

Abstract
A concise and selective synthesis of the dichlorinated meroterpenoid azamerone is described. The paucity of tactics for the synthesis of natural product-relevant chiral organochlorides motivated the development of unique strategies for accessing these motifs in enantioenriched forms. The route features a novel enantioselective chloroetherification reaction, a Pd-catalyzed cross-coupling between a quinone diazide and a boronic hemiester, and a late-stage tetrazine [4+2]-cycloaddition/oxidation cascade.
Graphical Abstract
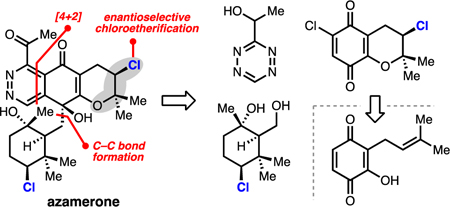
The napyradiomycins are a diverse class of halogenated meroterpenoids that have been isolated from terrestrial and marine actinomycetes (Figure 1A).1 Initial isolation efforts were driven by a desire to identify novel antibiotic scaffolds; the napyradiomycins have since demonstrated potent inhibition of gastric (H+-K+)-ATPase, nonsteroidal estrogen antagonism, cancer cell cytotoxicities, and activity against Gram-positive bacteria.1c-f Of the over 40 members within this class, only napyradiomycin A1 has succumbed to chemical synthesis (2, Figure 1A).2 Syntheses of more highly oxidized members of the napyradiomycins (e.g. 1 and 3, Figure 1A), which feature densely functionalized, chiral halocycle appendages, remain elusive, representing an exciting arena for synthetic development.
Figure 1.

(A) Representative napyradiomycin meroterpenoids. (B) Proposed biosynthesis of azamerone. (C) Retrosynthetic analysis of azamerone. (D) Methodological gap.
Azamerone is structurally unique among the napyradiomycins. It is the lone example of a phthalazinone-containing natural product.3 Moore has postulated that this nitrogen–nitrogen bond-containing heterocycle arises from oxidative rearrangement of SF2415A1 (5) via intermediate 6 (Figure 1B).3a,b In addition to its unusual hetereocycle, azamerone’s highly oxidized structure, diversity of heteroatoms, two stereogenic tertiary alcohols, and two distinct chlorine-bearing stereogenic centers pose significant synthetic challenges. We envisaged that azamerone could be retrosynthetically traced to a chlorobenzopyran, tetrazine, and chlorocyclohexane of general forms 7, 8, and 9 (Figure 1C). A challenging enantioselective chloroetherification on prenyl-containing hydroxyquinone 10 (a reaction with no enantioselective variants, Figure 1D), a hindered carbon–carbon bond formation between 9 and 7, and an electronically mismatched late-stage tetrazine Diels–Alder reaction were thus identified as key challenges for this chemical synthesis.
Our synthetic approach first required the assembly of enantioenriched chlorocycles 7 and 9. Biosynthetically, the chlorocycle motifs found in 7 and 9 are proposed to derive from chloronium-initiated cyclizations of the prenyl and geranyl fragments within 5 (Figure 1B,C).4 Neither of these transformations have enantioselective, synthetic parallels (Figure 1D).5 In fact, the use of enantioselective halogenation in natural product synthesis has been limited thus far to the enantioselective bromochlorination and dichlorination of olefins.2b,6 Cognizant of these methodological gaps and the challenge of chiral organochloride synthesis in general, we set out to develop a new method for enantioselective chloroetherification to produce a benzochloropyran akin to 7 and to discover a means for resolving the enantiomers of chlorocycle 9, which was previously made in racemic form (9, X = OAc, Figure 1C).9a
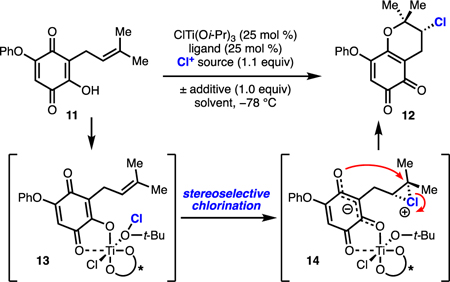
