

Abstract
Background
Cardiac ischemic/reperfusion (I/R) injury leads to brain damage. A new antihyperlipidemic drug is aimed at inhibiting PCSK9 (proprotein convertase subtilisin/kexin type 9), a molecule first identified in a neuronal apoptosis paradigm. Thus, the PCSK9 inhibitor (PCSK9i) may play a role in neuronal recovery following cardiac I/R insults. We hypothesize that PCSK9i attenuates brain damage caused by cardiac I/R via diminishing microglial/astrocytic hyperactivation, β‐amyloid aggregation, and loss of dendritic spine.
Methods and Results
Adult male rats were divided into 7 groups: (1) control (n=4); (2) PCSK9i without cardiac I/R (n=4); (3) sham (n=4); and cardiac I/R (n=40). Cardiac I/R rats were divided into 4 subgroups (n=10/subgroup): (1) vehicle; (2) PCSK9i (10 μg/kg, IV) before ischemia; (3) PCSK9i during ischemia; and (4) PCSK9i at the onset of reperfusion. At the end of cardiac I/R protocol, brains were removed to determine microglial and astrocytic activities, β‐amyloid aggravation, and dendritic spine density. The cardiac I/R led to the activation of the brain's innate immunity resulting in increasing Iba1+ microglia, GFAP + astrocytes, and CD11b+/CD45+high cell numbers. However, CD11b+/CD45+low cell numbers were decreased following cardiac I/R. In addition, cardiac I/R led to reduced dendritic spine density, and increased β‐amyloid aggregation. Only the administration of PCSK9i before ischemia effectively attenuated these deleterious effects on the brain following cardiac I/R. PCSK9i administration under the physiologic condition did not affect the aforementioned parameters.
Conclusions
Cardiac I/R injury activated microglial activity in the brain, leading to brain damage. Only the pretreatment with PCSK9i prevented dendritic spine loss via reduction of microglial activation and Aβ aggregation.
Keywords: amyloid, brain, ischemia/reperfusion injury/neuroprotection, proprotein convertase subtilisin/kexin type 9
Subject Categories: Basic Science Research, Inflammation, Neuroprotectants
Clinical Perspective
What Is New?
This is the first study showing that proprotein convertase subtilisin/kexin type 9 inhibitor given before cardiac I/R prevented dendritic spine loss via reducing brain inflammation, microglial activation, and Aβ aggregation.
What Are the Clinical Implications?
Regarding the clinical implication, this study suggests the possible neuroprotective advantages for the use of proprotein convertase subtilisin/kexin type 9 inhibitors in hyperlipidemic individuals and a potential new strategy for the improvement of neurological outcomes following cardiovascular events.
Introduction
Acute myocardial infarction (MI) is a serious cardiovascular event happening in response to the malperfusion of the heart muscle, which can be life threatening.1 MI decreases the pumping efficiency of the heart and cardiac output, leading to a potential reduction of systemic blood flow and causes damage to other vital organs, such as the brain.2 Immediate reperfusion therapy through primary percutaneous coronary intervention and fibrinolytic therapies is recommended for managing patients with acute MI in those presenting with ST‐segment–elevation myocardial infarction.3 However, percutaneous coronary intervention may cause further deleterious effects on both the heart and brain, effects that are known as ischemic/reperfusion injury (I/R). In the brain, cardiac I/R causes brain damage that is characterized by increased brain inflammation.4, 5 In addition, beta‐amyloid (Aβ) aggregation, tau hyperphosphorylation, reactive gliosis, the reduction of spine density, as well as decreased cognition and neurogenesis have been found in the brain following cardiac I/R injury.4, 6, 7
Although PCSK9 (proprotein convertase subtilisin/kexin type 9) is a protease primarily associated with lipoprotein homeostasis,8 it was first identified in a neuronal apoptosis paradigm, suggesting that it could be involved in neuronal injury and death.9 Studies exploring the alternative mechanisms of action of PCSK9 have found that it may be involved in inflammatory and apoptotic pathways by promoting NF‐kB activation,10 ApoER2 signaling,11 and/or by Bcl‐2/Bax‐caspase9/3 cascade activation.12, 13, 14 Recently, PCSK9 monoclonal antibodies have already started being used for treatment in patients with hyperlipidemia,15 but with inconclusive results regarding their neurocognitive side effects.16, 17, 18, 19, 20 Despite possible progress in this field, hyperlipidemic individuals have a significantly increased risk of cardiovascular events such as MI.21 Because its apparent involvement is in neuronal apoptotic pathways, it is important to define whether PCSK9 inhibitor (PCSK9i) may confer additional neuroprotection in the case of MI.
