

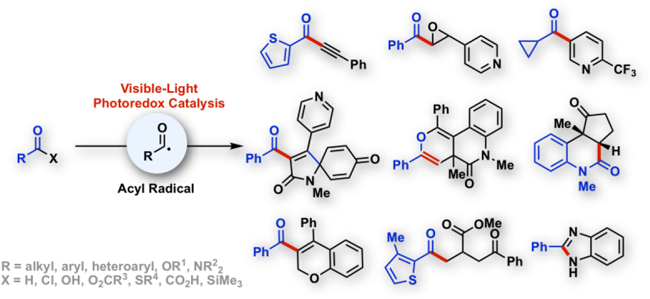
Abstract
Visible light photoredox catalysis has enabled easy access to acyl radicals under mild reaction conditions. Reactive acyl radicals, generated from various acyl precursors such as aldehydes, α-ketoacids, carboxylic acids, anhydrides, acyl thioesters, acyl chlorides, or acyl silanes, can undergo a diverse range of synthetically useful transformations, which were previously difficult or inaccessible. This review summarizes such recent progress of visible-light-driven acyl radical generation using transition metal photoredox catalysts, metallaphotocatalysts, hypervalent iodine catalysts or organic photocatalysts.
Keywords: Acylation, Photoredox Catalysis, Acyl Radical, Visible-Light, Single Electron Transfer, Oxidation, Reduction, Cross-Coupling
Graphical Abstract
Introduction
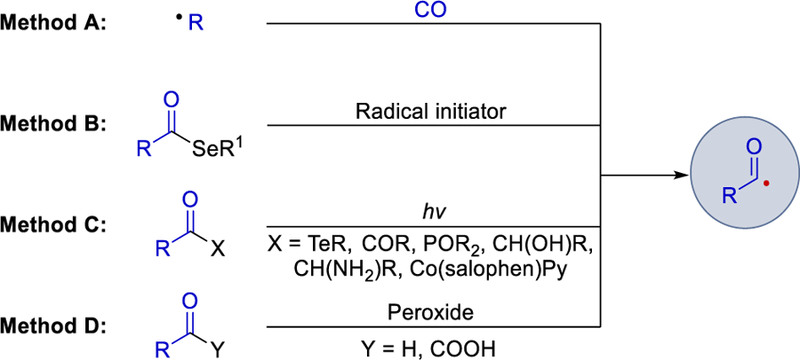
Acyl radicals are nucleophilic in nature1 and serve as versatile synthetic intermediates in Giese-type additions to activated alkenes,2 Minisci-type acylation of heteroarenes,3 and preparation of a wide range of natural and biologically active molecules.4, Conventional approaches to the generation of acyl radicals, however, generally require harsh reaction conditions such as UV irradiation or high temperatures.5 One way to access these intermediates is by generation of alkyl radicals from alkyl iodides through photochemical or thermal initiation and subsequent carbonylation by CO (Scheme 1, method A). The use of acyl selenides in the presence of an organotin reagent such as Bu3SnH together with a radical initiator4a, 6 is an alternative means of generating acyl radicals (method B). Through the photochemical cleavage of the RC(O)–X bond of acyl tellurides (X = Te-R’),7 benzoylphosphine oxides (X = P(O)Ph2)8 can also afford acyl radicals. α-Hydroxy or α-amino ketones RC(O)–X [X = CH(OH)R or CH(NH2)R]8a, 8b, 9 or cyclic ketones (via a Norrish type I cleavage)10 or esters (via the photo-Fries rearrangements)11 or acyl-cobalt salophen reagents [X = Co(salophen)Py]12 can all also deliver acyl radicals (method C). In addition, peroxide-mediated homolytic abstraction of a hydrogen atom from aldehydes and α-keto acids are the viable option for the generation of an acyl radical13 (method D). Many directed or non-directed C-H acylation processes catalyzed by transition-metals were developed in the last decades using this strategy.14 However, from a synthetic viewpoint, their utility was somewhat limited due to the requirement of high energy conditions such as high temperatures, UV irradiation, or stoichiometric amounts of toxic reagents or oxidants.
Scheme 1.
Recently, visible-light photoredox catalysis has emerged as a powerful tool for the synthesis of organic scaffolds that are difficult to prepare by traditional methods.15 Photoredox catalysts (PC) upon excitation with visible light generate an excited photocatalyst (*PC) which can act by single electron transfer (SET) processes not only as a one electron-oxidant or a one electron-reductant but also as an energy donor, activating acceptor molecules by an energy transfer (EnT) process.16 This photoredox strategy, therefore, provides a new platform for radical reactions by obviating the need for radical initiators and a stoichiometric amount of strong reducing agents. All these features make the photoredox catalysis a sustainable alternative from the viewpoint of radical reactions and one that can be utilized as an elegant way to access acyl radicals.
The scope of this review
Over the past decade, various research groups reported the visible light mediated generation of acyl radicals using photoredox catalysts, metallaphotocatalysts, hypervalent iodine photocatalysts, or organic photocatalysts. This review aims to provide an overview of recent progress in such visible-light driven acyl radical generation using aldehydes, α-ketocarboxylic acids, carboxylic acids, carboxylic acid anhydrides, acyl thioesters, acid chlorides, or acylsilanes as acyl radical precursors (Scheme 2). The scope, limitations, and the proposed reaction mechanism of each transformation are discussed. It is noteworthy that most of the reactions described in this review proceed through photoredox catalytic cycles, but the radical chain mechanism cannot be excluded.
Scheme 2.
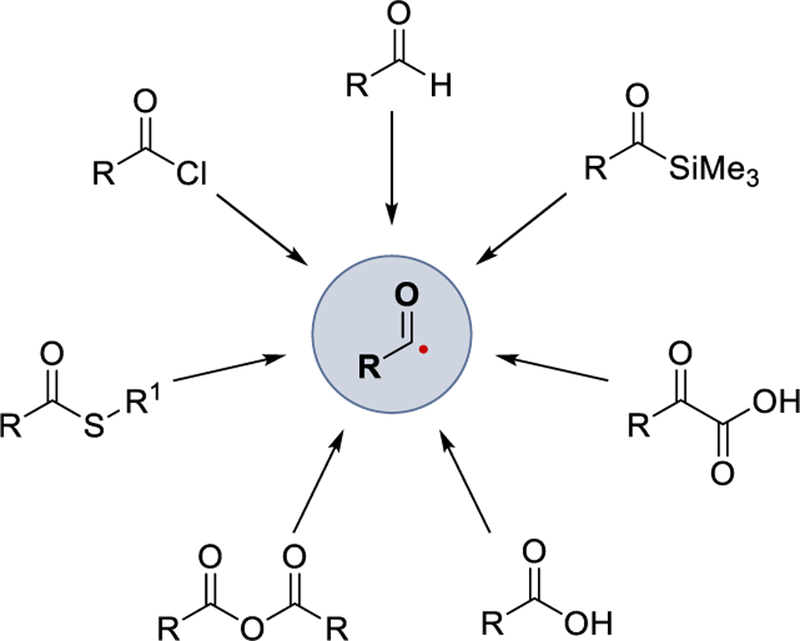
Various modes of acyl radical generation by visible light photocatalysis.
Aldehydes as a source of acyl radicals
Aldehydes are abundant, readily available, and versatile intermediates that are commonly converted to acyl radicals through a hydrogen atom transfer (HAT). In this process, a radical species generated from a HAT reagent abstracts the hydrogen atom of aldehydes forming an acyl radical. Common HAT reagents4b, 17 such as persulfates,18 or tert-butyl hydro-peroxide,19 which are used in the traditional thermal radical chemistry, are also applicable to photoredox catalysis. Compounds like quinuclidine20 and Eosin Y21 have also emerged as new HAT reagents for activation of aldehydes. Bond dissociation energy (BDE) of C−H of different aldehydes are strikingly similar within 88.0 kcal/mol to 89.4 kcal/mol, while the BDE of common HAT reagents could range from 88.2 kcal/mol to 106.3 kcal/mol (Scheme 3).20, 22
Scheme 3.
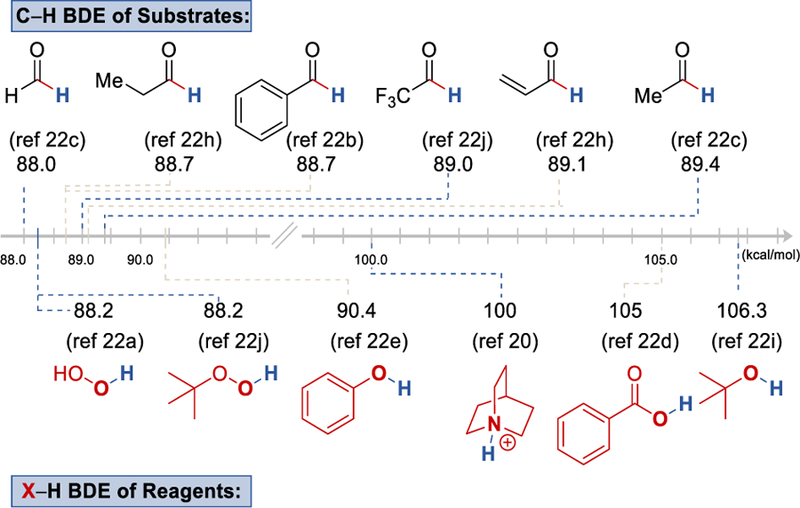
Selected Bond Dissociation Energy (BDE) of common aldehydes and HAT reagents.
In 2013, Cho et al. reported a synthesis of carboxylic acids through photocatalytic oxidation of aldehydes using molecular O2 as an oxidant and a HAT reagent (Scheme 4).23 Both electron deficient and electron-rich aromatic aldehydes, as well as aliphatic aldehydes, were oxidized to carboxylic acids in excellent yields (90% to 99%).
Scheme 4.
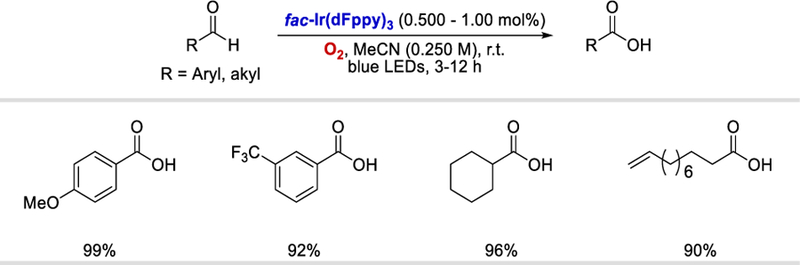
Formation of carboxylic acids via photo-induced energy transfer.23
It was proposed that the excited *Ir(dFppy)3 (ET = 60.1 kcal/mol)24 converts the triplet 3O2 into the singlet 1O2 via photo-induced energy transfer. The 1O2 then serves as a HAT reagent that abstracts an aldehyde hydrogen atom to form an acyl radical 5.2 and a hydroperoxyl radical (5.3). These two radicals recombine to give a peroxy acid 5.4, which reacts with another molecule of aldehyde to afford an adduct 5.5. This compound then undergoes Bayer-Villiger type rearrangement to form the desired carboxylic acid product 5.6.
In 2014, Zeng et al. published a photoredox catalysis method to generate benzoyl radicals from benzaldehydes for acylation of phenanthridine (Scheme 6).25 Although various benzaldehydes with halogen, alkyl, methoxy, and acetoxy substituents could serve as benzoyl radical donors, phenanthridine was the only radical acceptor reported in this work. The reaction afforded the desired products in yields ranging from 27 to 73%, but aldehydes with strong electron donating groups such as p-NMe2 or strong electron withdrawing groups such as p-NO2 failed to afford any desired product.
Scheme 6.
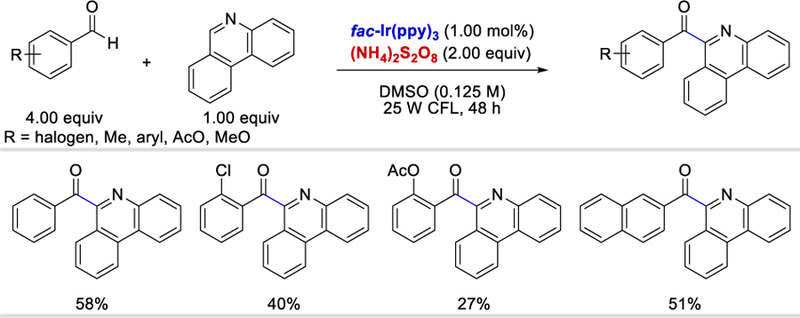
Photocatalytic aroylation of phenanthridine.25
The proposed mechanism of this reaction is shown in Scheme 7. The persulfate salt (7.1, E½red = +0.35 V vs. SCE)26 oxidizes the excited fac-*Ir(ppy)3 (E½IV/*III = −1.73 V vs. SCE) via single electron transfer (SET) to form a sulfate ion (7.3) and a sulfate radical anion (7.2). The resulting sulfate radical anion abstracts a benzaldehyde hydrogen atom via a HAT process to give the benzoyl radical (7.5). Subsequent addition of the benzoyl radical to phenanthridine generates an amidyl radical 7.8, which is deprotonated by SO42-to afford a radical anion 7.10. This radical anion is then oxidized by the IrIV(ppy)3 to form the final product 7.11.
Scheme 7.
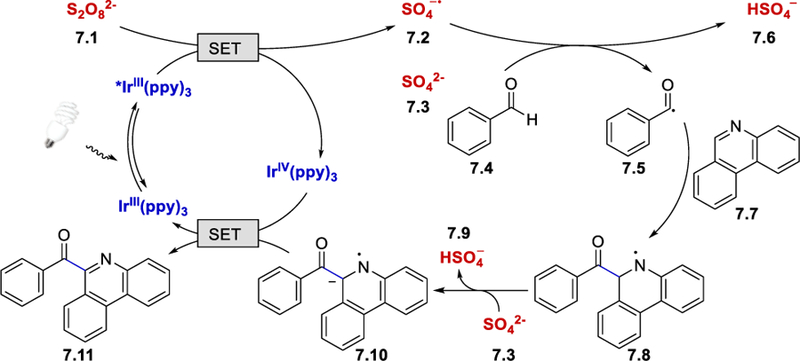
Persulfate salts as the HAT reagent under photocatalytic conditions. Adapted with permission from (25). Copyright (2014) Elsevier Ltd.25
t-Butyl hydroperoxide (TBHP) is a versatile HAT reagent. While single electron reduction of TBHP generates the t-butoxy radical, single electron oxidation of deprotonated TBHP affords the t-butyl peroxy radical (Scheme 8). Both of these radical species efficiently abstract an aldehyde hydrogen atom, giving an acyl radical.27 Consequently, combining TBHP with photoredox catalysis provides a useful and mild tool in the formation of acyl radicals from aldehydes.
Scheme 8.
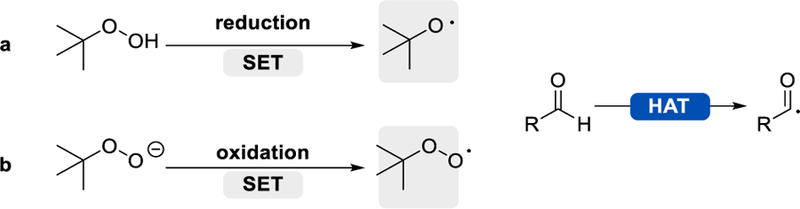
Abstraction of aldehyde hydrogen using TBHP
In 2015, Wang et al. reported a synthesis of α,β-epoxy ketones from styrenes and benzaldehydes under photocatalytic conditions using TBHP as a HAT reagent.28 Aromatic aldehydes with halogen, alkyl, and methoxy substituents and thiophenecarboxaldehyde reacted well, giving 61 to 83% yields. In terms of the styrene scope, arenes with halogen, alkyl, and methoxy substituents, pyridine, naphthalene, and 1,1-disubstituted styrenes including 1,1-diphenylethylene and α-methylstyrene coupled to afford the desired product in 51–85% yield.
Wang et al. proposed that the t-butyl hydroperoxide serves two roles in the reaction. First, TBHP oxidizes the excited *Ru(II) (E½III/*II = ‒0.81 V vs. SCE) to form a hydroxide and a t-butoxy radical tBuO•, which subsequently abstracts a hydrogen atom from an aldehyde to give a benzoyl radical. Second, deprotonation of the TBHP by the hydroxide ion –OH gives a t-butyl peroxide anion tBuOO−, which reduces the oxidized RuIII(bpy)32-+ regenerating the photocatalyst and forming a t-butyl peroxy radical tBuOO•. Recombination of the t-butyl peroxy radical with the radical intermediate 10.7 affords a β-peroxy ketone 10.8, which undergoes base promoted elimination of tBuO− to produce the final product 10.9.
In 2017, Hong et al. reported an intramolecular cyclization and epoxidation (Scheme 11), developed in analogy to the intermolecular method reported by Wang et al., using TBHP as the HAT reagent.29 The reaction conditions were applicable to both allyloxy and allylamino substrates to produce the desired spiroepoxy chroman-4-ones and enaminones. Interestingly, a three-step tandem process starting from benzyl alcohol and using the 8.00 equivalents of TBHP also occurred, albeit with lower yields.
Scheme 11.
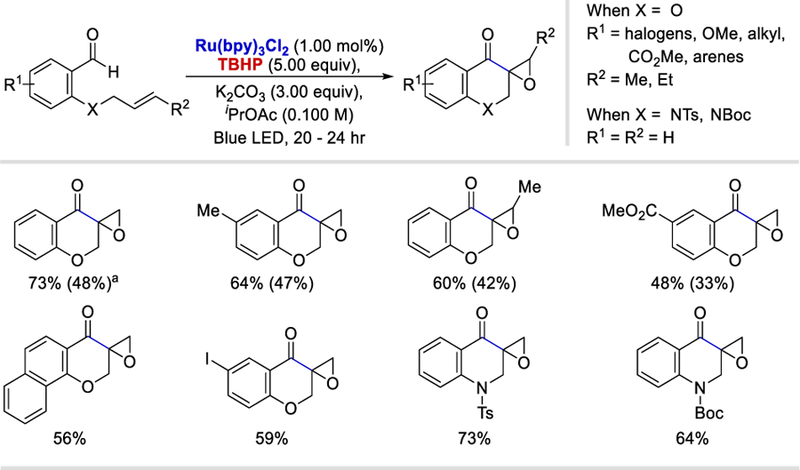
Intramolecular acyl radical addition/epoxidation.29 aYields in parentheses were obtained by the reaction of benzyl alcohol substrates using 8 equiv of TBHP.
It was reported that addition of TEMPO completely inhibits the reaction and forms the TEMPO adduct, which indicates the involvement of a radical intermediate. Exposure of the β-peroxy ketone 12.10 to a solution of K2CO3 in iPrOAc afforded the desired α-carbonyl epoxide 12.11, suggesting that the product is derived from the β-peroxy ketone intermediate. Based on these experimental results and the observation that benzylic alcohols also afforded the desired spiroepoxy chroman-4-ones, tandem oxidation of the benzylic alcohols to aldehydes followed by radical cyclization was proposed (Scheme 12). TBHP (12.1) oxidatively quenches the excited *RuII(bpy)32+ to generate RuIII(bpy)33+ and a t-butoxy radical tBuO• (12.2). This radical abstracts an α-hydrogen atom from the benzylic alcohol 12.3 to generate an α-hydroxy radical 7.5, which is oxidized and deprotonated to form benzaldehyde 12.7. Once the benzaldehyde is formed, the rest of the reaction mechanism is parallel to the catalytic cycle proposed by Wang et al.
Scheme 12.
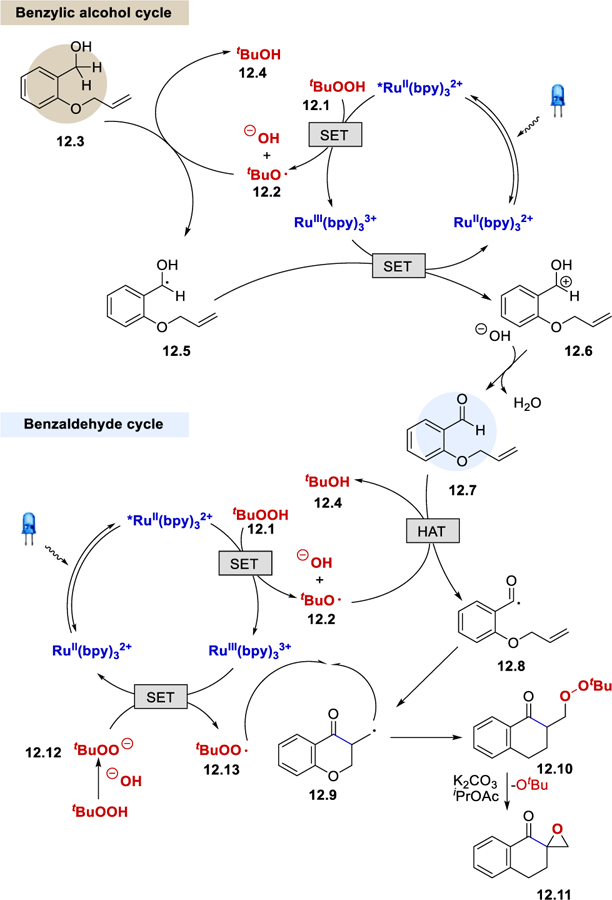
Photocatalytic cycle involving benzaldehyde and benzylic alcohol.29
Following Wang’s discovery using TBHP for the generation of acyl radicals, Cho and Iqbal, in 2016 disclosed a photocatalytic amide formation protocol from aldehydes and amines (Scheme 13).30 It was proposed that reduction of TBHP affords hydroxide and the t-butoxy radical, which undergoes HAT with benzaldehyde to form the benzoyl radical (13.5, path a). This radical abstracts the chlorine atom from N-chlorosuccinimide, forming benzoyl chloride (13.8), which reacts with an amine to afford the amide product. The reaction proceeds with a sub-stoichiometric quantity of TBHP, leading to speculation that other radical species such as the t-butyl peroxy radical (path b) and the succinimide radical (path c) generated in the reaction mixture might also be responsible for the HAT of benzaldehyde, generating the benzoyl radical.
Scheme 13.
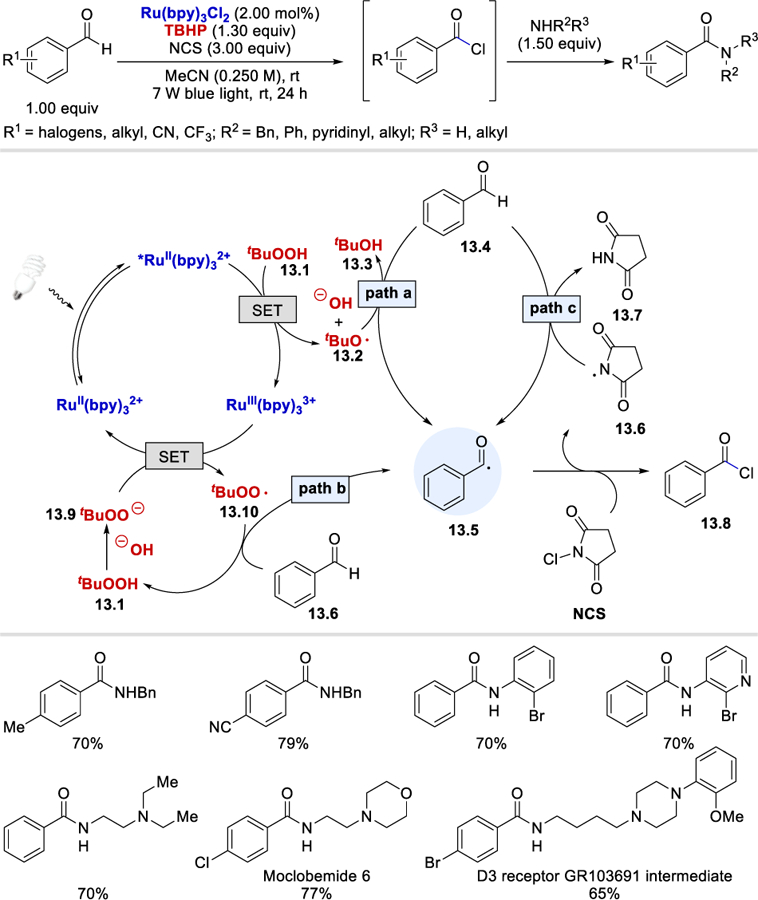
Photocatalytic amide synthesis from benzaldehydes using TBHP.30
This reaction has a broad substrate scope. Both electron rich and electron poor benzaldehydes with substituents, such as alkyl, halogens, a cyano, and a CF3 group coupled to afford the desired amides in 65–87% yields. Primary and secondary amines, anilines, and aminopyridines were all viable substrates giving the desired products in 58–82% yields. This strategy was used in the synthesis of the antidepressant Moclobemide 6 and the D3 Receptor GR103691 intermediate.
In 2017, Glorius et al. reported a photocatalytic alkynylation of aldehydes, formates, and formamides using alkynylbenziodoxolones as the alkyne source and the resulting benziodoxolonyl radical as a HAT reagent.31 This reaction has a broad substrate scope, and a wide range of alkyl, vinyl, aryl, and heteroaryl aldehydes, formates, and formamides reacted with various aryl or silyl alkynylbenziodoxolones to afford the desired alkynylated products in 46–90%. The carbonyl radicals generated under the optimized conditions fail to react with double bonds, and thus the method is compatible with α,β-unsaturated aldehydes. This transformation is also compatible with late-stage alkynylation of complex substrates such as cholesterol, lithocholic acid, probenecid, and adapalene derivatives giving the desired products in 48–78% yields.
Stern-Volmer quenching experiments showed that the 2-iodobenzoate was the only component that quenched the photocatalyst. In the presence of TEMPO, the formation of the alkynylated product was completely inhibited, and a TEMPO trapped adduct was obtained. On the basis of these results, the authors proposed that 2-iodobenzoate (15.1) reductively quenches the excited [*Ir(dF(CF3)ppy)2(5,5’-dCF3bpy)]PF6 (E½III*/II = +1.21 V vs. SCE)32 to form IrII and a 2-iodobenzoyloxyl radical (15.2) (Scheme 15). Hydrogen-atom abstraction of the aldehyde hydrogen by 15.2 provides a carbonyl radical 15.5 that reacts with alkynylbenziodoxolone (15.6) to form the desired product 15.8, simultaneously releasing the benziodoxolonyl radical (15.7). Reduction of 15.7 with IrII forms 15.1 and regenerates the ground state Ir photocatalyst. Alternatively, since 15.7 is in equilibrium with 15.2, it can undergo a HAT reaction directly.
Scheme 15.
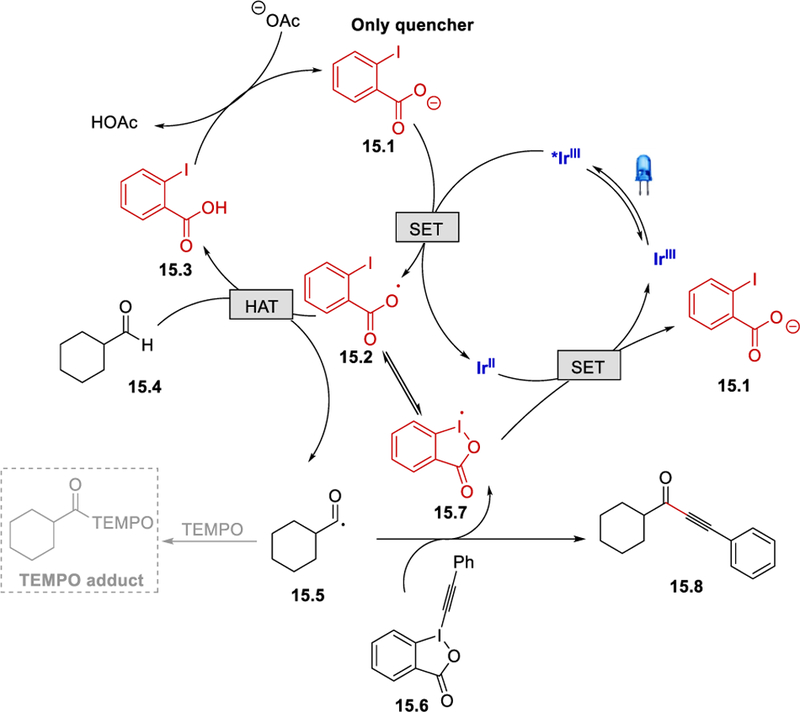
Proposed mechanism for photocatalytic alkynylation of aldehydes.31
In 2017, MacMillan et al. reported a triple catalytic protocol for arylation, vinylation, and alkylation of aldehydes via an acyl radical intermediate using aryl, vinyl, and alkyl bromides as coupling partners and quinuclidine, Ni(II), and a photocatalyst as catalysts (Scheme 16).33 This approach has a very broad scope and is insensitive to the electronic nature of the aryl bromides. Heteroaryl, cyclic and acyclic vinyl, and alkyl bromides are viable substrates and afforded the desired ketones in 50–90% yields. Regarding aldehyde scope, both alkyl and aryl aldehydes coupled well to afford the desired products in 70–90% yields, although 6–10 equivalents of aryl aldehydes were needed.
