type. *p < 0.05, **p < 0.01, ***p < 0.001 by unpaired Student’s t test with Welch’s correction compared to the respective DMSO-treated control. Data are represented as mean ± SEM. n = 4 experiments, each performed in triplicate.
(D) CSCs were plated in a subconfluent monolayer on Geltrex, treated with 2 mM clofazimine for 16 h, and microinjected with 2-NBDG (pseudocolored black) and a far-red fluorescently labeled immunoglobulin G (IgG) (pseudocolored magenta). Cells were imaged over 2 h, and the number of cells receiving 2-NBDG from each donor cell was quantified. *p < 0.05 by unpaired Student’s t test with Welch’s correction compared to the DMSO-treated control. Data are represented as mean ± SEM. n = 8 donors over 7 fields (DMSO) and n = 4 donors over 2 fields (clofazimine). Scale bar, 50 μm.
(E) Flow cytometry was used to measure the amount of fluorescent DCF produced from H2DCFDA as a measurement of ROS. CSCs were treated concurrently for 24 h with 50 mM temozolomide (TMZ) and for 16 h with 1 mM clofazimine, manually removed from the plate using a cell scraper, and subjected to flow cytometry r. Representative data from 1 of n = 3 experiments are shown.
(F) Cells were plated in 96-well plates and treated as in (E). Active caspase-3/7 was measured using Caspase-Glo. Data are normalized to the total number of cells at that time and are shown relative to the DMSO-treated control. *p < 0.05, ***p < 0.001 by two-way ANOVA with multiple comparisons compared to treatment with clofazimine alone. Data are represented as mean ± SEM. n = 4 experiments, each performed in triplicate.
See also Figure S3.
Based on our data that clofazimine inhibited dye coupling in HeLa cells expressing Cx46, but not other connexins (Figures 3B and 3C), we hypothesized that clofazimine was similarly acting through an inhibition of GJIC in CSCs. Treatment with clofazimine inhibited the spread of the fluorescent glucose analog 2-NBDG microinjected in CSCs compared to vehicle (Figure 4D), confirming that clofazimine is able to inhibit GJIC in CSCs. To test whether clofazimine induced additional off-target effects, we performed RNA sequencing on CSCs from xenograft specimen T4121 treated with 2 μM clofazimine for a short period of 6 h. Increases and decreases in transcript expression with treatment compared to vehicle were relatively modest, with changes falling within 3-fold of the value of the vehicle-treated samples (Figures S3B and S3C). We performed functional gene annotation and pathway enrichment analysis on the top differentially expressed genes (https://david.ncifcrf.gov/) and found no significant pathway enrichment within reported gene groups, suggesting limited off-target effects with clofazimine treatment. Clofazi-mine has also been reported to target GBM cells by affecting the function of the membrane potassium channel Kv1.3, which is highly expressed in many cancer cell lines compared to normal tissue (Leanza et al., 2015; Venturini et al., 2017). We therefore tested CSCs and non-CSCs to determine whether higher levels of Kv1.3 in the CSCs could be responsible for their sensitivity to clofazimine. However, GBM CSCs from the PDX T4121, which are more sensitive to clofazimine than their non-CSC counterparts, expressed approximately 4-fold less Kv1.3 transcript than non-CSCs (Figure S3D), suggesting that the enhanced sensitivity to clofazimine of CSCs is likely not due to Kv1.3 channels.
Inhibition of GJs has been reported to increase the cellular levels of reactive oxygen species (ROS) (Giardina et al., 2007; Le et al., 2014; Zündorf et al., 2007). As expected, treatment with 1 mM clofazimine for 3 days led to an increase in intracellular ROS, as measured by production of fluorescent 2’,7’-dichlorofluorescein (DCF) from 2’,7’-dichlorodihydrofluorescein diace-tate (H2DCFDA) and detected using flow cytometry (Figure 4E).Based on our observations that clofazimine is toxic to GBM CSCs, we combined clofazimine with temozolomide, the GBM standard-of-care chemotherapy. Temozolomide alone (50 μM) did not increase ROS compared to DMSO vehicle treatment, but a combination of temozolomide with clofazimine increased ROS above the level observed for clofazimine alone (Figure 4E). This increase in ROS was accompanied by a significant increase in apoptosis in cells treated with both temozolomide and clofazi-mine compared to either compound alone (Figure 4F), and this increase with the combination treatment was greater than an additive effect, suggesting that clofazimine sensitizes CSCs to chemotherapy. Altogether, these results indicate that clofazi-mine inhibits GBM CSC growth, survival, and self-renewal, likely through its effects on Cx46-mediated GJIC, and combines with GBM standard-of-care therapies to increase tumor cell death.
Clofazimine Decreases Tumor Growth In Vivo
Clofazimine is generally well tolerated in patients; a 19-year retrospective study of patients receiving multi-drug therapy for leprosy reported no adverse effects of clofazimine when given at the recommended dosage (Nair, 2018). Minor side effects include gastrointestinal intolerance and skin pigmentation; however, rare cases of cardiotoxicity have been observed (Choudhri et al., 1995). The current World Health Organization (WHO) dosing schedule of clofazimine for multibacillary leprosy includes one monthly dose of 300 mg and an additional 50 mg daily in combination with the drugs dapsone and rifampicin for a period of 12 months (Fischer, 2017).
To determine whether clofazimine inhibits tumor growth in vivo, we selected a dosage equivalent to the maximum recommended daily human dose (FDA, 2016), 200 mg/day (2.44 mg/kg based on an average human body weight of 80 kg), solubilized in corn oil. At this dose, the brains of animals treated intraperitoneally for 2 weeks contained less than 0.1 ng of clofazimine per milliliter of brain homogenate (Figure 5A). Even when animals were treated with the supraphysiological dose of 4.88 mg/kg for 2 weeks, the concentration of clofazi-mine in the brain (~ 0.55 ng/mL) was almost 2,000-fold less than the calculated IC50 for CSCs in vitro. We also observed low penetration of the blood-brain barrier by clofazimine in mice microscopically (Figure S4A). Based on this low brain penetration by clofazimine, rather than treating mice with intracranial tumors, we instead treated animals bearing subcutaneous flank tumors generated by implantation of CSCs from the PDX specimen T4121. Clofazimine administration began once all animals presented palpable tumors. Treatment with clofazimine at 2.44 mg/kg by intratumoral injection led to a significant decrease in tumor growth over time (Figure 5B) and a decrease in final tumor size (Figure 5C). A similar effect was observed when animals were treated intraperitoneally with clofazimine (Figure S4B). Because the normal tissue distribution of Cx46 is primarily in the lens, we also tested whether inhibition of Cx46 had an effect on animal vision and observed no significant changes compared to treatment with vehicle (Figure S4C). Altogether, our results indicate that clofazimine targeting of Cx46-mediated GJIC is able to slow tumor growth without affecting other major Cx46 functions, including vision.
