

Abstract
Caseinolytic protease P (ClpP) has emerged as a promising new target for antibacterial development. While ClpPs from single isoform expressing bacteria have been studied in detail, the function and regulation of systems with more than one ClpP homologue are still poorly understood. Herein, we present fundamental studies toward understanding the ClpP system in C. difficile, an anaerobic spore-forming pathogen that contains two chromosomally distant isoforms of ClpP. Examination of proteomic and genomic data suggest that ClpP1 is the primary isoform responsible for normal growth and virulence, but little is known about the function of ClpP2 or the context required for the formation of functional proteases. For the first time in a pathogenic bacterium, we demonstrate that both isoforms are capable of forming operative proteases. Interestingly, ClpP1 is the only homologue that possesses characteristic response to small molecule acyldepsipeptide activation. On the contrary, both ClpP1 and ClpP2 respond to cochaperone activation to degrade an ssrA-tagged substrate. These observations indicate that ClpP2 is less susceptible to acyldepsipeptide activation but retains the ability to interact with a known cochaperone. Homology models reveal no obvious characteristics that would allow one to predict less efficient acyldepsipeptide binding. The reported findings establish the uniqueness of the ClpP system in C. difficile, open new avenues of inquiry, and highlight the importance of more detailed structural, genetic, and biological characterization of the ClpP system in C. difficile.
Keywords: serine protease, bacterial pathogenesis, ATP-dependent protease, acyldepsipeptide, resistance
Graphical Abstract
As a key regulator of virulence and drug resistance in infectious bacteria, caseinolytic protease P (ClpP) has emerged as a new target for antibacterial development.1 Aberrant ClpP activity (inhibition or activation) induced by small molecules is detrimental to microbial fitness, reduces virulent phenotypes, and disrupts biofilm formation.2–4 Specifically, activation of ClpP has been validated and proven safe in vivo as an antibacterial strategy against systemic lethal infections of Enterococcus faecium, E. faecalis (vancomycin-sensitive and -resistant), Staphylococcus aureus (methicillin-resistant and -sensitive), and Streptococcus pneumonia. In each case, activation of ClpP with a small molecule acyldepsipeptide (ADEP) outperforms clinically utilized antibiotics, including linezolid and ampicillin.
There are several advantages to targeting ClpP: (l) Both inhibition and activation are possible, with each tactic affecting different aspects of bacterial pathogenicity.2,4 This provides an opportunity to determine the therapeutic potential of two orthogonal strategies on a single target, a rare phenomenon in antibacterial drug discovery. (2) Due to the diverse regulatory roles of ClpP (e.g., growth, motility, virulence, stress response, sporulation), disruption of its natural activity would have pleotropic effects that may not be easily resolved by compensatory mutations.1 (3) ClpP activation itself demonstrates efficacy against both actively growing and dormant persister cells,3 a necessity for improved antibacterial treatments. (4) In organisms investigated thus far, ClpP is essential for pathogenicity but not survival (with the exception of Mycobacterium tuberculosis).1 Therefore, inhibiting ClpP may provide a means to negatively affect the virulence of pathogens but preserve the viability of beneficial microbes. (5) Targeting nonessential virulence regulators like ClpP is likely to impair the organism’s infectivity without imparting typical selective pressures that drive resistance.
Despite the advancements in ClpP research, major questions regarding the clinical potential of targeting this protease persist. Although the general involvement of ClpP in bacterial virulence is well-established, the distinct attributes regulated by ClpP are organism dependent. Additionally, while resistance to ADEPs has been generated in laboratory settings (Escherichia coli, Bacillus subtilis, E. faecalis, S. pneumoniae, S. aureus), all studies have been conducted on single clpP containing organisms. While ClpPs from single isoform expressing bacteria have been studied in detail, the function and regulation of systems with more than one clpP gene are still poorly understood. Interrogating the behavior of ClpP systems in multi-isoform containing organisms is expected to reveal additional insight into mechanisms of resistance development that should be considered, both during the validation of a target and in its clinical exploitation. In these contexts, we are interested in elucidating the behavior, biological relevance, and therapeutic potential of the ClpP system in Clostridium difficile.
C. difficile infection (CDI) is a leading cause of hospital-acquired illness and presents a unique challenge to therapeutic development, as it is both caused by and clinically managed with traditional antibiotics that indiscriminately eradicate pathogenic and commensal bacteria.5 Importantly, C. difficile is unique from typical beneficial microflora in that it expresses two isoforms of ClpP (ClpP1 and ClpP2).6 To date, our understanding of multiple clpP gene expressing organisms is based primarily upon four microbes, Synechococcus elongatus,7,8 Listeria monocytogenes,9–13 Pseudomonas aeruginosa,14 and M. tuberculosis,15–21 all of which reveal structural disparities and distinct regulation profiles of ClpP (Figure 1).
Figure 1.

Composition and in vitro behavior of reported multi-ClpP systems. “Mixing” denotes that this result was obtained by mixing ClpP1 and ClpP2 homotetradecamers.
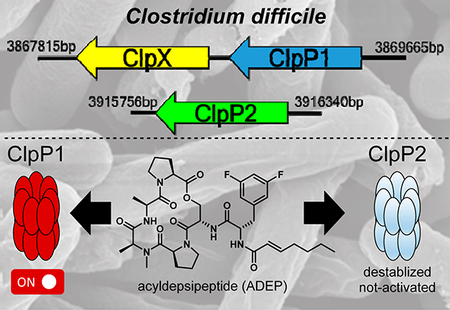
Gene location and expression characteristics of ClpP1 and ClpP2 in C. difficile are not only unique from commensal organisms but also distinct from any pathogenic system disclosed to date. Of the two ClpP isoforms contained within the C. difficile genome, clpP1 is located in an apparent operon with the cochaperone clpX. Interestingly, clpP2 is expressed in a chromosomally distant region of the genome without evidence of proximal cochaperones or adaptors. In previous studies, mass spectrometry-based proteomic evaluation and genomic characterization of sporulated and heat-shocked C. difficile 630 revealed the presence of ClpP1 and the cochaperones ClpC and ClpX but no evidence of ClpP2.22,23 On the contrary, a microarray analysis completed by Emerson et al. detected ClpP1 and ClpP2, with ClpP1 being the only isoform upregulated under pH-induced shock and antibiotic challenge (amoxicillin, clindamycin, and metronidazole).24 clpP2 transcripts have also been detected in NAP125 and ĈD38–2 prophage26 containing C. difficile variants, suggesting that clpP2 may have involvement in hypervirulent and/or resistant phenotypes. Taken together, ClpP1 has been detected in ribotype 630 and hypervirulent strains, while the presence of ClpP2 expression or protein has been reported in only hypervirulent strains. Nevertheless, these results suggest that contrary to other multi-ClpP organisms, which rely on mixtures of ClpP1 and ClpP2 monomers to form the functionally relevant protease (Figure 1), C. difficile ClpP1 and ClpP2 may operate in an uncoupled fashion and exhibit different biological roles. To evaluate the first portion of this hypothesis and begin to understand the behavior of the C. difficile ClpP system, we overexpressed, purified, and reconstituted the homomeric ClpP1 and ClpP2 complexes and assessed the activity of these proteases.