Scheme 16.
Alkylation, vinylation, and arylation of aldehydes via triple catalysis.
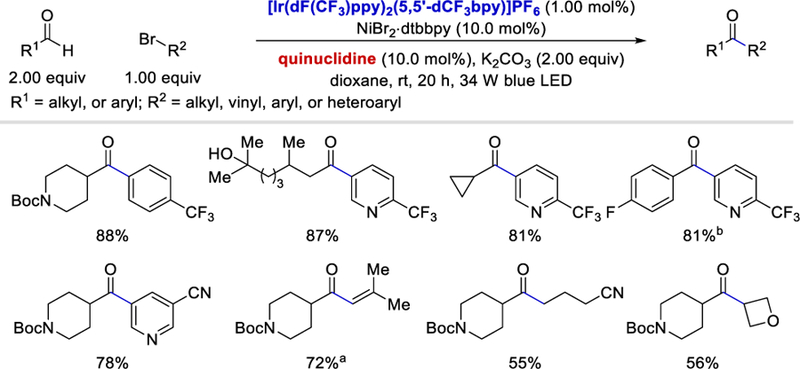
aK2CO3 (2.00 equiv). bp-fluorobenzaldehyde (10 equiv).33
The mechanistic hypothesis for this triple catalysis is shown in Scheme 17. An SET from quinuclidine (E½ox = +1.1 V vs. SCE in MeCN)20 to the excited [*Ir(dF(CF3)ppy)2(5,5’-dCF3bpy)]PF6 (E½III*/II = +1.21 V vs. SCE) generates an electrophilic quinuclidinium cation radical (17.2), which selectively abstracts the weak and hydridic aldehyde hydrogen atom to form the acyl radical 17.5. The α-amino C‒H bond also exhibits hydridic bond polarization and is subjected to hydrogen atom abstraction.34 The authors discovered that the reaction performed in 1,4-dioxane circumvented the unwanted competing α-amino C‒H activation affording exclusively the desired acylated product, whereas MeCN or DMSO solvents delivered the α-amino arylation product in addition to the desired product. Oxidative addition of arylbromide to Ni(0) on the other hand delivers an aryl-Ni(II) species 17.7, which is intercepted by the acyl radical 17.5 to form acyl-Ni (III) complex 17.8. Subsequently, reductive elimination forms the desired ketone product and a Ni(I) species 17.9. A single electron transfer between IrII and Ni(I) regenerates IrIII and Ni(0) catalysts and completes the catalytic cycle.
Scheme 17.
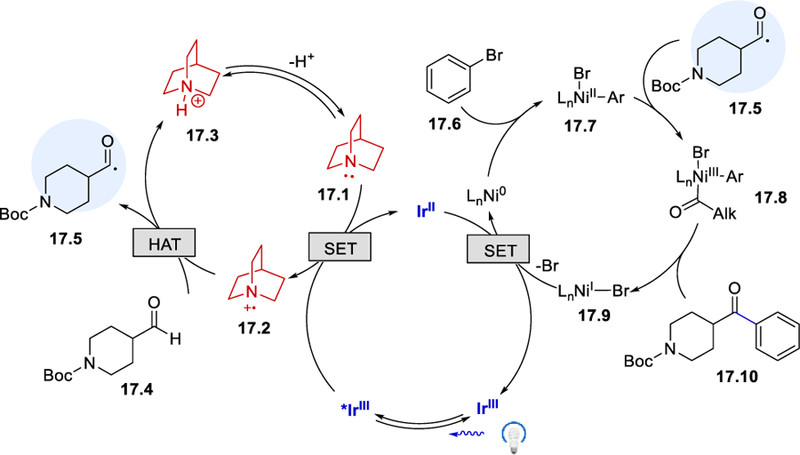
Proposed reaction mechanism for the triple catalysis.33 Adapted with permission from (33). Copyright (2017) American Chemical Society.
Also in 2017, Liu et al. used the quinuclidine catalyst to generate the acyl radical from aldehydes under photoredox conditions (Scheme 18).35 Both aromatic and aliphatic aldehydes were viable substrates under the dual-catalytic conditions. The synthetic utility was also well demonstrated by a variety of electron deficient olefin acceptors.
Scheme 18.
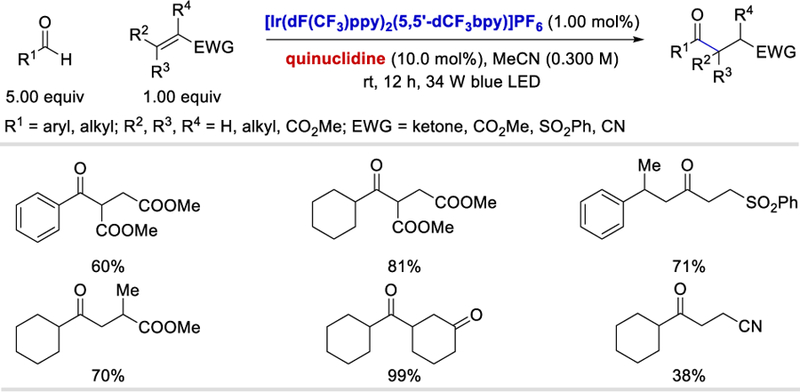
Photocatalytic hydroacylation using quinuclidine as the HAT reagent.35
With a mechanism similar to that described in Scheme 17, reductive quenching of the excited *IrIII catalyst by quinuclidine (19.1) forms a HAT reagent 19.2, which abstracts the aldehyde hydridic hydrogen to form an acyl radical. The acyl radical is then intercepted by an olefin to give the carbon radical intermediate 19.7. A single electron transfer from IrII to this intermediate, followed by protonation, delivers the desired product and regenerates the IrIII catalyst.
In the same paper, Liu et al. presented the preliminary result of a catalytic cross-coupling process using the Ir photocatalyst and NiCl2. In contrast to the MacMillan’s work,33 Liu et al. used a stoichiometric amount of quinuclidine, and the scope of the reaction is significantly limited as a result of the unwanted reduction of aryl bromides. (Scheme 20).
Scheme 20.
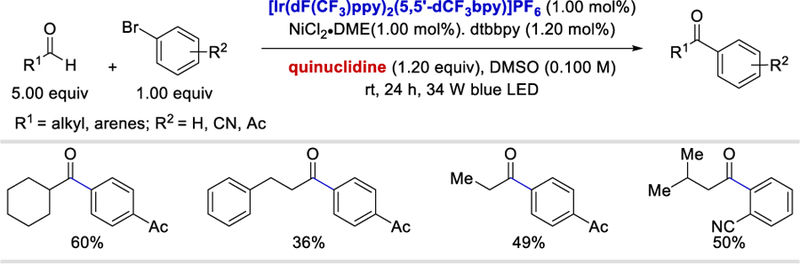
Preliminary results of metallophotocatalytic cross-coupling using NiCl2.35
In 2018, Itoh et al. used 2-t-butylanthraquinone (2-tBu-AQN) as a photocatalyst and benzoyl peroxide (BPO) as an oxidant in the presence of potassium carbonate to synthesize 3-acyl-4-arylcoumarin derivatives (Scheme 21).36 Alkyl, alkoxy, and acetoxy groups on both benzaldehydes and propynate substrates were well tolerated. The authors showed that many of their coumarin products have potential biological applications demonstrated by their effectiveness in inhibition of PSA secretion and proliferation of androgen-dependent prostate cancer.
Scheme 21.
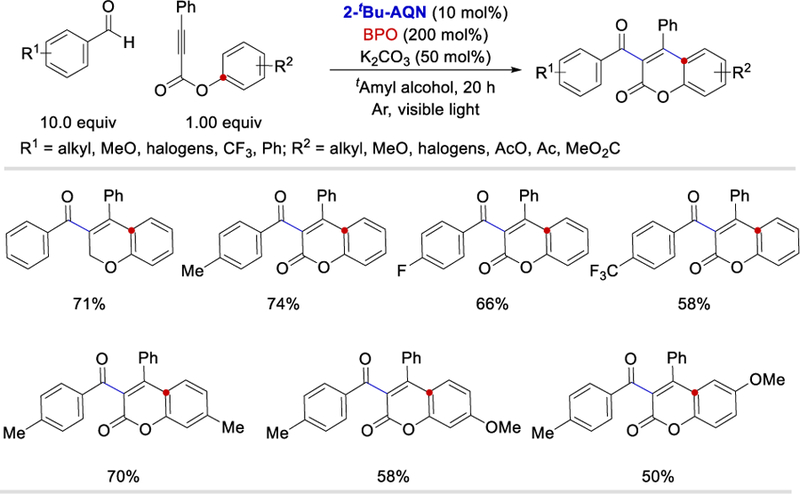
Photocatalytic formation of 3-acyl-4-arylcoumarin derivatives catalyzed by anthraquinone derivatives.36
The proposed mechanism for this reaction is depicted in Scheme 22. Photoexcited 2-t-butylanthraquinone (22.2) acts as a HAT reagent and abstracts the formyl hydrogen atom of benzaldehyde, forming the semiquinone radical37 AQH• and the benzoyl radical (22.5). Addition of the benzoyl radical to the propynate 22.6 generates a vinyl radical intermediate 22.7, which undergoes 5-exo-trig cyclization to afford a spirocyclic species 22.8. This spirocyclic intermediate is then oxidized either by either BPO or the benzoyloxyl radical (22.13) to form a carbocation 22.9. 1,2-Ester migration and deprotonation afford the desired product 22.11. Oxidation of AQH• by BPO, followed by deprotonation produce benzoic acid, benzoyloxyl radical and the ground state AQN catalyst. This reaction also proceeds under thermal conditions, where a 54% yield was obtained in the absence of the photocatalyst, indicating that benzoyloxy radical formed from the thermal decomposition of the benzoyl peroxide can also function as an alternative HAT reagent.
Scheme 22.
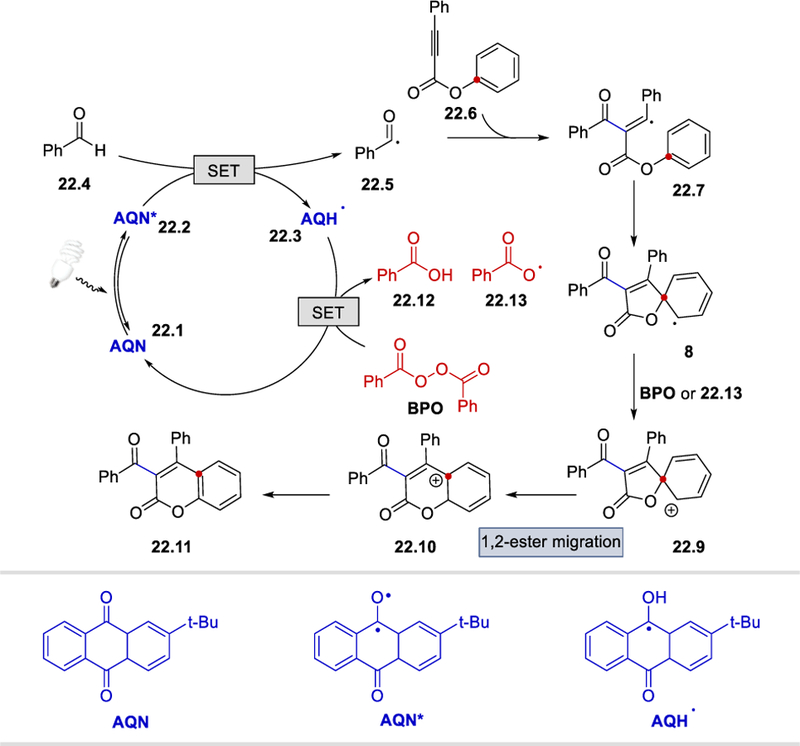
Photoredox-catalyzed coumarin formation from aldehydes via acyl radical intermediates.36
In 2018, Wu et al. reported that the excited Eosin Y, functioning as a HAT reagent, can abstract the aldehyde hydrogen atom under irradiation of an 18 W white LED light (Scheme 23).21 Alkyl, aryl and heteroaryl aldehydes reacted with electron deficient 1,1-dicyano-2-phenylethylene to afford the desired products in 83–94% yields. Benzaldehyde can react with a SOMOphile such as methyl 2-[(phenylsulfonyl)methyl]acrylate to form an allylation product in 48% yield. It should be noted that the major focus of their work was on the alkylation of a wide range of C–H bonds with electron deficient alkenes under photocatalytic conditions. The C–H partners include THF, thioethers, amides, alcohols, and cyclohexane. However, these coupling reactions are outside the scope of this review.
Scheme 23.
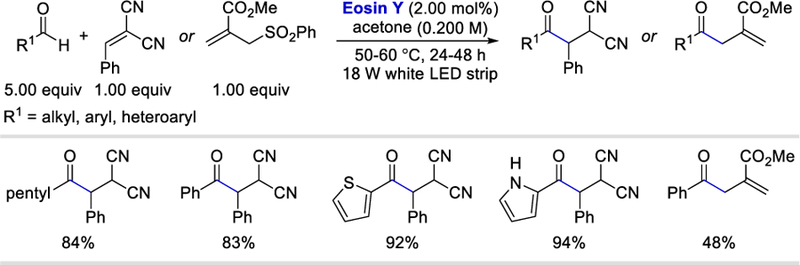
Scope of Eosin Y catalysed reductive acylation of electron deficient alkenes.21
Anionic Eosin Y is commonly engaged in SETs under photoredox catalyzed conditions. However, the neutral Eosin Y, which is used by Wu et al. is inactive in SET processes.38 It was shown that neither THF nor phenyl vinyl sulfone quenches the excited Eosin Y indicating that the reaction did not proceed through single electron transfer or energy transfer. Transient absorbance experiments showed that the excited *Eosin Y absorbs at both 329 nm and 543 nm, whereas the Eosin Y-H absorbs at 366 nm. In the presence of phenyl vinyl ketone, a radical acceptor, the decay of Eosin Y-H was much faster than took place in the absence of the alkene (Scheme 24), demonstrating the feasibility of HAT between Eosin Y-H and radical intermediates formed during the coupling process. The quantum yield of the reaction was estimated to be 0.40.
Scheme 24.
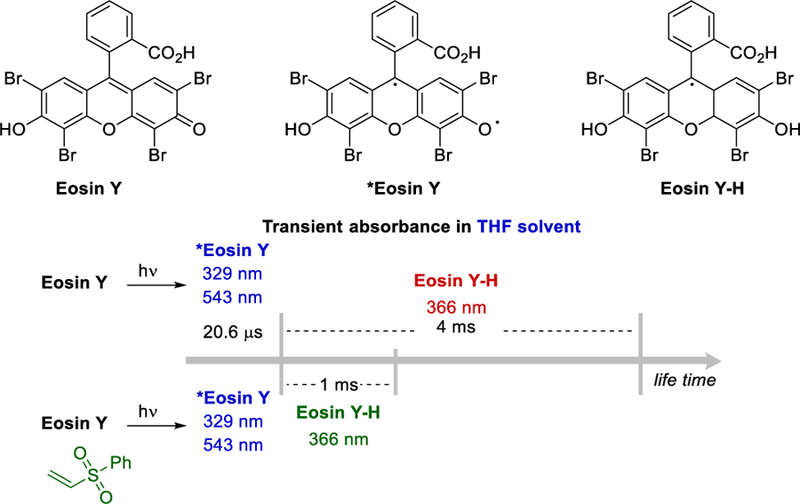
Lifetime of the transient absorbance of excited Eosin Y and the Eosin Y-H intermediate.21
On the basis of these mechanistic studies, the authors proposed the direct photocatalytic hydrogen atom transfer mechanism shown in Scheme 25.39 Photoexcitation of Eosin Y (25.1) affords excited *Eosin Y (25.2), which abstracts an aldehyde hydrogen atom to form an acyl radical species 25.5 and Eosin Y-H (25.3). The acyl radical is subsequently trapped by an electron deficient alkene to form the alkyl radical intermediate 25.7. At this stage, there are two reaction pathways accounting for the formation of the desired product and regeneration of the Eosin Y catalyst. In path a, a reverse hydrogen atom transfer (RHAT) between the Eosin Y-H and radical species 25.7 affords the final product and Eosin Y catalyst. In path b, aldehyde 25.4 undergoes HAT with the radical species 25.7 to form the desired product 25.8 and an acyl radical 25.5. The acyl radical reacts with Eosin Y-H via RHAT to afford an aldehyde and regenerate the Eosin Y catalyst. Although the authors performed computational and deuterium labeling studies, they could not distinguish between these two pathways.
Scheme 25.
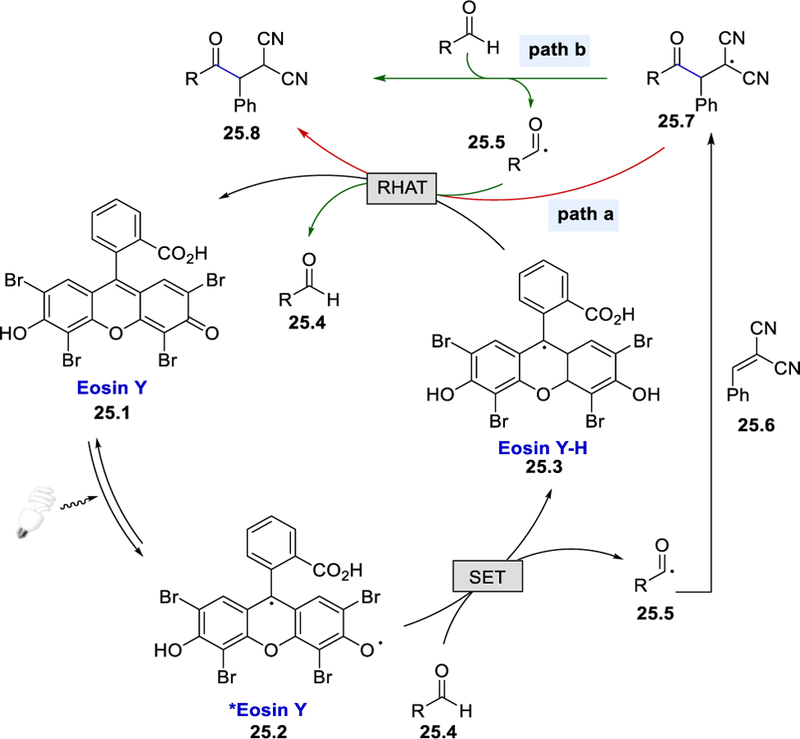
A direct HAT process photocatalyzed by Eosin Y.21
Salles et al., in 2018, achieved acyl-epoxylation and hydroacylation of alkenes using methylene blue (MB) as a photoredox catalyst and K2S2O8 as an oxidant in air-equilibrated water solution (Scheme 26).40 With high loading of the photocatalyst and oxidant (2.50 mol% and 2.00 equiv, respectively), simple benzaldehydes with arene substituted with methyl, Cl, F, MeO or CF3 reacted with styrenes or 2-vinylpyridine to afford the desired epoxide products in 75% to 92%. 1-Naphthaldehyde and cyclohexanecarboxyaldehyde were also viable substrates forming the final products in 84% and 79% yields. With lower loading of the photocatalyst and oxidant (0.500 mol% and 1.00 equiv, respectively), benzaldehydes coupled with long chain aliphatic alkenes to deliver the hydroacylated products in 67% to 89% yields.
Scheme 26.
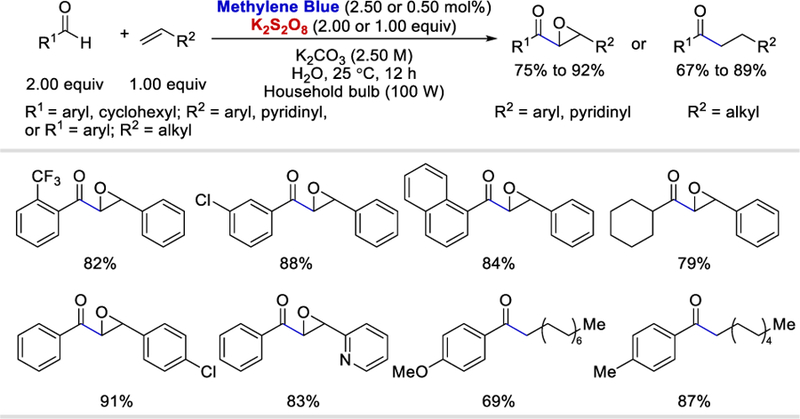
Epoxyacylation and hydroacylation using methylene blue as the photocatalyst and persulfate as the oxidant.40
The reaction of benzaldehyde and styrene using K2S2O8 in the absence of K2CO3 only afforded trace acyl-epoxylated product, whereas the reaction proceeded well using H2O2 in the presence of K2CO3. Based on these observations and literature precedents,41 a reaction mechanism involving hydroperoxide anion as a HAT reagent was proposed and is shown in Scheme 27. Persulfate anion (27.1) reacts water under basic conditions to generate the hydroperoxide anion (27.2) (E½red = –0.88 V vs SHE)42, which undergoes single electron oxidation with excited MB* (E½red = –1.21 V vs SHE) to form a hydroperoxyl radical (27.3) and methylene blue neutral radical MB•. Oxidation of MB• by the S2O82+ or O2 regenerates the methylene blue catalyst. The hydroperoxyl radical abstracts an aldehyde hydrogen atom to form an acyl radical 27.6, which adds onto an alkene to give the alkyl radical intermediate 27.8. With the lower loading of the photocatalyst and oxidant, the alkyl radical 27.8 undergoes a HAT with an aldehyde substrate to afford the desired hydroacylation product 27.9 via radical-chain propagation process. With a higher loading of the photocatalyst and persulfate anion, the alkyl radical 27.8 is trapped by the hydroperoxyl radical to deliver a β-peroxyl adduct 27.10. Base-promoted elimination affords the desired epoxide product 27.11.
Scheme 27.
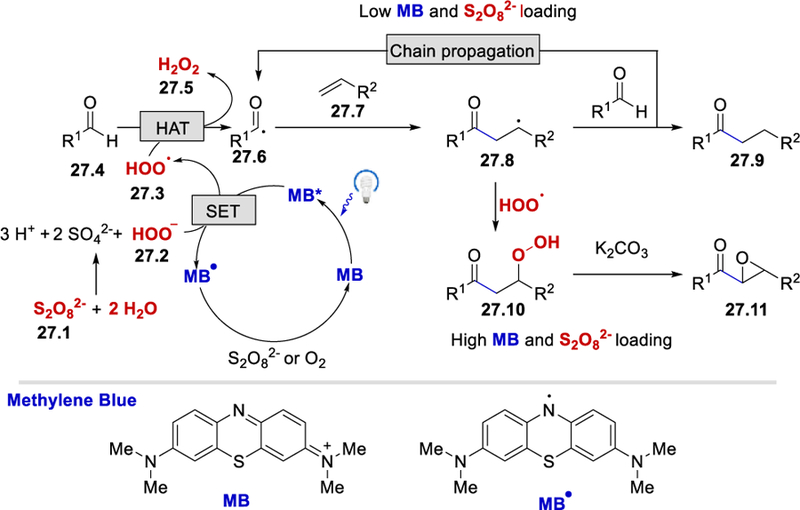
Proposed mechanism for methylene blue-catalyzed acyl radical formation.40
α-Keto acids as a source of acyl radicals
Conversion of α-keto acids to the acyl radical equivalents can be achieved via single electron transfer (SET) of the corresponding carboxylate by photocatalytic oxidation and subsequent decarboxylation. Alternatively, installation of a good leaving group (X) as in N-hydroxyphthalimide, generates a keto-ester that can be reduced by a photoredox catalyst (PC) and forms an acyl radical after further decarboxylation (Scheme 28).
Scheme 28.
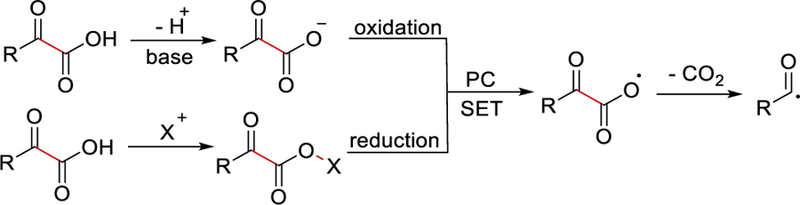
Generation of an acyl radical from α-ketoacids by photoredox catalysis.
In 2014, Lei and Lan reported the first visible light mediated photocatalytic oxidative decarboxylation of α-ketoacids in the synthesis of amides.43 This efficient radical decarboxylative coupling was catalyzed by the photocatalyst [Ru(phen)3]Cl2 (E½II*/I = +0.82 V vs. SCE)44 and molecular oxygen as an oxidant. A wide range of electron-donating and withdrawing aromatic α-keto acids reacted smoothly under these reaction conditions to afford the desired amides in good yields (64–85%). (Scheme 29). Heteroaromatic and aliphatic α-keto acids all provided desired products in 48 and 40% yields. Various electron-rich aromatic and aliphatic amines provided the corresponding products in 40–79% and 25–77% yields, respectively.
Scheme 29.
Visible-light-mediated decarboxylation/oxidative amidation of α-ketoacids with amines.43
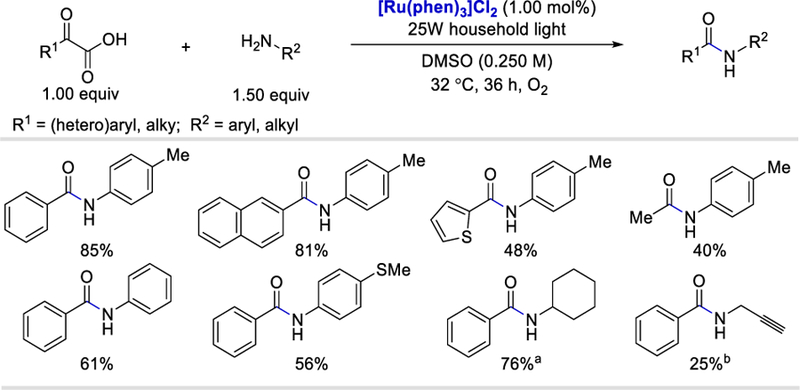
aUsing 10 equiv of amine. bUsing 5 equiv of amine.