Figure 5. Clofazimine Decreases Tumor Growth In Vivo.

(A) Male and female (n = 4 each) NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice were treated with clofazimine (CFZ) at 2.44 or 4.88 mg/kg in 200 mL of corn oil by intraperitoneal (IP) injection daily for 2 weeks under a treatment plan of 5 days on, 2 days off, and 5 days on the treatment. On day 12, animals were euthanized, and brains were homogenized in PBS and subjected to mass spectrometry for clofazimine. Data are shown as mean ± SEM.
(B) (B and C) Male NSG mice (n = 4 per arm) were injected with 5 3 105 T4121 CSCs into their right flanks. Four weeks later, when tumors became palpable, animals were treated daily with clofazimine at 2.44 mg/kg in 10 mL of corn oil injected directly into the tumor for 8 days. Tumor size was measured using digital calipers, and the change in tumor volume over time (B) and the final tumor volume (C) are provided. *p < 0.05 by two-way ANOVA to test for differences between the curves in (B). The p value for (C) was generated using Student’s t test with Welch’s correction.
The data are shown as the mean ± SEM (B) or SD (C), and for (C), all data points are shown. See also Figure S4.
DISCUSSION
Connexin proteins serve three main cellular functions: exchange of small molecules between cells, exchange of small molecules between cells and the extracellular space, and mediation of intracellular protein-protein interactions. We previously showed that Cx46 is required for GBM CSC proliferative ability, survival, self-renewal, and tumor formation (Hitomi et al., 2015). Here, using point mutations that disrupt specific functions of the protein, we show that the essential function of Cx46 in these cells is the formation of functional Cx46 GJs. It remains an open question as to the key tumor cell and CSC mediators that pass through GJs, which likely include a combination of ions (K+, Ca2+, and Na+), ROS and antioxidants, metabolites such as glucose, cyclic AMP (cAMP), and non-coding and miRNAs (Lim et al., 2011; Loewenstein and Kanno, 1964; Patel et al., 2016). Our results contrast with the hypothesis that aberrant hemichannel activity of connexins underlies their role in pathologies (Kim et al., 2016; Leybaert et al., 2017) and suggest that therapies designed to target GJIC mediated by specific connexins may be valuable for certain diseases, including GBM.
To identify Cx46-specific inhibitors, we screened FDA-approved compounds for Cx46 GJIC inhibitors and identified the anti-leprosy drug clofazimine, which inhibited GBM CSC cell-cell communication; decreased CSC growth, survival, and self-renewal; and decreased tumor growth in a subcutaneous tumor model. Although pan-GJ inhibitors are available clinically and have shown efficacy in our models (Hitomi et al., 2015), specific inhibitors for connexin isoforms have yet to be identified or developed. Most connexin modulators developed so far, the majority of which are designed to target Cx43 or multiple connexin isoforms, are peptide mimetics that interrupt a specific binding activity of the molecule—either within the molecule or between molecules—and thus affect protein or channel function (Jaraíz-Rodríguez et al., 2017; Naus and Giaume, 2016). Although little is known about precisely how these mimetics modulate connexin activity, they possess varying efficiencies at inhibiting and/or stimulating both GJ and hemichannel activity (Evans et al., 2012; Wang et al., 2013). However, due to the homology among connexin isoforms, many of these mimetics fail to exhibit specificity for a specific connexin. In contrast, we show that the small molecule clofazi-mine is more specific for Cx46 than Cx43, Cx45, and Cx37. Few small molecules have been identified to target connexins; those that have been developed increase GJIC in astrocytes or specifically target hemichannels, neither of which are relevant to blocking Cx46-mediated GJIC in GBM CSCs (Naus and Giaume, 2016).
Previous studies described an inhibitory role for clofazimine in GBM cells. Significant apoptosis has been observed in conventional GBM cell lines treated with clofazimine, and this cell death was attributed to inhibition of the mitochondrial membrane ion channel Kv1.3 (Venturini et al., 2017). We observed similar cell death of GBM CSCs upon treatment with clofazimine, with little effect on non-CSCs. However, we also detected 4-fold higher levels of Kv1.3 transcript in the clofazimine-resistant non-CSC population, suggesting that clofazimine does not act through Kv1.3 inhibition in our hands. Clofazimine was also previously identified in a screen to inhibit growth of the conventional GBM cell line U87 (Jiang et al., 2014). In contrast, rather than screening for compounds that inhibit GBM cell growth in culture, we identified a CSC essential process, Cx46-mediated GJIC, and screened for inhibitors specifically targeting this cellular process. Our future work will investigate the mechanism by which clofazimine blocks Cx46-mediated intercellular communication. Based on our observations that the cysless mutant inhibits CSC maintenance similarly to clofazimine and that few transcripts were altered by short-term treatment, we speculate that the drug could act extracellularly to physically block the channel opening or hemi-channel-hemichannel docking. However, it remains possible that clofazimine functions in another manner, for example, by altering membrane permeability, mitochondrial function, or cell signaling.