RESULTS AND DISCUSSION
Significantly Higher Transcript Levels of clpP1 Are Observed in Exponential and Stationary Growth Phases
To determine the expression profile of clpP1 and clpP2 during exponential and stationary growth phases, we quantified transcript levels utilizing quantitative reverse-transcriptase polymerase chain reaction (qRT-PCR). Total RNA extracts were isolated from C. difficile 630 cells in biological triplicate from both exponential (OD600 = 0.6) and stationary (OD600 = 1.2) growth populations in brain heart infusion-supplemented (BHIS) media. As shown in Figure 2 (Ct values, Table S2), clpP1 and clpP2 transcripts were detected during both phases of growth, albeit with clpP2 transcripts significantly less prevalent than clpP1.
Figure 2.

Relative quantification of clpP1 and clpP2 mRNA expression during exponential and stationary growth phases in BHIS broth. Transcript levels were normalized to the reference gene rpoB, with clpP2 transcript level reported relative to clpP1. The resulting data were analyzed via multiple comparison two-way ANOVA and corrected by the Sidak method. ****, P ≤ 0.0001.
The observed difference in relative mRNA expression levels, paired with previously reported proteomic and transcriptomic data, suggests that clpP1 is the major contributor to homeostasis in C. difficile. The detection of clpP2, albeit at a much lower transcript level, indicates that clpP2 likely carries some function within the organism but is likely not tied to a large role in general homeostasis. While this speculation is not particularly ground-breaking, it does demonstrate that clpP2 is transcribed in both phases, a detail that is not obvious from studies described previously. The disparity in transcript levels adds confidence to our original hypothesis that unlike other pathogens investigated to date, ClpP isoforms in C. difficile are capable of uncoupled activity and are responsible for disparate biological roles. The RTPCR results compelled us to investigate the oligomerization behavior and catalytic profile of reconstituted ClpP1 and ClpP2.
ClpP1 and ClpP2 Share Highly Conserved Sequence Homologies and Retain Key Structural Motifs
Within the last 5 years, elegant biophysical studies have revealed ClpP operation to be governed by inter- and intramolecular conformational switches that propagate regulation signals through the complex.12,27–29 The presence or absence of these hotspots may provide an indicator for how uncharacterized ClpP isoforms can be expected to function. Therefore, to determine if critical motifs are conserved, we aligned the primary sequences of C. difficile ClpP1 and ClpP2 to B. subtilis ClpP (BsClp; identity: ClpP1 74% and ClpP2 63%), an evolutionarily related organism with a well-characterized ClpP homologue. As shown in Figure 3, the primary sequence alignment reveals that key hotspot regions, including the Ser-His-Asp catalytic triad, Tyr63 activation trigger, Asp(Glu)/Arg oligomerization sensor domains, and the Gly-rich heptamer dimerization domain, are highly conserved in both C. difficile ClpP isoforms. Although there seems to be significant variation in the E-helix domain, a motif involved in heptamer dimerization to form the operative tetradecamer, there is no obvious evidence to suggest that either isoform would have difficulty oligomerizing or exhibiting stability in the tetradecameric form as a homomeric complex.
Figure 3.

Primary sequence alignment of C. difficile ClpP isoforms (CdClpP1 and CdClpP2) and B. subtilis ClpP (BsClpP). Alignment and graphic generated with Clustal Omega. Green circles indicate amino acids shown to interact with acyldepsipeptides in B. subtilis.
ClpP1 Forms a More Stable Tetradecameric Complex
Because ClpP1 and ClpP2 share high sequence identity (~72%) and retain key structural motifs, we hypothesized that both homologues would form active tetradecameric complexes capable of peptide/protein degradation. This, however, cannot be assumed, as has been demonstrated by the S. elongates, L. monocytogenes, P. aeruginosa, and M. tuberculosis studies. To begin the assessment of possible oligomeric configurations of C. difficile ClpP1 and ClpP2, we separately expressed and purified each isoform to evaluate homogeneous oligomeric reconstitution. Initially, C. difficile ClpP1 and ClpP2 were expressed with C-terminal 6x His-tags in E. coli Rosetta cells and purified via affinity chromatography before size exclusion chromatography (SEC) was performed to buffer exchange and confirm oligomeric status.
Assessment of purified fractions by SDS-PAGE and SEC suggested pure, completely tetradecameric ClpP complexes. Identities of ClpP1 and ClpP2 were confirmed by highresolution electrospray mass spectrometry (HR-ESIMS) and peptide digest and sequencing experiments. To our surprise, however, E. coli ClpP (EcClpP) was detected in ~10–30% relative abundance within our purified ClpP1 and ClpP2 preparations. We suspect that endogenous EcClpP from the expression cell line copurified with the C. difficile ClpPs and was incorporated into the isolated tetradecameric complex. Given the high sequence homology of EcClpP and C. difficile ClpPs, this seems plausible and should not be overlooked when reconstituting other complexes prepared from recombinant techniques. One might assume that such an impurity would be evident by SDS-PAGE analysis; however, no such contamination was evident in our experiments, probably due to very similar molecular weights and suboptimal protein loading concentrations and/or gel gradients. Unless one is specifically looking for a host-ClpP band and varying experimental conditions in attempts to reveal this, it will not likely be evident in typical preparations. As such, we suggest utilizing a variety of methods to verify a pure and monodisperse preparation. To ensure that subsequent biochemical evaluations of these complexes were not influenced by contaminating EcClpP, each C. difficile ClpP isoform was overexpressed and purified from cells lacking EcClpP (ΔEcClpP, graciously provided by Dr. Robert Sauer’s group, MIT).30 Unless otherwise noted, all data presented herein utilize C. difficile ClpP prepared from this AClpP E. coli strain.
ClpP1 rapidly tetradecamerizes prior to SEC purification, as indicated by the single peak corresponding to the molecular weight of tetradecameric ClpP1 (Figure 4A). The heptameric ClpP1 oligomer was not observed over the course of multiple purifications. ClpP2, however, elutes as a mixture of oligomeric species. If ClpP2 is allowed to equilibrate ≥48 h, nearly complete oligomerization to the tetradecameric complex occurs (Figure 4B). HR-ESIMS confirmed the identity of both isoforms (Figure S1) and the absence of any contaminating EcClpP. Dynamic light scattering (DLS) experiments confirmed tetradecameric assembly for each isoform after purification (Figures S2 and S3).
Figure 4.

Tetradecameric assembly and thermal stability of ClpP1 and ClpP2 homomeric complexes. (A) size exclusion chromatograph of purified recombinant ClpP1 and ClpP2 following Ni-affinity purification. B. subtilis ClpP (BsClpP) is included for comparison. (B) time-dependent oligomerization of ClpP2. An aliquot of ClpP2 was injected onto a S300 SEC (blue, solid) and again 14 days later (blue, dashed). (C and D) SYPRO Orange TSA of tetradecameric ClpP1 (C) and ClpP2 (D) in the presence and absence of varying concentrations of an ADEP activator.
To evaluate the relative stability of ClpP1 and ClpP2, both were exposed to thermal shift analysis (TSA). As shown in Figure 4C, ClpP1 exhibits a Tm = 53.2 °C and thus is significantly more stable to thermal denaturation than ClpP2 (Tm = 46.5 °C). Additionally, the presence of a synthetic acyldepsipeptide (100 μM, ADEP, Figure 7A) known to stabilize other ClpP homologues, results in stabilization of ClpP1 (ΔTm = +11.2 °C, Figure 4C) but an apparent destabilization of ClpP2 (ΔTm = −1.6 °C, Figure 4D). ADEP destabilization of tetradecameric ClpP complexes has been reported previously31 and is attributed to substoichiometric occupation of ligand binding sites, which leads to disruption of the tetradecameric complex. These results hint at a rather drastic difference in ADEP interactions between the two isoforms. Nonetheless, both isoforms are capable of forming stable homotetradecamers, a feature only observed thus far in M. tuberculosis but has recently been disputed32
Figure 7.