A series of studies were conducted to examine the reaction mechanism. For example, cyclic voltammetry (CV) experiments of an α-ketoacid salt (PhCOCO2K) suggest an oxidative decarboxylation path, TEMPO trapping experiments confirm the formation of an acyl radical, and DFT calculations support the reaction between aroyl radical and amines followed by deprotonation leading to an amide radical anion. Interestingly, in the absence of the α-keto carboxylic acid, EPR studies of this photocatalytic reaction detected two peaks; [Ru(phen)3]+ and an amine radical. Addition of an α-keto carboxylic acid diminishes the organic radical peak and only the [Ru(phen)3]+ peak remains. Thus, the authors proposed that the oxidation of [Ru(phen)3]+ by molecular oxygen is crucial and probably the rate-determining step of this process. Based on these results, a possible reaction mechanism was proposed and is shown in Scheme 30. Reductive quenching of the excited photocatalyst *[Ru(phen)3]2+ by amines forms an amine radical cation 30.2 and [Ru(phen)3]+. Single electron oxidation of [Ru(phen)3]+ by molecular oxygen affords the ground state photoredox catalyst, [Ru(phen)3]Cl2, and a superoxide radical anion, which deprotonates and oxidizes the ammonium α-ketocarboxylate salt 30.3 to produce the dicarbonyl radical intermediate 30.5. Decarboxylation of 30.5 gives acyl radical 30.6, which is then trapped by an amine 30.1 followed by deprotonation in the presence of hydrogen peroxide anion HO2‒ (30.4) to produce the amide radical anion 30.7. Oxidation of 30.7 via a SET process affords the desired amide product.
Scheme 30.
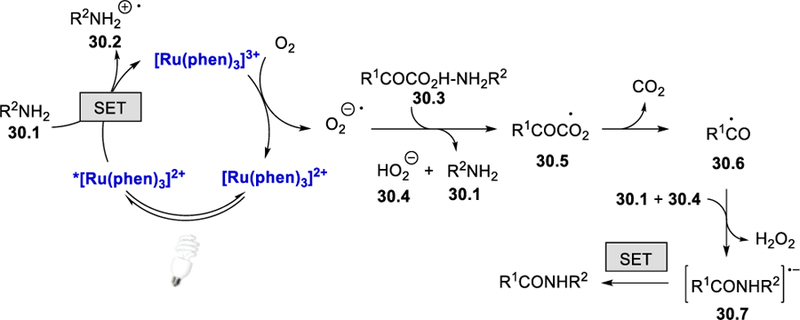
Mechanism for amidation from α-ketoacids and amines.43
This method was also applied to anilines, o-substituted with NH2, OH, and SH groups, to construct important heterocyclic compounds such as benzimidazoles (68–93%), benzoxazoles (39–46%), and benzothiazole (32%) in good yields (Scheme 31).
Scheme 31.
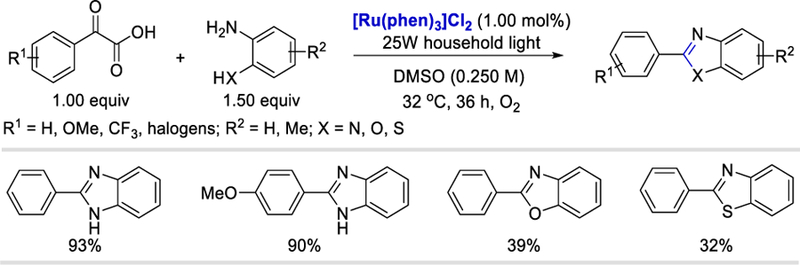
Benzoyl radical from α-ketoacids for the synthesis of 2-arylbenzazoles.43
Following initial studies by Lei and Lan on photoredox-catalyzed decarboxylative amidation, the acyl radical generation from α-keto acids has been employed in several radical coupling reactions. For example, in 2015, Macmillan et al. reported the first decarboxylative arylation of α-keto acids using aryl halides via a merger of visible-light photoredox catalysis and nickel catalysis.45 Substituted aromatic and aliphatic α-keto acids are compatible under the reaction conditions and provide moderate to excellent yields (57–92%) of the desired ketones (Scheme 32). A sterically hindered o-substituted α-keto acid was also efficiently coupled under the same reaction conditions and provided the desired product in high yield (92%). Halo-arenes (halo = Br, I) and halo-heteroarenes containing electron donating and withdrawing substituents also provided the desired ketones in 70–90% and 64–85% yields, respectively.
Scheme 32.
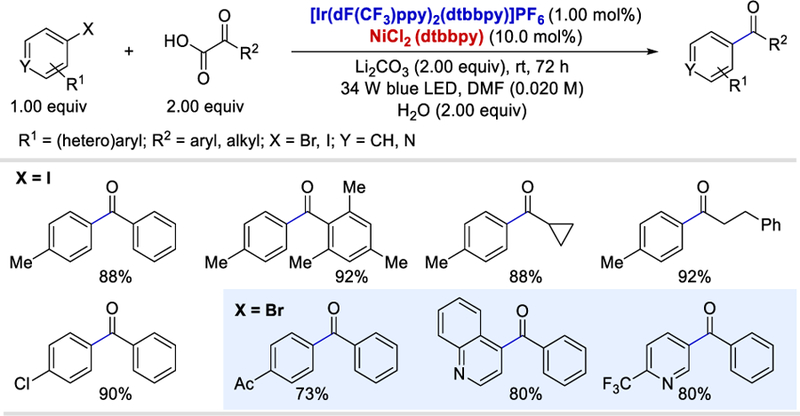
Decarboxylative arylation of α-ketoacids using metallaphotoredox.45
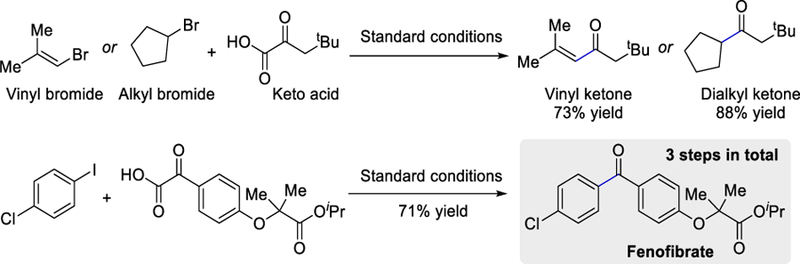
Further generalization of this method was demonstrated by successfully coupling hindered vinyl halides and cyclopentyl bromide with α-keto-acids under the optimized reaction conditions to generate a vinyl ketone and a dialkyl ketone in good yields (73–88%). This strategy was also elaborated for the synthesis of fenofibrate, a cholesterol-controlling drug (Scheme 33).
Scheme 33.
Scope of the decarboxylative arylation strategy.45
The authors proposed that the strongly oxidizing excited *[Ir(dF(CF3)ppy)2(dtbbpy)]+ (34.2) (E½III*/II = +1.21 V vs. SCE in CH3CN)46 generated upon visible-light irradiation reacts with the α-ketocarboxylate via single-electron oxidation to generate the corresponding carboxyl radical species and the reduced photocatalyst 34.4. This carboxyl radical species subsequently delivers the acyl radical species 34.5 via decarboxylation. SET from the strong reducing IrII species 34.4 (E½III/II = –1.37 V vs. SCE in CH3CN)46 to the in-situ generated NiI–dtbbpy complex would afford the Ni0 catalyst 34.6, which initiates the second catalytic cycle through oxidative addition to the aryl halide to generate the NiII aryl complex 34.8. The addition of the nucleophilic acyl radical 34.5 to 34.8 produces the nickel acyl complex 34.9. Final reductive elimination from this NiIII complex provides the desired ketone 34.10 with the regeneration of the NiI–dtbbpy complex 34.11 (Scheme 34 ).
Scheme 34.
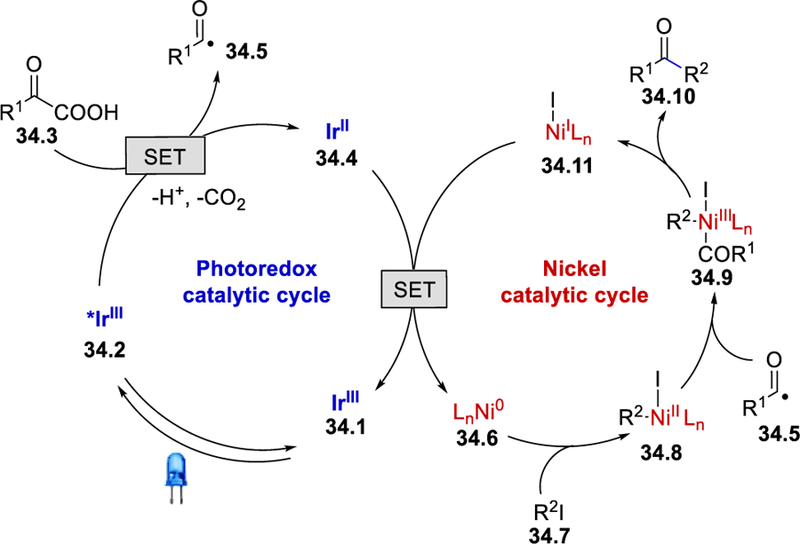
Proposed mechanism of ketone formation from α-ketoacids and iodoarenes.45
In the last decades, palladium has been widely used in various transition metal-catalyzed decarboxylative cross-coupling reactions.47 A typical example is the decarboxylative acylation of aryl halides by using α-keto acids.48 In 2015, Shang and Fu, combined photoredox and palladium catalysis and achieved such decarboxylative cross-coupling reactions for the first time.49 The reported decarboxylative coupling strategy of aryl halides with α-ketocarboxylic acids provides access to various unsymmetrical ketones. The electronic and steric effects of the substituents present in aryl halides have no substantial effects on the yields of the products (74–96%). Interestingly, aryl iodides containing t-butoxycarbonyl (Boc-)-protected phenylalanine ester and aryl boronic pinacol ester both provided the corresponding products in 88% and 70% yields. This reaction is compatible with electron donating aryl groups (82–95%) as well as heteroaryl (46–89%) and alkyl α-ketocarboxylic acids (60–89%), but the presence of electron-withdrawing substituents on the aryl ring of α-ketocarboxylic acids sharply decreased the product yield to ~10%. The use of an external phosphine ligand (Ni-xantphos) in case of bromoarenes is due to its relatively slow oxidative addition to the Pd0 catalyst when compared to iodoarenes (Scheme 35 ).
Scheme 35.
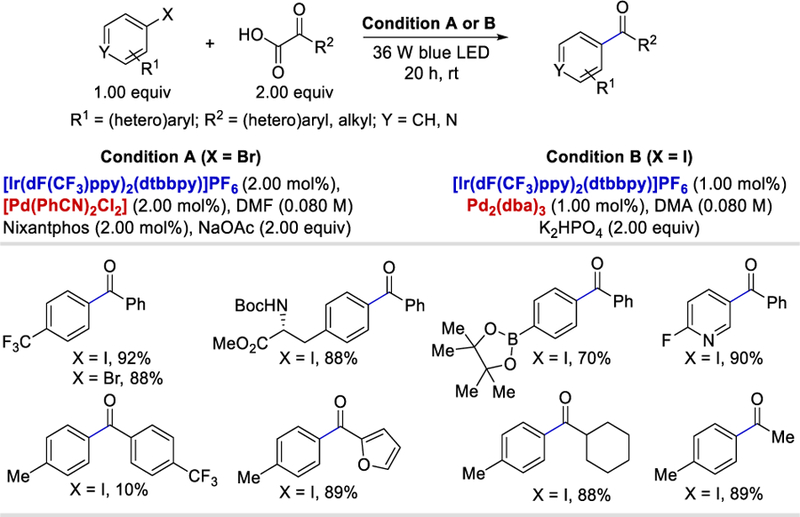
Ir-Pd metallophotoredox catalysis for ketone synthesis using α-keto acids.49
Radical trap experiments showed that addition of 4 equivalents of TEMPO completely shut down the reaction and a stoichiometric amount of TEMPO-adduct was isolated (Scheme 36 ). These experiments strongly suggest a radical mechanism in which the excited iridium catalyst (*IrIII) oxidizes the α-ketocarboxylate to generate the acyl radical and IrII species. Although in this context, the oxidation of IrII is possible by either the PdI, PdII or PdIII species, DFT calculations support the involvement of the PdIII species 36.9.
Scheme 36.
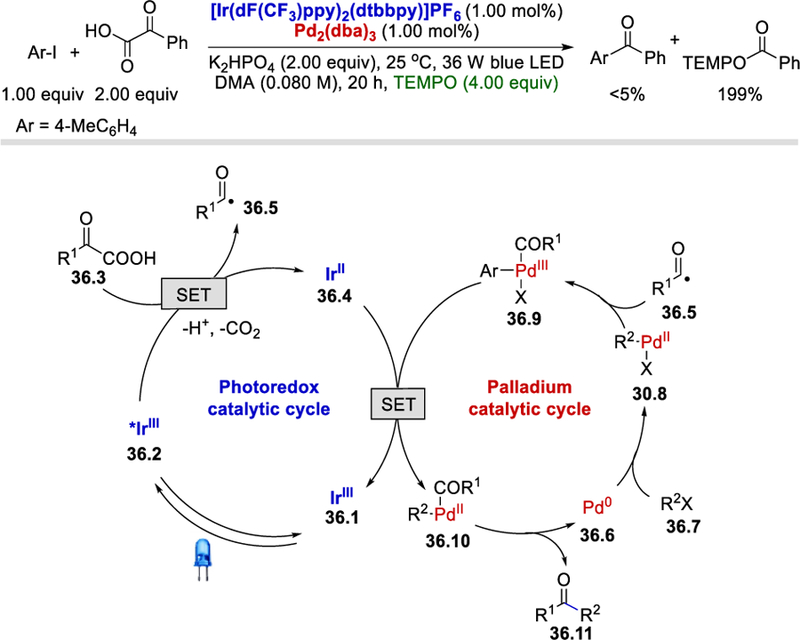
Mechanism for ketone synthesis using Ir-Pd metallophotoredox catalysis.49
Merging transition metal-catalyzed o-directed C–H bond activation with photochemically generated radical intermediates in a dual-catalytic fashion is an attractive strategy with which to form the desired bonds in a shortened and mild reaction pathway. In 2015, Wang and Li disclosed a protocol for room-temperature decarboxylative o-acylation of acetanilides with α-ketocarboxylic acids via a novel Eosin Y/Pd dual catalysis.50 Various aromatic and aliphatic α-ketocarboxylic acids all coupled with acetanilide and provided the desired products in 70–84% and 51–62% yields, respectively (Scheme 37 ). o-Substituted aryl α-ketocarboxylic acids are also compatible with these reaction conditions providing good yields of o-acylated products (78–85%). A range of electron-donating and withdrawing acetanilide derivatives all afforded desired products in good yields (68–82%) when coupled with α-ketophenylacetic acid. However, more hindered o-substituted acetanilides such as o-ethyl, o-(isopropyl), and (o-t-butyl) acetanilides failed to afford the o-acylated products under the reported conditions.
Scheme 37.
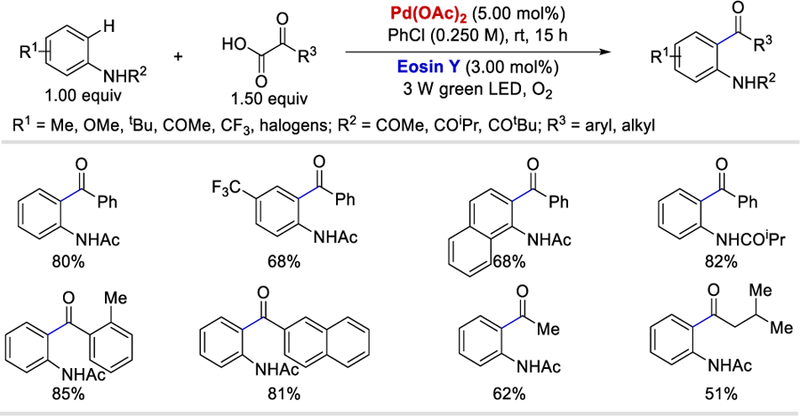
Anilide directed decarboxylative o-acylation using Pd-organic photoredox.50
On the basis of TEMPO trapping experiments and confirmation of a superoxide radical anion (O2•−) by electron paramagnetic resonance (EPR), the authors proposed a plausible reaction pathway shown in Scheme 38. The excited *Eosin Y (38.2) oxidizes arylglyoxylic acid 38.3 to form (Eosin Y)•− (38.5) and arylglyoxylic radical cation 38.4, which undergoes deprotonation and decarboxylation to give aroyl radical 38.8. (Eosin Y)•− is oxidized by molecular oxygen regenerating Eosin Y and producing the superoxide radical anion O2•− (38.6) The Pd-catalytic cycle is initiated by C−H activation of acetanilide forming a palladacyclic intermediate 38.7, which subsequently reacts with the nucleophilic aroyl radical 38.8 to afford a PdIII intermediate 38.9. This PdIII intermediate is further oxidized by the superoxide radical anion to generate a PdIV intermediate 38.10 along with the formation of O22− and H2O2. Finally, reductive elimination of the PdIV intermediate affords the desired product 38.11 and an active PdII catalyst for the next catalytic cycle.
Scheme 38.
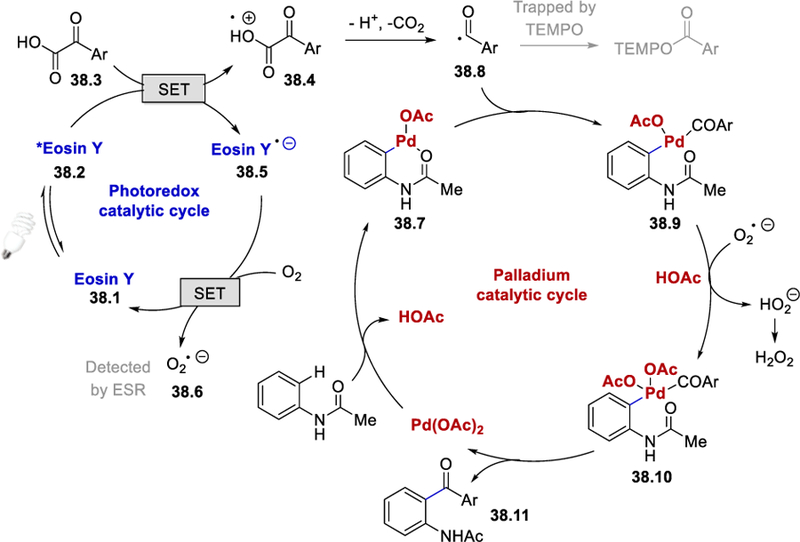
Proposed mechanism for photocatalytic decarboxylative o-acylation.50
By merging Pd-catalysis with organic photocatalysis, the same group further developed a protocol for efficient ortho C‒H acylation of azo and azoxybenzenes by α-ketoacids in 2016 (Scheme 39 ).51 A range of aromatic as well as heteroaromatic α-ketocarboxylic acids reacted with azobenzene under the optimized reaction conditions and provided coupled products in 66–79% and 62% yields, respectively. Naphthyl α-ketocarboxylic acids gave the desired products in 77–84% yields. However, no reaction was observed when aliphatic α-oxocarboxylic acid was used as the acyl surrogate. Disubstituted azo-and azoxybenzenes also afforded desired o-acylated products in decent yields (57–78%) regardless of their electronics, however, o-disubstituted azo-and azoxybenzene derivatives gave lower yields (37–56%) due to steric problems.
Scheme 39.
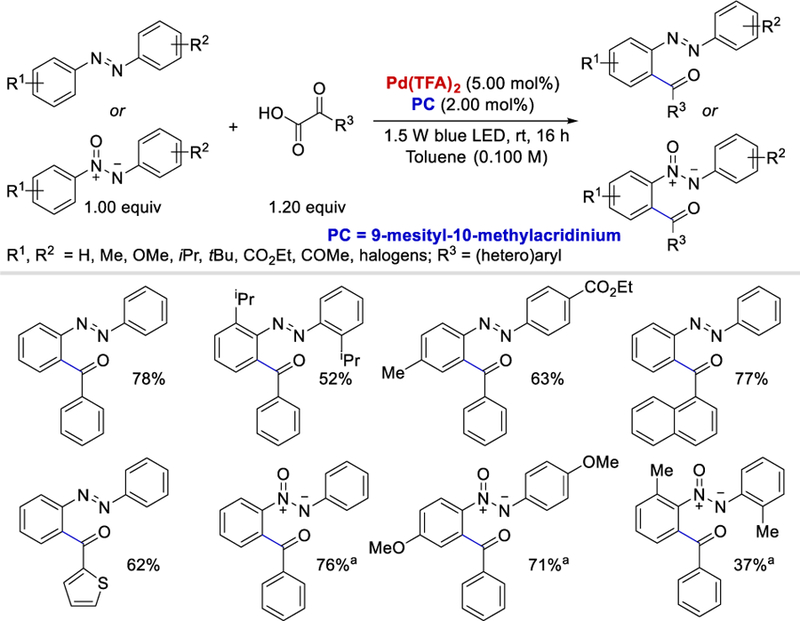
Decarboxylative o-acylation of azo/azoxybenzenes using Pd-organic photoredox. aDCE used as the solvent.51
A series of mechanistic studies were conducted to establish the reaction mechanism. A TEMPO trapping experiment suggests the generation of acyl radicals. The intermolecular kinetic isotopic effect observed (kH/kD = 3.7) in the reaction identifies the C–H activation as the rate-determining step, and an EPR spectroscopic data confirms the generation of superoxide radical anion (O2•−) in the reaction medium. On the basis of these results, the authors proposed a mechanism that begins with the photoexcitation of mesityl acridinium catalyst (PC) 40.1 generating the excited photocatalyst (*PC) 40.2. Oxidation of 40.2 by the molecular oxygen affords PC•+ 40.3 and a superoxide radical anion. Subsequently, 40.3 oxidizes the α-ketocarboxylic acid to regenerate the ground state photocatalyst PC along with the corresponding carboxyl radical species that leads to the formation of an aroyl radical species 40.5. On the other side, a Pd-catalytic C–H activation of the azobenzene forms the palladacyclic intermediate 40.6. Addition of the in-situ generated radical 40.5 to this palladacyclic intermediate 40.6 affords the PdIV or PdIII species 40.7 in an analogy to the observation of Wang and Li in Scheme 38. At this stage, reductive elimination from 40.7 affords the desired acylated product 40.8 with the formation of the PdI intermediate 40.9, which is further re-oxidized by the superoxide radical anion to regenerate PdII catalyst, completing the catalytic cycle.
Acyl radicals, generated by photocatalytic decarboxylation, were also reported by Shang and Fu in 2015 for acyl reductive Michael addition with various Michael acceptors. Various aromatic and heteroaromatic α-ketocarboxylic acids were decarboxylated in the presence of photoredox catalysts to form (hetero)aroyl radicals. These radicals underwent Michael addition to various α,β-unsaturated esters, ketones, amides, aldehydes, nitriles, and sulfones to afford the corresponding products in 45–94% yields (Scheme 41 ).52
Scheme 41.
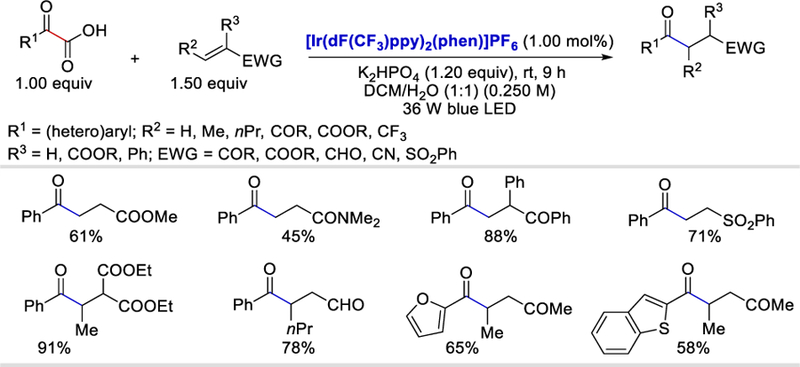
Photocatalytic decarboxylative reductive acyl Michael addition of olefins.52
The authors reported that the product yield was significantly decreased by reducing the loading of the base to the catalytic amount, which demonstrates the role of the carboxylate anion for quenching the excited photoredox catalyst. The reaction has been proposed to proceed via a reductive quenching cycle of photoexcited *[Ir(dF(CF3)ppy)2(phen)]PF6 42.2 by α-keto-carboxylate as depicted in Scheme 42. The acyl radical 42.4, generated via the decarboxylation of an α-ketocarboxylate radical, was subsequently trapped by a Michael acceptor affording the radical 42.6. This radical oxidizes the IrII catalyst regenerating the photocatalyst Ir[(dF(CF3)ppy)2(phen)]PF6 and 42.8, which readily protonates to provide the 1,4-addition product 42.8 (Scheme 42 ). Acrylic acid is an ineffective Michael acceptor under these reaction conditions due to its possible reductive quenching in competition with 2-ketocarboxylate. β-Dimethylated and β-phenyl-substituted alkene substrates are also incompatible Michael acceptors due to their ability to form stable tertiary or benzylic radicals with a lower oxidation potential to oxidize IrII.
Scheme 42.
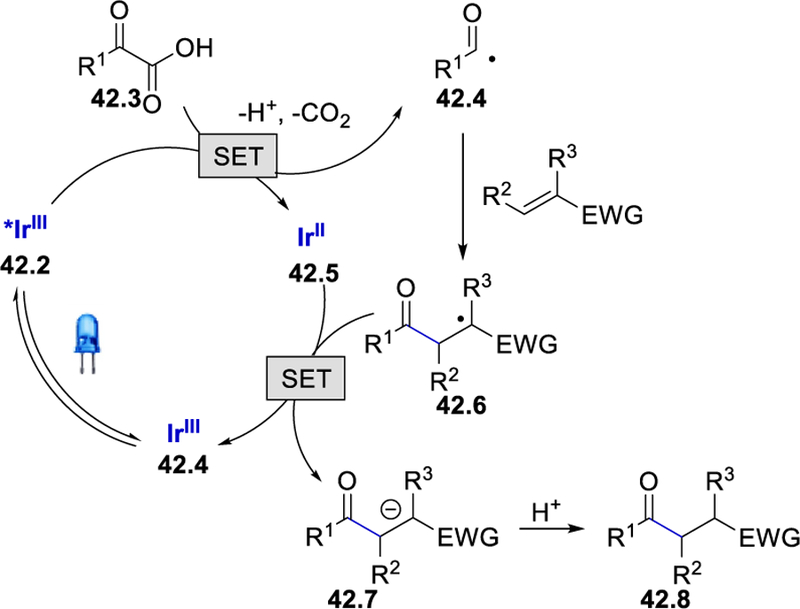
Proposed mechanism for decarboxylative reductive acyl Michael addition of olefins.52 Adapted with permission from (52). Copyright (2015) American Chemical Society.
By strategically using the oxidant Selectfluor, Zhu et al. developed a photocatalytic strategy for the synthesis of α,β-unsaturated ketones using α-ketoacids and styrene derivatives in 2017.53 With a variety of substituents tolerated on both reactants, this domino fluorination−protodefluorination strategy provides access to the construction of α,β-unsaturated ketones in good yields (50–84%) (Scheme 43). 2-Furanyl-, 2-thienyl-, and 2-naphthyl-substituted α-ketoacids all reacted smoothly as the acyl surrogate and gave corresponding α,β-unsaturated ketones in 50–57% yields.
Scheme 43.