Although clofazimine shows promise for targeting GBM CSCs, there are several challenges to its therapeutic use. Here, we show that clofazimine exhibits minimal penetration of the blood-brain barrier, and its low solubility and high lipophilicity are also barriers to translation for brain tumors. There has been conflicting evidence for whether clofazimine is able to penetrate the blood-brain barrier; while some studies have reported no detectable levels in the brain (Baik et al., 2013; Holdiness, 1989), other studies detected a level of 156 ng/mL of clofazimine in the brain of mice treated with 25 mg/kg of the drug (Baijnath et al., 2015) and an effect on Kv1.3 channels in the brain of animals treated with clofazimine at 50 mg/kg after traumatic brain injury (Reeves et al., 2016). In contrast, using the equivalent of twice the maximum tolerated human dose (4.88 mg/kg in mice), we detected only 0.55 ng/mL of clofazimine in the brain, a level approximately 2,000-fold lower than the IC50 value for CSCs, using mass spectrometry. This difference may be due to differences in concentration, delivery route, or solvent. In a previous report, clofazimine failed to inhibit growth of intracranial syngeneic mouse gliomas (Venturini et al., 2017), which is supported by our observations that clofazimine at human-relevant doses does not effectively cross the blood-brain barrier. These challenges would preclude the direct use of clofazimine to treat patients with brain tumors. However, future medicinal chemistry derivatization of clofazi-mine to optimize solubility and blood-brain barrier penetration may allow us to develop a more optimal analog based on the clofazimine scaffold for further pre-clinical and clinical testing. These optimized compounds could lead to improved next-generation therapies with reduced side effects for patients with GBM.
STAR+METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Requests for reagents should be directed to the Lead Contact, Dr. Justin D. Lathia, at lathiaj@ccf.org.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Origin of Cells
Established GBM xenografts T4121, T3691, and T387 were previously reported (Alvarado et al., 2016; Bao et al., 2006; Schonberg et al., 2015) and were obtained via a material transfer agreement from Duke University. L2 cells were obtained from the University of Florida (Deleyrolle et al., 2011; Siebzehnrubl et al., 2013). All human GBM samples were originally established under an IRB-approved protocol that facilitated the generation of xenografts in a de-identified manner from excess tissue taken from consented patients. GBM cells were passaged in immune-deficient NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice (obtained from The Jackson Laboratory,Bar Harbor, ME, USA) and dissociated from established mouse xenografts under Cleveland Clinic-approved protocols. Six-week-old female mice were unilaterally injected subcutaneously in the flank with freshly dissociated human GBM cells, and animals were sacrificed by CO2 asphyxiation and secondary cervical dislocation when tumor volume exceeded 5% of the animal’s body weight. HeLa and NIH 3T3 cells were obtained from ATCC.
Subcutaneous Tumors
Six- to eight-week-old immunocompromised male or female NSG mice were injected with either 5×105 or 1×106 CSCs from the patient-derived xenograft T4121 into their right flank as specified in the figure legends. Three to four weeks later, when tumors were palpable, mice were treated with clofazimine. Clofazimine was solubilized in corn oil, and mice received either 100 mL via intraperitoneal injection or 10 mL by intratumoral injection. Tumor dimensions were measured using digital calipers, and tumor volume was calculated assuming that the tumors were ellipsoid using the formula: tumor volume = (4/3)p(width/2)2(height/2). Animals were sacrificed when they reached endpoint. All animal experiments were performed under Cleveland Clinic-approved Institutional Animal Care and Use Committee-approved protocols.
Cell Culture
Xenograft tumors were dissociated using papain (Worthington Biochemical Corporation, Lakewood, NJ) and cultured overnight in supplemented neurobasal medium (neurobasal medium (Life Technologies) with 2% B27 (Life Technologies), 1% penicillin/streptomycin (Life Technologies), 1 mM sodium pyruvate (Life Technologies), 2 mM L-glutamine, 20 ng/mL EGF (R&D Systems, Minneapolis, MN, USA), and 20 ng/mL FGF-2 (R&D Systems)). T4121, T3691, and T387 xenografts were sorted for CD133+ (CSC) and CD133-(non-CSC) populations using the CD133 Magnetic Bead Kit for Hematopoietic Cells (CD133/2; Miltenyi Biotech, San Diego, CA, USA). CD133+ cells were maintained in supplemented neurobasal. CD133- cells were maintained in DMEM with 10% FBS and 1% pen/strep. L2 cells were maintained in these divergent media conditions without sorting.
HeLa and NIH 3T3 cells were maintained in DMEM with 10% FBS and 1% pen/strep. The HeLa-Cx46 stable cell line was cultured with the addition of 400 mg/mL G418. All cells were grown in a humidified incubator at 37 C with 5% CO2.
METHOD DETAILS
Plasmids and DNA Constructs
The Cx46 expression vector was created by inserting the Cx46 cDNA (catalog# RDC0535, R&D Systems) between the HindIII and XbaI sites of pEGFP-N3, excising the GFP tag. This backbone was used for site-directed mutagenesis to introduce the L11S, T19M, and cysless mutations, using the primers shown in Table S2. The primers for the cysless mutant were designed so that the PCR reactions must be performed sequentially from the N-terminus to the C-terminus.
pLPCX-Cx43-IRES-GFP was obtained from Addgene (#65433). pcDNA3.1/Hygro(+)-GJC1 (Cx45; cloneID: OHu04829) and pcDNA3.1/Hygro(+)-GJA4 (Cx37; cloneID: OHu33346) were obtained from GenScript.
Transfections and Establishment of HeLa-Cx46 Stable Cell Line
For GBM CSC transfections, 1×106 cells were plated per well of a 6-well plate adherently on Geltrex (Thermo Fisher Scientific) to obtain a confluence of approximately 75%–80%. Six hours later, cells were transfected with Cx46 or its mutant forms using FuGENE HD (Promega) according to the manufacturer’s protocol. Briefly, cells were transfected with 5 mg total DNA (4 mg of connexin and 1 mg pEGFP-N3 to track transfection efficiency) using 15 ml FuGENE per well. The following day, cells were removed from the plate using Accutase (BioLegend) and plated for downstream assays. pEGFP-N3 was used as a vector control.
HeLa cells were seeded at 400,000 cells per well in a 6-well plate and transfected using XtremeGene HP (Roche) according to the manufacturer’s protocol. In brief, each well received 2 ug of DNA and 6 uL of XtremeGene reagent. Dye-transfer recipients were plated 24 hours after transfection, and donors were plated and images taken at 48 hours post-transfection. Stable HeLa-Cx46 cells were derived by transfecting HeLa cells with Cx46 (without the GFP tag). Cells were selected with G418 (400 mg/mL), and single-cell clones were tested for the ability to exhibit dye coupling.