De novo development of an enantioselective chloroetherification required the identification of a substrate that was not only amenable to asymmetric halogenation but also could be parlayed into a synthesis of azamerone. Although considerable advances have been made in the area of intramolecular catalytic, enantioselective chlorolactonization and chloroetherification, high selectivity has been achieved only for styrenyl substrates; use of non-stabilized olefins is accompanied by a precipitous drop in enantioselectivity.5,7 After extensive investigation of potential substrate structures and chiral catalysts, we discovered that prenylated hydroxyquinone 11 could be chlorocyclized to a benzochloropyran by a TADDOL-ligated titanium complex and tert-butyl hypochlorite in modest yield and 10% ee (entry 1, Table 1). Application of other common systems for enantioselective chlorofunctionalization, including cinchona alkaloids, returned racemic product.7 Unexpectedly, under the conditions of entry 1, ortho-quinone 12 was formed as the major product. Significant enol chlorination was observed with formation of a racemic α-chlorinated triketone byproduct. Initial formation of intermediate 13 is proposed, as this is consistent with our hypothesis in a related dihalogenation system that a coordinatively saturated octahedral titanium complex is necessary for high selectivity.6d Chloroether 12 likely arises via trapping of the chloronium by the vinylogous carboxylate carbonyl oxygen in 14; however, titanium is not necessary for such regioselectivity, as the racemic product could be produced in 52% yield solely by the action of tert-butyl hypochlorite (see Supporting Information). We anticipated that 12 could be leveraged as a precursor to a para-quinone intermediate for the synthesis of azamerone and, therefore, pursued optimization of this chloroetherification.
Table 1.
Enantioselective chloroetherification optimization
 | |||
|---|---|---|---|
| entrya | conditions | yield (%) | ee (%) |
| 1 | A, t-BuOCI, DCM | 29 | 10 |
| 2 | B, t-BuOCI, DCM | 39 | 7 |
| 3 | B, t-BuOCI, hexanes | 11 | <1 |
| 4 | B, t-BuOCI, PhMe | 38 | 16 |
| 5 | B, t-BuOCI, Et20 | 36 | 14 |
| 6 | B, t-BuOCI, t-BuOMe | 25 | 16 |
| 7 | B, t-BuOCI, THF | 18 | 22 |
| 8 | B, t-BuOCI, 2-Me-THF | 33 | 57 |
| 9 | B, NCS, 2-Me-THF | <5 | - |
| 10 | B, DCDMH, 2-Me-THF | 42 | 33 |
| 11 | B, Palau’Chlor, 2-Me-THF | 28 | 29 |
| 12 | B, t-BuOCI, 2-Me-THF, pyridine | 20 | 78 |
| 13 | B, t-BuOCI, 2-Me-THF, quinoline | 24 | 81 |
| 14b | B, t-BuOCI, 2-Me-THF, quinoline | 68 | 84 |
| 15c | B, t-BuOCI, 2-Me-THF, quinoline | 45 | 79 |
| 16c,d | B, t-BuOCI, 2-Me-THF, quinoline | 40e | 84 |
 | |||
Reactions were conducted on 0.035–0.176 mmol scale, and 1H-NMR yields are reported based on 1,4-dinitrobenzene as internal standard;
100 mol % ClTi(Oi-Pr)3, 100 mol % B
1.3 equiv t-BuOCl
reaction conducted on 4.0 grams 11
isolated yield.
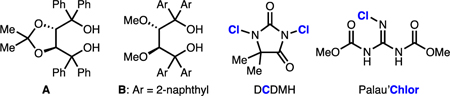
A survey of chiral ligands afforded optimal TADDOL ligand B,8 which produced chloropyran 11 in increased yield but with similarly low enantioselectivity (entry 2, Table 1). Although comparable to ligand A under the conditions of entries 1 and 2, acyclic ligand B proved to be more selective and higher yielding across a broader range of conditions and was selected for further optimization efforts. Screening of reaction solvents revealed that 2-methyltetrahydrofuran increased the selectivity of the chloroetherification to 57% ee (entries 3–8, Table 1). Use of other electrophilic chlorine sources led to a reduction in the yield or enantioselectivity of the process (entries 9–11, Table 1). Hypothesizing that adventitious acid could be promoting a racemic background reaction, we found that inclusion of heterocyclic base additives, such as pyridine and quinoline, increased the enantioselectivity of chlorocyclization (entries 12, 13, Table 1); the precise role of these additives is unclear, but they could serve as general bases, activating agents for transfer of electrophilic chlorine, or ligands on titanium. Employing a stoichiometric amount of titanium and chiral ligand delivered only a modest increase in selectivity but a dramatic improvement in yield (entry 14, Table 1). Increasing the amount of tert-butyl hypochlorite to 1.3 equivalents provided an improvement in yield when using 25 mol % titanium and ligand (entry 15, Table 1). Gratifyingly, performing the reaction on 4-gram scale under these conditions resulted in an isolated 40% yield (entry 16, Table 1) which was deemed sufficient for completing the synthesis.