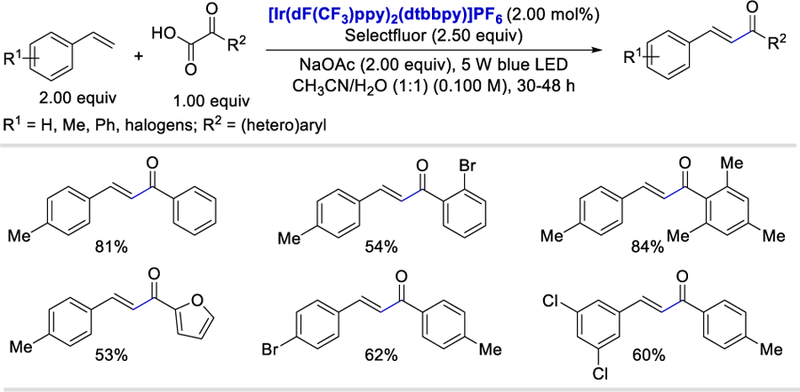
Photocatalyzed domino fluorination−protodefluorination decarboxylative cross-coupling of α-ketoacids with styrene derivatives.53
The use of TEMPO under the standard reaction conditions completely inhibited the product formation, indicating a radical process. Use of the oxidant t-butyl hydroperoxide under the standard reaction conditions was unproductive which rules out the possibility of benzylic oxidation. The absence of the base in the optimized reaction conditions provided the desired enone product in 10% yield along with the detection, by NMR analysis, of the fluoro-acylated product. From their observations in control experiments, the authors proposed a mechanism which starts with the visible-light irradiation of the photocatalyst [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 generating a long-lived excited state *IrIII 44.2 (Scheme 44). With the aid of Selectfluor, *IrIII can oxidize an α-ketoacid to give the aroyl radical species 44.5, which reacts with styrene derivatives to deliver the benzylic radical 44.6. Fluorination of 44.6 by Selectfluor produces aroylfluorinated species 44.8 and the corresponding radical dication 44.7 which oxidizes the reduced IrII to the ground state IrIII species, completing the photoredox cycle. Finally, the acylfluorinated product delivers the final α,β-unsaturated ketone product 44.9 via a base-mediated elimination process.
Scheme 44.
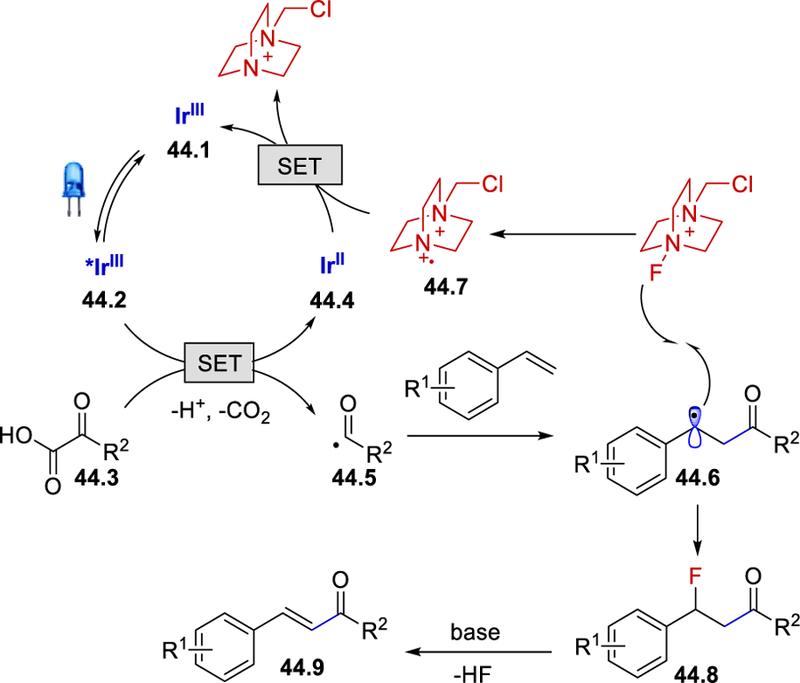
Proposed mechanism for enone synthesis.53
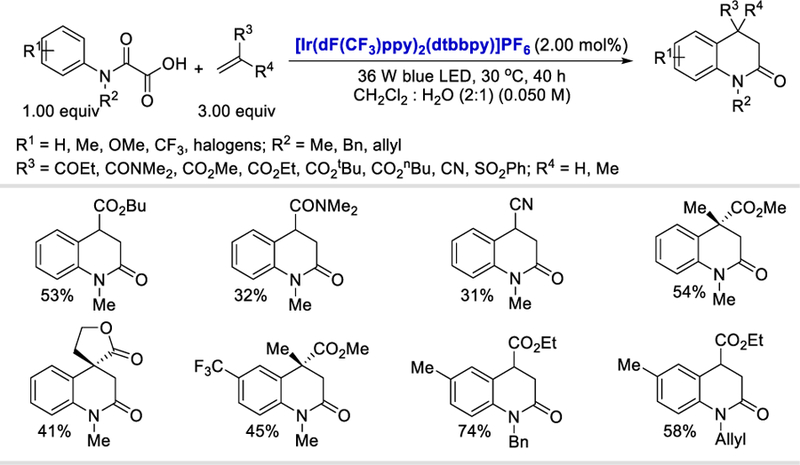
In 2016, Overman’s group reported the use of N-hydroxyphthalimido esters for the efficient generation of alkyl as well as methoxycarbonyl radicals via a photoredox-catalyzed reductive SET.54 Based on this idea, in 2017, Taylor and Donald reported the generation of carbamoyl radicals from N-hydroxyphthalimido oxamides and used this for the synthesis of 3,4-dihydroquinolin-2-ones under mild photocatalytic conditions (Scheme 45).55 This method provides access to a diverse collection of o-, m-or p-substituted oxamides for the successful generation of 3,4-dihydroquinolin-2-ones in good yields (41–80%). However, a clear trend of increasing yields was observed for oxamides containing electron-withdrawing substituents compared to their electron-donating counterparts. Several mono-and di-substituted alkenes as well as exocyclic alkenes all afforded the desired 3,4-dihydroquinolin-2-ones containing fused cyclic (38–81%) and spirocyclic systems (43–71%).
Scheme 45.
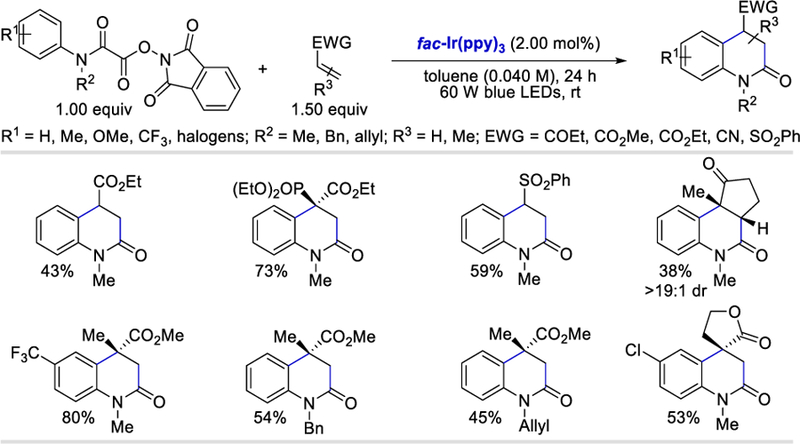
Photocatalytic intermolecular addition/cyclization for the synthesis of 3,4-dihydroquinolin-2-ones.55
The proposed mechanistic cycle begins with the irradiation of the photocatalyst fac-Ir(ppy)3 with visible light, which could lead to the photoexcited state fac-*Ir(ppy)3 46.2. Excited fac-*Ir(ppy)3 (E½IV/III* = –1.73 V vs. SCE)56 reduces N-hydroxyphthalimido oxamide to form the radical anion 46.5, which rapidly fragments, releasing CO2, N-Phth–, and the carbamoyl radical 46.6. The carbamoyl radical undergoes Michael addition to an electron-deficient olefin followed by cyclization to produce cyclohexadienyl radical 46.8. Oxidation of 46.8 by the photocatalyst (E½IV/III = +0.77 V vs. SCE)56 forms cyclohexadienyl cation 46.9, which is deprotonated and rearomatized to give 3,4-dihydroquinolin-2-one 46.10 and the ground state IrIII catalyst simultaneously regenerated.
In contrary to the earlier reductive approach to the generation of carbamoyl radical from oxamide (Scheme 45), Feng et al. reported in 2018 a photocatalytic oxidative decarboxylation approach by taking oxamic acids as the carbamoyl precursor for the synthesis of analogous 3,4-dihydroquinolin-2(1H)-ones (Scheme 47).57 A variety of electron-donating and withdrawing oxamic acids are compatible with the reaction under the optimized conditions and afforded a wide variety of 3,4-dihydroquinolin-2(1H)-ones in moderate (41–74%) yields. A range of mono, di-substituted alkenes, and exocyclic alkenes are compatible with these reaction conditions and afforded the desired products containing fused cyclic (31–60%) and spirocyclic systems (41%).
Scheme 47.
Photocatalytic synthesis of 3,4-dihydroquinolin-2(1H)-ones using oxamic acids.57
From the reaction of N-methyl-N-phenyloxamic acid with ethyl acrylate under the standard conditions, a trace amount of N-methyl-N-phenylformamide was isolated, and the addition of the radical scavenger TEMPO to the standard reaction mixtures gave only a trace amount of the desired product. Both of these results indicate the formation of a carbamoyl radical in the reaction medium. On the basis of these results, the authors proposed a mechanistic cycle in which the photoexcited catalyst *IrIII 48.2 undergoes a reductive quenching to generate IrII and carbamoyl radical intermediate 48.5 (Scheme 48). Analogous to Scheme 46, the addition of carbamoyl radical to the electron-deficient olefins followed by intramolecular cyclization forms a cyclohexadienyl radical intermediate 48.7. Final oxidation of reduced IrII by molecular oxygen affords the ground state IrIII and superoxide radical anion, which abstracts a hydrogen atom from cyclohexadienyl radical 48.7 to deliver the desired 3,4-dihydroquinolin-2(1H)-one 48.8.
Scheme 48.
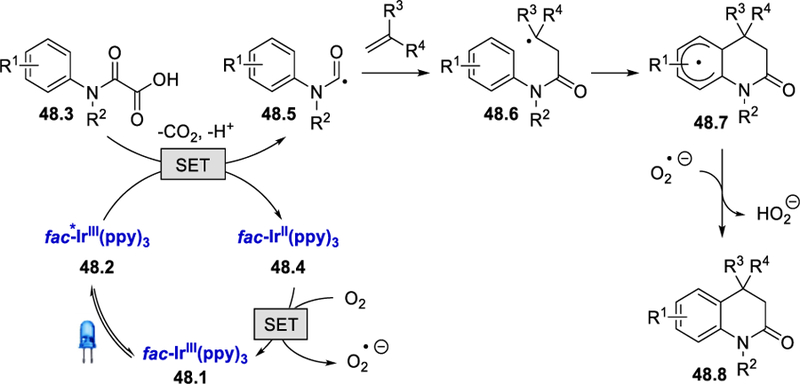
Proposed mechanism for the synthesis of 3,4-dihydroquinolin-2(1H)-ones.57
Scheme 46.
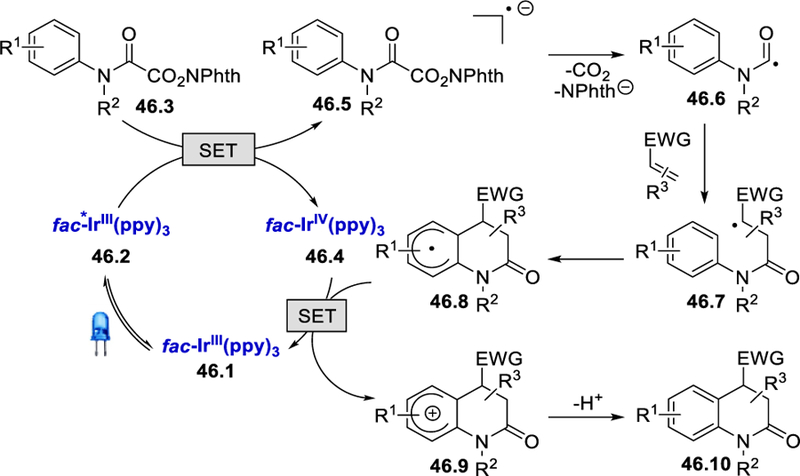
Proposed mechanism for the photocatalytic synthesis of 3,4-dihydroquinolin-2-ones.55 Adapted with permission from (55). Copyright (2017) American Chemical Society.
The combination of visible light photocatalysis with hypervalent iodine reagents (HIR) was successfully employed by Chen et al. in 2015 for the generation of an acyl radical.58 This chemoselective decarboxylative alkynylation strategy shows a broad substrate scope in terms of HIR-bound alkynes as well as the α-keto acids. Aryl substituted benziodoxolonyl-alkynes (BI-alkynes) containing electron-donating and withdrawing substituents as well as alkyl bound BI-alkynes all reacted well to deliver the desired alkynylated products in 65–93% and 65–85% yields, respectively (Scheme 49). The triisopropylsilyl (TIPS) substituted BI-alkynes which can be easily deprotected to generate terminal alkynones also provided good (61–70%) yields of the desired products. Irrespective of steric and electronic effects, substituted alkyl, aryl, and heteroaryl α-ketoacids were well tolerated and afforded the desired product in 57–87% yields. Aryl α-ketoacid substrates bearing sensitive functional groups such as allyl esters, propargyl esters, alcohols, and azides are also well tolerated in these reaction conditions and gave desired alkynylated products in 62–74% yields.
Scheme 49.
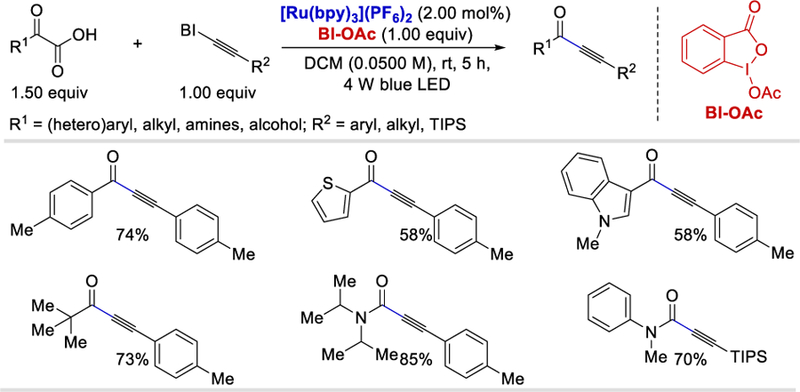
Photocatalytic decarboxylative alkynylation using α-ketoacids.58
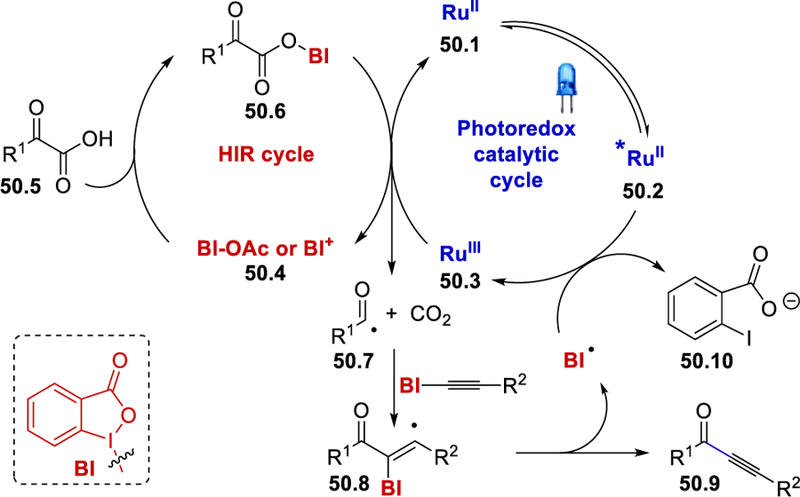
NMR studies and formation of alkynylation products using a BI-ketoacid complex in place of α-ketoacids and BI-OAc, under otherwise identical reaction conditions confirms the formation of a BI-ketoacid complex in the reaction medium. The luminescence quenching of [Ru(bpy)3]2+ was much weaker in the presence of the α-ketoacid compared to BI-OAc, indicating that BI-OAc is primarily the oxidative quencher in this reaction. In accordance with observations from control experiments, the authors proposed that the benziodoxole-oxoacid complex 50.6 (BI–O2CCOR1), generated in-situ from an α-ketoacid and BI-OAc, is oxidized by RuIII to liberate carbon dioxide, benziodoxole cation (BI+ or BI-OAc), [Ru(bpy)3]2+ and the aroyl radical 50.7 (Scheme 50). The aroyl radical further undergoes α-addition to BI-alkyne followed by the elimination of benziodoxolonyl radical (BI•) to yield the alkynone 50.9. The BI• oxidizes the photoexcited *[Ru(bpy)3]2+ to complete the photoredox cycle.
Scheme 50.
Mechanism for alkynylation using HIR and photoredox catalysis.58 Adapted with permission from (58). Copyright (2015) John Wiley and Sons.
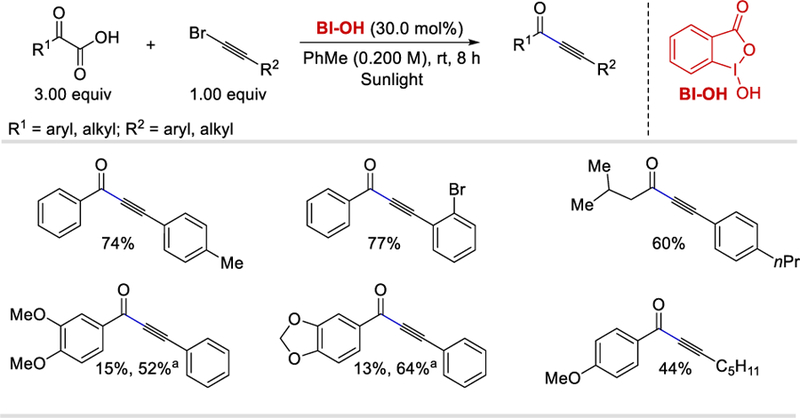
In 2015, Li and Wang used a BI-OH reagent instead of BI-OAc to activate α-ketocarboxylic acids for the successful development of sunlight-driven decarboxylative alkynylation.59 A range of aromatic α-ketoacids reacted with (bromoethynyl)-benzene under the optimized conditions to afford the corresponding alkynones in 44–76% yields. 4-Methyl-2-oxopentanoic acid, an aliphatic α-ketoacids, reacted with a series of o-, m-and p-substituted 2-aryl-1-bromoethynes to provide the desired products in good yields (60–74%). Irrespective of steric and electronic effects, substituted bromoacetylenes, when reacting with 2-keto-2-phenylacetic acid, form the desired products in 65–80% yields (Scheme 51). The presence of two strongly electron-donating groups (-OR) on the aromatic ring of 2-keto-2-arylacetic acid such as 2-(3,4-dimethoxyphenyl)-2-ketoacetic acid and 2-(benzo[d][1,3]-dioxol-5-yl)-2-oxoacetic acid when reacted with (bromo-ethynyl)-benzene afforded the desired product in only 15% and 13% yields, respectively. Replacing the catalytic BI-OH with a stoichiometric amount of PhI(OAc)2 however improved the yields of these two products to 52% and 64%, respectively.
Scheme 51.
Decarboxylative alkynylation of α-ketoacids with bromoacetylenes using sunlight.59
aUsing 2 equiv of PhI(OAc)2 in place of BI-OH.
The trapping of a TEMPO-adduct in the presence of TEMPO and trapping of the benzoyl as well as the benziodoxolonyl radical by BHT in the standard reaction conditions confirms the formation of benziodoxolonyl and benzoyl radicals in the medium. Using an equivalent amount of BI-alkyne and α-ketoacid, the desired product was obtained in 66% yield only in the presence of 30% of BI-OH (HIR) catalyst. Separately synthesized BI-ketoacid (BI-OCOCOPh), when reacted with bromoacetylenes, gave a good yield of the desired product. These results indicate the involvement of BI-alkynes and a BI-ketoacid complex in this reaction. From these mechanistic studies, it was proposed that BI-OH (52.1) reacts with the ketocarboxylic acid 52.2 to form the BI-oxoacid complex 52.3 (Scheme 52). This complex generates the benziodoxolonyl radical (52.4) and a ketocarboxyl radical 52.5 under irradiation of sunlight. Subsequently, 52.4 reacts with bromoacetylene to give the BI-alkyne 52.7 and releases a Br radical. At the same time, decarboxylation of 52.5 produces aroyl radical that adds to BI-alkyne to form 52.8, which releases the alkynone product 52.9 and the benziodoxolonyl radical. This radical recombines with Br radical, and this is followed by hydrolysis to regenerate BI-OH and complete the catalytic cycle.
Scheme 52.
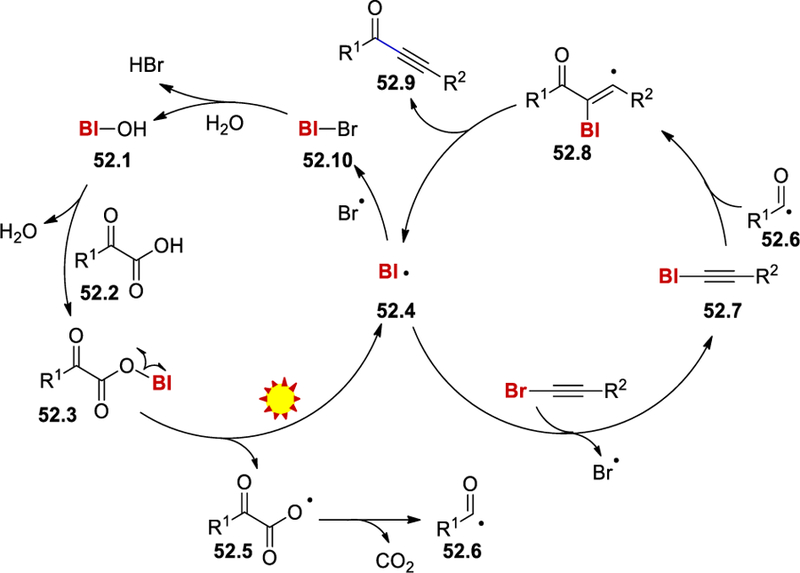
Mechanism for photocatalytic alkynylation of α-ketoacids by HIR-catalysis.59
In 2016, Wang et al. reported BI-OAc as an efficient HIR-catalyst for the decarboxylative 1,2-acylarylation/tandem cyclization of acrylamides with α-ketocarboxylic acids in the presence of only visible light (Scheme 53).60 Regardless of steric and electronic effects, various N-methyl-N-arylmethacrylamides as well as aromatic ketocarboxylic acids afforded the desired products in moderate to good (58–78%) yields. Methyl substitution at the m-position of N-methyl-N-arylmethacrylamide provided a regioisomeric product (70%) in 3:2 ratio, but acrylamides containing a free amine or alcohol failed to a couple under the reaction conditions. Rather than N-methyl-N-arylmethacrylamides, other acrylamides such as N-ethyl-N-arylmethacrylamide and N-benzyl-N-arylmethacryl-amide form the desired products in 70–84% yields. However, N-phenyl-N-arylmethacrylamide was unproductive under the optimized reaction conditions.
Scheme 53.
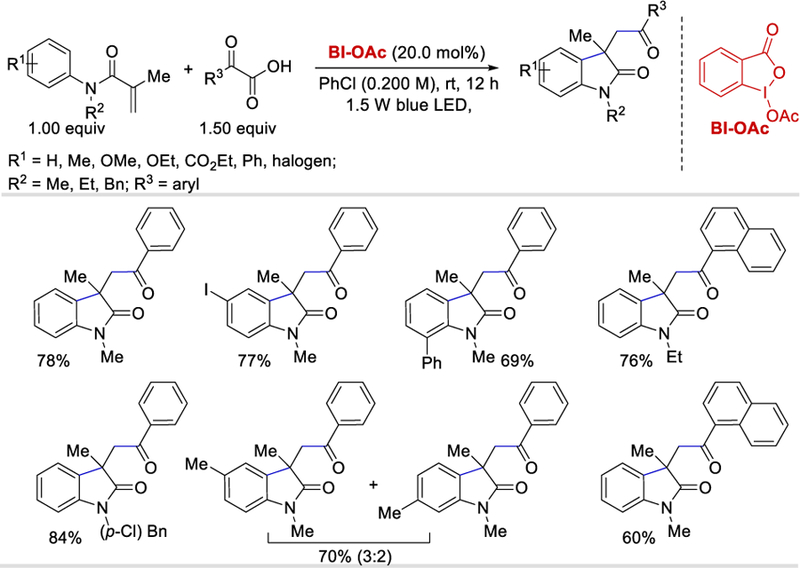
Photocatalytic acyl-arylation of olefins using α-ketocarboxylic acids.60
The catalytic cycle is initiated by the reaction of BI-OAc and the ketocarboxylic acid 54.1 to generate the BI-ketoacid intermediate 54.2. Visible light-mediated homolytic cleavage of the BI‒O bond of 54.2 liberates CO2, a benzoyl radical 54.3, and a benziodoxolonyl radical (54.4). Addition of 54.3 across the double bond of arylmethacrylamides 54.5 followed by cyclization gives intermediate 54.7, which undergoes hydrogen atom abstraction by 54.4 to deliver the desired 3,3-disubstituted 2-oxindole 54.8 and BI-H (54.9). The reaction of 54.9 with the ketocarboxylic acid 54.1 liberates hydrogen gas and generates the BI-oxoacid intermediate 54.2 for the next cycle.
Carboxylic acids as a source of acyl radicals
Simple and inexpensive carboxylic acids can be an alternate source from which to generate the acyl radical by visible light photoredox catalysis (PC). Base-mediated photocatalytic oxidation, decarboxylation and further carbonylation of the in-situ generated alkyl radical by carbon monoxide (CO) can afford acyl radicals. Alternatively, the carboxylic acid is converted to its redox-active ester by the use of an activating agent (X = active electrophile) which can be reduced by photoredox catalyst (PC) to form the acyl radical (Scheme 55). Redox properties of some carboxylates and anhydrides are listed in the Scheme 55.61,62
Scheme 55.
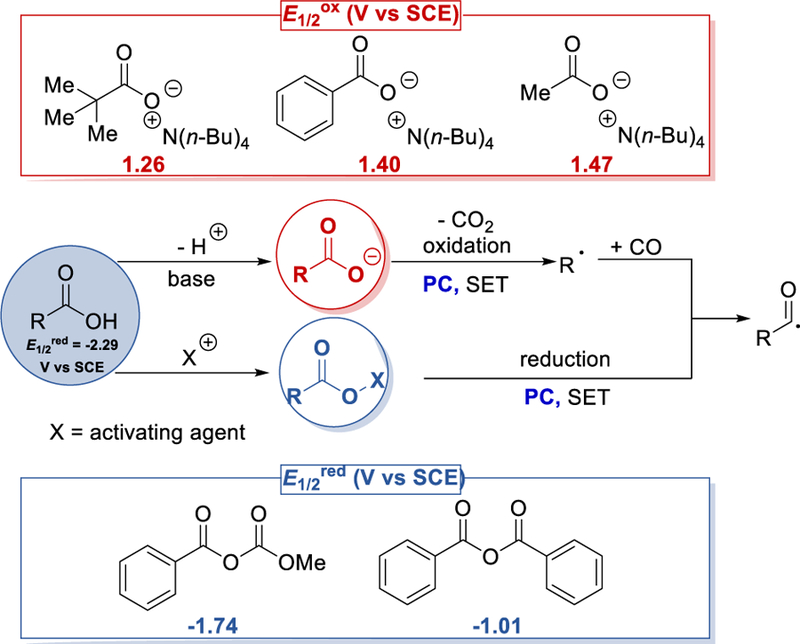
General scheme for photocatalytic acyl radical formation from carboxylic acids.61,62
In 2015, Lu and Xiao reported a visible light photocatalytic decarboxylative-carbonylative-alkynylation of carboxylic acids with hypervalent iodine bound alkynes in the presence of carbon monoxide.63 A range of cyclic and acyclic aliphatic carboxylic acids exhibited good reactivity in this radical carbonylative-alkynylation giving the desired products in 42–90% yield. Rather than aromatic BI-alkynes (BI = benziodoxolonyl), tri-isopropylsilyl-and t-butyl-substituted BI-alkynes also provided the desired products in moderate (27–56%) yields when reacted with cyclohexyl carboxylic acid (Scheme 56). Benzoic acids fail to react, which is likely due to the slower decarboxylation of the benzoates to aryl radicals under the reaction conditions.64 (rate constants for the decarboxylation (s-−1): aryl carboxylic radical65: 1.4±0.3×106; alkyl carboxylic radical64: 2.2×109 ). α-Amino-or α-oxygen-substituted carboxylic acids are also incompatible with the reported reaction conditions.