Compounds
Clofazimine was obtained from Sigma-Aldrich (catalog # C8895) and solubilized in DMSO at a concentration of 10 mM for in vitro experiments and in corn oil for in vivo experiments.
Proliferation and Apoptosis
For proliferation, IC50, and apoptosis assays, 2,000 cells were plated in growth media per well of a white-walled 96-well plate in trip-licate. The number of cells was measured using CellTiter-Glo (Promega) on days 0, 1, 3, 7, and 10 according to the manufacturer’s protocol, using ATP content as a surrogate of cell number, and apoptosis was measured using CaspaseGlo 3/7 (Promega) on days 1 and 3 according to the manufacturer’s protocol. For the proliferation of GBM CSCs in the presence of Cx46 and Cx46 mutants, similar results were obtained using the DNA-based CyQUANT Direct Cell Proliferation Assay Kit (Thermo Fisher Scientific). For drug treatments, cells were seeded in triplicate at 2,000 cells per well of a 96-well plate, and the appropriate concentration of drug was added 6–24 hours later. Cells were analyzed both at 0 and 72 h after treatment with drug.
Limiting Dilution Analysis
CSCs were dissociated using Accutase and plated in a 96-well plate at increasing cell numbers (1, 5, 10, and 20 cells/well) with 24 replicates per cell number. Cells were plated into drug-containing media, and the number of wells containing spheres was counted after 10–14 days. An online algorithm (http://bioinf.wehi.edu.au/software/elda/) (Hu and Smyth, 2009) was used to calculate stem cell frequency.
cDNA and qPCR
For qPCR, RNA was extracted from cells using TriZOL (Life Technologies) according to the manufacturer’s protocol. A total of 1 mg of RNA was used for reverse transcription using a qScript cDNA Synthesis Kit (QuantaBio) according to the manufacturer’s recommendations. Equal volumes of cDNA were amplified using Fast SYBRâ Green Master Mix (Applied Biosystems) on a Step-One Plus Real-Time PCR system (Applied Biosystems). Data were analyzed using the ΔΔCt method to calculate relative levels of product. qPCR primers are provided in the Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological Samples | ||
| GBM xenograft T4121 | Duke University | T4121 |
| GBM xenograft T3691 | Duke University | T3691 |
| GBM xenograft T387 | Duke University | T387 |
| GBM xenograft L2 | University of Florida | hGBM L2 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Clofazimine | Sigma-Aldrich | C8895 |
| Geltrex | Life Technologies | A1413201 |
| FuGENE HD | Promega | E2311 |
| X-tremeGene HP | Roche | 6366244001 |
| Vybrant DiD | Thermo Fisher | V22887 |
| 2-NBDG | Thermo Fisher | N13195 |
| Temozolomide | Santa Cruz | sc-203292A |
| H2DCFDA | Thermo Fisher | D399 |
| Calcein AM | Thermo Fisher | C3099 |
| Calcein red-orange AM | Thermo Fisher | C34851 |
| Critical Commercial Assays | ||
| CellTiter-Glo | Promega | G7572 |
| CaspaseGlo 3/7 | Promega | G8090 |
| Papain Dissociation Kit | Worthington Biochemical | LK003150 |
| qSCRIPT cDNA Supermix | Quanta Biosciences | 95048-100 |
| SYBR-Green Mastermix | SA Biosciences | 330523 |
| Experimental Models: Cell Lines | ||
| HeLa cells | ATCC | CCL-2 |
| NIH3T3 cells | ATCC | CRL-1658 |
| Experimental Models: Organisms/Strains | ||
| Mouse: NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ | The Jackson Laboratory | 005557 |
| Oligonucleotides | ||
| qPCR primer Kv1.3 F: CAAAACGGGCAATTCCACTG | This paper | N/A |
| qPCR primer Kv1.3 R: TGAGCACAGCATGTCACTTG | This paper | N/A |
| qPCR primer Cx46 F: TGCACAGGAGCACTCCA | This paper | N/A |
| qPCR primer Cx46 R: GCGTGGACACGAAGATGAT | This paper | N/A |
| Primers for creating Cx46 mutants, see Table S2. | This paper | |
| Recombinant DNA | ||
| Cx46 cDNA | R&D Systems | RDC0535 |
| pLPCX-Cx43-IRES-GFP | Addgene | 65433 |
| pcDNA3.1/Hygro(+)-GJC1 | GenScript | cloneID: OHu04829 |
| pcDNA3.1/Hygro(+)-GJA4 | GenScript | cloneID: OHu33346 |
| Cx46 L11S | This paper | N/A |
| Cx46 T19M | This paper | N/A |
| Cx46 cysless | This paper | N/A |
| Software and Algorithms | ||
| Extreme limiting-dilution analysis | Hu and Smyth, 2009 | http://bioinf.wehi.edu.au/software/elda/ |
| GlioVis | Bowman et al., 2017 | http://gliovis.bioinfo.cnio.es |
| DAVID | Jiao et al., 2012 | https://david.ncifcrf.gov/ |
Screen of the NIH Clinical Collection for Cx46 Inhibitors Non-labeled Cx46-HeLa cells were seeded at 20,000 cells per well in a 96-well plate in DMEM with 10% FBS and 1% pen/strep. The following morning, drugs were added to a concentration of 10 μM to 80 of the wells, leaving 16 for positive and negative inhibition controls. CBX (200 mM) was used a positive control for dye transfer inhibition, while negative control wells were left untreated. Separately, a population of calcein AM/Vybrant DiD dual-labeled Cx46-HeLa cells was generated. These cells were incubated in serum-free DMEM containing calcein AM (resuspended in 50 μL of DMSO and used at 1:1000; Thermo Fisher Scientific) and Vybrant DiD (1:500, Thermo Fisher Scientific) at 37 C for 1 h. Following a 3 h incubation of the unlabeled recipients with drug, the dual-labeled donor population was added at a concentration of 3,000 cells/well. These cells were incubated together at 37 C for 5 h and then imaged. Each plate contained 80 drugs and 16 controls, accounting for 9 experimental runs. Each drug was screened one time per drug as a cursory screen. Following the identification of possible targets, a secondary screen of a selection of top hits that were visually verified and readily available was performed at drug concentrations of 10 μM, 1 μM, and 0.1 μM.