Susceptibility of ClpP1 and ClpP2 to ADEP activation. (A) Synthetic ADEP utilized in these experiments. (B) Dose-dependent ADEP-induced degradation of the decapeptide Abz-DFAPKMALVPYNO2. (C) Time-dependent ADEP (100 μM) induced degradation of FITC β-casein; ClpP1 (black, triangles) and ClpP2 (blue, squares).
ClpP1 Exhibits Prototypical Peptidolysis
When in an extended conformation, the catalytic triad of ClpP is aligned and capable of degrading small peptides (<5–6 amino acids) without the requirement of a cochaperone or ATP.33 As exemplified by studies on M. tuberculosis ClpP isoforms, formation of a stable tetradecamer does not signify catalytic competency. To determine whether or not the assembled homo-oligomeric ClpP1 and ClpP2 complexes exist in the extended confirmation and have active (i.e., aligned) catalytic triads, we evaluated the capability of each ClpP tetradecamer to degrade two fluorescently labeled peptides, Suc-Leu-Tyr-aminomethylcoumarin (dipeptide, SLY-AMC)33 and Z-Gly-Gly-Leu-AMC (tripeptide, Z-GGL-AMC).15 The small peptides freely diffuse into the proteolytic chamber of ClpP where, in the presence of an active catalytic Ser-His-Asp triad, the peptides are hydrolyzed, releasing quantifiable fluorescence. As shown in Figure 5A, ClpP1 degrades SLY-AMC in a time dependent manner and exhibits Michaelis-Menten kinetics (Figure 5B), while ClpP2 fails to induce appreciable degradation. In fact, neither increased time (<24 h) nor alterations in substrate concentration produced measurable ClpP2 mediated degradation of this substrate (data not shown). Both ClpP1 and ClpP2 are, however, able to hydrolyze Z-GGL-AMC, although ClpP2 is not nearly as efficient as ClpP1 and requires extended coincubation periods (>30 h) with this substrate to see appreciable hydrolysis (Figures 5C and 5D). Mixing equal concentrations of homomeric ClpP1 and ClpP2 resulted in less peptidolysis for both SLY-AMC and Z-GGL-AMC (Figure S6) than ClpP1 alone. Native gel assessment of the mixture revealed no evidence of a new heteromeric species (Figure S7) While this experiment is a rather crude assessment, it provides evidence that disassembly of homomeric complexes and reassembly into more active heteromeric complexes is not thermodynamically favored and does not occur in vitro.
Figure 5.

Peptidolytic activity of ClpP1 and ClpP2. (A) Time-dependent degradation of SLY-AMC by ClpP1 expressed from ΔEcClpP cells (black, solid, solid triangle), ClpP1 from Rosetta DE3 (black, dashed, hollow triangle), ClpP2 from ΔEcClpP (blue, solid, solid square), and ClpP2 from Rosetta DE3 (blue, dashed, hollow square). Experiments conducted with 1 μM ClpP tetradecamer and 0.5 mM SLY-AMC. (B) Michaelis-Menten analysis of ClpP1 and ClpP2 degradation of SLY-AMC. ClpP1 (DE3) Vmax = 1213 ± 38.0; ClpP1 (ΔEcClpP) Vmax = 1357 ± 39.9. ClpP2 Vmax values were not determined due to relatively negligible cleavage of the substrate. (C) Time-dependent (0–30 min) degradation of Z-GGL-AMC by ClpP isoforms. Experiments conducted with 1 μM ClpP tetradecamer and 0.1 mM Z-GGL-AMC. (D) Degradation of Z-GGL-AMC by ClpP isoforms after extended periods (30 and 46 h) of incubation. Means of each data set were compared to the control (Z-GGL-AMC alone) at each time point for statistical significance within a 95% confidence interval by two-way ANOVA analysis with Dunnett’s multiple comparison test. (**, P < 0.05). (E) Serine-protease active site labeling with ActivX TAMRA-FP visualized in an SDS-PAGE gel with chymotrypsin (CHY) as a positive control.
The Z-GGL-AMC results suggest, however, that the catalytic triad for ClpP2 is aligned to some extent under the evaluated conditions but that experimental (e.g., buffer composition) and/or structural nuances dictating substrate specificity may exist for each ClpP isoform. Further evidence supporting that both complexes exhibit an aligned catalytic triad arises upon incubation of each with ActivX TAMRA-FP, a fluorescent probe that selectively tags active serine hydrolases.34 As shown in Figure 5E, TAMRA-FP labels ClpP1 efficiently, while reacting to a lower extent with ClpP2. It is worth noting that in all three of these experiments, C. difficile ClpP samples prepared from Rosetta DE3 cells (contaminated with EcClpP) were assessed in parallel with samples prepared from the ΔEcClpP strain, and no significant differences in behavior were observed. Likewise, we failed to notice any difference in thermal stability of ClpP complexes prepared from either strain. So, while EcClpP contamination exists in the Rosetta DE3 preparations, its presence does not seem to affect the activity of either complex. We imagine this may not be the case for other ClpP homologues, especially those that exhibit lower homology, and caution others utilizing endogenous ClpP expressing cell lines for the characterization of recombinant ClpP complexes.
While investigating potential reasons for the lack of ClpP2 proteolytic activity, we determined through HR-ESIMS that ClpP2 was capable of forming disulfide bonds (reversible with DTT addition) that may affect enzymatic efficiency. DiANNA, a web server developed by Boston College to define cysteine oxidation states and predict disulfide bond partners, identified two potential disulfide bonds (i.e., Cys86-Cys92 and Cys92-Cys113, Figure S4) for ClpP2. The algorithm reveals disulfide bond formation to be unique to ClpP2, as ClpP1 contains only a single cysteine. Because the two predicted disulfide bonds in ClpP2 are located in proximity to the catalytic triad, we could not discount that disulfide bond formation may dictate proteolytic activity or cleavage specificity by misshaping the topology around serine catalytic site or locking ClpP2 in an inactive conformation. Therefore, we also evaluated the activity of ClpP2 in the presence of varying concentrations of dithiothreitol (DTT) but found that this failed to stimulate proteolytic activity, even at incubation times exceeding 48 h (Figure S5). It is also worth noting that reduced conditions (expected for an anaerobic environment) had no significant effect on the thermal stability of ClpP1 or ClpP2.
ClpP1 and ClpP2 Respond to ClpX Activation
As mentioned previously, ClpP proteolysis of targeted substrates is tightly regulated via coordination between ATP-dependent cochaperones (e.g., ClpX, ClpA, and ClpC). These cochaperones recognize degron sequences (e.g., ssrA) that tag proteins for degradation by ClpP.35 Because clpP1 and clpX are situated in an apparent operon in C. difficile, we anticipated that ClpP1 and ClpX would readily combine to form an operative heteroprotein complex capable of degrading ssrA-tagged substrates. We, however, did not know what to expect with ClpP2, as the lack of proteolytic activity in the SLY-AMC assay is not necessarily indicative of the capability to function with natural cochaperones. To determine the ability of each C. difficile ClpP homologue to form the ClpP:ClpX proteolytic machinery and degrade protein substrates we expressed and purified C. difficile ClpX and ssrA-tagged green-fluorescent protein (ssrA-GFP) to allow for reconstitution of the “natural” proteolytic machinery.