In this study, we investigated for the first time the association between PCSK9 inhibition and neuronal responses in a rat model of cardiac I/R injury. We hypothesized that PCK9i attenuates brain damage caused by cardiac I/R via decreasing microglial and astrocytic hyperactivation, β‐amyloid aggregation, and the reduction of dendritic spine density. In addition, we evaluated whether the administration of PCSK9i before cardiac I/R, during cardiac ischemia, or at the onset of reperfusion provided similar beneficial effects to the brain in cases of cardiac I/R injury.
Methods
Ethics Approval
The authors declare that all supporting data are available within the article. The study was approved by the Institutional Animal Care and Use Committee of the Faculty of Medicine, Chiang Mai University (permit number: 31/2560). It is in accordance with the Guide for the Care and Use of Laboratory Animals (8th edition, 2011) published by the National Institutes of Health (NIH).
Animals
Adult male Wistar rats (250–400 g, n=52) were obtained from the National Laboratory Animal Center, Mahidol University, Salaya Campus, Bangkok, Thailand. The animals were housed in a temperature‐ and humidity‐controlled environment with a 12:12 light–dark cycle and were allowed ad libitum access to a standard laboratory rat diet and water. Rats were blindly randomized into 3 groups: (1) nonoperation (n=8); (2) sham operation (n=4); and (3) cardiac I/R (n=40).
In the nonoperation group, rats were subdivided into 2 subgroups (n=4/subgroup); (1) Control (saline) and (2) PCSK9i (P‐PCSK9, Prep2‐8 trifluoroacetate salt, Sigma‐Aldrich, Missouri, 10 μg/kg in NSS, IV). Rats were euthanized after 180 minutes of drug injection.
In the sham operation group (n=4), rats were subjected to open chest surgery, but the left anterior descending (LAD) coronary artery was not ligated, and the rats were euthanized after 180 minutes of operation.
In the cardiac I/R group, rats were randomly assigned to 4 treatment subgroups (n=10/subgroup): (1) Vehicle (0.5 mL normal saline solution, administered 15 minutes before ischemia); (2) Pretreatment with PCSK9i (P‐PCSK9, Prep2‐8 trifluoroacetate salt, Sigma‐Aldrich, Missouri, 10 μg/kg in NSS, IV, administered 15 minutes before ischemia); (3) PCSK9i administration during cardiac ischemia (D‐PCSK9, 10 μg/kg in NSS, IV, administered 15 minutes after LAD ligation); (4) PCSK9i administration at the onset of reperfusion (A‐PCSK9, 10 μg/kg in NSS, IV, administered at the onset of reperfusion). The total sample size was calculated using the G‐power program,6 and the effect size is 0.7.
At the end of the reperfusion period or after 180 minutes of operation/injection, the animals were deeply anesthetized and decapitated. The brains were quickly removed. The brain tissue was used for the following analyses: microglial activity, morphological assessments of microglia and astrocytes, dendritic spine density determination, and immunoblotting.
For the control group, rats were deeply anesthetized with isoflurane and decapitated. The brains were quickly removed and used for the same analyses performed on the treatment rats. The experimental protocol is shown in Figure 1.
Figure 1.