For screen quantification, calcein fluorescence was used to create a mask to eliminate any cells left entirely unlabeled and any background fluorescence. The Vybrant DiD fluorescence image was used to create another binary mask to define DiD-positive donor cells. These mask images were given values of 0 (no dye present) or 1 (dye present) and then multiplied by the calcein image. ImageJ particle analysis of the resulting product images provided us with the raw integrated density (RID) of the total calcein dye per imaged cell. The sum of the particle analysis of the product of the calcein mask and the calcein image gave the total calcein amount, and that of the product of the DiD mask and the calcein image gave the amount of calcein retained in the donor cells. Percent transfer was calculated by ((total calcein – retained calcein)x100)/total calcein.
For hemichannel function assessment, labeled populations were generated as described above and seeded at 3,000 cells per well. Cells were given an hour to adhere and then imaged every 15 minutes for 5 hours. Loss of calcein through hemichannels was quantified as the percent of dye that was lost at 5 h compared to time 0.
For HeLa cells expressing different connexin proteins, cells were prepared and imaged as stated above. Images were quantified as the number of unlabeled cells (recipients) receiving calcein dye per donor cell.
For microinjection of CSCs, subconfluent monolayers of cells plated on Geltrex-coated glass coverslips in 35 mm dishes were pre-treated for 16 h with the indicated concentration of clofazimine in growth media. Cells were then injected with far-red fluorescent IgG and the fluorescent glucose analog 2-(N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino)-2-deoxyglucose (2-NBDG) as described (Hitomi et al., 2015) and imaged as above. Images were again quantified as the number of unlabeled cells (recipients) receiving calcein dye per donor cell.
GlioVis Analysis of Connexins in GBM
The Cancer Genome Atlas (TCGA) dataset was interrogated using GlioVis (http://gliovis.bioinfo.cnio.es) (Bowman et al., 2017) for microarray (Agilent-4502A) and RNA-seq levels of all available connexin genes. Relative levels of non-tumor and GBM tissues were analyzed, and the fold change is represented as a heatmap.
RNA Sequencing
T4121 CSCs were treated with clofazimine at 2 mM for 6 hours and lysed for RNA using a Nucleospin RNA isolation kit (Macherey-Nagel, Duren, Germany). RNA-seq libraries were prepared using ~10,000 ng of total RNA. Briefly, the protocol included PolyA+ RNAselection, cDNA synthesis, end repair, A-base addition, and ligation of the Illumina-indexed adapters according to previously published methods (Zhang et al., 2012). Total transcriptome libraries were prepared as previously described. Library quality and quantity were measured on an Agilent 2100 Bioanalyzer for product size and concentration. Libraries were also precisely quantified by using a KAPA Library Quantification kit prior to loading on the sequencer and pooled at equimolar quantities between samples. Single-end libraries were sequenced with the Illumina HiSeq 2500 (1×5 read length), with sequence coverage up to 20 M total reads.
Single-end transcriptome sequencing reads were aligned to the human reference genome (GRCh37/hg19) using the spliced read mapper TopHat2 (TopHat 2.0.4) (Kim et al., 2013). Gene expression, as fragments per kilobase of exon per million fragments mapped (FPKM; normalized measure of gene expression), was calculated using Cufflinks (Trapnell et al., 2012). We considered differential expression of the gene when the calculated p < 0.01 and there was a 1.5-fold difference (increase or decrease).
The database for annotation, visualization and integrated discovery (DAVID) analysis was used for functional clustering and annotation of differentially expressed genes (https://david.ncifcrf.gov/) (Jiao et al., 2012). DAVID is a web-based online bioinformatics resource that aims to provide tools for pathway mining and the subsequent functional interpretation of large lists of genes/proteins using a comprehensive and exhaustive set of knowledge-based libraries. The publication on the DAVID webserver suggests investigating clusters with an enrichment score ≥ 1.3, while our highest enrichment score was 1.06, suggesting no major disturbance of any functional pathway/gene ontology group.
Reactive Oxygen Species (ROS)
To measure intracellular ROS, CSCs were concurrently treated with 50 mM temozolomide for 24 h and 1 mM clofazimine for 16 h. Cells were then collected and incubated with 1 mM H2DCFDA (Life Technologies) for 15 min at 37 C. Cells were then washed twice in PBS, and the green fluorescent DCF produced was analyzed on a BDFortessa flow cytometer. DAPI exclusion was used to gate for live cells, and H2O2 was used as a positive control for ROS production.
Blood-Brain Barrier
To assess the permeation of clofazimine into normal brain tissue, mice were intraperitoneally injected with 100 mL of a 25 mg/mL suspension of clofazimine in corn oil or vehicle. After 10 minutes of circulation, mice were euthanized, and brains were extracted, snap frozen in isopentane, and sliced into 20 mm sections. Slides were analyzed using a MVX10 MacroView microscope (Olympus) equipped with an ORCA_Flash4.0 v2 sCMOS fluorescent camera (Hamamatsu). A linear range of standards in the brain was developed with varying concentrations (16 μg/mg to 2.5 μg/mg).