ClpX and ssrA-GFP were incubated with each ClpP isoform to determine if ssrA-GFP degradation could be mediated by ClpX. We observed reductions in GFP gel-band intensity for both ClpP isoforms with ClpX and ATP present (Figure 6A), with ClpP1 once again outperforming ClpP2. Incubation of ssrA-GFP with each ClpP isoform in the absence of ATP revealed no degradation of the substrate, indicating that the observed degradation is induced by ClpX (Figure 6B). It is well-known that ClpX ATP hydrolysis is repressed upon engagement with ClpP.36,37 As such, we determined ATP hydrolysis rates for ClpX in the presence of either ClpP1 or ClpP2. As seen in Figure 6C, ClpX ATPase hydrolysis decreases when incubated with either ClpP1 (~18% rate reduction) or ClpP2 (~7% rate reduction).
Figure 6.

ClpXP mediated degradation of ssrA-GFP and ClpP induced reduction in ATP hydrolysis. (A) ClpXP1 and ClpXP2 mediated degradation of ssrA-GFP. Experiment was performed in triplicate, and SDS-PAGE results were quantified with ImageJ (NIH) to provide the quantified results (graph). (B) Fraction of ssrA-GFP remaining when incubated with ClpP variants in the absence of ClpX. (C) Rate of C. difficile ClpX ATP hydrolysis in the presence or absence of C. difficile ClpP1 or ClpP2. C. difficile ClpX hydrolyzed ATP at a rate of 149.2 ± 6.0 min−1·ClpX6−1. In the presence of ClpP1, ClpX hydrolyzed ATP at a rate of 122.5 ± 0.3 min−1·ClpX6−1. In the presence of ClpP2, ClpX hydrolyzed ATP at a rate of 138.7 ± 0.4 min−1·ClpX6−1. Km values for ClpX, ClpXP1, ClpXP2 are 375.2, 318.9, and 402.9 μM, respectively.
In an attempt to elicit ClpP2 peptidolytic activity, we added SLY-AMC to an aliquot of the ClpX:ClpP2 reaction. The rationale was that perhaps the ClpX interaction may trigger an autocatalytic-processing, or other maturation event, giving rise to peptidolytic activity. However, no degradation of SLY-AMC was observed.
ClpP1 and ClpP2 Exhibit Significant Differences in Susceptibility to Chemoactivation
The ADEPs (Figure 7A) bind and activate bacterial ClpP in a cochaperone and ATP-independent manner, resulting in unselective proteolysis and bacterial cell death.2 Similar to the natural cochaperones, ADEP binding results in a large conformational shift and a subsequent widening of the proteolytic chamber.38 In the case of chemoactivation, however, the proteolysis is uncontrolled and unregulated and thus detrimental to bacterial survival.39 Analysis of cocrystal structures reveals that the ADEPs mimic the natural isoleucine/leucine-glycine-phenylalanine (I/LGF) loop utilized by AAA+ cochaperones for ClpP binding and thus bind competitively to the same pocket.38–40 As such, activators of ClpP operate via two mechanisms of action: (1) activating the ClpP protease and inducing unselective degradation and (2) inhibiting the interaction of AAA+ cochaperones with ClpP, thus disrupting the ability of cochaperones to deliver natural substrates for degradation. Therefore, bacterial cells treated with ClpP activators suffer from simultaneous self-digestion and a buildup of toxic substrates, resulting in a dual attack on microbes. Recent interest in pursuing ADEPs for CDI treatment has appeared in literature, but no experimental assessment has followed.41
To determine the susceptibility of homomeric ClpP1 and ClpP2 to ADEP activation, we incubated both isoforms with serially diluted concentrations of a synthetic ADEP (Figure 7A) in the presence of a self-quenching decapeptide (Abz-DFAPKMALVPYNO2) or FITC-β-casein. Upon ADEP mediated activation, ClpP will degrade the self-quenching substrate and release quantifiable fluorescence. As shown in Figure 7, ClpP1 displays a prototypical response to ADEP activation and efficiently degrades both the decapeptide (Figure 7B) and protein (Figure 7C) substrates. ClpP2, on the other hand, demonstrates a much lower susceptibility to ADEP activation and fails to produce significant levels of peptidolysis or proteolysis, even at ADEP concentrations as high as 1 mM.
Homology Models Reveal No Obvious Preclusions in ClpP2 Activating Pocket
Intrigued by the response of ClpP2 to cochaperone activation while exhibiting significantly less susceptibility to ADEP activation, we generated homology models to aid in visualization of the ADEP binding pocket and elucidate any amino acid substitutions that may preclude ADEP binding. ClpP from B. subtilis exhibits 74 and 63% sequence identity to C. difficile ClpP1 and ClpP2, respectively, and was thus utilized to generate the models (SWISS-MODEL). Assessment of the primary sequences (Figure 3) and models (Figure 8), however, failed to reveal any obvious amino acid substitutions that would allow one to predict less efficient ADEP binding. This leaves us to speculate that alterations in the primary sequence outside of the cochaperone/ADEP binding pocket may lead to subtle perturbations of higher order ClpP structures that result in ADEP insensitivity while maintaining natural function. Adding intrigue to this speculation is an observation that in hypervirulent strains of C. difficile, the primary sequence of ClpP1 and ClpP2 retain complete sequence identity to C. difficile 630 but include three FT amino acid insertions. Studies are ongoing to determine the effect of those insertions on ClpP1 and ClpP2 functionality. Systematic insertions of this type in other ClpP isoforms has not been previously reported, nor do they occur in characteristic structural motifs relevant to ADEP activation.
Figure 8.

Depiction of the (A) ClpP1 and (B) ClpP2 ADEP binding pockets based on homology modeling with B. subtilis ClpP. Prime numbers indicate one ClpP subunit and nonprime numbers another. Gray indicates the culled surface of ClpP with a solvent radius set to 2.0 Å.
CONCLUSION
Reported mechanisms of evolved resistance to ADEP activation include efflux pump upregulation and point mutations close to the active site (e.g., T182A), resulting in impaired proteolytic activity. Both of these observations were obtained from E. coli. Unspecified point mutations that eliminate proteolytic activity have also been generated in a laboratory setting at a frequency range of l06 in E. faecalis and S. pneumoniae. All of these studies have been conducted in single ClpP gene containing organisms. We believe that more complex resistance or redundancy mechanisms may exist in multi-ClpP organisms and that studying these systems will reveal new insights into ClpP modulation as a therapeutic strategy and possible resistance evolution. Analysis of previous C. difficile genomic and proteomic studies indicated that the ClpP system in this organism may exhibit a behavior distinct from that of other multi-ClpP organisms interrogated to date and thus inspired us to begin studying this system in C. difficile.
The data demonstrate that the proteolytic activity and susceptibility to chemoactivation differ between C. difficile ClpP1 and ClpP2 and that these two isoforms are capable of functioning in an uncoupled fashion. This has yet to be observed in other multi-clpP organisms (e.g., M. tuberculosis, L. monocytogenes, S. elongatus), which require both ClpP1 and ClpP2 to form functionally relevant heterotetradecameric proteolytic complexes. Recently, it was reported that ClpP1 and ClpP2 of P. aeruginosa exhibit different expression profiles and disparate biological roles, but reconstitution of an active or fully assembled ClpP2 homomeric complex could not be obtained in vitro. Thus, to the best of our knowledge, for the first time in a multi-clpP organism, we demonstrate that both isoforms are capable of forming operative proteases. These results clearly differentiate the ClpP system of C. difficile and highlight the possibilities of (l) uncoupled function of ClpP isoforms in pathogenic bacteria and (2) selective or specific modulation of one isoform over another.