With an enantioselective chloroetherification in hand, we commenced our synthesis of azamerone. Prenylquinone 11, which can be made in two steps from commercially available materials, was cyclized to chloropyran 12 using our optimized conditions (Scheme 1). Treatment of this ortho-quinone with aqueous acid induced hydrolysis and isomerization to an intermediate 2-hydroxy-para-quinone, which was smoothly converted to its corresponding ortho-chloro-para-quinone 15 with oxalyl chloride and DMF. Pyranoquinone 15 is embedded within nearly all members of the napyradiomycins (e.g. 1–4, Figure 1).
Scheme 1.
Short enantioselective synthesis of azamerone.a
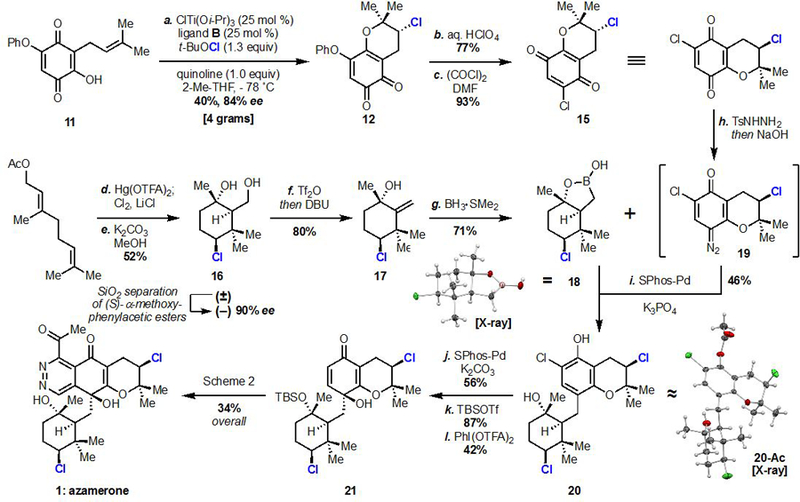
a Reagents and conditions: (a) ClTi(Oi-Pr)3 (0.25 equiv), B (0.25 equiv), quinoline (1.0 equiv), t-BuOCl (1.3 equiv), 2-Me-THF, –78 ˚C, 40%, 84% ee; (b) aq. HClO4 (1.3 equiv), Et2O, 77%; (c) oxalyl chloride (1.1 equiv), DMF (1.4 equiv), MeCN, 0 ˚C, 93%; (d) Hg(OTFA)2 (1.1 equiv), MeNO2; Cl2, LiCl (3.0 equiv), pyridine; (e) K2CO3, MeOH, 50 ˚C, 52% overall; (f) Tf2O (1.05 equiv), 2,6-lutidine (1.2 equiv), then DBU (2.5 equiv), DCM, –78 ˚C to RT, 80%; (g) BH3·SMe2 (2.0 equiv), THF, 0 ˚C to RT, 71%; (h) TsNHNH2 (1.1 equiv), MeOH, then 1M aq. NaOH, DCM; (i) (SPhos)Pd-G3: (SPhos)[2-(2′-amino-1,1′-biphenyl)]palladium(II) methanesulfonate (0.1 equiv), K3PO4 (1.3 equiv), dioxane, 60 ˚C, 46% over 2 steps, 10:1 dr; (j) (SPhos)Pd-G3 (0.1 equiv), K2CO3 (2.0 equiv), i-PrOH, 90 ˚C 56%; (k) TBSOTf (3.0 equiv), (i-Pr)2NEt (4.0 equiv), DCM then 3M aq. NaOH (7.5 equiv), i-PrOH, 87%; (l) PhI(OTFA)2 (1.1 equiv), 3:1 MeCN/H2O, 42%.