Scheme 56.
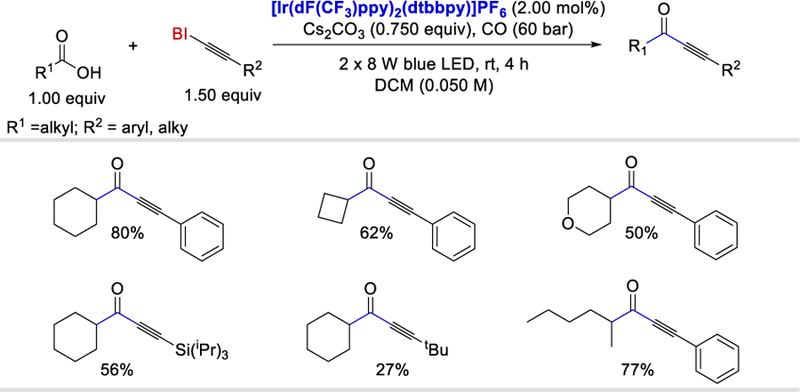
Visible-light-induced decarboxylative carbonylative alkynylations of carboxylic acids.63
The use of 2-cyclopropylacetic acid as the carboxylic acid substrate afforded a ring-opened alkene-alkyne product in 52% yield under the standard reaction conditions, which confirms the intermediacy of an alkyl radical. On the basis of this radical clock experiment, the author proposed a tentative mechanism, in which the excited state *IrIII photocatalyst (E½III*/II = +1.21 V vs. SCE)46 57.2 reacts with carboxylic acid 57.3 [E½red (cyclohexyl carboxylate) = +1.18 V vs. SCE]63 to form the alkyl radical 57.5 with the release of CO2. This alkyl radical further produces acyl radical 57.6 in the presence of carbon monoxide. Addition of acyl radical to the BI-alkyne 57.7 followed by the release of BI radical (57.9) provides the alkynone product 57.10. The BI radical oxidizes IrII to regenerate the ground state IrIII photocatalyst and form the carboxylate 57.11.
In 2015, Wallentin et al. employed for the first time the activating agent dimethyl dicarbonate (DMDC) to generate the acyl radical from carboxylic acids using visible light photocatalysis (Scheme 58).62 A range of substituted methacrylamides, when reacted with benzoic acid in the standard reaction conditions, afforded the 3,3-disubstituted 2-oxindoles derivatives in good to excellent yields (74–95%). Benzoic acids bearing substituents at the o-, m-or p-position as well as carboxylic acids with extended aromatic systems all reacted very well (76–97%) under the optimized reaction conditions. Surprisingly, o-and p-methyl-, as well as p-hydroxy and p-trifluoromethyl benzoic acids reacted very poorly under the standard conditions, however after replacing DMDC with Boc2O and 1 equiv of MgCl2 and increasing loading of fac-Ir(ppy)3 to 2.5 mol%, the desired products were produced in good (72–99%) yields. These new reaction conditions [Boc2O, 1 equiv of MgCl2 and 2.5 mol% of fac-Ir(ppy)3] were very effective for heteroaromatic carboxylic acids and afforded the 1,2-acylarylation products in good (33–78%) yields.
Scheme 58.
Visible-light 1,2-acylarylation of methacrylamides using carboxylic acids.62
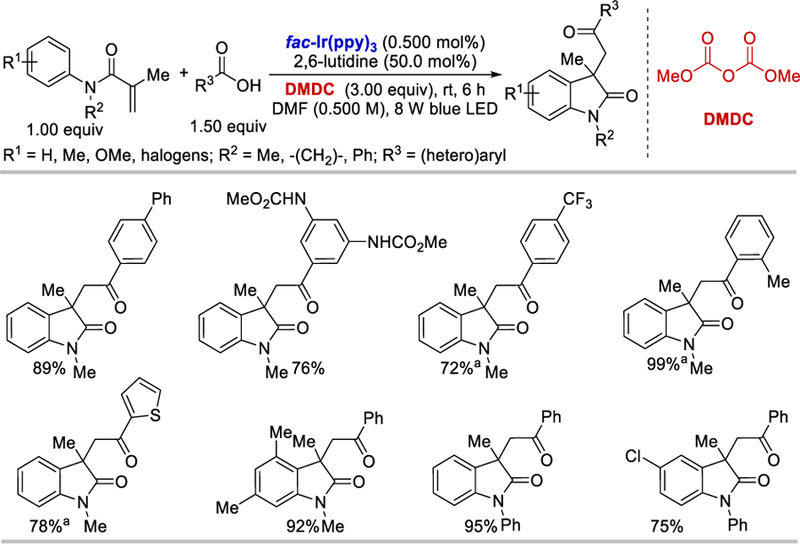
aUsing 2.5 mol% of fac-Ir(ppy)3, 3 equiv of Boc2O and 1 equiv of MgCl2 over 48 h.
When a mixed anhydride synthesized separately from the reaction of benzoic acid and DMDC reacted with methacrylamides under the standard reaction conditions, the desired product was formed in a good yield. Stern-Volmer fluorescence quenching studies clearly revealed that anhydrides quench the excited fac-*Ir(ppy)3. From these mechanistic studies, a plausible mechanism was derived and is depicted in Scheme 59. The photoexcited fac-*Ir(ppy)3 59.2 (E½ IV/*III = –1.73V vs. SCE) reduces the anhydride 59.4, generated in situ from the carboxylic acid 59.3 in the presence of DMDC and 2,6-lutidine, to produce fac-IrIV(ppy)3 (59.5) and a radical anion 59.6. Decarboxylation of 59.6 gives the acyl radical 59.7, which adds to the double bond of the methacrylamide 59.8 and subsequently undergoes cyclization to afford intermediate 59.10. Oxidation of 59.10 by fac-IrIV(ppy)3 provides the desired product 59.11 with regeneration of the photocatalyst fac-Ir(ppy)3 to close the catalytic cycle.
Scheme 59.
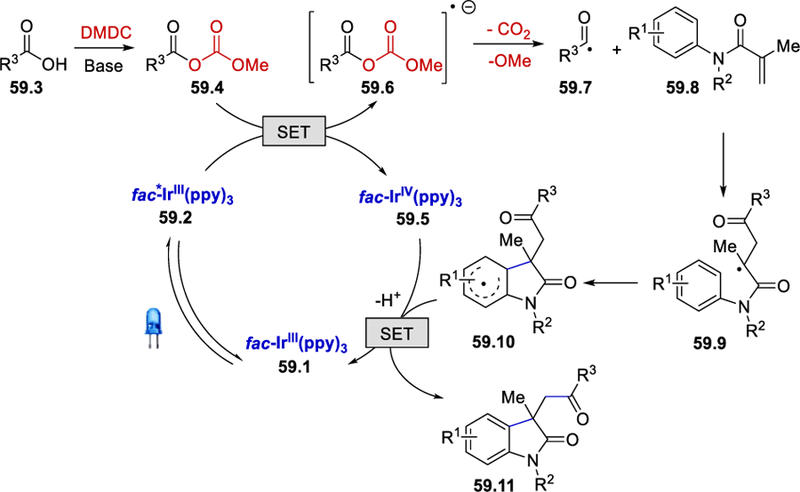
Mechanism of visible-light catalysed 1,2-acylarylation of methacrylamides.62
In 2017, the Wallentin group merged the concept of photocatalytic acyl radical formation from carboxylic acid with multicomponent reactions (Scheme 60).66 Strategically combining electron-poor and electron-rich alkenes with acyl radicals generated from mixed anhydride of DMDC and carboxylic acids; this method provides access to the synthetically challenging 1,2-dicarbofunctionalization of alkenes. This strategy yielded 1,2-difunctionalized products in good (33–97%) yields when reacted with substituted aromatic as well as heteroaromatic carboxylic acids with different silanol ethers and methyl acrylate. The scope of the reaction was further extended to other electron-poor olefins beyond methyl acrylates. They also afforded the desired difunctionalized products in 41–66% yields.
Scheme 60.
Photoredox-catalyzed intermolecular three-component synthesis of β-functionalized δ-diketones.66
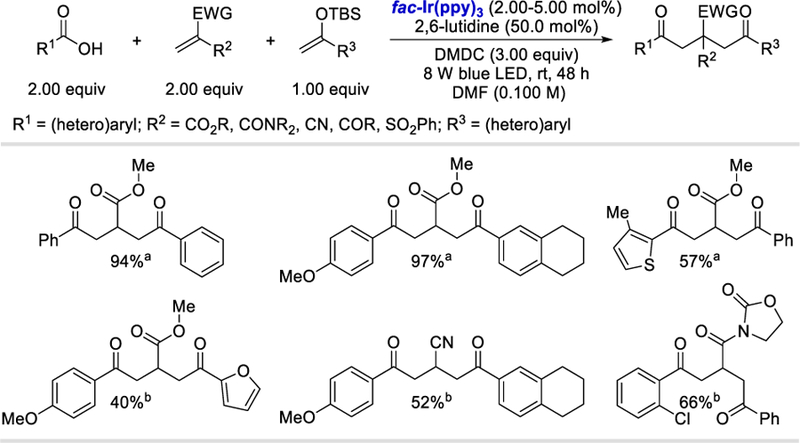
aUsing 5.00 mol% of fac-Ir(ppy)3. bUsing 2.00 mol% of fac-Ir(ppy)3.
The reaction pathway for the formation of the acyl radical 61.5 is the same as that proposed in Scheme 59. This nucleophilic acyl radical first adds to the electron-poor olefin 61.6 forming the radical intermediate 61.7 (Scheme 61). This electron deficient radical species reacts selectively with silyl enol ether 61.8 to deliver 61.9, which is oxidized by fac-IrIV(ppy)3 and this is followed by desilylation to produce the desired product 61.10.
Scheme 61.
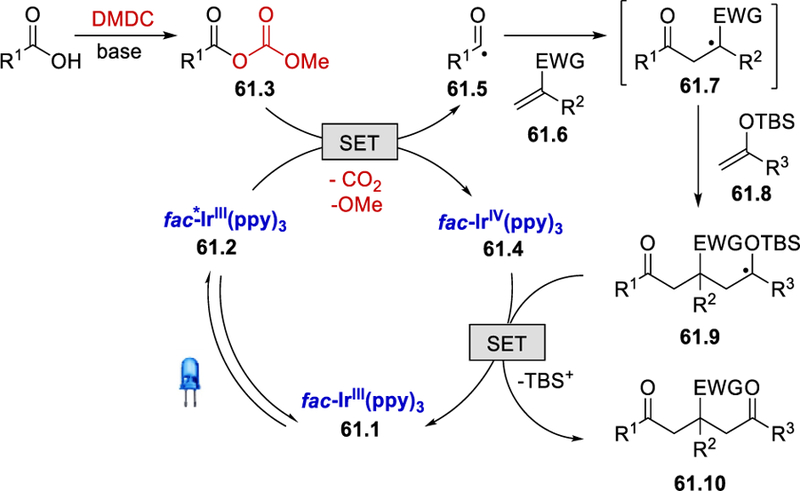
Proposed mechanism for the photoredox-catalyzed intermolecular three-component synthesis of β-functionalized δ-diketones.66
Using the idea of in-situ generation of anhydrides as the acyl radical precursors, Zhu group reported three photocatalytic reactions in 2017.67,68,69 First, reported the photoredox catalyzed hydroacylation reaction of olefins using carboxylic acids as acyl radical precursors and tris(trimethylsilyl)silane (TMS3Si-H) as a hydrogen atom source (Scheme 62).67 A range of electron-donating or withdrawing substituted styrenes, as α,β-substituted or unsubstituted vinyl esters, vinyl sulfone, and aliphatic olefin all reacted well with benzoic acid to afford the corresponding hydroacylated products in 36–84% yields. Both aromatic and heteroaromatic carboxylic acids were viable substrates and coupled with styrene to give the desired products in 53–70% yields.
Scheme 62.
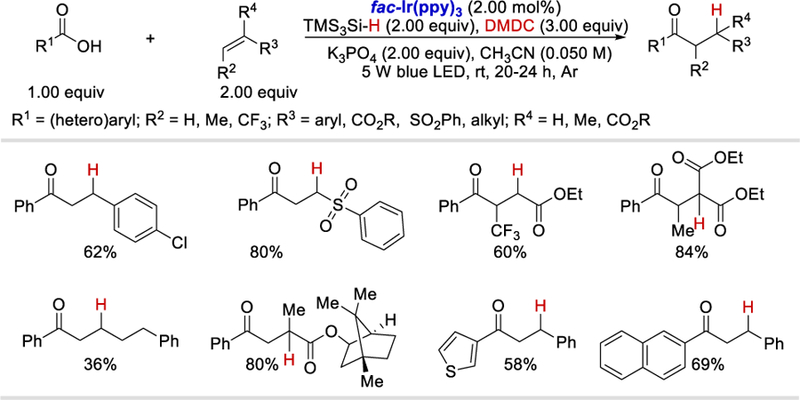
Photocatalytic hydroacylation of olefins using carboxylic acids and hydrosilanes.67
TEMPO additives completely shut down this hydroacylation reaction, which indicates the radical nature of this process (Scheme 63). The mechanism starts with the excitation of fac-Ir(ppy)3 63.1 to excited fac-*Ir(ppy)3 63.2 by visible light irradiation. Single electron reduction of the mixed anhydride 63.3, generated by reacting carboxylic acid with DMDC, by this excited fac-*Ir(ppy)3 forms acyl radical 63.5 with the extrusion of CO2 and OMe. Subsequent addition of acyl radical to the activated olefins forms radical intermediate 63.7. This radical intermediate then quickly grabs the proton from TMS3Si-H 63.8 to give the desired hydroacylation product 63.9. The final SET process between fac-IrIV(ppy)3 and 63.10 regenerates the ground state fac-Ir(ppy)3 for the next cycle.
Scheme 63.
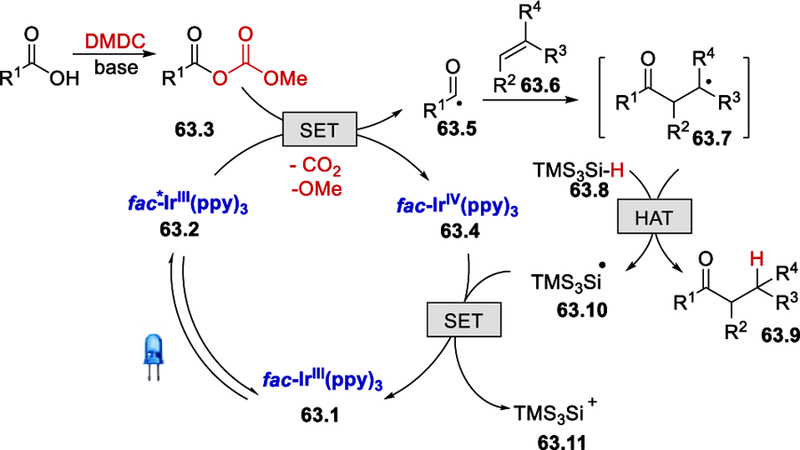
Proposed mechanism for photocatalytic hydroacylation of olefin.67 Adapted with permission from (67). Copyright (2017) American Chemical Society.
Shortly after photocatalytic hydroacylation of activated alkenes, they reported the selective photocatalytic reduction of aromatic carboxylic acids to corresponding aldehydes using fac-Ir(ppy)3 and tris(trimethylsilyl)silane (TMS3Si-H) system (Scheme 64).68 Aromatic carboxylic acids, regardless of position and electronic properties of substituents including free alkynyl, amide, and ester groups, reacted smoothly under the optimized reaction conditions to afford the corresponding aldehydes in 82–92% yields. Heteroaromatic carboxylic acids and a few complex aryl carboxylic acids were successfully reduced to the desired aldehydes as well. However, aliphatic carboxylic acids such as 3-phenylpropanoic acid, cyclohexane carboxylic acid, and N-Boc-glycine all were unproductive under the standard reaction conditions.
Scheme 64.
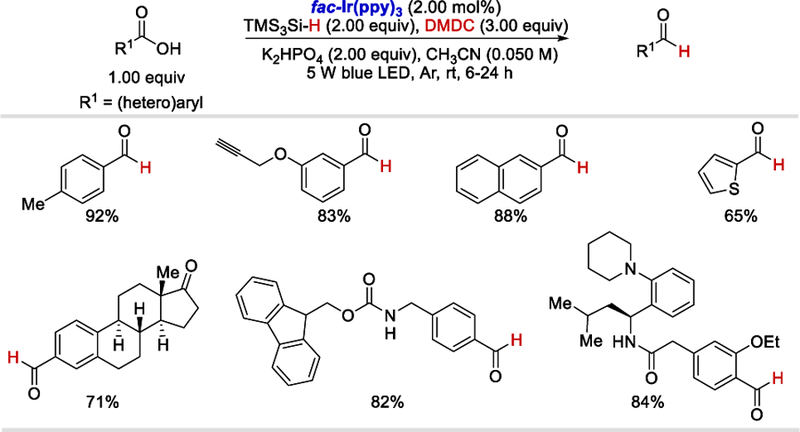
Photocatalytic reduction of carboxylic acids to aldehydes.68
The proposed mechanism up to the formation of an acyl radical 65.5 by excited fac-*Ir(ppy)3 65.2 (Scheme 65) is the same as the hydroacylation reaction mechanism using carboxylic acids (Scheme 63). Once the reactive acyl radical 65.5 is generated, it rapidly reacts with TMS3Si-H (65.6), forming the corresponding aldehyde 65.7. Regeneration of fac-Ir(ppy)3 occurs in the final stage by the reaction of fac-IrIV(ppy)3 and 65.8.
Scheme 65.
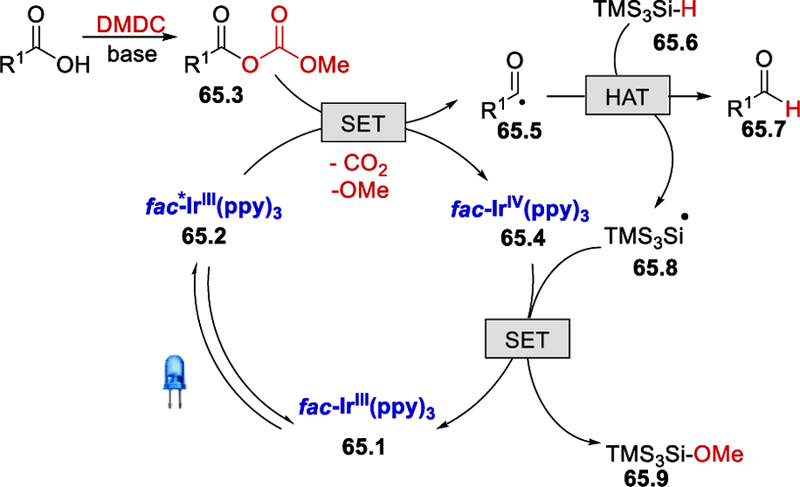
Proposed mechanism for photocatalytic reduction of carboxylic acids. 68
The third reaction developed by Zhu group in 2017 was an efficient deoxygenative intramolecular acylation/radical cyclization via photoredox catalysis for the synthesis of valuable fluorenone products.69 A variety of functionalized biarylcarboxylic acids containing o-and p-substituents on the 2-aryl ring afforded fluorenones in 54–85% yields while the m-substituted aromatics gave regioisomeric products. Electron-donating or withdrawing substituents on the aromatic moiety of carboxylic acid were well tolerated under the standard reaction conditions (61–86%) (Scheme 66).
Scheme 66.
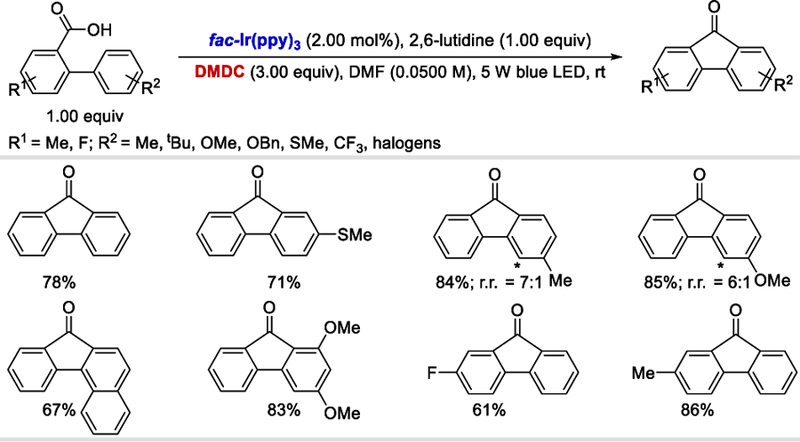
Photocatalytic intramolecular acylation/radical cyclization of biarylcarboxylic acids.69
A plausible mechanism for this intramolecular radical cyclization process is shown in Scheme 67. The aroyl radical 67.6 generated from the mix-anhydride 67.4 adds to the ortho position of the 2-aryl ring gives intermediate 67.7. Oxidation of intermediate 67.7 by fac-IrIV(ppy)3 followed by deprotonation regenerates the ground state Ir(ppy)3 and affords the desired product 67.8.
Scheme 67.
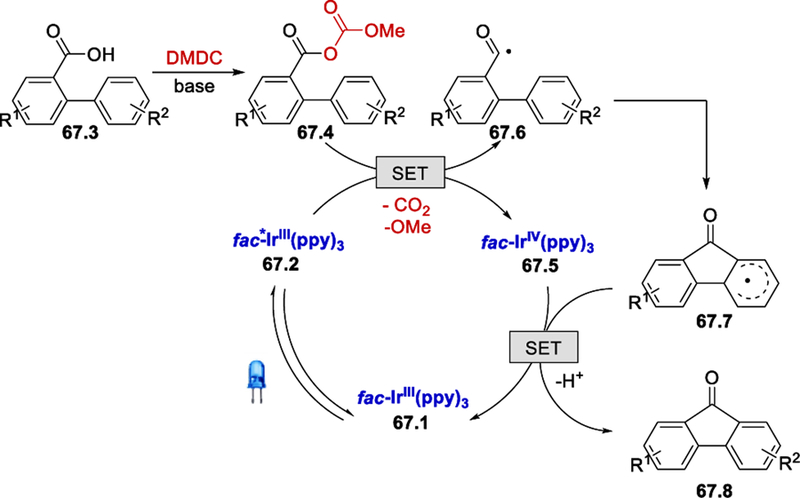
Mechanism for intramolecular acylation/radical cyclization.69
In 2018, Zhu et al. merged the phosphoranyl radical chemistry with photoredox catalysis to form acyl radicals from carboxylic acids via C−O cleavage using Ph3P or Ph2POEt. These acyl radicals added to alkenes and imines to give the desired hydroacylation products (Scheme 68).70 The scope of the reaction is very broad. Various aryl and heteroaryl carboxylic acids reacted with a wide array of electronically diverse alkenylpyridines, styrenes, and Michael acceptors such as acrylate, phenyl vinyl sulfone, diethyl vinylphosphonate, cyclohexanone and lactones to give the desired ketones in 38–89% yields. However, alkyl, alkenyl, and alkynyl carboxylic acids failed to give the desired product. Notably, the reaction is amenable to late-stage functionalization of complex carboxylic acids and alkenes in 40–76% yields. The reaction is also applicable to hydroacylation of imines generated in situ under the reaction conditions to afford α-amino-ketone products.
Scheme 68.
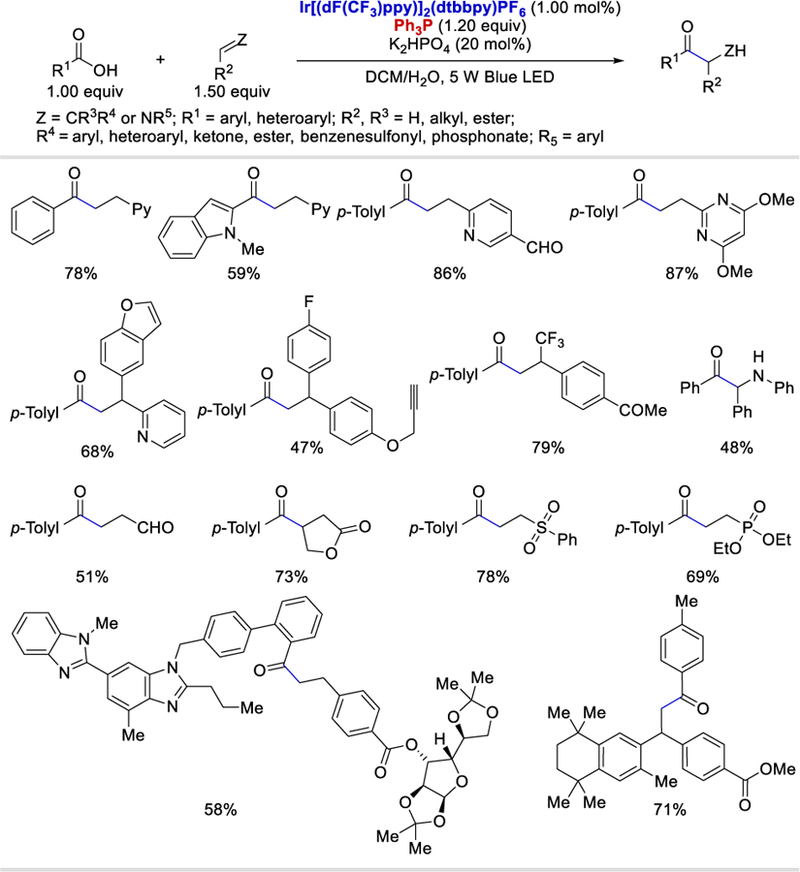
Substrates scope of the hydroacylation of alkenes and imines under photocatalytic conditions using Ph3P as a deoxygenating reagent.70
Based on a series of mechanistic studies including radical trap, deuterium-and O18-labeling, and Stern-Volmer quenching experiments, a proposed reaction mechanism is depicted in (Scheme 69). Photoexcited Ir*[(dF(CF3)ppy)]2(dtbbpy)PF6 (E½III*/II = +1.21 V vs. SCE)46 undergoes reductive quenching by PPh3 (E½red = +0.98 V vs. SCE)71 to form Ir(II) and triphenylphosphine radical cation 69.1. Deprotonation of an aryl or heteroaryl carboxylic acid by K2HPO4 forms carboxylate, which reacts with 69.1 to form a phosphoryl radical 69.3. β-scission of the phosphoryl radical 69.3 liberates triphenylphosphine oxide and aroyl radical 69.4, which adds to olefin 69.5 to deliver the alkyl radical intermediate 69.6. Reduction of 69.6 by Ir(II) (E½III*/II = ‒1.37 V vs. SCE)15a regenerates the Ir(III) photoredox catalyst and anion 69.7, which undergoes protonation to give the desired ketone 69.8.
Scheme 69.
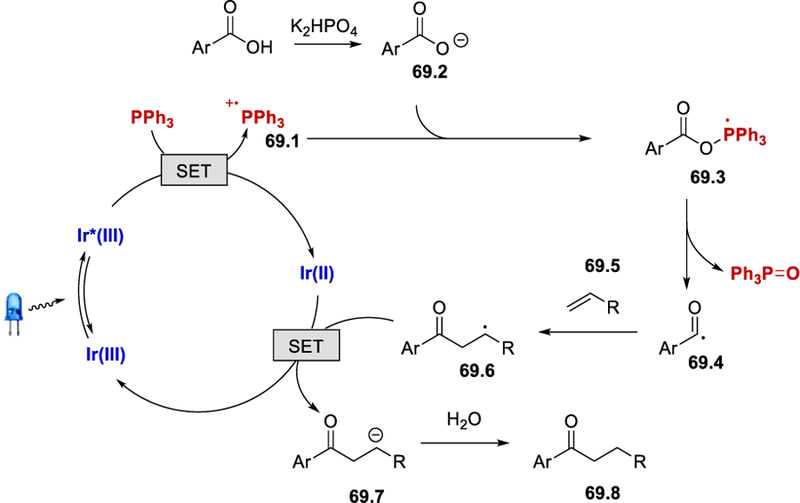
Proposed mechanism for the photoredox-catalyzed aroylation of alkenes using Ph3P as a deoxygenating reagent.70
Concurrently and independently, Doyle et al. (Scheme 70) applied the same concept for converting alkyl, aryl, and heteroaryl carboxylic acids to the corresponding acyl radicals,72 which abstract hydrogen atom from arylthiols to form the desired aldehydes. The scope of the reaction is quite general, electron rich and deficient aryl carboxylic acids, heteroaryl carboxylic acids with indole, benzothiophene and quinolone, and aliphatic carboxylic acids all reacted under optimized conditions to form the corresponding aldehydes in 34–94% yields.