Mass Spectrometry
Brains from mice treated IP with 2.44 mg/kg and 4.88 mg/kg clofazimine were excised and homogenized in PBS. For mass spec-trometry, clofazimine was used as the internal standard. Brain homogenate (50 μL) was mixed with 150 μL methanol and then centrifuged at 12,000 x g for 10 min. The supernatant (100 μL) was transferred to an HPLC vial. For LC/MS/MS analysis of clofazimine, 2 μL supernatant was injected into a Shimadzu LCMS-8050 for quantitation of clofazimine. A gradient with a flow rate of 0.3 μL/min was used to separate clofazimine by reverse-phase chromatography using a Prodigy C18 column (2.1 ×50 μm, 5 μm) from Phenomenex.The mobile phases were A (water containing 5 mM ammonium acetate) and B (methanol containing 5 mM ammonium acetate). The run started with 70% mobile phase B from 0 to 2 min. Solvent B was then increased linearly to 100% B from 2 to 6 min and held at 100% B from 6 to 12 min. The column was finally re-equilibrated with 70% B for 7 min. The HPLC eluent was directly injected into a triple quadrupole mass spectrometer (Shimadzu LCMS-8050), and the clofazimine was ionized at ESI positive mode, using selected Reaction monitoring (SRM). The SRM transitions (m/z) were 474 to 432. For data analysis, the software Labsolutions was used to process the data and obtain the peak areas for clofazimine. The external standard calibration curve was used to calculate the concentration of clofazimine in the brain homogenate samples.
Retinal Imaging Procedures
Animal preparation and imaging procedures have been previously described (Bell et al., 2015). Briefly, mice were anesthetized using an IP injection of sodium pentobarbital (68 mg/kg). Mydriasis was induced using a 0.5 mL of 0.5% tropicamide phenylephrine mixture.Topical anesthesia was induced using 0.5% proparacaine. Cornea hydration and ocular media opacities were minimized using frequent applications of hydrating drops and topical eye shields (Bell et al., 2014). Following the procedure, eyes were covered with puralube ointment. Mice recovered in a warmed Plexiglas chamber with supplemental oxygen.
Confocal scanning laser ophthalmoscope (cSLO) imaging was performed using an HRA2 system (Heidelberg Engineering, Inc). A wide-field objective (55) was used to image the retina with the optic nerve disk centrally located within the image frame. Imaging modes of infrared reflectance (IR-cSLO) at 800 nm and blue peak autofluorescence (BAF-cSLO) at 488 nm were used to image the retina and vitreoretinal interface.
Spectral-domain optical coherence tomography was performed following cSLO to examine and compare the in-depth retinal morphology between treatment groups. Orthogonal B-scans (1000 a-scans/b-scan c 15 frames) were collected through the optic disk from the horizontal and vertical meridians. The 15 frames from each meridian were co-registered and averaged using ImageJ and StackReg and TurboReg Plugins (Schneider et al., 2012; Thévenaz et al., 1998).
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical tests were performed using GraphPad Prism 6.0. Information regarding the numbers of experimental replicates, statistical tests performed, and significance values can be found in the figure legend for each figure panel. p % 0.05 was considered significant.
Supplementary Material
Highlights.
Cx46-mediated cell-cell communication is essential for GBM self-renewal
A communication-based screening platform identifies clofazimine as a Cx46 inhibitor
Clofazimine targets CSCs and has little effect on non-stem tumor cells
Clofazimine treatment of tumor-bearing mice extends survival in vivo
ACKNOWLEDGMENTS
We thank the members of the Lathia laboratory and the Reizes laboratory for insightful discussion and constructive comments on the manuscript. We thank Kevin Stoltz for assistance setting up the screening platform, Joseph Gerow and Eric Schultz for flow cytometry assistance, Amanda Mendelsohn for the illustrations included in this manuscript, and Earl Poptic and Melanie Hoffner in the LRI Molecular Screening Core for assistance with small-molecule screening. This work was funded by the NIH (grant NS089641), the Cleveland Clinic VeloSano Bike Race, and Cleveland Clinic Innovations (to M.H. and J.D.L.). D.J.S. is supported by an NIH Kirschstein NRSA (F32CA213727). D.B. was supported by a Case Comprehensive Cancer Center training grant (T32CA059366). The Lathia laboratory also receives funding from the NIH (grants NS083629 and CA157948), a Distinguished Scientist Award from the Sontag Foundation, and the Case Comprehensive Cancer Center.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j. celrep.2019.03.079.
DECLARATIONS OF INTERESTS
The Cleveland Clinic has filed a pending patent on using clofazimine and similar compounds to target connexin 46 in GBM and other conditions: U.S. provisional application 62/560,251 filed September 19, 2017, and provisional application 62/644,687 filed March 19, 2018.
REFERENCES
- Aasen T, Mesnil M, Naus CC, Lampe PD, and Laird DW (2016). Gap junctions and cancer: communicating for 50 years. Nat. Rev. Cancer 16, 775–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarado AG, Thiagarajan PS, Mulkearns-Hubert EE, Silver DJ, Hale JS, Alban TJ, Turaga SM, Jarrar A, Reizes O, Longworth MS, et al. (2016). Glioblastoma cancer stem cells evade innate immune suppression of self-renewal through reduced TLR4 expression. Cell Stem Cell 6, 450–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baijnath S, Naiker S, Shobo A, Moodley C, Adamson J, Ngcobo B, Bester LA, Singh S, Kruger HG, Naicker T, and Govender T (2015). Evidence for the presence of clofazimine and its distribution in the healthy mouse brain. J. Mol. Histol 46, 439–442. [DOI] [PubMed] [Google Scholar]
- Baik J, Stringer KA, Mane G, and Rosania GR (2013). Multiscale distribution and bioaccumulation analysis of clofazimine reveals a massive immune system-mediated xenobiotic sequestration response. Antimicrob. Agents Chemother 57, 1218–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao X, Chen Y, Reuss L, and Altenberg GA (2004). Functional expression in Xenopus oocytes of gap-junctional hemichannels formed by a cysteine-less connexin 43. J. Biol. Chem 279, 9689–9692. [DOI] [PubMed] [Google Scholar]