Our synthesis of the chlorocyclohexane of azamerone focused on first establishing a means to resolve racemic 16 (Scheme 1). In 2010, the Snyder group reported the direct chlorocyclization of geranyl acetate with chlorodiethylsulfonium hexachloroantimonate (see Scheme S2 in Supporting Information).9a This reaction provides the primary acetate of diol 16 in racemic form. Important recent work by Gulder,9b along with a protocol used here by Snyder9c on a mercury-based two-step mimic for halopolyene cyclizations, provided strategies for accessing racemic chlorocycle 16 (Scheme 1). Attempts to catalytically resolve diol 16 by either chemical or enzymatic means were met with limited success. Investigations into resolution by chiral derivatization revealed the (S)-α-methoxyphenylacetic ester of diol 16 to be uniquely competent in providing chromatographic resolution of diastereomers on silica gel.10 This result is noteworthy given the challenges associated with resolving primary alcohols,11 and we anticipate that access to enantioenriched diol 16 will enable syntheses of other chlorinated natural products. Using this approach, multi-gram quantities of racemic diol 16 can be separated into its constituent enantiomers.
We next investigated the coupling of quinone 15 and an appropriate derivative of chlorocyclohexane (–)-16. Initial studies attempted to unite these pieces through a 1,2-addition of generalized organometallic 9 (X = metal, Figure 1C) into the more electrophilic carbonyl of 15. These efforts were hampered due to Grob-type fragmentation of organometallic 9, which occurs at temperatures above –78 ˚C, and due to preferential reduction of the quinone by organometallic reagents. Owing to these obstacles, a cross-coupling approach was adopted for the union of these fragments (Scheme 1). Chemoselective dehydration of the primary alcohol in 16 with triflic anhydride and DBU provided olefin 17, which was hydroborated to form boronic hemiester 18 in good yield and diastereoselectivity. Boronic hemiester 18 is stable to column chromatography, allowing for facile removal of a minor diastereomer; X-ray crystallographic analysis of racemic 18 unambiguously verified its relative configuration. This cross-coupling partner has precedent in the synthesis of sclareolide derivatives and was chosen to attenuate Grob-type fragmentation.12
Quinone 15 was joined with boronic hemiester 18 through a quinone diazide-based coupling strategy (Scheme 1). Chemoselective condensation of tosyl hydrazide with quinone 15 followed by treatment with base provided intermediate quinone diazide 19, which was directly used as a cross-coupling electrophile without purification. Screening of conditions revealed that SPhos-ligated palladium was able to catalyze Csp3–Csp2 bond formation between sterically hindered boronic hemiester 18 and quinone diazide 19, providing phenol 20 in 46% overall yield from 15. Quinone diazide 19 was completely consumed in this reaction, and no elimination of either alkly chloride was observed. The structure and relative configuration of phenol 20 was confirmed via X-ray crystallography of its acetate. Small amounts of another diastereomer were produced due to the presence of minor enantiomers of each substrate. Limited precedent exists for Suzuki reactions with quinone diazides, and to the best of our knowledge, this represents the first example of this reaction type with a Csp3 nucleophile.13
Subsequent dechlorination, silylation, and hypervalent iodine-mediated para oxidation14 of phenol 20 provided the desired stereoisomer of para-quinol 21 as the major product in 42% yield (Scheme 1). Protection of the tertiary alcohol of 21 was necessary to prevent two undesired pathways: (1) the unprotected tertiary alcohol in 21 acting as a nucleophile during arene oxidation to exclusively form a diastereomeric mixture of spirocycles; (2) in oxidized quinol structures such as 21, free tertiary alcohols are prone to oxa-Michael additions into the proximal enone (see Schemes S11, S15, S16 in Supporting Information). Two diastereomeric products from the latter of these undesired pathways were verified via X-ray crystallography (see Supporting Information), enabling relative stereochemical assignments.