Scheme 70.
Substrates scope of the photocatalytic deoxygenative approach for aldehydes formation from carboxylic acids.72
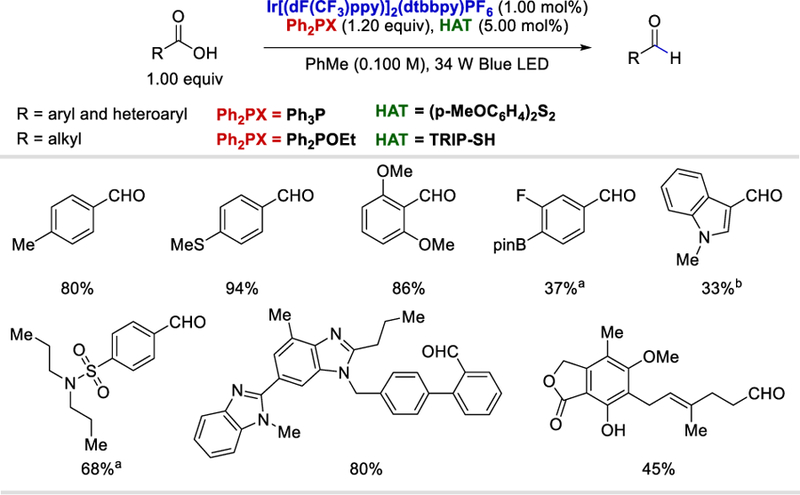
a 2,6-lutidine was added (1.00 equiv); bN-Methyl-2-pyrrolidone(NMP) as the solvent.
When a radical acceptor such as carbonyl and iminyl derivatives and alkenes was ortho to the carboxylic acid group on a benzene ring, intramolecular acyl radical addition took place to form cyclized products in 50–93% yields (Scheme 71). Aliphatic carboxylic acids could cyclize to form five-member lactone and ketone products in 43% and 44% yields, respectively.
Scheme 71.
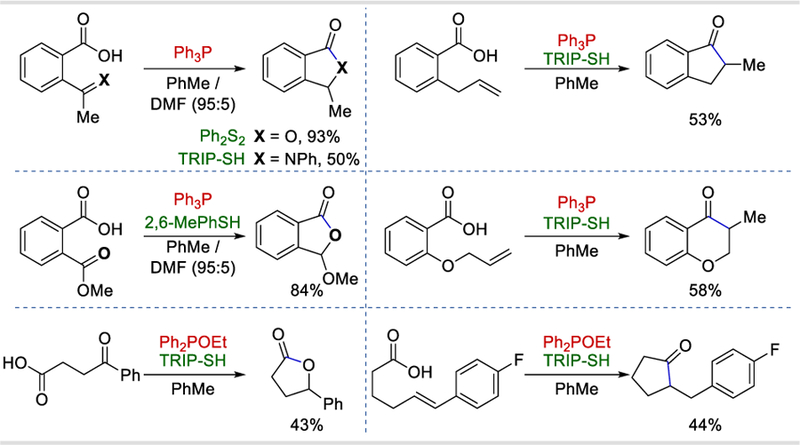
Intramolecular cyclization of acyl radicals and radical acceptors under the photocatalytic conditions.72
Generation of the acyl radical is similar to the mechanism proposed by Zhu (Scheme 69). Once the acyl radical 72.3 is formed, it abstracts a hydrogen atom from an arylthiol 72.4 to give the desired aldehyde 72.5 and an arylthiyl radical 72.6 (Scheme 72). Reduction of 72.6 by Ir(II) regenerates the Ir(III) photoredox catalyst and thiolate 72.7, which upon protonation forms the arylthiol 72.4.
Scheme 72.
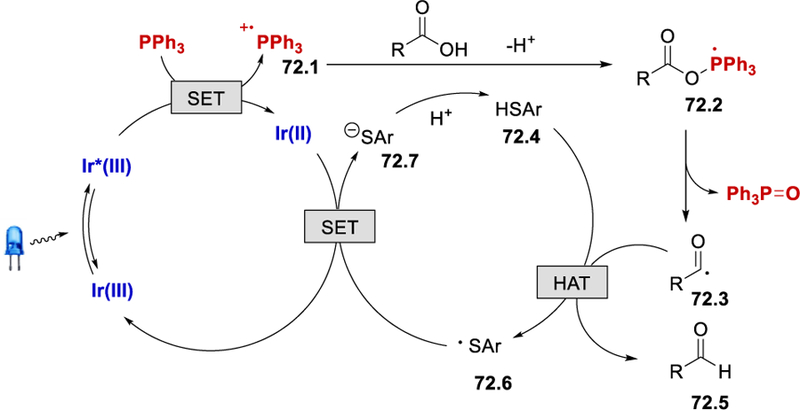
Proposed mechanism for the photocatalytic acyl radical formation using Ph3P as a deoxygenating reagent.72
Anhydrides as a source of acyl radicals
Rather than the in-situ formation of anhydrides from carboxylic acids in the presence of DMDC or Boc2O, the direct use of anhydrides is an alternate means of generating acyl radicals using visible light photoredox catalysis. In 2016, Wallentin et al. employed aromatic carboxylic anhydride as the direct acyl radical source for olefinic radical acylarylation under photocatalytic conditions.73 The method efficiently yielded substituted 3,3-disubstituted 2-oxindoles (45–98%) using a variety of symmetrical electron-withdrawing aromatic anhydrides. This protocol was also applied to variously substituted N-phenylacrylamides, obtaining products in 80–98% yields (Scheme 73). Interestingly, the Lewis acid activation of more challenging electron-rich aromatic and heteroaromatic carboxylic anhydrides was found to be necessary for the generation of the corresponding carbonyl radicals and the efficient synthesis of the desired products in 31–95% yields.
Scheme 73.
Photocatalytic 1,2-acylarylation of olefins using anhydrides.73
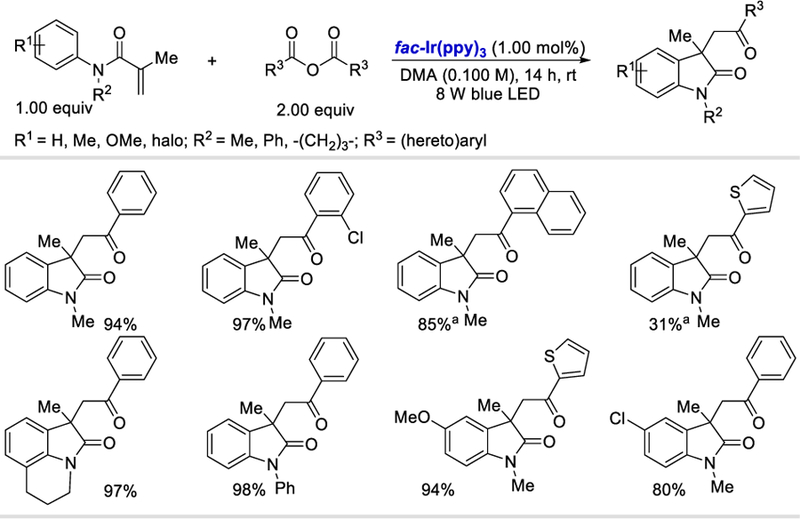
aReaction performed by adding 1 equiv of MgCl2; reaction time=60 h.
Stern-Volmer experiments indicate that the emission intensity of excited fac-*Ir(ppy)3 [E½ IV/*III = ‒1.73V vs. SCE] was quenched significantly in the presence of symmetrical anhydrides (benzoic anhydride: E½red = ‒1.01V vs. SCE).61 The plausible mechanism is analogous to Wallentin’s previous mechanism for 1,2-acylarylation of methacrylamide (Scheme 59) except the anhydride is directly employed here rather than being generated in-situ from a carboxylic acid.
Following the Wallentin group’s report on the direct use of the anhydrides as the acyl radical surrogate under photoredox conditions, Ye et al. in 2017, reported the construction of 1,4-dicarbonyl compounds using analogous symmetrical carboxylic anhydrides or mixed anhydrides.74 A wide range of symmetrical anhydrides containing electron-withdrawing or moderately electron-donating groups worked efficiently in these reaction conditions and gave the desired products in 62–79% yields. However, strongly electron-donating groups like -OMe lower the product yield to 58% (Scheme 74). Bromo-substituted symmetrical anhydrides gave lower yields (42–48%) of the desired products due to their poor solubility. Various olefin acceptors reacted smoothly (40–87%) with benzoic anhydrides although the α-or β-substituted olefins gave lower yields for steric or electronic reasons.
Scheme 74.
Photocatalytic hydroacylation using carboxylic acid anhydrides.74
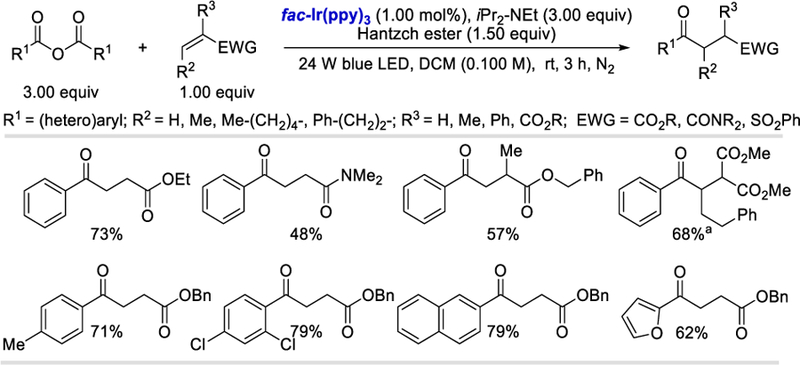
aReaction time was 24 h.
Radical trapping experiments with TEMPO confirms the formation of an aroyl radical. Stern-Volmer experiments, on the other hand, indicate that Hantzsch ester is the main quencher of the photoexcited fac-*Ir(ppy)3 at low concentrations. However, the same excited photocatalyst can be quenched by higher concentrations of carboxylic anhydride and iPr2NEt. Based on the above experiments, a plausible mechanism is proposed in Scheme 75 which begins with the photoexcitation of fac-Ir(ppy)3 with visible light. The photoexcited fac-*Ir(ppy)3 75.2 undergoes single electron transfer with the Hantzsch ester (E½red = +0.887 V vs. SCE)75 or iPr2NEt forming IrII species 75.3 and Hantzsch ester/iPr2NEt radical cation (75.4 or 75.5). Anhydride 75.6 is reduced by 75.3 (E½III/II = −2.19 V vs. SCE in MeCN) and then fragments to provide the aroyl radical 75.7, which reacts with alkene 75.8 to form α-carbonyl radical 75.9. This α-carbonyl radical 75.9 abstracts a hydrogen atom from 75.4 or 75.5 or Hantzsch ester to deliver the desired product 75.10.
Scheme 75.
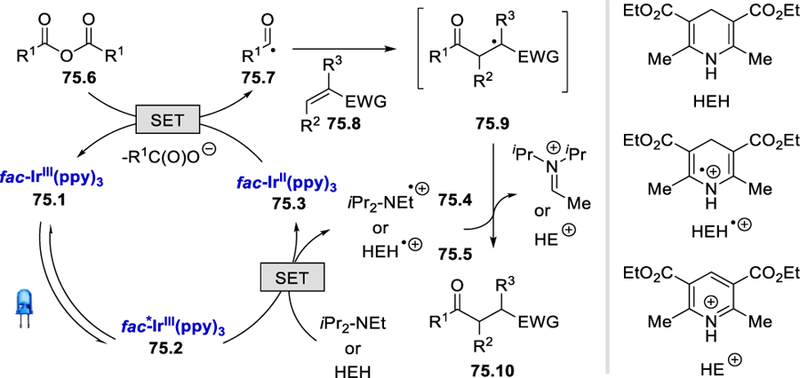
Mechanism for hydroacylation of electron deficient olefins.74
Acyl thioesters as a source of acyl radicals
In 2017, Gryko et al. reported the visible light driven vitamin B12 catalyzed generation of an acyl radical from 2-S-pyridyl thioesters (acyl-X reagent) via a single electron reduction and the subsequent reaction of an acyl radical with electron-deficient olefins.76 Scheme 76 demonstrates the superiority of pyridyl thioesters compared to other acylating reagents employed in this reaction. The higher activity of thioesters compared with active esters results from the stronger electrophilic character of the carbonyl group in the thioester derivatives.
Scheme 76.
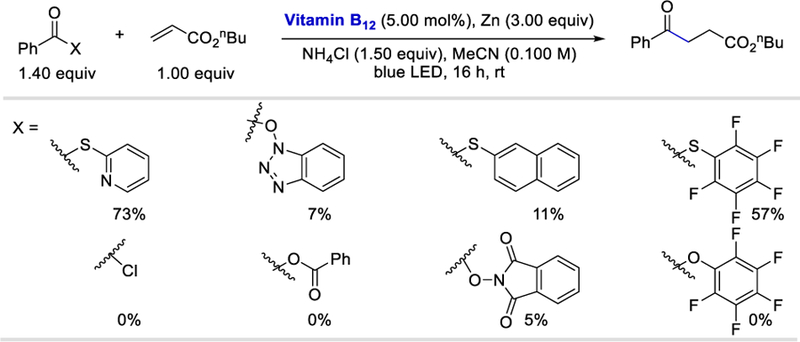
Investigation of various acyl derivatives.76
Olefins bearing electron-withdrawing groups such as esters, nitriles, sulfones, amides or ketones all produced the desired ketones in 58–99% yields (Scheme 77). While α-substituted olefins afforded the desired products in good (62–82%) yields, β-substituted olefins gave lower yields (27–47%) probably for steric reasons. Aryl, heteroaryl, and alkyl thioesters reacted equally well under the reaction conditions and formed the desired products in 62–97% yields. In the case of aryl thioesters containing strongly electron-withdrawing groups such as -CN or -CF3, the desired products were obtained in lower yields (26–60%) presumably due to their higher susceptibility to reduction [for the compound with a Me substituent Epc= ‒1.77 V, CF3 Epc= ‒1.49 V, CN Epc= ‒1.36 V vs. Ag/AgCl].76
Scheme 77.
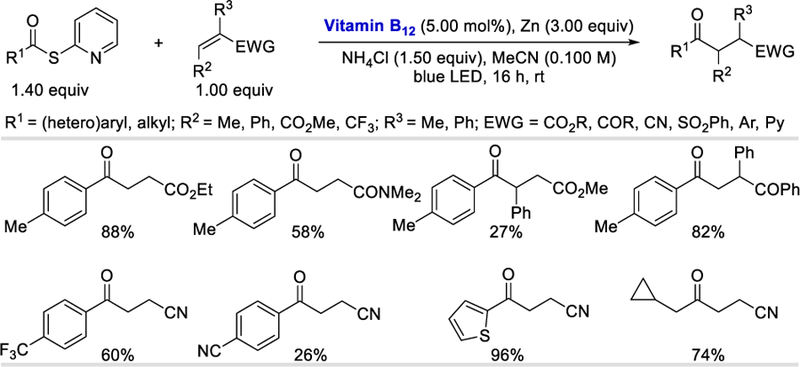
Vitamin B12-catalyzed radical acylation of electron deficient alkenes.76
LCMS detection of the reaction aliquot showed the presence of the acyl-cobalt complex 78.4 in the medium and TEMPO trapping experiments indicates the acyl radical formation. In the absence of the reducing agent zinc, no reaction was observed using CoIII-or CoII-vitamin B12, which confirms the formation of acyl-cobalt complex by the reaction of CoI-vitamin B12 generated in-situ, with thioesters. Addition of ND4Cl in place of NH4Cl showed deuterium incorporation of α-position to the electron-withdrawing group, indicating the role of NH4Cl as a proton source at the final step of the reaction. Light ON/OFF experiments support the formation of an acyl radical under constant irradiation of light. The proposed mechanism starts with the reduction of thioesters by CoI-vitamin B12 (78.1) and the generation of acyl-cobalt complex 78.4 which provides the acyl radical 78.6 under irradiation with visible light (Scheme 78). Subsequent addition of nucleophilic acyl radical to activated olefins 78.7 generates 78.8, which is reduced by Zn and protonated by NH4Cl to deliver the desired product (78.9). The CoII-vitamin B12 catalyst is reduced to CoI-vitamin B12 by Zn to complete the catalytic cycle.
Scheme 78.
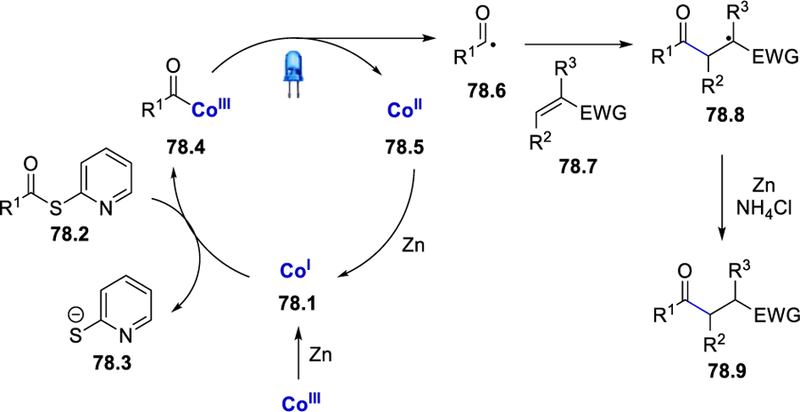
Mechanism for vitamin B12 catalyzed reductive acylation of olefins.76
In 2018, McErlean et al. used a thioester as the acyl radical precursor for hydro-acylation of olefins under photocatalytic conditions (Scheme 79).77 With 10 equivalents of tri-butylamine and formic acid, intramolecular acyl radical-olefin addition can react to form the desired chromanone and indanone derivatives in 18%−71% yields (Scheme 79a). When 2.00 equivalents of tri-butylamine and alkenes such as cyclohexene and allyltrimethylsilane were employed in the absence of formic acid (based on their SI), intermolecular coupling reactions took place to afford the desired products in 10%−37% yields (Scheme 79b).
Scheme 79.
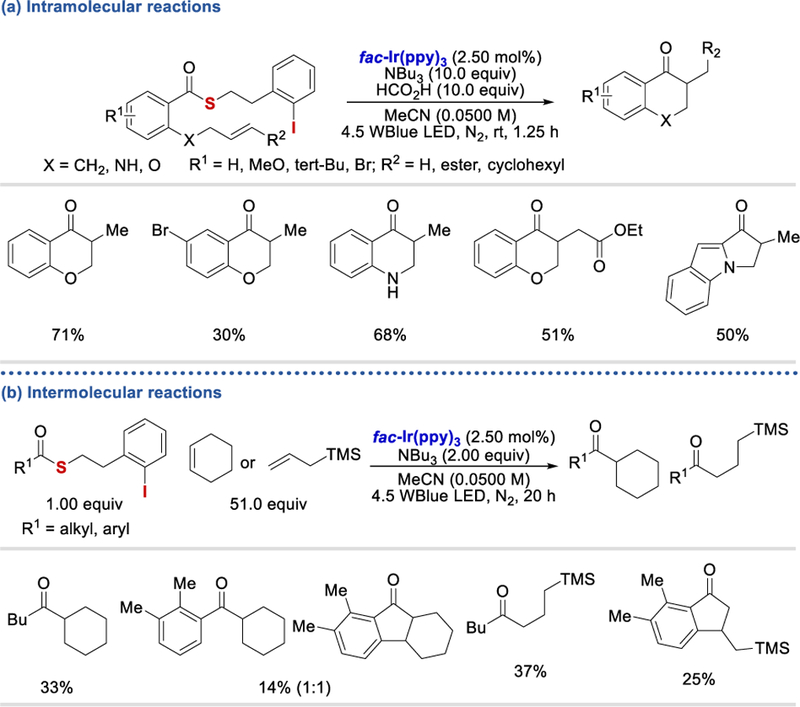
Photocatalytic reductive intra-and intermolecular hydroacylation of olefins.77
When 2.06 equivalents of tri-butyl amine and formic acid (based on their SI) were used, the ketone products were further reduced to form ketyl radicals, which dimerized, forming pinacol-type products (Scheme 80a). The authors also conducted a one-pot intramolecular cyclization-intermolecular radical-olefin addition reaction, which afforded the two-and three-component coupling products in 29% and 24% yields, respectively (Scheme 80b).
Scheme 80.
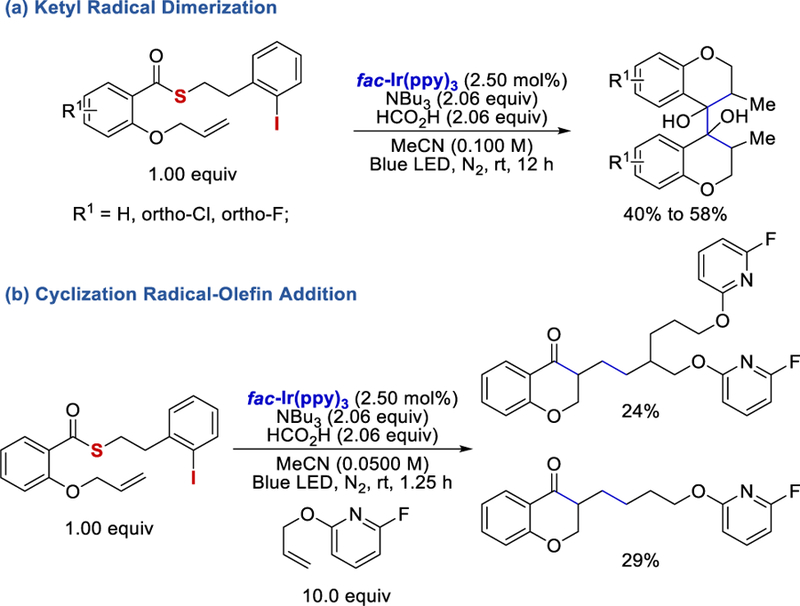
Photocatalytic ketyl radical dimerization and cyclization-radical olefin addition reactions.77
The unique feature of this reaction is the formation of an acyl radical from the thioester, triggered by the generation of an aryl radical. The idea was inspired by the work of Crich et al.78 It is proposed that reductive quenching of the excited *Ir(ppy)3 (E½III*/II = +0.31 V vs. SCE)56 by tributylamine (triethylamine: E½ox = +0.83 V vs. SCE)61 produces tributylamine radical cation 81.1 and IrII(ppy)3‒ (E½III/II = ‒2.19 V vs. SCE) (Scheme 81).56 This strongly reducing species reduces the iodobenzene moiety of thioester 81.2 (iodobenzene: E½red = ‒1.59 V vs. SCE)79 via SET to form the aryl radical 81.3, which attacks the sulfur to release dihydrobenzothiophene (81.4) and an acyl radical 81.5. Trapping of 81.5 with olefins forms an alkyl radical intermediate 81.6, which abstracts a hydrogen atom from tributylamine radical cation 81.1, liberating the desired product 81.7.
Scheme 81.
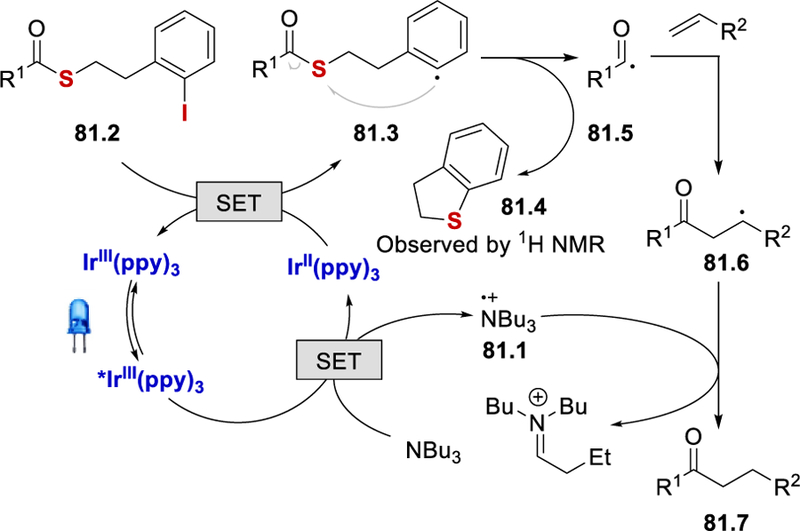
Proposed mechanism for photocatalytic reductive intra-and intermolecular hydroacylation of olefins.77
Acyl chlorides as a source of acyl radicals
Acyl chlorides are versatile intermediates and have been widely used as electrophilic acylating reagents in organic synthesis. Their electrophilic property can be reversed by a single electron reduction to generate acyl radicals. An early example of formation of acyl radical intermediate from acyl chloride was reported by Van Der Kerk et al. using triphenyltin hydride in 1957.80 Under their conditions, triphenyltin chloride and benzaldehyde were formed. A systematic study of the reaction between triphenyltin hydride and benzoyl chloride was conducted by Kuivila in 1960.81 A mechanism that involved the generation of an acyl radical was further demonstrated by Kuivila in 1966.82 The formation of acyl radical from acyl chloride via SET with SmI2 was reported by Kagan et al. in 1981, and the acyl radical was further reduced to an acyl anion under their reaction conditions.83 Only recently, photoredox catalysis was used for acyl radical formation from acyl chloride.84 In 2017, Xu et al. reported the first protocol to convert a benzoyl chloride to a benzoyl radical, which then reacts with 1,7-enynes to form fused pyran derivatives (Scheme 82).84a The reaction starts with excitation of fac-Ir(ppy)3 by blue LED and then the excited fac-*Ir(ppy)3 (E½IV/III* = ‒1.73 V vs. SCE)56 is involved in single electron reduction of a benzoyl chloride (Ep = –1.02 V vs. SCE)84b forming fac-IrIV(ppy)3 and benzoyl radical (82.2). The benzoyl radical then attacks the carbon-carbon double bond of 82.3 to afford an alkyl radical intermediate 82.4, which undergoes radical cyclization with the alkyne triple bond to form a vinyl radical intermediate 82.5. Oxidation of the vinyl radical intermediate by fac-IrIV(ppy)3 gives a vinyl cation species 82.6. The acyl carbonyl oxygen attacks the vinyl cation carbon center to form an oxonium ion 82.7, which is then deprotonated to give the desired product 82.8.
Scheme 82.