The studies presented herein lay a foundation for more detailed characterization of the structure, function, and behavior of the ClpP system in C. difficile, as many questions remain to be answered. For example, is it possible for ClpP1 and ClpP2 to form operable heterogeneous tetradecamers like those exhibited by S. elongates, M. tuberculosis, and L. monocytogenes? If so, how does the activity and susceptibility to chemo-modulation compare to the homogeneous complexes? This question is not one that can be answered merely by mixing the two isoforms together. Rather, extensive protein science and analytical work are necessary to assess heteromeric formation and to determine the stoichiometric ratio of each isoform. What are the structural nuances that differentiate operative ClpP complexes and their responses to ADEP activation? This question is particularly interesting because no obvious characteristics emerge during the analysis of the primary sequences. Answering this question will likely require structural characterization of these complexes. While continued biochemical and biophysical evaluation of reconstituted C. difficile ClpP complexes will undoubtedly lead to new insights about enzyme kinetics and structure, genetic manipulation of the ClpP system in C. difficile is essential to understanding the biological significance and therapeutic potential. These studies are ongoing, and results will be presented in due course.
EXPERIMENTAL PROCEDURES
Unless otherwise noted, all ClpP concentrations are expressed as the tetradecameric (ClpP14) protease.
Expression and Purification of ClpP1 and ClpP2
clpP1 (YP_001089821.1) and clpp2 (YP_001089868.1) were synthesized with a Poly-His(6x) C-terminal tag, codon optimized, and cloned into pET28a by Genscript USA (Piscataway, NJ). clpX (YP_001089820.1) with a TEV-cleavable Poly-His(6x) N-terminal tag was also synthesized by Genscript USA, codon optimized, and cloned into pET21a. All plasmids were transformed into Rosetta (DE3) and ΔEcClpP (DE3) cells (a gracious gift from Dr. Robert Sauer’s laboratory) for expression through standard techniques. M4100 (DE3) cells (ΔEcClpX) possessing ssrA-tagged (LAA)-GFP with a Poly-His(6x) N-terminal tag was provided as a gracious gift from Dr. Tania A. Baker’s laboratory and was expressed and purified as previously described.42 All purified proteins were flash frozen in liquid N2 and stored at −80 °C until needed.
To express ClpP1, ClpP2, and ClpX, a 4 L culture was inoculated with an overnight stock (1:100) and grown to an OD600 of 0.5–0.7, shaking at 250 rpm at 37 °C. Prior to induction with 1 mM IPTG, the temperature was decreased to 18 °C, and shaking was reduced to 180 rpm. Overexpression was carried out over 16–18 h at 18 °C, shaking at 180 rpm. The bacteria were pelleted via centrifugation (5000g, 4 °C, 15 min). The pellet was washed with ice-cold lysis buffer and stored at −80 °C until lysis. For isolation and purification of ClpP1 and ClpP2, the pellet was resuspended in ice-cold lysis buffer (50 mM Tris pH 8.0, 200 mM NaCl, 10% glycerol) before being loaded into an Emulsiflex cell-disruption system for four-rounds of lysis at ~15 000 psi while being cooled on ice. The lysate was clarified via centrifugation (28 500g, 4 °C, 45 min) and loaded onto a 5 mL HF His-Trap column (GE Healthcare) via an NGC Explorer FPLC (Bio-Rad) at 4 °C. The column was washed with 5% His-elution buffer (Tris pH 8.0, 200 mM NaCl, 10% glycerol, 500 mM imidazole) for 20 column volumes (CV). His-tagged protein was eluted with a stepwise increase of elution buffer (10/15/30/70/100%) in reverse-flow. Highly pure fractions were collected in 5 mL intervals and concentrated to ~5 mL before injection onto a HiPrep 16/60 Sephacryl S-300 HR Size Exclusion Column (GE Life Sciences). ClpP was exchanged into activity buffer (25 mM HEPES, 100 mM KCl, 5 mM MgCl2, 1 mM DTT, 10% glycerol) with an NGC Explorer FPLC at 4 °C. ClpP was concentrated with a 50 kDa MW-cutoff concentrator and flash frozen until needed. All proteins expressed in the ΔEcClpP cell line were purified identically, except for induction of expression was accomplished with 0.5 mM IPTG.
Both isoforms express well in ΔEcClpP cells and are stable at 4 °C for >1-month. Typical ClpP1 and ClpP2 protein expression yields range from 15 to 25 mg/mL and 7.5–12.5 mg/mL, respectively. ClpP1 can be concentrated to ~60 mg/mL without any sign of precipitation, while ClpP2 exhibits less stability at concentrations exceeding 12.5 mg/mL. Samples were flash frozen in activity buffer (25 mM HEPES, 100 mM KCl, 5 mM MgCl2, 1 mM DTT, 10% glycerol) and stored at −80 °C until further use.
For isolation and purification of ClpX, the pellet was resuspended in ice-cold ClpX lysis buffer (50 mM Tris-Cl pH 8.8, 200 mM NaCl, 100 mM KCl, 20 mM imidazole, 10% glycerol). Cells were lysed, and the lysate was clarified in a manner identical to that of ClpP1 and ClpP2. The clarified lysate was then loaded onto a 5 mL HF His-Trap column and washed/eluted similar to ClpP1 and ClpP2, except the concentration of imidazole in the His-elution buffer was decreased to 300 mM. Fractions were pooled and concentrated to ~5 mL and buffer exchanged via SEC into ClpX activity buffer (50 mM HEPES pH 7.6, 200 mM KCl, 100 mM NaCl, 20 mM MgCl2, 1 mM DTT, 10% glycerol). ClpX fractions were concentrated with a 10 kDa MW-cutoff Amicon Ultra concentrator. All assay protein concentrations were obtained via A280 nm readings and are reported as tetradecameric concentrations unless otherwise stated.
RNA Extraction, cDNA Preparation, Quantitative Reverse-Transcriptase PCR, and Genomic DNA Isolation
Freezer stocks of C. difficile 630 were struck onto BHIS agar plates and grown at 37 °C in a Coy vinyl anaerobic chamber (85% N2, 5% H2, and 10% CO2). In biological triplicate, single colonies were picked and used to inoculate ~5 mL of BHIS media. When the absorbance reached OD600 of ~0.6 (exponential phase) and ~1.2 (stationary phase), the cultures were harvested. Samples were added directly to 2× volume of RNAprotect Bacteria Reagent (Qiagen) centrifuged at 5000g, 25 °C for 10 min, decanted, then stored at −20 °C until further use. RNeasy Mini kit (Qiagen) was used to isolate the total RNA as described by the manufacturer. Contaminating genomic DNA was removed from the total RNA samples by two rounds of treatment using TURBO DNA-free kit (Invitrogen). cDNA was generated with the SuperScript IV VILO cDNA synthesis kit (Thermo) from 1 μg of total RNA in 20 μL reactions. Control samples lacking RT or templates were performed alongside each biological replicate. One microliter of the cDNA reaction mixture was added to 12.5 μL 2× qPCR iTaq Universal SYBR Green Supermix (BioRad), 10 nM of the forward and reverse primers, and DNase/ RNase free water was added to bring the total PCR reaction volume to 25 μL. A 7500 Fast Real-Time PCR System (Applied Biosystems) was used to perform qPCR (See Table S1 for primers) for 40 cycles of amplification at 51.5 °C. qRT-PCR primers were designed utilizing the PrimerQuest tool by Integrated DNA Technologies. Each primer was then put into the NCBI BLAST program to confirm no off-target hits. Primer efficiencies were calculated for each set of primers and are as follows rpoB = 99.1%, clpP1 = 97.1%, and clpP2 = 97.8%. Expression of clpP1 and clpP2 was normalized to the reference gene rpoB, with the results calculated via the comparative cycle threshold method.43
As previously stated, freezer stocks of C. difficile 630 were struck onto BHIS agar plates and grown at 37 °C in an anaerobic chamber. Then, a single colony was picked and used to inoculate ~5 mL of BHIS media. Genomic DNA was isolated using GenElute Bacterial Genomic DNA kit and purified using Zymo Genomic DNA Clean and Concentrator kit, following manufacturer’s protocols.