Inspired by Boger, we envisioned that quinol 21 could be expediently translated to azamerone through a [4+2]-cycloaddition with an appropriately functionalized tetrazine.15 Although it is a direct means for phthalazinone construction, we recognized that electron-deficient enone 21 was ill-matched to participate in this inverse electron-demand cycloaddition. After discovering that acyltetrazine 8 (Figure 1C) had limited stability, we chose its reduced variant 22 for further study (Scheme 2). Unfortunately, initial attempts to thermally induce reactivity between 21 and tetrazine 22 were unsuccessful. During our studies, Wegner and coworkers reported that bisboron complex 23 is able to catalyze the cycloaddition of tetrazines with naphthoquinones.16 Encouraged by their auspicious results, we applied complex 23 to our system and were delighted to observe formation of silyl-azamerone 1-TBS. This reaction was surprisingly regioselective, and the alternate constitutional isomer was not observed. This cascade is presumed to occur through initial [4+2]-cycloaddition, subsequent expulsion of dinitrogen via [4+2]-retrocycloaddition (to provide dihydropyridazine 24), and both in situ oxidative aromatization and benzyl alcohol oxidation to 1-TBS. The alcohol oxidation occurred to a varying extent in some experiments but could also be accomplished with Dess–Martin periodinane (see Supporting Information). Treatment with hydrochloric acid affected desilylation and provided azamerone 117 in 34% overall yield from 21.
Scheme 2.
Tetrazine cycloaddition sequence.a
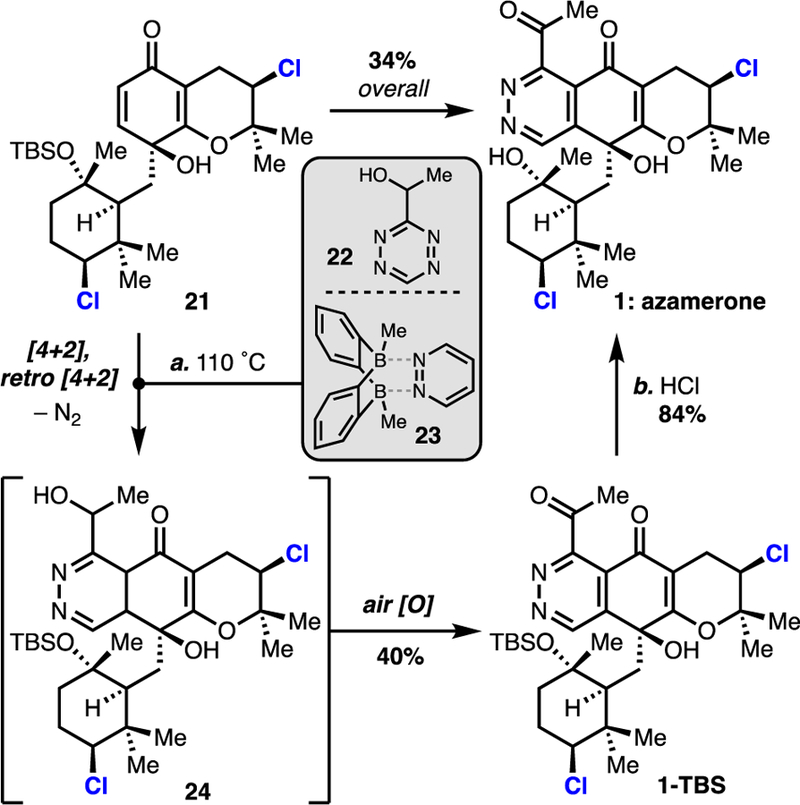
a Reagents and conditions: (a) 22 (15.0 equiv), 23 (2.0 equiv), CF3Ph, 110 ˚C, 40%; (b) 12M aq. HCl, MeOH, 84%, 34% from 21.
The first synthesis of azamerone has been reported. Our successful route relied on development of an enantioselective chloroetherification with a substrate suitable for elaboration into the target. Identification of a scalable resolution process provided access to an enantioenriched chlorocyclohexane that had been made previously in racemic form. A convergent cross-coupling between a quinone diazide and a hindered boronic hemiester, followed by a late-stage arene oxidation and bisboron-mediated tetrazine [4+2]-annulation installed the fully-oxidized framework of azamerone. We anticipate that this sequence and the strategies leveraged herein will find use in syntheses of other members of the napyradiomycin meroterpenoids.
Supplementary Material
ACKNOWLEDGMENT
This work is dedicated to Prof. Dale Boger on the occasion of his 65th birthday. We are grateful to Dr. A. Oliver (Notre Dame) and Dr. N. Settineri (UC Berkeley) for X-ray crystallographic analysis, Dr. S. Lynch (Stanford University) for assistance with NMR spectroscopy, Prof. B. Moore (Scripps Institute for Oceanography) for providing an authentic sample of 1, Prof. Dr. H. Wegner (Justus Liebig University Giessen) for providing an initial amount of bisboron complex 23, Sajan Patel (Stanford University) for the synthesis of additional 23, and Prof. Y. Xia (Stanford University) for assistance with LCMS.