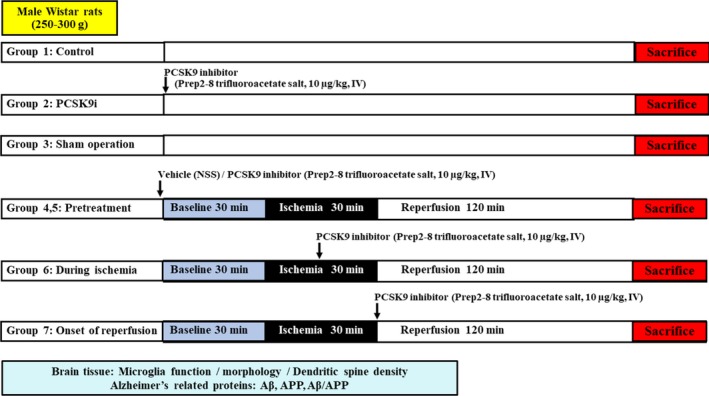
Experimental protocol of the study. APP indicates amyloid beta precursor protein; Aβ, amyloid beta; PCSK9, proprotein convertase subtilisin/kexin type 9.
Cardiac I/R
In the cardiac I/R group, rats underwent 30 minutes of LAD ligation followed by 120 minutes of reperfusion as previously described.22 Briefly, rats were anesthetized with an intramuscular injection of tiletamine/zolazepam (Zoletil) (50 mg/kg; Virbac Laboratories, Bangkok, Thailand) and xylazine (0.15 mg/kg; LBS Labs, Bangkok, Thailand). A tracheostomy was performed, and the rats were ventilated with room air from a rodent–ventilator (SAR‐830 Series, CWE Inc, Pennsylvania), while a cannula in the left femoral vein facilitated quick drug and saline administration. A left‐sided thoracotomy was performed at the fourth intercostal space to expose the heart; the LAD was identified, and ligated at a distance 2 mm distal to its origin. A small vinyl tube was passed through the ligature, which was used to occlude the LAD by pulling on the thread. Data from the ECG lead II were recorded continuously throughout the experiment and an ST elevation together with a change in myocardium color were used to confirm ischemia.
Isolation of Brain Microglia
Microglia were isolated from the brain tissue using the Percoll gradient technique. The brain tissue was homogenized in Hank's balanced salt solution, and the homogenate was centrifuged at 500g, 25°C for 6 minutes. The supernatant was discarded, and the pellet resuspended in a solution of 70% Percoll in PBS, followed by the gentle addition of 50% Percoll, 30% Percoll, and PBS. The gradient was centrifuged at 2000g, 25°C for 20 minutes, and the microglia were collected from the interface between the 70% and 50% Percoll layers. The cells were washed with PBS at 1500 rpm, 4°C for 10 minutes, followed by the addition of 1% BSA in PBS to resuspend the pellet. Microglia (3×104 cells) were stained with FITC‐CD11b (1:50 dilution, Abcam™) and PE‐CD45 (1:20 dilution, BD Pharmigen) antibodies for 30 minutes at 4°C, and the % CD11b+, %CD45+ cells were counted by flow cytometry (FACS Celesta, BD Biosciences, California).
Identification of Brain Microglia Using Confocal Laser Scanning Microscopy
The lower left hemispheres of the brains were incubated with 4% paraformaldehyde for 1 week at room temperature, then moved into a 30% sucrose solution and kept at 4°C until further analysis. Brain slices of 20‐μm thickness were cut by cryosectioning and incubated with 3% peroxide for 1 hour, followed by an overnight incubation with a solution of BSA and primary antibody against Iba‐1 (Abcam, Cambridge, UK), glial fibrillary acidic protein (Abcam, Cambridge, UK), and DAPI (TOCRIS, Bristol, UK). PBS was used to wash the brain slices clear of unbound primary antibody, then AlexaFluor 488 anti‐goat Iba1 and AlexaFluor 647 anti‐rabbit glial fibrillary acidic protein conjugated secondary antibodies were incubated with the samples for 1 hour. A FV3000 confocal laser scanning microscope (Olympus Corp., Tokyo, Japan) was used to acquire images of the brain slices every 0.5 μm in the z‐plane. The images were analyzed using Imaris Image Analysis Software (Bitplane, Belfast, Northern Ireland).