FITC-β-Casein Proteolysis Assay
ClpP in activity buffer (260 nM) was incubated with ADEP (serially diluted from 500 μM) at 37 °C for 15 min in flat bottom, nonbinding, nonsterile, white polystyrene 96-well plates (Corning 3990). After the preincubation period, 1 μL of 100× FITC-β-casein stock was added to each well to give a final concentration of 4.5 μM FITC-β-casein and final well volume of 100 μL. Assay plates were then incubated at 37 °C, and hydrolysis of the fluorogenic substrate was monitored via an i-TECAN Infinite M200 plate reader (excitation: 485 nm; emission: 538 nm). Readings were taken every 30 min for 6 h.
SLY-AMC and Z-GGL-AMC Peptidolysis Assays
All reactions were performed in activity buffer. Reactions contained 1 uM ClpP and were performed at 30 °C unless otherwise specified. SLY-AMC was dissolved in DMSO at a stock concentration of 5 mM that was aliquoted into a 2× SLY-AMC master mix containing nanopure water and 10% DMSO. Reactions of 200 μL were assembled in 2× concentrations, and three 50 μL 2× reactions were dispensed into black 96-well plates. Reactions were started by adding 50 μL of 2× SLY-AMC, and a 0 h reading was taken (excitation: 380 nm/emission: 440 nm) before incubation at 30 °C. Hydrolysis of the fluorogenic substrate was monitored via an i-TECAN Infinite M200 plate reader, and readings were taken at the time points indicated in the reported data.
Decapeptide Degradation Assay
Assays were performed with 25 nM ClpP in activity buffer supplemented with 5% DMSO. ADEP was serially diluted and pipetted into the plate at 10× concentration, before a ClpP master mix was added, and both were incubated at 30 °C for 15 min in flat bottom, nonbinding, nonsterile, black polystyrene 96-well plates (Grenier). After the preincubation period, 1 μL of 1.5 mM Abz-DFAPKMALVPY–NO2 (Biomatik) solution was added to each assay well to give a final assay concentration of 15 μM fluorogenic decapeptide and final assay volume of 100 μL. Assay plates were incubated at 30 °C, and hydrolysis of the fluorogenic peptide was monitored via an i-TECAN Infinite M200 plate reader (excitation: 320 nm; emission: 420 nm). Readings were taken at the time points indicated in the reported data. The data were normalized to a 5% DMSO + peptide negative control.
ssrA-GFP Degradation Assay
ClpP1 and ClpP2 were separately coincubated with ClpX at 25 °C in ATPase buffer for 1 h prior to the start of the assay. An ATP-regeneration system containing 75 μg/mL creatine kinase and 5 mM creatine phosphate was used. All components were freshly prepared prior to addition to the reaction. The final concentrations of ClpP and ClpX were 400 and 100 nM, respectively. Two micromolar ssrA-GFP was added prior to the addition of ATP. All controls were incubated for 6 h at 30 °C. The reactions were initiated by addition of ATP in ATPase buffer to a final concentration of 4 mM into a master reaction mixture which was distributed into 50 μL aliquots, placed onto a 30 °C heat block, and covered in foil. Reactions were then quenched each hour with 95 °C Laemmli buffer. Ten microliters of each reaction was loaded into individual wells of a 4–20% SDS-PAGE gel. Experiments were performed in triplicate, and intensities were analyzed with ImageJ. The fraction of ssrA-GFP remaining was calculated at each time point by dividing the intensity ssrA-GFP band by the intensity of the ssrA-GFP + ClpP control.
ssrA-GFP ATPase Hydrolysis Assay
ClpP1 and ClpP2 were separately coincubated with ClpX at 25 °C in ATPase buffer (50 mM HEPES pH 7.6, 10 mM Tris pH 7.6, 100 mM KCl, 20 mM MgCl2, 1 mM DTT, 10% glycerol, 0.032 NP-40, 0.01% Triton X-100) for 1 h prior to the start of the assay. An ATP-regeneration system44 was utilized to monitor ATP-hydrolysis by ClpX via absorbance measurements taken at 340 nm in clear 96-well plates. Final concentrations of the ATP-regeneration assay were: 1 mM NADH, 20 U lactate dehydrogenase/pyruvate kinase, and 7.5 mM phosphoenolpyruvate. All components were freshly prepared prior to addition to the reaction. The final concentration of ClpP and ClpX were 200 and 50 nM, respectively. The reaction was initiated by addition of ATP in ATPase buffer, and measurements were taken every 2 min over the course of 4 h to ensure assay completion. Initial rates were processed in Microsoft Excel and transferred to GraphPad Prism for Michaelis-Menten and statistical analysis.
Thermal Shift Assay
The procedure was performed as previously described31 with slight modifications. One micromolar ClpP1 in activity buffer was incubated in the presence or absence of ADEP for 15 min at 37 °C prior to addition of SYPRO Orange (2× final concentration), final reaction volume 200 μL. The reaction was then allowed to equilibrate at room temperature in the dark for 15 min before dispensing 50 μL aliquots into 96-well plates in triplicate, sealed with optically clear microplate tape, and centrifuged briefly to remove bubbles. Thermal shift analysis was carried out on a CFX96 Real-Time System (Bio-Rad), heating from 25 to 85 °C in 0.3 °C increments, measuring fluorescence every 1 min in FRET mode. The data were then processed in GraphPad Prism and fit to a Boltzmann distribution curve after normalization.
Mass Spectrometry Analysis of Oligomeric Species
ClpP proteins in solution were reduced with DTT, alkylated with iodoacetamide, and digested with trypsin according to standard protocols. Liquid chromatography tandem mass spectrometry was performed by coupling a nanoAcquity UPLC (Waters Corp., Manchester, UK) to a Q-TOF SYNAPT G2S instrument (Waters Corp., Manchester, UK). Each protein digest (about 100 ng of peptide) was delivered to a trap column (300 μm × 50 mm nanoAcquity UPLC NanoEase Column 5 μm BEH C18, Waters Corp, Manchester, UK) at a flow rate of 2 μL/min in 99.9% solvent A (10 mM ammonium formate, pH 10, in HPLC grade water). After 3 min of loading and washing, peptides were transferred to another trap column (180 μm × 20 nanoAcquity UPLC 2G-V/MTrap 5 μm Symmetry C18, Waters Corp., Manchester, UK) using a gradient from 1 to 60% solvent B (100% acetonitrile). The peptides were then eluted and separated at a flow rate of 200 nL/min using a gradient from 1 to 40% solvent B (0.1% formic acid in acetonitrile) for 60 min on an analytical column (7.5 μm × 150 mm nanoAcquity UPLC 1.8 μm HSST3, Waters Corp, Manchester, UK). The eluent was sprayed via PicoTip Emitters (Waters Corp., Manchester, UK) at a spray voltage of 3.0 kV, a sampling cone voltage of 30 V, and a source offset of 60 V. The source temperature was set to 70 °C. The cone gas flow was turned off; the nano flow gas pressure was set at 0.3 bar, and the purge gas flow was set at 750 mL/h. The SYNAPT G2S instrument was operated in data-independent mode with ion mobility (HDMSe). Full scan MS and MS2 spectra (m/z 50–2000) were acquired in resolution mode (20 000 resolution fwhm at m/z 400). Tandem mass spectra were generated in the trapping region of the ion mobility cell by using a collisional energy ramp from 20 V (low mass, start/end) to 35 V (high mass, start/end). A variable IMS wave velocity was used. Wave velocity was ramped from 300 to 600 m/s (start to end), and the ramp was applied over the full IMS cycle. A manual release time of 500 μs was set for the mobility trapping and a trap height of 15 V with an extract height of 0 V. The pusher/ion mobility synchronization for the HDMSe method was performed using MassLynx V4.1 and DriftScope v2.4. LockSpray of glufibrinopeptide-B (m/z 785.8427) was acquired every 60 s, and lock mass correction was applied postacquisition.