Funding Sources
This work was supported by Stanford University, the National Institutes of Health (R01 GM114061), the National Science Foundation (DGE-114747 GRF to MLL), and the Center for Molecular Analysis and Design at Stanford (GRF to GMM).
Footnotes
ASSOCIATED CONTENT
Supporting Information. Experimental procedures, char-acterizations, spectral data, and CIF files. The Supporting Information is available free of charge on the ACS Publica-tions website
No competing financial interests have been declared.
REFERENCES
- (1).(a) Shiomi K; Iinuma H; Hamada M; Naganawa H; Manabe M; Matsuki C; Takeuchi T; Umezawa H; Matsuki C; Takeuchi T Novel antibiotics napyradiomycins production, isolation, physico-chemical properties and biological activities. J. Antibiot 1986, 39, 487–493. [DOI] [PubMed] [Google Scholar]; (b) Shiomi K; Nakamura H; Iinuma H; Naganawa H; Isshiki K; Takeuchi T; Umezawa H Structures of new antibiotics napyradiomycins. J. Antibiot 1986, 39, 494–501. [DOI] [PubMed] [Google Scholar]; (c) Dantzig AH; Minor PL; Garrigus JL; Fukuda DS; Mynderse JS Studies on the mechanism of action of A80915A, a semi-naphthoquinone natural product, as an inhibitor of gastric (H+-K+)-ATPase. Biochem. Pharmacol 1991, 42, 2019–2026. [DOI] [PubMed] [Google Scholar]; (d) Hori Y; Abe Y; Shigematsu N; Goto T; Okuhara M; Kohsaka M Napyradiomycins A and B1: Non-steroidal estrogen-receptor antagonists produced by a Streptomyces. J. Antibiot 1993, 46, 1890–1893. [DOI] [PubMed] [Google Scholar]; (e) Cheng Y-B; Jensen PR; Fenical W Cytotoxic and antimicrobial napyradiomycins from two marine-derived Streptomyces strains. Eur. J. Org. Chem 2013, 3751–3757. [DOI] [PMC free article] [PubMed]; (f) Farnaes L; Coufal NG; Kauffman CA; Rheingold AL; DiPasquale AG; Jensen PR; Fenical W Napyradiomycin derivatives, produced by a marine-derived actinomycete, illustrate cytotoxicity by induction of apoptosis. J. Nat. Prod 2014, 77, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).(a) Tatsuta K; Tanaka Y; Kojima M; Ikegami H The first total synthesis of (±)-napyradiomycin A1. Chem. Lett 2002, 14–15.; (b) Snyder SA; Tang ZY; Gupta R Enantioselective total synthesis of (–)-napyradiomycin A1 via asymmetric chlorination of an isolated olefin. J. Am. Chem. Soc 2009, 131, 5744–5745. [DOI] [PubMed] [Google Scholar]; (c) Miles ZD; Diethelm S; Pepper HP; Huang DM; George JH; Moore BS A unifying paradigm for naphthoquinone-based meroterpenoids biosynthesis. Nat. Chem 2017, 9, 1235–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) McKinnie SM; Miles ZD; Jordan PA; Awakawa T; Pepper HP; Murray LAM; George JH; Moore BS Total enzyme synthesis of napyradiomycins A1 and B1. J. Am. Chem. Soc 2018, 140, 17840–17845. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) While this manuscript was in review, a synthesis of two fragments of azamerone was reported: Schnell SD; Linden A; Gademann K Synthesis of two key fragments of the complex polyhalogenated marine meroterpenoid azamerone. Org. Lett 2019. DOI: 10.1021/acs.orglett.9b00090. [DOI] [PubMed]
- (3).(a) Cho JY; Kwon HC; Williams PG; Jensen PR; Fenical W Azamerone, a terpenoid phthalazinone from a marine-derived bacterium related to the genus Streptomyces (actinomycetales). Org. Lett 2006, 8, 2471–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Using 13C- and 15N-labeled precursors, Moore was able to show that both nitrogen atoms in azamerone originate from the diazo group in 5: Winter JM; Jansma AL; Handel TM; Moore BS Formation of the pyridazine natural product azamerone by biosynthetic rearrangement of an aryl diazoketone. Angew. Chem. Int. Ed 2009, 48, 767–770. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) For a review, see: Blair LM; Sperry J Natural Products Containing a Nitrogen–Nitrogen Bond. J. Nat. Prod 2013, 76, 794–812. [DOI] [PubMed] [Google Scholar]