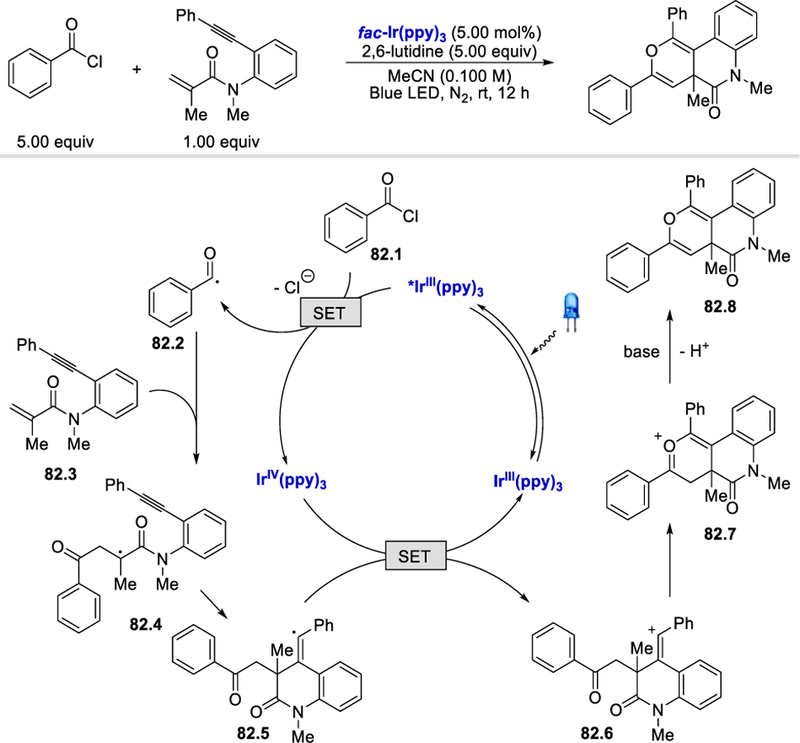
Proposed catalytic cycle for the photocatalytic synthesis of fused pyran derivatives from benzoyl chlorides and 1,7-enynes.84a Adapted with permission from (84a). Copyright (2017) American Chemical Society.
In terms of the acyl chloride substrates scope (Scheme 83), both electron deficient (e.g., CF3) and electron rich (e.g., OMe and Me) substituted benzoyl chlorides coupled to afford the desired product in 55–90% yields. Enyne substrates with alkynyl 3-thiophene and arenes bearing halogen, methyl, and methoxy substituents were tolerated and formed the desired fused pyran derivatives in 63–92% yields (Scheme 83). However, alkynyl alkyl substituents such as t-butyl and cyclopropyl groups gave only a trace amount of the desired products. The reaction tolerates substituents such as Cl, CN, and CF3 on the arene ring of anilides. Anilides with different N-protecting groups such as benzyl, acyl, and tosyl groups were competent, delivering the products in 67–70% yields. Aryl ester linked 1,7-enynes were also viable substrates.
Scheme 83.
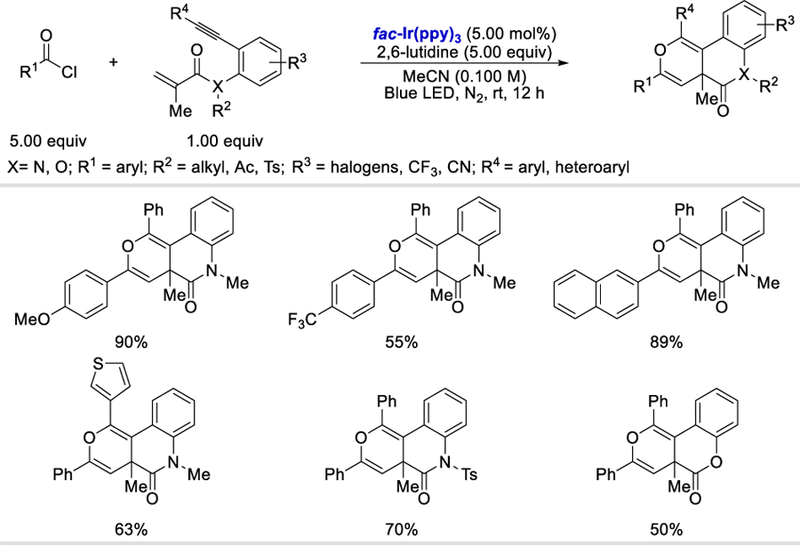
Photocatalytic synthesis of fused pyran derivatives from benzoyl chlorides and 1,7-enynes.84a
In the same year, Xu et al. extended the scope of the reaction to substrates without an alkyne motif (Scheme 84).84b In this reaction, the α-carbonyl radical intermediate 84.4 generated from the reaction of an N-phenyl methacrylamide (84.3) and the acyl radical 84.2 undergoes intramolecular cyclization to form intermediate 84.5, which is oxidized by IrIV then deprotonated to give a 3,3-dialkyl 2-oxindole derivative. The reaction scope is similar to that described in their previous report (Scheme 83).
Scheme 84.
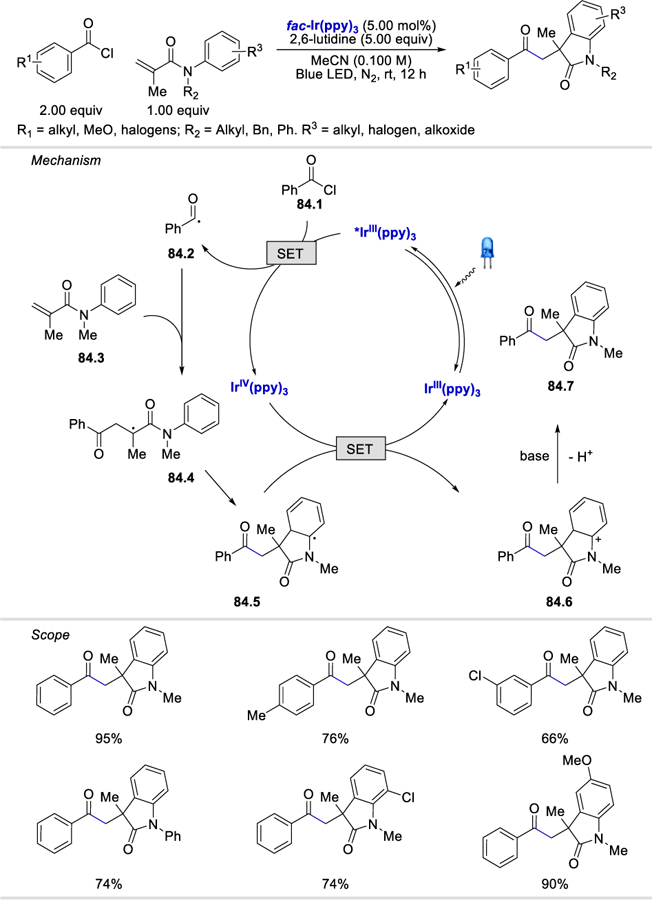
Photocatalytic formation of 2-oxindoles from acyl chlorides.84b
Acyl radicals formed from acyl chlorides can add directly to alkynes. In 2017, Tang et al. synthesized a diverse group of 3-acylspiro trienones via ipso-carboacylation of N-(p-methoxy aryl)propiolamides with acyl chloride.84c The p-methoxy group on the arene ring is critical to formation of the desired products. The reaction conditions worked well with benzoyl chlorides bearing alkyl, methoxy, and halogen substituents and thiophenecarbonyl chloride, affording the desired products in 60–86% yields. N-(p-methoxyaryl)propiolamides, containing a benzyl, 2-iodobenzyl acyl, and allylic groups on the nitrogen atom, were tolerated, providing the 3-acylspiro trienones in 69–73% yields. In terms of the alkynyl substituents (R4), pentyl, thiophene, pyridine, naphthalene, and arenes bearing alkyl, methoxy, acyl, CF3, or halogen groups were compatible and afforded the desired products in 57–88% yields. Mechanistically, the authors proposed that after the initial photocatalytic acyl radical formation followed by a two-step tandem acyl radical-alkyne coupling and radical cyclization, a cyclic radical species 85.1 is generated. An O18-labeling experiment suggests that 85.1 is attacked by an exogenous H2O molecule with assistance from 2,6-lutidine to give a radical anion intermediate 85.2. Removal of the methoxy group gives 85.3, which is then oxidized by IrIV to afford the desired product 85.4.
In 2018, Tang et al. further expanded this type of acyl radical chemistry to the synthesis of 3-acylcoumarins (Scheme 86).85 Although thiophenecarbonyl chloride and benzoyl chlorides bearing methyl, methoxy, and halogen substituents were well tolerated, alkyl, vinyl, and electron deficient (e.g., p-NO2Ph) acyl chlorides failed to produce the desired products. Aryl 3-phenylpropiolates possessing methyl, halogen, methoxy, Ac and CF3 groups at the p-position of the phenoxy ring underwent the reaction smoothly, affording the products in 68% to 80% yields. With respect to the scope of alkynyl substituents, arenes bearing electron donating and electron withdrawing groups were compatible under the reaction conditions.
Scheme 86.
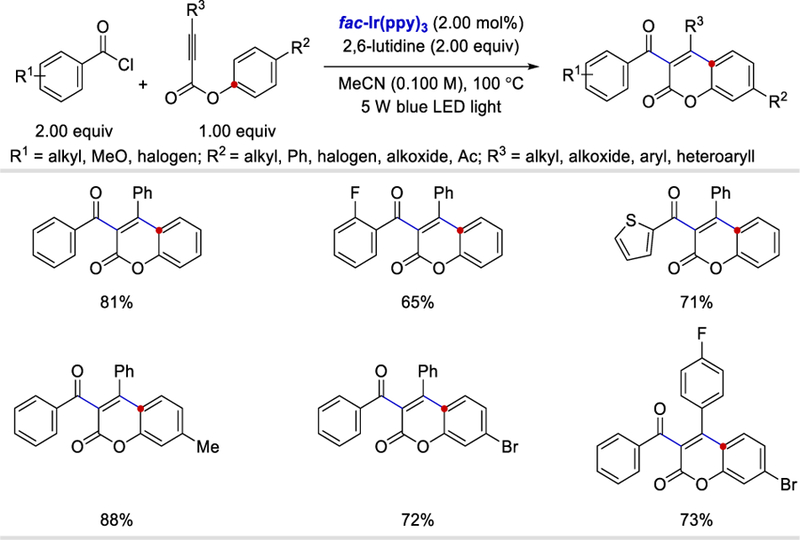
Photocatalytic synthesis of 3-acylcoumarin via cascade difunctionalization/cyclization of alkynoates and acyl chlorides. 85
The proposed reaction mechanism is depicted in Scheme 87. Once the radical intermediate 87.4 is formed, it undergoes an intramolecular 5-exo-trig cyclization followed by oxidation, giving the cationic species 87.6. 1,2-ester migration leads to 87.7, which upon deprotonation affords the desired product 87.8. Notably, both Tang’s and Itoh’s (Scheme 22) methods involve the formation of a 5-exo-trig cyclic intermediate, which is distinct from the transition metal-catalyzed acyl radical addition reported by Wu86 and Wang87 in which a 6-endo-trig cyclization process was proposed.
Scheme 87.
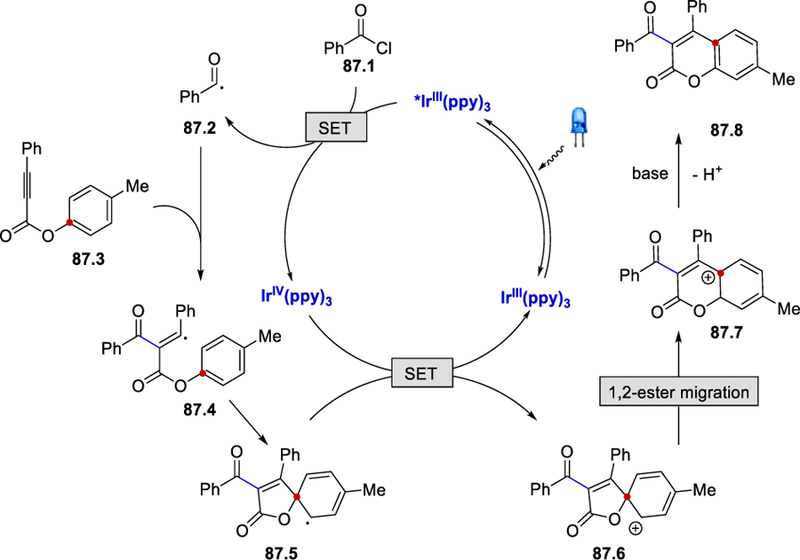
Proposed mechanism for the photocatalytic 3-acylcoumarin synthesis. 85
Acyl silanes as a source of acyl radicals
The first isolation of acyl silanes was reported by Brook in 1957.88 Acylsilanes are often regarded as unusual carbonyl compounds, as a result of the unique feature of an sp2 carbon that is bonded to both a silicon and oxygen atom.89 In 1969, Brook and Duff reported that photolysis of acylsilanes led to the formation of acyl radical intermediate via Norrish type I cleavage of the acyl-silicon bond.90 Electrochemical oxidation of acylsilanes, leading to the formation of acyl radical was described extensively by Keiji and Yoshida between 1986 and 1992.91 Application of acylsilanes in the formation of acyl radicals under photocatalytic conditions was described by Fagnoni et al. in 2017 (Scheme 88).92 Acyl-and benzoyltrimethylsilanes (AcylTMS and benzoylTMS) have higher oxidation potentials (E½ox = +1.26 to +1.51 V vs. SCE )92 than most of the common transition metal photocatalysts, such as fac-Ir(ppy)3 (E½III*/II = +0.31 V vs. SCE)56 and Ru(bpy)32+ (E½II*/I = +0.77 V vs. SCE).93 Thus, photocatalysts with stronger oxidizing power are needed to oxidize acylTMSs to acyl radicals. Fagnoni et al. discovered that photoexcited tetrabutylammonium decatungstate (TBADT)94 and 9-mesityl-10-methyl acridinium tetrafluoroborate (Acr+-Mes)95 (Scheme 88) could be used to enable the acylation of electron deficient alkenes. The authors proposed that the excited photocatalyst oxidizes an acylTMS to an acylTMS radical cation 88.2, which then loses the TMS group to form a nucleophilic acyl radical 88.3. The formation of the acyl radical was supported by their TEMPO trap experiments. The alkyl radical intermediate 88.5 formed by the addition of acyl radical to alkene 88.4 accepts an electron from the reduced photocatalyst to give a carbon anion 88.5, which is then protonated to form the final product 88.7.
Scheme 88.
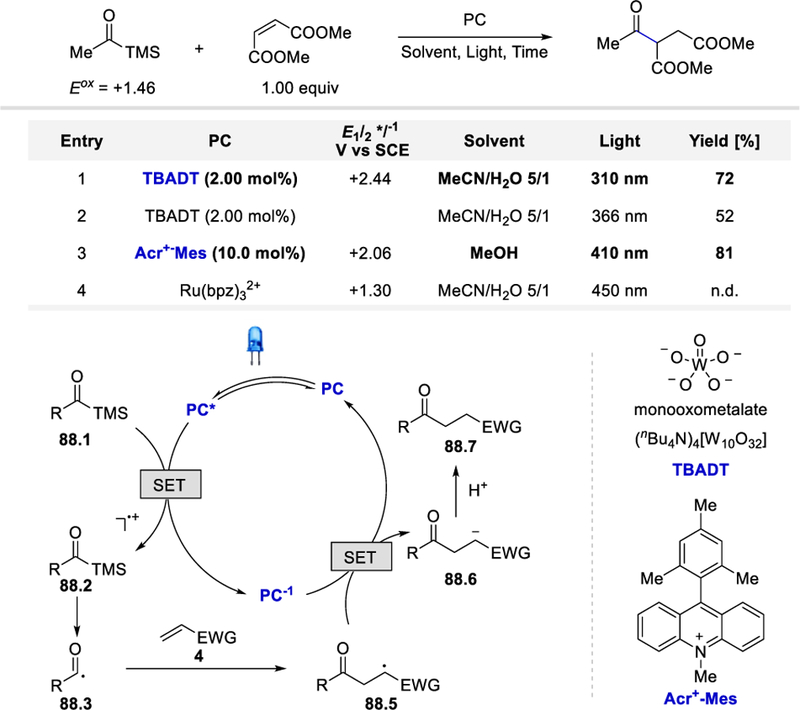
Optimization and reaction mechanism of photocatalytic hydroacylation using acylTMS as an acyl radical source.92
Regeneration of the TBADT catalyst through oxidation by the carbon radical intermediate was reported by Fagnoni.96 Although the absorption spectrum of TBDAT is not in the visible light region, it overlaps with the spectrum of sunlight, supporting the catalyst’s capacity as a “window-ledge” catalyst. For the Acr+-Mes-catalyzed reaction, *Acr+-Mes oxidizes the acylTMS to form an acyl radical and Acr•-Mes (E½red = –0.57 V.vs. SCE), the reduction potential of which is within reach of that of the carbon radical intermediate 88.5 (E½red ≈ –0.63 V .vs. SCE). The photocatalytic conditions operate under a different wavelength of light, making it suitable for a range of alkene substrates such as dimethyl maleate, electron poor styrenes, and acrylonitrile.
Conclusions and Future Outlook
Photoredox catalysis has emerged as a powerful tool in organic synthesis and has revolutionized how chemists tackle difficult bond forming challenges. In this review, we have provided an overview of the development of acyl radical chemistry under photocatalytic conditions. The unique reactivity of photoredox catalysts offers unprecedentedly mild reaction conditions for the generation of versatile acyl radicals from a wide range of precursors including aldehydes, α-ketocarboxylic acids, carboxylic acids and anhydrides, acyl thioesters, acyl chlorides, and acylsilanes. The mild conditions also enable the reactions of acyl radicals with a diverse set of coupling partners to construct molecular architectures that are otherwise difficult to prepare. Since the use of photoredox catalysis in acyl radical chemistry only began in 2013 and is still in its infancy, we anticipate that more innovative transformations involving acyl radical intermediates will continue to emerge in the near feature.
Scheme 5.
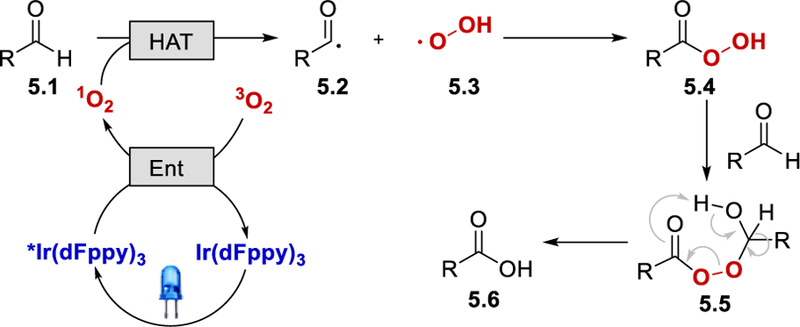
Proposed mechanism for aerobic oxidation of aldehydes to acyl radicals via photo-induced energy transfer. Adapted with permission from (23). Copyright (2013) Elsevier Ltd.23
Scheme 9.
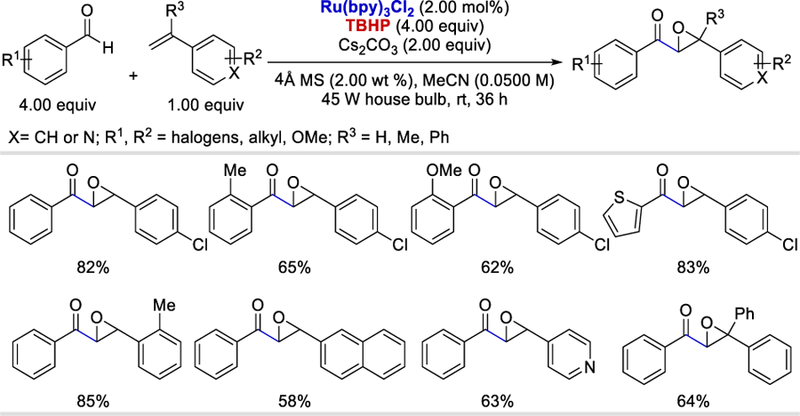
Photocatalytic synthesis of α,β-epoxy ketones via acyl radical addition using TBHP.28
Scheme 10.
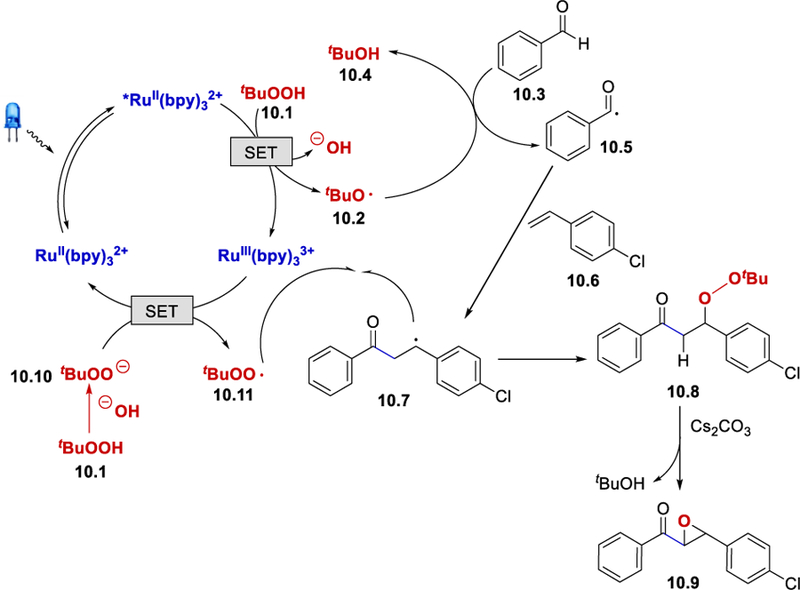
Proposed mechanism for the photocatalytic synthesis of α,β-epoxy ketones via an acyl radical intermediate.28 Adapted with permission from (28). Copyright (2015) American Chemical Society
Scheme 14.
Photocatalytic alkynylation of aldehydes.
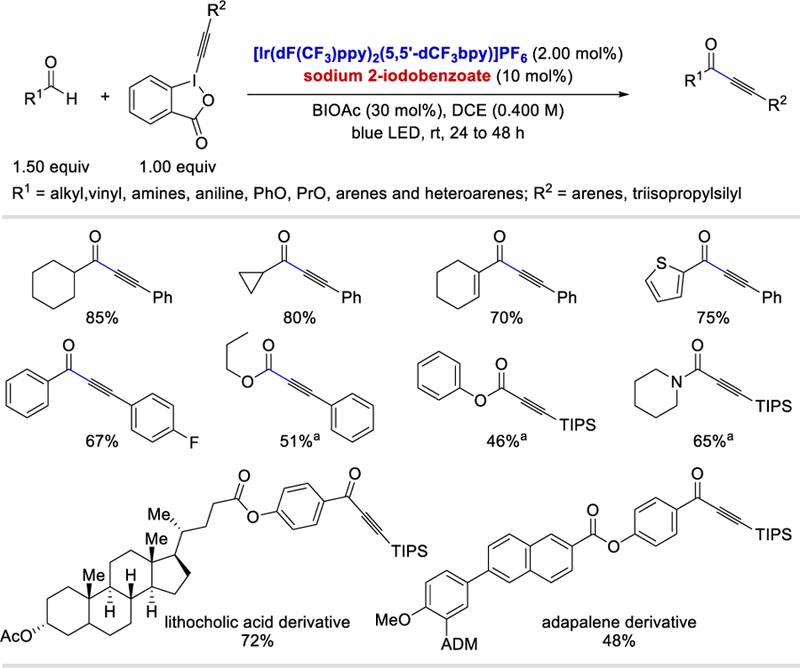
a48 h. BI = benziodoxolonyl group.31
Scheme 19.
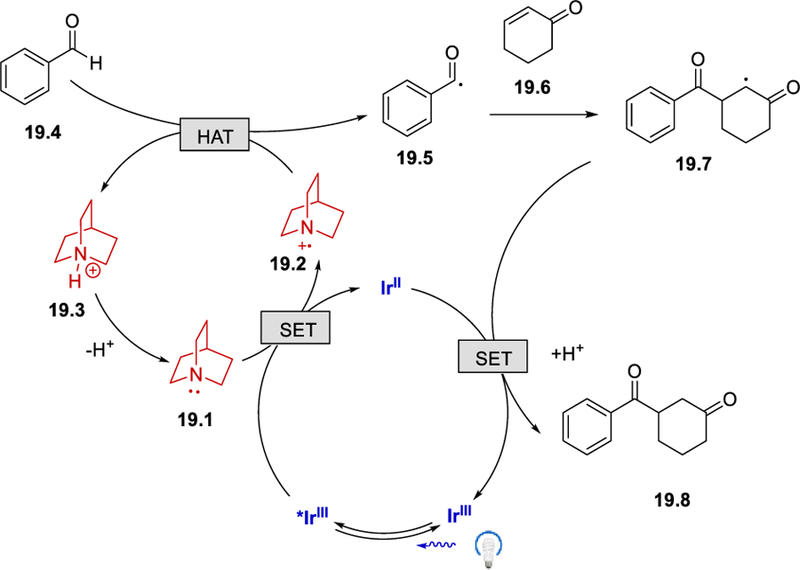
Photocatalytic hydroacylation of alkenes promoted by quinuclidine.35
Scheme 40.
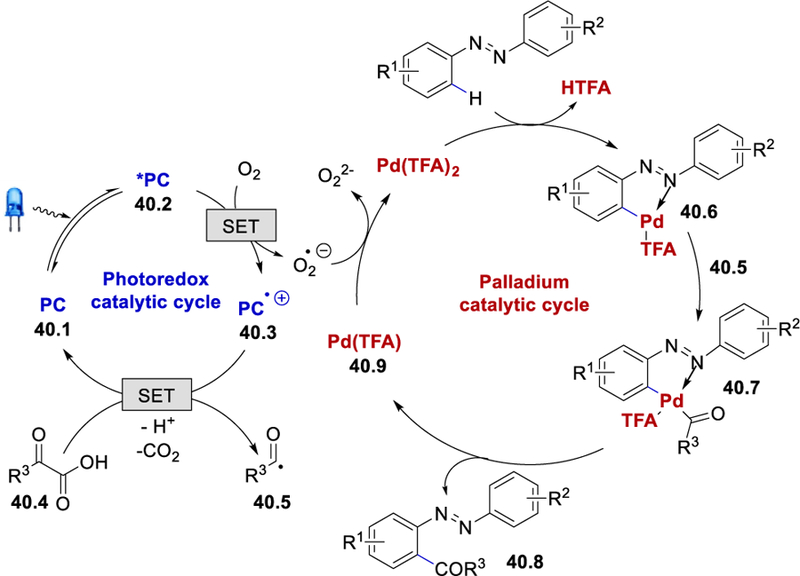
Proposed mechanism for photocatalytic decarboxylative o-acylation of azobenzene.51 Adapted with permission from (51). Copyright (2016) John Wiley and Sons.
Scheme 54.
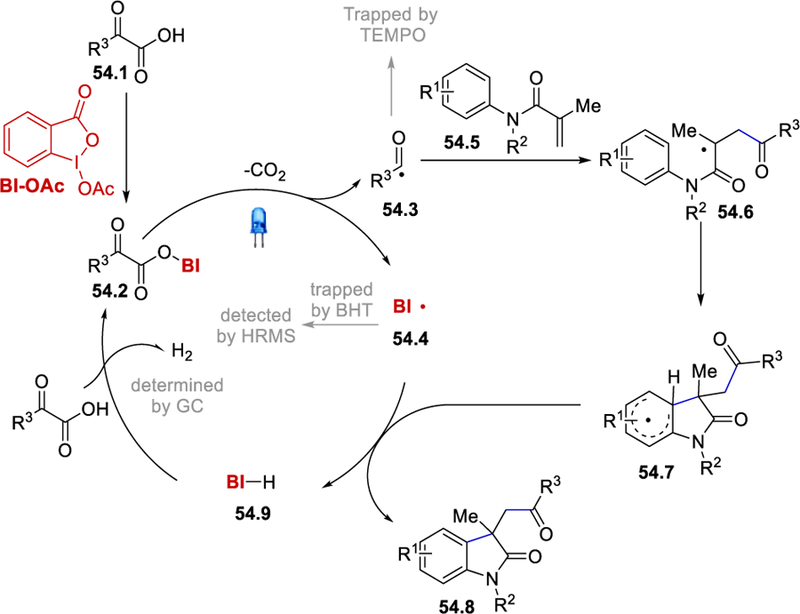
Proposed mechanism for photocatalytic acyl-arylation of olefins.60
Scheme 57.