Raw MS data were processed by PLGS (ProteinLynx Global Server, Waters Corp., Manchester, UK) for peptide and protein identification. MS/MS spectra were searched against the Uniprot E. coli database and the C. Difficile database (4322 reviewed proteins and 7753 unreviewed proteins, respectively) and with the following search parameters: full tryptic specificity up to two missed cleavage sites, carbamidomethylation of cysteine residues set as a fixed modification, and N-terminal protein acetylation and methionine oxidation.
Supplementary Material
ACKNOWLEDGMENTS
The authors gratefully acknowledge the OU Protein Production Core for protein purification services and instrument support. We thank Professor H. Zgurskaya for helpful discussions. The ΔEcClpP JK10 cell line was donated by Professor R. Sauer (MIT), and the ssrA-GFP was donated by Professor T. Baker (MIT). We are thankful for their willingness to share these materials. The authors also thank Dr. Fares Najar for discussions regarding genetic evolution and Dr. Virginie Sjoeland for the mass spectrometry analyses. Preliminary studies were made possible by a Health Research Grant from the Oklahoma Center for the Advancement of Science and Technology (OCAST, HR15-161, ASD). Research reported in this publication was supported by an Institutional Development Award (IDeA) from the National Institute General Medical Sciences of the National Institutes of Health under Grant P20GM103640 (ASD and JDB). Additional funding was provided by the National Institute of Infectious Disease under Grant R01AI119048 (JDB). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsinfecdis.8b00199.
qRTPCR primers, Ct counts, DLS data, HR-ESIMS digest and sequencing, DiANNA predictions, redox data on ClpP isoforms, mixing data, ssrA-GFP SDS-PAGE gel, and raw thermal shift data (PDF)
REFERENCES
- (1).Bhandari V, Wong KS, Zhou JL, Mabanglo MF, Batey RA, and Houry WA (2018) The role of ClpP protease in bacterial pathogenesis and human diseases. ACS Chem. Biol 13, 1413–1425. [DOI] [PubMed] [Google Scholar]
- (2).Brotz-Oesterhelt H, Beyer D, Kroll HP, Endermann R, Ladel C, Schroeder W, Hinzen B, Raddatz S, Paulsen H, Henninger K, Bandow JE, Sahl HG, and Labischinski H (2005) Dysregulation of bacterial proteolytic machinery by a new class of antibiotics. Nat. Med 11, 1082–1087. [DOI] [PubMed] [Google Scholar]
- (3).Conlon BP, Nakayasu ES, Fleck LE, LaFleur MD, Isabella VM, Coleman K, Leonard SN, Smith RD, Adkins JN, and Lewis K (2013) Activated ClpP kills persisters and eradicates a chronic biofilm infection. Nature 503, 365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Gersch M, Gut F, Korotkov VS, Lehmann J, Bottcher T, Rusch M, Hedberg C, Waldmann H, Klebe G, and Sieber SA (2013) The mechanism of caseinolytic protease (ClpP) inhibition. Angew. Chem. Int. Ed. 52, 3009–3014. [DOI] [PubMed] [Google Scholar]
- (5).Jarrad AM, Karoli T, Blaskovich MA, Lyras D, and Cooper MA (2015) Clostridium difficile drug pipeline: Challenges in discovery and development of new agents. J. Med. Chem 58, 5164–5185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Yu AYH, and Houry WA (2007) ClpP: A distinctive family of cylindrical energy-dependent serine proteases. FEBS Lett. 581, 3749–3757. [DOI] [PubMed] [Google Scholar]
- (7).Mikhailov VA, Stahlberg F, Clarke AK, and Robinson CV (2015) Dual stoichiometry and subunit organization in the ClpP1/P2 protease from the cyanobacterium synechococcus elongatus. J. Struct. Biol 192, 519–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Stanne TM, Pojidaeva E, Andersson FI, and Clarke AK (2007) Distinctive types of ATP-dependent Clp proteases in cyanobacteria. J. Biol. Chem 282, 14394–14402. [DOI] [PubMed] [Google Scholar]
- (9).Gaillot O, Bregenholt S, Jaubert F, Di Santo JP, and Berche P (2001) Stress-induced ClpP serine protease of Listeria monocytogenes is essential for induction of listeriolysin o-dependent protective immunity. Infect. Immun 69, 4938–4943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Gaillot O, Pellegrini E, Bregenholt S, Nair S, and Berche P (2000) The ClpP serine protease is essential for the intracellular parasitism and virulence of Listeria monocytogenes. Mol. Microbiol 35, 1286–1294. [DOI] [PubMed] [Google Scholar]
- (11).Zeiler E, Braun N, Bottcher T, Kastenmuller A, Weinkauf S, and Sieber SA (2011) Vibralactone as a tool to study the activity and structure of the ClpP1P2 complex from Listeria monocytogenes. Angew. Chem. Int. Ed 50, 11001–11004. [DOI] [PubMed] [Google Scholar]
- (12).Zeiler E, List A, Alte F, Gersch M, Wachtel R, Poreba M, Drag M, Groll M, and Sieber SA (2013) Structural and functional insights into caseinolytic proteases reveal an unprecedented regulation principle of their catalytic triad. Proc. Natl. Acad. Sci. U. S. A 110, 11302–11307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Dahmen M, Vielberg MT, Groll M, and Sieber SA (2015) Structure and mechanism of the caseinolytic protease ClpP½ heterocomplex from Listeria monocytogenes. Angew. Chem. Int. Ed 54, 3598–3602. [DOI] [PubMed] [Google Scholar]
- (14).Hall BM, Breidenstein EB, de la Fuente-Nunez C, Reffuveille F, Mawla GD, Hancock RE, and Baker TA (2017) Two isoforms of Clp peptidase in Pseudomonas aeruginosa control distinct aspects of cellular physiology. J. Bacteriol 199, E00568–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Akopian T, Kandror O, Raju RM, Unnikrishnan M, Rubin EJ, and Goldberg AL (2012) The active ClpP protease from M. tuberculosis is a complex composed of a heptameric ClpP1 and a ClpP2 ring. EMBO J. 31, 1529–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Famulla K, Sass P, Malik I, Akopian T, Kandror O, Alber M, Hinzen B, Ruebsamen-Schaeff H, Kalscheuer R, Goldberg AL, and Brotz-Oesterhelt H (2016) Acyldepsipeptide antibiotics kill mycobacteria by preventing the physiological functions of the ClpP1P2 protease. Mol. Microbiol 101, 194–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Li M, Kandror O, Akopian T, Dharkar P, Wlodawer A, Maurizi MR, and Goldberg AL (2016) Structure and functional properties of the active form of the proteolytic complex, ClpP1P2, from Mycobacterium tuberculosis. J. Biol. Chem 291, 7465–7476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Ollinger J, O’Malley T, Kesicki EA, Odingo J, and Parish T (2012) Validation of the essential ClpP protease in Mycobacterium tuberculosis as a novel drug target. J. Bacteriol 194, 663–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Raju RM, Unnikrishnan M, Rubin DH, Krishnamoorthy V, Kandror O, Akopian TN, Goldberg AL, and Rubin EJ (2012) Mycobacterium tuberculosis ClpP1 and ClpP2 function together in protein degradation and are required for viability in vitro and during infection. PLoS Pathog. 