- (4).Bernhardt P; Okino T; Winter JM; Miyanaga A; Moore BS A stereoselective vanadium-dependent chloroperoxidase in bacterial antibiotic biosynthesis. J. Am. Chem. Soc 2011, 133, 4268–4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Denmark SE; Kuester WE; Burk MT Catalytic, asymmetric halofunctionalization of alkenes–a critical perspective. Angew. Chem. Int. Ed 2012, 51, 10938–10953. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Castellanos A; Fletcher SP Current methods for asymmetric halogenation of olefins. Chem. Eur. J 2011, 17, 5766–5776. [DOI] [PubMed] [Google Scholar]; (c) Chung WJ; Vanderwal CD Stereoselective halogenation in natural product synthesis. Angew. Chem. Int. Ed 2016, 55, 4396–4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Bucher C; Deans RM; Burns NZ Highly selective synthesis of halomon, plocamenone, and isoplocamenone. J. Am. Chem. Soc 2015, 137, 12784–12787. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Landry ML; Hu DX; McKenna GM; Burns NZ Catalytic enantioselective dihalogenation and the selective synthesis of (−)-deschloromytilipin A and (−)-danicalipin A. J. Am. Chem. Soc 2016, 138, 5150–5158. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Burckle AJ; Vasilev V; Burns NZ A unified approach for the enantioselective synthesis of the brominated chamigrene sesquiterpenes. Angew. Chem. Int. Ed 2016, 55, 11476–11479. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Landry ML; Burns NZ Catalytic enantioselective dihalogenation in total synthesis. Acc. Chem. Res 2018, 51, 1260–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) Whitehead DC; Yousefi R; Jaganathan A; Borhan B An organocatalytic asymmetric chlorolactonization. J. Am. Chem. Soc 2010, 132, 3298–3300. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhang W; Liu N; Schienebeck CM; Decloux K; Zheng S; Werness JB; Tang W Catalytic enantioselective halolactonization of enynes and alkenes. Chem. Eur. J 2012, 18, 7296–7305. [DOI] [PubMed] [Google Scholar]; (c) Zeng X; Miao C; Wang S; Xia C; Sun W Asymmetric 5-endo chloroetherification of homoallylic alcohols toward the synthesis of chiral β–chlorotetrahydrofurans. Chem. Commun 2013, 49, 2418–2420. [DOI] [PubMed] [Google Scholar]; (d) Han X; Dong C; Zhou HB C3-Symmetric cinchonine-squaramide-catalyzed asymmetric chlorolactonization of styrene-type carboxylic acids with 1,3-dichloro-5,5-dimethylhydantoin: an efficient method to chiral isochroman-1-ones. Adv. Synth. Catal 2014, 356, 1275–1280. [Google Scholar]; (e) Gieuw MH; Ke Z; Yeung Y-Y Lewis Base Catalyzed Stereo- and Regioselective Bromocyclization. Chem. Rec 2017, 17, 287–311. [DOI] [PubMed] [Google Scholar]
- (8).Teller H; Corbet M; Mantilli L; Gopakumar G; Goddar R; Thiel W; Fürstner A One-point binding ligands for asymmetric gold catalysis: phosphoramidites with a TADDOL-related but acyclic backbone. J. Am. Chem. Soc 2012, 134, 15331–15342. [DOI] [PubMed] [Google Scholar]