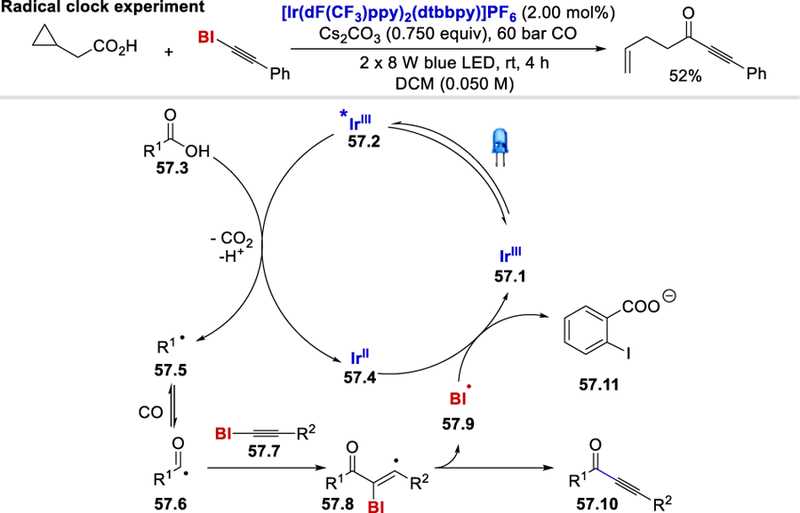
Mechanism for photocatalytic decarboxylative carbonylative alkynylations of carboxylic acids.63
Scheme 85.
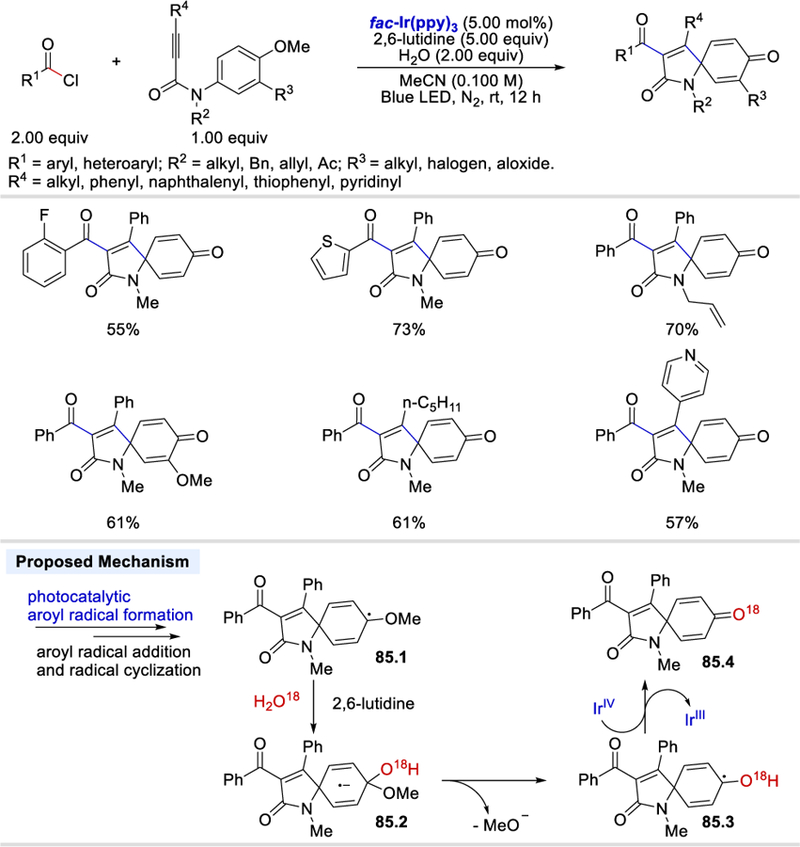
Visible light-mediated ipso-carboacylation of alkynes.84c
Scheme 89.
Photocatalytic hydroacylation using acylTMS as an acyl radical source.92
Acknowledgments
Funding Information
We thank the National Institute of General Medical Sciences (R35GM119652) for supporting our research.
Biosketches

Arghya Banerjee was born in West Bengal, India. He obtained his B.Sc. degree in chemistry from Ramakrishna Mission Vidyamandira Belur in 2008. After completing his M.Sc. from IIT Guwahati India in 2010, he continued his PhD. degree in the same institute under the guidance of Prof. Bhisma K. Patel. In 2017, he joined as a postdoctoral associate at Stony Brook University under the supervision of Prof. Ming Yu Ngai. His current research is focused on the development of novel acylation strategies using photoredox catalysis.

Zhen Lei was born in Xi’an, China. In 2011, he graduated with BSc degree in chemistry from Sichuan University in China. He continued his study at the State University of New York at Binghamton, where he worked with Prof. Susan L. Bane on borazine containing bioorthogonal reactions and he was awarded MSc degree in 2015. In the same year, he began his PhD study at Stony Brook University under the guidance of Prof. Ming-Yu Ngai. His current research focuses on the development of photoredox catalyzed C‒H functionalization and asymmetric reactions.

Ming-Yu Ngai (right) was born in Fuching, China and graduated with BSc degree from the University of Hong Kong in 2003. After he received his PhD degree with honors in chemistry from the University of Texas at Austin under the guidance of Prof. Michael J. Krische in 2008, he worked with Prof. Barry M. Trost at Stanford University as the Croucher post-doctoral fellow (2009–2011) and with Prof. Tobias Ritter at Harvard University as a postdoctoral associate (2011–2013). In 2013, he was appointed as an Assistant Professor in the Department of Chemistry at Stony Brook University. His research focuses on the development of photoredox catalysis and fluorine chemistry.
References
- (1).Caronna T; Fronza G; Minisci F; Porta O; Gardini GP J. Chem. Soc., Perkin Trans. 2 1972, (10), 1477. [Google Scholar]
- (2).(a) Bugaut X; Glorius F Chem. Soc. Rev 2012, 41, (9), 3511. [DOI] [PubMed] [Google Scholar]; (b) Enders D; Niemeier O; Henseler A Chem. Rev 2007, 107, (12), 5606. [DOI] [PubMed] [Google Scholar]; (c) Stetter H; Kuhlmann H Org. React 2004, 40, 407. [Google Scholar]
- (3).Duncton MA J. MedChemComm 2011, 2, (12), 1135. [Google Scholar]
- (4).(a) Boger DL; Mathvink RJ J. Org. Chem 1992, 57, (5), 1429 [Google Scholar]; (b) Chatgilialoglu C; Crich D; Komatsu M; Ryu I Chem. Rev 1999, 99, (8), 1991. [DOI] [PubMed] [Google Scholar]
- (5).(a) Liu W; Li Y; Liu K; Li Z J. Am. Chem. Soc 2011, 133, (28), 10756. [DOI] [PubMed] [Google Scholar]; (b) Benati L; Calestani G; Leardini R; Minozzi M; Nanni D; Spagnolo P; Strazzari S Org. Lett 2003, 5, (8), 1313. [DOI] [PubMed] [Google Scholar]; (c) Bath S; Laso NM; Lopez-Ruiz H; Quiclet-Sire B; Zard SZ Chem. Commun 2003, 0, (2), 204. [DOI] [PubMed] [Google Scholar]
- (6).Boger DL; Mathvink RJ J. Org. Chem 1989, 54, (8), 1777. [Google Scholar]
- (7).(a) Chen C; Crich D; Papadatos A J. Am. Chem. Soc 1992, 114, (21), 8313 [Google Scholar]; (b) Crich D; Chen C; Hwang J-T; Yuan H; Papadatos A; Walter RI J. Am. Chem. Soc 1994, 116, (20), 8937. [Google Scholar]
- (8).(a) Colley CS; Grills DC; Besley NA; Jockusch S; Matousek P; Parker AW; Towrie M; Turro NJ; Gill PMW; George MW J. Am. Chem. Soc 2002, 124, (50), 14952. [DOI] [PubMed] [Google Scholar]; (b) Hristova D; Gatlik I; Rist G; Dietliker K; Wolf J-P; Birbaum J-L; Savitsky A; Moebius K; Gescheidt G Macromolecules 2005, 38, (18), 7714 [Google Scholar]; (c) Sluggett GW; Turro C; George MW; Koptyug IV; Turro NJ J. Am. Chem. Soc 1995, 117, (18), 5148. [Google Scholar]
- (9).Yagci Y; Pappas SP; Schnabel WZ Naturforsch., A: Phys. Sci 1987, 42, (12), 1425. [Google Scholar]
- (10).(a) Brown CE; Neville AG; Rayner DM; Ingold KU; Lusztyk J Aust. J. Chem 1995, 48, (2), 363 [Google Scholar]; (b) McGimpsey WG; Scaiano JC J. Am. Chem. Soc 1987, 109, (7), 2179 [Google Scholar]; (c) Neville AG; Brown CE; Rayner DM; Lusztyk J; Ingold KU J. Am. Chem. Soc 1991, 113, (5), 1869 [Google Scholar]; (d) Davies AG; Sutcliffe R J. Chem. Soc., Perkin Trans. 2 1980, (5), 819. [Google Scholar]
- (11).(a) Rosenthal I; Mossoba MM; Riesz P Can. J. Chem 1982, 60, (12), 1486 [Google Scholar]; (b) Miranda MA; Galindo F In Photo-Fries Reaction and Related Processes, 2004; CRC Press LLC: 2004; pp 42/1. [Google Scholar]
- (12).(a) Coveney DJ; Patel VF; Pattenden G Tetrahedron Lett 1987, 28, (47), 5949 [Google Scholar]; (b) Coveney DJ; Patel VF; Pattenden G; Thompson DM J. Chem. Soc., Perkin Trans. 1 1990, (10), 2721. [Google Scholar]
- (13).(a) Wang H; Guo L-N; Duan X-H Adv. Synth. Catal 2013, 355, (11–12), 2222 [Google Scholar]; (b) Chaubey NR; Singh KN Tetrahedron Lett 2017, 58, (24), 2347 [Google Scholar]; (c) Meng M; Wang G; Yang L; Cheng K; Qi C Adv. Synth. Catal 2018, 360, (6), 1218. [Google Scholar]
- (14).Wu X-F Chem. -Eur. J 2015, 21, (35), 12252. [DOI] [PubMed] [Google Scholar]
- (15).(a) Prier CK; Rankic DA; MacMillan DWC Chem. Rev 2013, 113, (7), 5322. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Skubi KL; Blum TR; Yoon TP Chem. Rev 2016, 116, (17), 10035. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Shaw MH; Twilton J; MacMillan DWC. J. Org. Chem 2016, 81, (16), 6898. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Romero NA; Nicewicz DA Chem. Rev 2016, 116, (17), 10075. [DOI] [PubMed] [Google Scholar]; (e) Karkas MD; Porco JA; Stephenson CRJ Chem. Rev 2016, 116, (17), 9683. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Douglas JJ; Sevrin MJ; Stephenson CRJ Org. Process Res. Dev 2016, 20, (7), 1134 [Google Scholar]; (g) Twilton J; Le C; Zhang P; Shaw MH; Evans RW; MacMillan DWC Nat. Rev. Chem 2017, 1, (7). [Google Scholar]
- (16).Xiao WJ; Zhou QQ; Zou YQ; Lu LQ Angew. Chem. Int. Ed 2018, 10.1002/anie.201803102. [DOI]
- (17).Luca C; Davide R Eur. J. Org. Chem 2017, 2017, (15), 2056. [Google Scholar]
- (18).Shi Z; Glorius F Chem. Sci 2013, 4, (2), 829. [Google Scholar]
- (19).Jhuang H-S; Reddy DM; Chen T-H; Lee C-F Asian J. Org. Chem 2016, 5, (12), 1452. [Google Scholar]
- (20).Jeffrey JL; Terrett JA; MacMillan DWC Science 2015, 349, (6255), 1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Fan XZ; Rong JW; Wu HL; Zhou Q; Deng HP; Tan JD; Xue CW; Wu LZ; Tao HR; Wu J Angew. Chem. Int. Ed 2018, 57, (28), 8514. [DOI] [PubMed] [Google Scholar]
- (22).(a) Shum LG; Benson SW Int. J. Chem. Kinet 1983, 15, (5), 433 [Google Scholar]; (b) Simoes JM; Griller D Chem. Phys. Lett 1989, 158, (1–2), 175 [Google Scholar]; (c) Berkowitz J; Ellison GB; Gutman DJ Phys. Chem 1994, 98, (11), 2744 [Google Scholar]; (d) Bordwell FG; Satish A J. Am. Chem. Soc 1994, 116, (20), 8885 [Google Scholar]; (e) Bordwell FG; Liu W-Z J. Am. Chem. Soc 1996, 118, (44), 10819 [Google Scholar]; (f) Lund H; Daasbjerg K; Ochiallini D; Pedersen S ChemInform 1996, 27, (4), no [Google Scholar]; (g) Liu W-Z; Bordwell FG J. Org. Chem 1996, 61, (14), 4778. [DOI] [PubMed] [Google Scholar]; (h) Atkinson R; Baulch D; Cox R; Hampson R Jr; Kerr J; Rossi M; Troe J J. Phys. Chem. Ref. Data 2000, 29, (2), 167 [Google Scholar]; (i) Ervin KM; DeTuri VF J. Phys. Chem. A 2002, 106, (42), 9947 [Google Scholar]; (j) Luo Y-R, Handbook of Bond Dissociation Energies in Organic Compounds ed.; CRC press: 2002. [Google Scholar]
- (23).Iqbal N; Choi S; You Y; Cho EJ Tetrahedron Lett 2013, 54, (46), 6222. [Google Scholar]
- (24).Teegardin K; Day JI; Chan J; Weaver J Org. Process Res. Dev 2016, 20, (7), 1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Cheng P; Qing Z; Liu S; Liu W; Xie H; Zeng J Tetrahedron Lett 2014, 55, (49), 6647. [Google Scholar]
- (26).White HS; Bard AJ J. Am. Chem. Soc 1982, 104, (25), 6891. [Google Scholar]
- (27).Natascia T; Ulrich N; Alfons B; Jozef P; Ive H Chem. -Eur. J 2010, 16, (44), 13226.20945308 [Google Scholar]
- (28).Li J; Wang DZ Org. Lett 2015, 17, (21), 5260. [DOI] [PubMed] [Google Scholar]
- (29).Jung S; Kim J; Hong S Adv. Synth. Catal 2017, 359, (22), 3945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Iqbal N; Cho EJ J. Org. Chem 2016, 81, (5), 1905. [DOI] [PubMed] [Google Scholar]
- (31).Mukherjee S; Garza‐Sanchez RA; Tlahuext‐Aca A; Glorius F Angew. Chem. Int. Ed 2017, 56, (46), 14723. [DOI] [PubMed] [Google Scholar]
- (32).Choi GJ; Zhu Q; Miller DC; Gu CJ; Knowles RR Nature 2016, 539, 268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Zhang X; MacMillan DWC J. Am. Chem. Soc 2017, 139, (33), 11353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Shaw MH; Shurtleff VW; Terrett JA; Cuthbertson JD; MacMillan DWC Science 2016, 352, (6291), 1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Vu MD; Das M; Liu XW Chem. -Eur. J 2017, 23, (63), 15899. [DOI] [PubMed] [Google Scholar]
- (36).Kawaai K; Yamaguchi T; Yamaguchi E; Endo S; Tada N; Ikari A; Itoh A J. Org. Chem 2018, 83, (4), 1988. [DOI] [PubMed] [Google Scholar]
- (37).Allen NS; Hurley JP; Bannister D; Follows GW; Navaratnam S; Parsons BJ J. Photochem. Photobiol., A 1992, 68, (2), 213. [Google Scholar]
- (38).Majek M; Filace F; Wangelin A. J.v. Beilstein J. Org. Chem 2014, 10, 981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39). We used the benzaldehyde as the hydrogen donor to illustrate the mechanism.
- (40).de Souza GF; Bonacin JA; Salles AG Jr J. Org. Chem 2018, 83, (15), 8331. [DOI] [PubMed] [Google Scholar]
- (41).Furman OS; Teel AL; Watts RJ Environ. Sci. Technol 2010, 44, (16), 6423. [DOI] [PubMed] [Google Scholar]
- (42).(a) Tanielian C; Mechin RJ Photochem. Photobiol., A 1997, 107, (1), 291 [Google Scholar]; (b) Erickson PR; Walpen N; Guerard JJ; Eustis SN; Arey JS; McNeill K J. Phys. Chem. A 2015, 119, (13), 3233. [DOI] [PubMed] [Google Scholar]
- (43).Liu J; Liu Q; Yi H; Qin C; Bai R; Qi X; Lan Y; Lei A Angew. Chem. Int. Ed 2014, 53, (2), 502. [DOI] [PubMed] [Google Scholar]
- (44).Young RC; Meyer TJ; Whitten DG J. Am. Chem. Soc 1976, 98, (1), 286. [Google Scholar]
- (45).Chu L; Lipshultz JM; MacMillan DWC Angew. Chem. Int. Ed 2015, 54, (27), 7929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Lowry MS; Goldsmith JI; Slinker JD; Rohl R; Pascal RA Jr.; Malliaras GG; Bernhard S Chem. Mater 2005, 17, (23), 5712. [Google Scholar]
- (47).(a) Tsuji J; Yamada T; Minami I; Yuhara M; Nisar M; Shimizu I J. Org. Chem 1987, 52, (14), 2988 [Google Scholar]; (b) Goossen LJ; Zimmermann B; Knauber T Angew. Chem. Int. Ed 2008, 47, (37), 7103. [DOI] [PubMed] [Google Scholar]; (c) Shang R; Yang Z-W; Wang Y; Zhang S-L; Liu L J. Am. Chem. Soc 2010, 132, (41), 14391. [DOI] [PubMed] [Google Scholar]; (d) Weaver JD; Ka BJ; Morris DK; Thompson W; Tunge JA J. Am. Chem. Soc 2010, 132, (35), 12179. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Goossen LJ; Lange PP; Rodriguez N; Linder C Chem. -Eur. J 2010, 16, (13), 3906. [DOI] [PubMed] [Google Scholar]; (f) Wang C; Rakshit S; Glorius F J. Am. Chem. Soc 2010, 132, (40), 14006. [DOI] [PubMed] [Google Scholar]; (g) Shang R; Ji D-S; Chu L; Fu Y; Liu L Angew. Chem. Int. Ed 2011, 50, (19), 4470. [DOI] [PubMed] [Google Scholar]; (h) Hu P; Zhang M; Jie X; Su W Angew. Chem. Int. Ed 2012, 51, (1), 227. [DOI] [PubMed] [Google Scholar]; (i) Fromm A; van Wuellen C; Hackenberger D; Goossen LJ J. Am. Chem. Soc 2014, 136, (28), 10007. [DOI] [PubMed] [Google Scholar]
- (48).Goossen LJ; Rudolphi F; Oppel C; Rodriguez N Angew. Chem. Int. Ed 2008, 47, (16), 3043. [DOI] [PubMed] [Google Scholar]
- (49).Cheng W-M; Shang R; Yu H-Z; Fu Y Chem. -Eur. J 2015, 21, (38), 13191. [DOI] [PubMed] [Google Scholar]
- (50).Zhou C; Li P; Zhu X; Wang L Org. Lett 2015, 17, (24), 6198. [DOI] [PubMed] [Google Scholar]
- (51).Xu N; Li P; Xie Z; Wang L Chem. -Eur. J 2016, 22, (7), 2236. [DOI] [PubMed] [Google Scholar]
- (52).Wang G-Z; Shang R; Cheng W-M; Fu Y Org. Lett 2015, 17, (19), 4830. [DOI] [PubMed] [Google Scholar]
- (53).Zhang M; Xi J; Ruzi R; Li N; Wu Z; Li W; Zhu C J. Org. Chem 2017, 82, (18), 9305. [DOI] [PubMed] [Google Scholar]
- (54).(a) Jamison CR; Overman LE Acc. Chem. Res 2016, 49, (8), 1578. [DOI] [PubMed] [Google Scholar]; (b) Slutskyy Y; Overman LE Org. Lett 2016, 18, (11), 2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Petersen WF; Taylor RJK; Donald JR Org. Lett 2017, 19, (4), 874. [DOI] [PubMed] [Google Scholar]
- (56).Flamigni L; Barbieri A; Sabatini C; Ventura B; Barigelletti F Top. Curr. Chem 2007, 281, 143. [Google Scholar]
- (57).Bai Q-F; Jin C; He J-Y; Feng G Org. Lett 2018, 20, (8), 2172. [DOI] [PubMed] [Google Scholar]
- (58).Huang H; Zhang G; Chen Y Angew. Chem. Int. Ed 2015, 54, (27), 7872. [DOI] [PubMed] [Google Scholar]
- (59).Tan H; Li H; Ji W; Wang L Angew. Chem. Int. Ed 2015, 54, (29), 8374. [DOI] [PubMed] [Google Scholar]
- (60).Ji W; Tan H; Wang M; Li P; Wang L Chem. Commun 2016, 52, (7), 1462. [DOI] [PubMed] [Google Scholar]
- (61).Roth HG; Romero NA; Nicewicz DA Synlett 2016, 27, (5), 714. [Google Scholar]
- (62).Bergonzini G; Cassani C; Wallentin C-J Angew. Chem. Int. Ed 2015, 54, (47), 14066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Zhou Q-Q; Guo W; Ding W; Wu X; Chen X; Lu L-Q; Xiao W-J Angew. Chem. Int. Ed 2015, 54, (38), 11196. [DOI] [PubMed] [Google Scholar]
- (64).Candish L; Freitag M; Gensch T; Glorius F Chem. Sci 2017, 8, (5), 3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Chateauneuf J; Lusztyk J; Ingold KU J. Am. Chem. Soc 1988, 110, (9), 2886. [Google Scholar]
- (66).Pettersson F; Bergonzini G; Cassani C; Wallentin C-J Chem. -Eur. J 2017, 23, (31), 7444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Zhang M; Ruzi R; Xi J; Li N; Wu Z; Li W; Yu S; Zhu C Org. Lett 2017, 19, (13), 3430. [DOI] [PubMed] [Google Scholar]
- (68).Zhang M; Li N; Tao X; Ruzi R; Yu S; Zhu C Chem. Commun 2017, 53, (73), 10228. [DOI] [PubMed] [Google Scholar]
- (69).Ruzi R; Zhang M; Ablajan K; Zhu C J. Org. Chem 2017, 82, (23), 12834. [DOI] [PubMed] [Google Scholar]
- (70).Zhang M; Xie J; Zhu C Nat. Commun 2018, 9, (1), 3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Pandey G; Pooranchand D; Bhalerao UT Tetrahedron 1991, 47, (9), 1745. [Google Scholar]
- (72).Erin S; Alyssa B,E; Tomislav R; Abigail G,D, Generation of Phosphoranyl Radicals via Photoredox Catalysis Enables Voltage–Independent Activation of Strong C–O Bonds ed.; 2018; 10.26434/chemrxiv.6349253.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Bergonzini G; Cassani C; Lorimer-Olsson H; Hoerberg J; Wallentin C-J Chem. -Eur. J 2016, 22, (10), 3292. [DOI] [PubMed] [Google Scholar]
- (74).Dong S; Wu G; Yuan X; Zou C; Ye J Org. Chem. Front 2017, 4, (11), 2230. [Google Scholar]
- (75).Zhu X-Q; Li H-R; Li Q; Ai T; Lu J-Y; Yang Y; Cheng J-P Chem. -Eur. J 2003, 9, (4), 871. [DOI] [PubMed] [Google Scholar]
- (76).Ociepa M; Baka O; Narodowiec J; Gryko D Adv. Synth. Catal 2017, 359, (20), 3560. [Google Scholar]
- (77).McErlean C; Norman AR; Yousif MN Org. Chem. Front 2018, 10.1039/C8QO00867A. [DOI]
- (78).Crich D; Yao Q J. Org. Chem 1996, 61, (10), 3566. [Google Scholar]
- (79).Fry AJ; Krieger RL J. Org. Chem 1976, 41, (1), 54. [Google Scholar]
- (80).M., v. d. K. G. J.; G., N. J.; A., L. J. G. J. Appl. Chem 1957, 7, (7), 356. [Google Scholar]
- (81).Kuivila HG J. Org. Chem 1960, 25, 284. [Google Scholar]
- (82).Kuivila HG; Walsh EJ Jr. J. Am. Chem. Soc 1966, 88, (3), 571. [Google Scholar]
- (83).Girard P; Couffignal R; Kagan HB Tetrahedron Lett 1981, 22, (40), 3959 [Google Scholar]; Souppe J; Namy JL; Kagan HB Tetrahedron Lett 1984, 25, (27), 2869. [Google Scholar]
- (84).(a) Li C-G; Xu G-Q; Xu P-F Org. Lett 2017, 19, (3), 512. [DOI] [PubMed] [Google Scholar]; (b) Xu S-M; Chen J-Q; Liu D; Bao Y; Liang Y-M; Xu P-F Org. Chem. Front 2017, 4, (7), 1331 [Google Scholar]; (c) Liu Y; Wang Q-L; Zhou C-S; Xiong B-Q; Zhang P-L; Yang C.-a.; Tang K-W J. Org. Chem 2018, 83, (4), 2210. [DOI] [PubMed] [Google Scholar]; (d) Wang CM; Song D; Xia PJ; Wang J; Xiang HY; Yang H Chem. -Asian. J 2018, 13, (3), 271. [DOI] [PubMed] [Google Scholar]
- (85).Liu Y; Wang Q-L; Zhou C-S; Xiong B-Q; Zhang P-L; Kang S-J; Yang C-A; Tang K-W Tetrahedron Lett 2018, 59, (21), 2038. [Google Scholar]
- (86).Mi X; Wang C; Huang M; Wu Y; Wu Y J. Org. Chem 2015, 80, (1), 148. [DOI] [PubMed] [Google Scholar]
- (87).Yan K; Yang D; Wei W; Wang F; Shuai Y; Li Q; Wang H J. Org. Chem 2015, 80, (3), 1550. [DOI] [PubMed] [Google Scholar]
- (88).Brook AG J. Am. Chem. Soc 1957, 79, (16), 4373. [Google Scholar]
- (89).(a) Page PCB; Klair SS; Rosenthal S Chem. Soc. Rev 1990, 19, (2), 147 [Google Scholar]; (b) Zhang H-J; Priebbenow DL; Bolm C Chem. Soc. Rev 2013, 42, (21), 8540. [DOI] [PubMed] [Google Scholar]
- (90).(a) Brook AG; Duff JM J. Am. Chem. Soc 1969, 91, (8), 2118 [Google Scholar]; (b) Brook AG; Dillon PJ; Pearce R Can. J. Chem 1971, 49, (1), 133. [Google Scholar]
- (91).(a) Kunio M; Shuko O; Kyoko I; Osamu K; Tohru T; Keiji Y Chem. Lett 1986, 15, (5), 805 [Google Scholar]; (b) Yoshida J.-i.; Matsunaga S.-i.; Isoe S Tetrahedron Lett 1989, 30, (39), 5293 [Google Scholar]; (c) Yoshida J; Itoh M; Matsunaga S; Isoe S J. Org. Chem 1992, 57, (18), 4877. [Google Scholar]
- (92).Capaldo L; Riccardi R; Ravelli D; Fagnoni M ACS Catal 2017, 8, (1), 304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Rillema DP; Allen G; Meyer TJ; Conrad D Inorg. Chem 1983, 22, (11), 1617. [Google Scholar]
- (94).Waele VD; Poizat O; Fagnoni M; Bagno A; Ravelli D ACS Catal 2016, 6, (10), 7174. [Google Scholar]
- (95).Tsudaka T; Kotani H; Ohkubo K; Nakagawa T; Tkachenko NV; Lemmetyinen H; Fukuzumi S Chem. -Eur. J 2017, 23, (6), 1306. [DOI] [PubMed] [Google Scholar]
- (96).Ravelli D; Protti S; Fagnoni M Acc. Chem. Res 2016, 49, (10), 2232. [DOI] [PubMed] [Google Scholar]