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, and Rich JN (2006). Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444, 756–760. [DOI] [PubMed] [Google Scholar]
- Bell BA, Kaul C, and Hollyfield JG (2014). A protective eye shield for prevention of media opacities during small animal ocular imaging. Exp. Eye Res 127, 280–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell BA, Kaul C, Bonilha VL, Rayborn ME, Shadrach K, and Hollyfield JG (2015). The BALB/c mouse: Effect of standard vivarium lighting on retinal pathology during aging. Exp. Eye Res 135, 192–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman RL, Wang Q, Carro A, Verhaak RG, and Squatrito M (2017). GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro-oncol. 19, 139–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, and Parada LF (2012). A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 488, 522–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhri SH, Harris L, Butany JW, and Keystone JS (1995). Clofazi-mine induced cardiotoxicity—a case report. Lepr. Rev 66, 63–68. [DOI] [PubMed] [Google Scholar]
- Deleyrolle LP, Harding A, Cato K, Siebzehnrubl FA, Rahman M, Azari H, Olson S, Gabrielli B, Osborne G, Vescovi A, and Reynolds BA (2011). Evidence for label-retaining tumour-initiating cells in human glioblastoma. Brain 134, 1331–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfgang C, Eckert R, Lichtenberg-Fraté H, Butterweck A, Traub O, Klein RA, Hulser DF, and Willecke K (1995). Specific permeability and selective formation of gap junction channels in connexin-transfected HeLa cells. J. Cell Biol 129, 805–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans WH, Bultynck G, and Leybaert L (2012). Manipulating connexin communication channels: use of peptidomimetics and the translational outputs. J. Membr. Biol 245, 437–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FDA. (2016). LAMPRENE (clofazimine). Novartis. https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/019500s013lbl.pdf. [Google Scholar]
- Fischer M (2017). Leprosy—an overview of clinical features, diagnosis, and treatment. J. Dtsch. Dermatol. Ges 15, 801–827. [DOI] [PubMed] [Google Scholar]
- Giardina SF, Mikami M, Goubaeva F, and Yang J (2007). Connexin 43 confers resistance to hydrogen peroxide-mediated apoptosis. Biochem. Biophys. Res. Commun 362, 747–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodenough DA, and Paul DL (2003). Beyond the gap: functions of un-paired connexon channels. Nat. Rev. Mol. Cell Biol 4, 285–294. [DOI] [PubMed] [Google Scholar]
- Goodenough DA, and Paul DL (2009). Gap junctions. Cold Spring Harb. Perspect. Biol 1, a002576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen L, Yao W, Eiberg H, Funding M, Riise R, Kjaer KW, Hejtmancik JF, and Rosenberg T (2006). The congenital “ant-egg” cataract phenotype is caused by a missense mutation in connexin46. Mol. Vis 12, 1033–1039. [PubMed] [Google Scholar]
- Hitomi M, Deleyrolle LP, Mulkearns-Hubert EE, Jarrar A, Li M, Sinyuk M, Otvos B, Brunet S, Flavahan WA, Hubert CG, et al. (2015). Differential connexin function enhances self-renewal in glioblastoma. Cell Rep. 11, 1031–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holdiness MR (1989). Clinical pharmacokinetics of clofazimine. A review. Clin. Pharmacokinet 16, 74–85. [DOI] [PubMed] [Google Scholar]
- Hu Y, and Smyth GK (2009). ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays.J. Immunol. Methods 347, 70–78. [DOI] [PubMed] [Google Scholar]
- Jaraíz-Rodríguez M, Tabernero MD, González-Tablas M, Otero A, Orfao A, Medina JM, and Tabernero A (2017). A Short Region of Connexin43 Reduces Human Glioma Stem Cell Migration, Invasion, and Survival through Src, PTEN, and FAK. Stem Cell Reports 9, 451–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P, Mukthavaram R, Chao Y, Bharati IS, Fogal V, Pastorino S, Cong X, Nomura N, Gallagher M, Abbasi T, et al. (2014). Novel anti-glioblastoma agents and therapeutic combinations identified from a collection of FDA approved drugs. J. Transl. Med 12, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao X, Sherman BT, Huang da W., Stephens R, Baseler MW, Lane HC, and Lempicki RA (2012). DAVID-WS: a stateful web service to facilitate gene/protein list analysis. Bioinformatics 28, 1805–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Davidson JO, Gunn KC, Phillips AR, Green CR, and Gunn AJ (2016). Role of Hemichannels in CNS Inflammation and the Inflammasome Pathway. Adv. Protein Chem. Struct. Biol 104, 1–37. [DOI] [PubMed] [Google Scholar]
- Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, and Rich JN (2015). Cancer stem cells in glioblastoma. Genes Dev. 29, 1203–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le HT, Sin WC, Lozinsky S, Bechberger J, Vega JL, Guo XQ, Sáez JC, and Naus CC (2014). Gap junction intercellular communication mediated by connexin43 in astrocytes is essential for their resistance to oxidative stress. J. Biol. Chem 289, 1345–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leanza L, Venturini E, Kadow S, Carpinteiro A, Gulbins E, and Becker KA (2015). Targeting a mitochondrial potassium channel to fight cancer. Cell Calcium 58, 131–138. [DOI] [PubMed] [Google Scholar]
- Leithe E, Mesnil M, and Aasen T (2018). The connexin 43 C-terminus: A tail of many tales. Biochim. Biophys. Acta Biomembr 1860, 48–64. [DOI] [PubMed] [Google Scholar]
- Leybaert L, Lampe PD, Dhein S, Kwak BR, Ferdinandy P, Beyer EC, Laird DW, Naus CC, Green CR, and Schulz R (2017). Connexins in Cardiovascular and Neurovascular Health and Disease: Pharmacological Implications. Pharmacol. Rev 69, 396–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim PK, Bliss SA, Patel SA, Taborga M, Dave MA, Gregory LA, Greco SJ, Bryan M, Patel PS, and Rameshwar P (2011). Gap junction-mediated import of microRNA from bone marrow stromal cells can elicit cell cycle quiescence in breast cancer cells. Cancer Res. 71, 1550–1560. [DOI] [PubMed] [Google Scholar]
- Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR, Lu L, Irvin D, Black KL, and Yu JS (2006). Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol. Cancer 5, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewenstein WR, and Kanno Y (1964). Studies on an Epithelial (Gland) Cell Junction. I. Modifications of Surface Membrane Permeability. J. Cell Biol 22, 565–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGirt MJ, Chaichana KL, Gathinji M, Attenello FJ, Than K, Olivi A, Weingart JD, Brem H, and Quiñones-Hinojosa AR (2009). Independent association of extent of resection with survival in patients with malignant brain astrocytoma. J. Neurosurg 110, 156–162. [DOI] [PubMed] [Google Scholar]
- Nair SP (2018). A 19-Year Retrospective Study of Adverse Drug Reactions to Multidrug Therapy in Leprosy Requiring a Change in Regime. Indian Dermatol. Online J 9, 33–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naus CC, and Giaume C (2016). Bridging the gap to therapeutic strategies based on connexin/pannexin biology. J. Transl. Med 14, 330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyamada M, Oyamada Y, and Takamatsu T (2005). Regulation of connexin expression. Biochim. Biophys. Acta 1719, 6–23. [DOI] [PubMed] [Google Scholar]
- Patel JS, Hu M, Sinha G, Walker ND, Sherman LS, Gallagher A, and Rameshwar P (2016). Non-coding RNA as mediators in microenvironment-breast cancer cell communication. Cancer Lett. 380, 289–295. [DOI] [PubMed] [Google Scholar]
- Reeves TM, Trimmer PA, Colley BS, and Phillips LL (2016). Targeting Kv1.3 channels to reduce white matter pathology after traumatic brain injury. Exp. Neurol 283 (Pt A), 188–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santhiya ST, Kumar GS, Sudhakar P, Gupta N, Klopp N, Illig T, Söker T, Groth M, Platzer M, Gopinath PM, and Graw J (2010). Molecular analysis of cataract families in India: new mutations in the CRYBB2 and GJA3 genes and rare polymorphisms. Mol. Vis 16, 1837–1847. [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonberg DL, Miller TE, Wu Q, Flavahan WA, Das NK, Hale JS, Hubert CG, Mack SC, Jarrar AM, Karl RT, et al. (2015). Preferential Iron Trafficking Characterizes Glioblastoma Stem-like Cells. Cancer Cell 28, 441–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siebzehnrubl FA, Silver DJ, Tugertimur B, Deleyrolle LP, Siebzehnrubl D, Sarkisian MR, Devers KG, Yachnis AT, Kupper MD, Neal D, et al. (2013). The ZEB1 pathway links glioblastoma initiation, invasion and chemoresistance. EMBO Mol. Med 5, 1196–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinyuk M, Alvarado AG, Nesmiyanov P, Shaw J, Mulkearns-Hubert EE, Eurich JT, Hale JS, Bogdanova A, Hitomi M, Maciejewski J, et al. (2015). Cx25 contributes to leukemia cell communication and chemosensitivity. Oncotarget 6, 31508–31521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stout C, Goodenough DA, and Paul DL (2004). Connexins: functions without junctions. Curr. Opin. Cell Biol 16, 507–512. [DOI] [PubMed] [Google Scholar]
- Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, et al. ; European Organisation for Research and Treatment of Cancer Brain Tumour and Radiation Oncology Groups; National Cancer Institute of Canada Clinical Trials Group (2009). Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 10, 459–466. [DOI] [PubMed] [Google Scholar]
- Stupp R, Taillibert S, Kanner AA, Kesari S, Steinberg DM, Toms SA, Taylor LP, Lieberman F, Silvani A, Fink KL, et al. (2015). Maintenance Therapy With Tumor-Treating Fields Plus Temozolomide vs Temozolomide Alone for Glioblastoma: A Randomized Clinical Trial. JAMA 314, 2535–2543. [DOI] [PubMed] [Google Scholar]
- Thévenaz P, Ruttimann UE, and Unser M (1998). A pyramid approach to subpixel registration based on intensity. IEEE Trans. Image Process 7, 27–41. [DOI] [PubMed] [Google Scholar]
- Thiagarajan PS, Sinyuk M, Turaga SM, Mulkearns-Hubert EE, Hale JS, Rao V, Demelash A, Saygin C, China A, Alban TJ, et al. (2018). Cx26 drives self-renewal in triple-negative breast cancer via interaction with NANOG and focal adhesion kinase. Nat. Commun 9, 578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong D, Li TY, Naus KE, Bai D, and Kidder GM (2007). In vivo analysis of undocked connexin43 gap junction hemichannels in ovarian granulosa cells. J. Cell Sci 120, 4016–4024. [DOI] [PubMed] [Google Scholar]
- Tong JJ, Sohn BC, Lam A, Walters DE, Vertel BM, and Ebihara L (2013). Properties of two cataract-associated mutations located in the NH2 terminus of connexin 46. Am. J. Physiol. Cell Physiol 304, C823–C832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong JJ, Minogue PJ, Kobeszko M, Beyer EC, Berthoud VM, and Ebihara L (2015). The connexin46 mutant, Cx46T19M, causes loss of gap junction function and alters hemi-channel gating. J. Membr. Biol 248, 145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, and Pachter L (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venturini E, Leanza L, Azzolini M, Kadow S, Mattarei A, Weller M, Tabatabai G, Edwards MJ, Zoratti M, Paradisi C, et al. (2017). Targeting the Potassium Channel Kv1.3 Kills Glioblastoma Cells. Neurosignals 25, 26–38. [DOI] [PubMed] [Google Scholar]
- Wang N, De Bock M, Decrock E, Bol M, Gadicherla A, Bultynck G, and Leybaert L (2013). Connexin targeting peptides as inhibitors of voltage- and intracellular Ca2+-triggered Cx43 hemichannel opening. Neuropharmacology 75, 506–516. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Theurkauf WE, Weng Z, and Zamore PD (2012). Strand-specific libraries for high throughput RNA sequencing (RNA-Seq) prepared without poly(A) selection. Silence 3, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang A, Hitomi M, Bar-Shain N, Dalimov Z, Ellis L, Velpula KK, Fraizer GC, Gourdie RG, and Lathia JD (2015). Connexin 43 expression is associated with increased malignancy in prostate cancer cell lines and functions to promote migration. Oncotarget 6, 11640–11651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziambaras K, Lecanda F, Steinberg TH, and Civitelli R (1998). Cyclic stretch enhances gap junctional communication between osteoblastic cells. J Bone Miner. Res 13, 218–228. [DOI] [PubMed] [Google Scholar]
- Zundorf G, Kahlert S, and Reiser G (2007). Gap-junction blocker carbenoxolone differentially enhances NMDA-induced cell death in hippocampal neurons and astrocytes in co-culture. J. Neurochem 102, 508–521. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.