- (9).(a) Snyder SA; Treitler DS; Brucks AP Simple reagents for direct halonium-induced polyene cyclizations. J. Am. Chem. Soc 2010, 132, 14303–14314. [DOI] [PubMed] [Google Scholar]; (b) Arnold AM; Pöthig A; Drees M; Gulder T NXS, morpholine, and HFIP: the ideal combination for biomimetic haliranium-induced polyene cyclizations. J. Am. Chem. Soc 2018, 140, 4344–4353. [DOI] [PubMed] [Google Scholar]; (c) Snyder SA; Treitler DS; Schall A A two-step mimic for direct, asymmetric bromonium- and chloronium-induced polyene cyclizations. Tetrahedron 2010, 66, 4796–4804. [Google Scholar]
- (10).(a) Whitesell JK; Reynolds D Resolution of chiral alcohols with mandelic acid. J. Org. Chem 1983, 48, 3548–3551. [Google Scholar]; (b) Trost BM; Belletire JL; Godleski S; McDougal PG; Balkovec JM; Baldwin JJ; Christy ME; Ponticello GS; Varga SL; Springer JP On the use of the O-methylmandelate ester for establishment of absolute configuration of secondary alcohols. J. Org. Chem 1986, 51, 2370–2374. [Google Scholar]
- (11).(a) Vedejs E; Jure M Efficiency in nonenzymatic kinetic resolution. Angew. Chem. Int. Ed 2005, 44, 3974–4001. [DOI] [PubMed] [Google Scholar]; (b) Terakado D; Koutaka H; Oriyama T Organocatalytic kinetic resolution of racemic primary alcohols using a chiral 1,2-diamine derived from (S)-proline. Tetrahedron: Asymmetry 2005, 16, 1157–1165. [Google Scholar]; (c) Qayed WS; Aboraia AS; Abdel-Rahman HM; Youssef AF Lipases-catalyzed enantioselective kinetic resolution of alcohols. J. Chem. Pharm. Res 2015, 7, 311–322. [Google Scholar]
- (12).Dixon DD; Lockner JW; Zhou Q; Baran PS Scalable, divergent synthesis of meroterpenoids via “borono-sclareolide.” J. Am. Chem. Soc 2012, 134, 8432–8435. [DOI] [PubMed] [Google Scholar]
- (13).(a) Schmidt B; Hölter F Suzuki-Miyaura cross coupling reactions with phenoldiazonium salts. Org. Biomol. Chem 2011, 9, 4914–4920. [DOI] [PubMed] [Google Scholar]; (b) For related precedent, see: Xia Y; Xia Y; Liu Z; Zhang Y; Wang J Palladium-catalyzed cross-coupling reaction of diazo compounds and vinyl boronic acids: an approach to 1,3-diene compounds. J. Org. Chem 2014, 79, 7711–7717. [DOI] [PubMed] [Google Scholar]; (c) Roglans A; Pla-Quintana A; Moreno-Mañas M Diazonium salts as substrates in palladium-catalyzed cross-coupling reactions. Chem. Rev 2006, 106, 4622–4643. [DOI] [PubMed] [Google Scholar]
- (14).Magdziak D; Meek SJ; Pettus TRR Cyclohexadienone ketals and quinols: four building blocks potentially useful for enantioselective synthesis. Chem. Rev 2004, 104, 1383–1430, and references cited therein. [DOI] [PubMed] [Google Scholar]
- (15).For reviews, see: (a) Boger DL Diels-Alder reactions of azadienes. Tetrahedron 1983, 39, 2869–2939. [Google Scholar]; (b) Boger DL Diels-Alder reactions of heterocyclic aza dienes. Scope and applications. Chem. Rev 1986, 86, 781–793. [Google Scholar]; (c) For studies on cycloadditions of unsubstituted 1,2,4,5-tetrazine, see: Sauer J; Heldmann DK; Hetzenegger J; Krauthan J; Sichert H; Schuster J 1,2,4,5-Tetrazine: synthesis and reactivity in [4+2] cycloadditions. Eur. J. Org. Chem 1998, 2885–2896, and references cited therein.
- (16).(a) Hong L; Ahles S; Strauss MA; Logemann C; Wegner HA Synthesis of 2,3-diaza-anthraquinones via the bidentate Lewis acid catalyzed inverse electron-demand Diels-Alder (IEDDA) reaction. Org. Chem. Front 2017, 4, 871–875. [Google Scholar]; (b) Hong L; Ahles S; Heindl AH; Tiétcha G; Petrov A; Lu Z; Logemann C; Wegner HA An air-stable bisboron complex: a practical bidentate Lewis acid catalyst. Beilstein J. Org. Chem 2018, 14, 618–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17). Synthetic 1: [α]D23 = –7 (c = 0.1, MeOH); ref. 3a: [α]D = –8.8 (c = 0.0025, MeOH).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.