8, e1002511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Schmitz KR, Carney DW, Sello JK, and Sauer RT (2014) Crystal structure of Mycobacterium tuberculosis ClpP1P2 suggests a model for peptidase activation by AAA+ partner binding and substrate delivery. Proc. Natl. Acad. Sci. U. S. A 111, E4587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Personne Y, Brown AC, Schuessler DL, and Parish T (2013) Mycobacterium tuberculosis ClpP proteases are co-transcribed but exhibit different substrate specificities. PLoS One 8, e60228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Jain S, Graham C, Graham RL, McMullan G, and Ternan NG (2011) Quantitative proteomic analysis of the heat stress response in Clostridium difficile strain 630. J. Proteome Res 10, 3880–3890. [DOI] [PubMed] [Google Scholar]
- (23).Lawley TD, Croucher NJ, Yu L, Clare S, Sebaihia M, Goulding D, Pickard DJ, Parkhill J, Choudhary J, and Dougan G (2009) Proteomic and genomic characterization of highly infectious Clostridium difficile 630 spores. J. Bacteriol 191, 5377–5386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Emerson JE, Stabler RA, Wren BW, and Fairweather NF (2008) Microarray analysis of the transcriptional responses of Clostridium difficile to environmental and antibiotic stress. J. Med. Microbiol 57, 757–764. [DOI] [PubMed] [Google Scholar]
- (25).Chong PM, Lynch T, McCorrister S, Kibsey P, Miller M, Gravel D, Westmacott GR, and Mulvey MR (2014) Canadian Nosocomial Infection Surveillance, P. Proteomic analysis of a nap1 Clostridium difficile clinical isolate resistant to metronidazole. PLoS One 9, e82622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Sekulovic O, and Fortier LC (2015) Global transcriptional response of Clostridium difficile carrying the cd38 prophage. Appl Environ. Microbiol 81, 1364–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Gersch M, List A, Groll M, and Sieber SA (2012) Insights into structural network responsible for oligomerization and activity of bacterial virulence regulator caseinolytic protease p (ClpP) protein. J. Biol. Chem 287, 9484–9494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Ni T, Ye F, Liu X, Zhang J, Liu H, Li J, Zhang Y, Sun Y, Wang M, Luo C, Jiang H, Lan L, Gan J, Zhang A, Zhou H, and Yang CG (2016) Characterization of gain-of-function mutant provides new insights into ClpP structure. ACS Chem. Biol 11, 1964–1972. [DOI] [PubMed] [Google Scholar]
- (29).Stahl M, and Sieber SA (2017) An amino acid domino effect orchestrates Clpp’s conformational states. Curr. Opin. Chem. Biol 40, 102–110. [DOI] [PubMed] [Google Scholar]
- (30).Kenniston JA, Baker TA, Fernandez JM, and Sauer RT (2003) Linkage between ATP consumption and mechanical unfolding during the protein processing reactions of an AAA+ degradation machine. Cell 114, 511–520. [DOI] [PubMed] [Google Scholar]
- (31).Gersch M, Famulla K, Dahmen M, Gobl C, Malik I, Richter K, Korotkov VS, Sass P, Rubsamen-Schaeff H, Madl T, Brotz-Oesterhelt H, and Sieber SA (2015) AAA+ chaperones and acyldepsipeptides activate the ClpP protease via conformational control. Nat. Commun 6, 6320. [DOI] [PubMed] [Google Scholar]
- (32).Leodolter J, Warweg J, and Weber-Ban E (2015) The Mycobacterium tuberculosis ClpP1P2 protease interacts asymmetrically with its ATPase partners ClpX and Clpc1. PLoS One 10, e0125345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Woo KM, Chung WJ, Ha DB, Goldberg AL, and Chung CH (1989) Protease ti from Escherichia coli requires ATP hydrolysis for protein breakdown but not for hydrolysis of small peptides. J. Biol. Chem 264, 2088–2091. [PubMed] [Google Scholar]
- (34).Liu Y, Patricelli MP, and Cravatt BF (1999) Activity-based protein profiling: The serine hydrolases. Proc. Natl. Acad. Sci. U. S. A 96, 14694–14699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Gottesman S, Roche E, Zhou Y, and Sauer RT (1998) The ClpXP and ClpAP proteases degrade proteins with carboxy-terminal peptide tails added by the ssrA-tagging system. Genes Dev. 12, 1338–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Martin A, Baker TA, and Sauer RT (2007) Distinct static and dynamic interactions control ATPase-peptidase communication in a AAA+ protease. Mol. Cell 27, 41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Kim YI, Levchenko I, Fraczkowska K, Woodruff RV, Sauer RT, and Baker TA (2001) Molecular determinants of complex formation between Clp/Hsp100 Atpases and the ClpP peptidase. Nat. Struct. Biol 8, 230–233. [DOI] [PubMed] [Google Scholar]
- (38).Lee BG, Park EY, Lee KE, Jeon H, Sung KH, Paulsen H, Rubsamen-Schaeff H, Brotz-Oesterhelt H, and Song HK (2010) Structures of ClpP in complex with acyldepsipeptide antibiotics reveal its activation mechanism. Nat. Struct. Mol. Biol 17, 471–478. [DOI] [PubMed] [Google Scholar]
- (39).Kirstein J, Hoffmann A, Lilie H, Schmidt R, Rubsamen-Waigmann H, Brotz-Oesterhelt H, Mogk A, and Turgay K (2009) The antibiotic ADEP reprogrammes ClpP, switching it from a regulated to an uncontrolled protease. EMBO Mol. Med 1, 37–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Li DH, Chung YS, Gloyd M, Joseph E, Ghirlando R, Wright GD, Cheng YQ, Maurizi MR, Guarne A, and Ortega J (2010) Acyldepsipeptide antibiotics induce the formation of a structured axial channel in ClpP: A model for the ClpX/ClpA-bound state of ClpP. Chem. Biol 17, 959–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Gil F, and Paredes-Sabja D (2016) Acyldepsipeptide antibiotics as a potential therapeutic agent against Clostridium difficile recurrent infections. Future Microbiol. 11, 1179–1189. [DOI] [PubMed] [Google Scholar]
- (42).Bolon DN, Grant RA, Baker TA, and Sauer RT (2004) Nucleotide-dependent substrate handoff from the SspB adaptor to the AAA+ ClpXP protease. Mol. Cell 16, 343–350. [DOI] [PubMed] [Google Scholar]
- (43).Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, 45e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Norby JG (1988) Coupled assay of Na+,K+-ATPase activity. Methods Enzymol 156, 116–119. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.