Immunoblotting
Protein was extracted from the brain tissue, and 40 μg of the tissue lysate was loaded onto on a 10% SDS–polyacrylamide gel and then transferred onto nitrocellulose membranes. The membranes were blocked for 1 hour with 5% nonfat dry milk in Tris‐buffered saline (pH 7.4) containing 0.1% Tween 20, then incubated overnight at 4°C with primary antibodies against amyloid beta precursor protein (APP) (1:1000 dilution, Cell Signaling Technology), Aβ (1:1000 dilution, Cell Signaling Technology), PCSK9 (1:1000 dilution, Abcam), p‐NFκB (1:1000 dilution, Cell Signaling Technology), NFκB (1:1000 dilution, Cell Signaling Technology), Bax (1:1000 dilution, Abcam), Bcl2 (1:1000 dilution, Abcam), and a loading control, β‐actin (1:2000 dilution, Santa Cruz Biotechnology). All immunoblots were incubated with a horseradish peroxidase conjugated anti‐rabbit secondary antibody for 1 hour. The membranes were exposed to ECL Western blotting substrate, and densitometric analysis was carried out using a ChemiDoc Touch Imaging system (Bio‐Rad Laboratories, California). The Western blot bands were analyzed using Image J (NIH image) analysis software.
Golgi Impregnation and Analysis
Dendritic spine density was determined using a commercially available kit (FD Neurotechnologies kit, PK 401, Ellicott City) as described previously.6 A IX‐81 microscope (Olympus, Tokyo, Japan) was used for the analysis of dendritic spine density. Tertiary dendrites of 3 neurons in the CA1 hippocampus area were randomly chosen and the 20‐nm area from the apical end of the dendrite was used for the dendritic spine count using Xcellence software (Olympus, Tokyo, Japan).
Terminal Deoxynucleotidyl Nick‐End Labeling Staining and Analysis
Brain apoptosis was determined using terminal deoxynucleotidyl nick‐end labeling (TUNEL)–positive cells (Roche, Basel, Switzerland). For in situ labeling, the brain slices were placed in PBS for 10 minutes after dehydration, then they were covered with 50 μL of Proteinase k solution (1:50) for 30 minutes followed by 50 μL of Cytonin for 120 minutes. For positive control, the samples were covered with TACS nuclease 1:50 in TACS nuclease buffer. TUNEL‐positive cells were detected with a fluorescence microscope (Nikon, Tokyo, Japan) at λex 494 nm and λem 512 nm. DAPI was detected at λex 358 nm and λem 461 nm. The apoptosis index was calculated as a percentage of TUNEL‐positive apoptotic cells over the total number of nucleated cells (DAPI staining).
Statistical Analysis
Data from each experiment are expressed as mean±SEM and processed using GraphPad Prism software (version 7, GraphPad Software, Inc, California). Data were analyzed using a 1‐way ANOVA followed by post hoc Tukey's analysis or unpaired t test, with 2‐tailed test. A P value of <0.05 was considered statistically significant.
Results
PCSK9 Inhibition Reduced Both the Number of CD11b+/CD45high Microglia and Microglial/Astrocytic Hyperactivity in Response to Cardiac I/R Injury
Microglia activity and morphology were determined by flow cytometry and confocal microscopy, respectively. Data from the flow cytometry showed that the number of CD11b+/CD45+low microglia in the brains of rats with I/R injury just with the vehicle was significantly reduced (P<0.05), when compared with that of the sham group (Figure 2A). However, the number of CD11b+/CD45+high microglia in the brains of rats with I/R injury with vehicle was significantly increased (P<0.05), when compared with that of the sham group (Figure 2B). Treatment with the PCSK9i did not affect the number of CD11b+/CD45+low (Figure 2A), but it significantly restored the number of CD11b+/CD45high microglia (P<0.01) in all treatment groups to the level of the sham group (Figure 2B).
Figure 2.
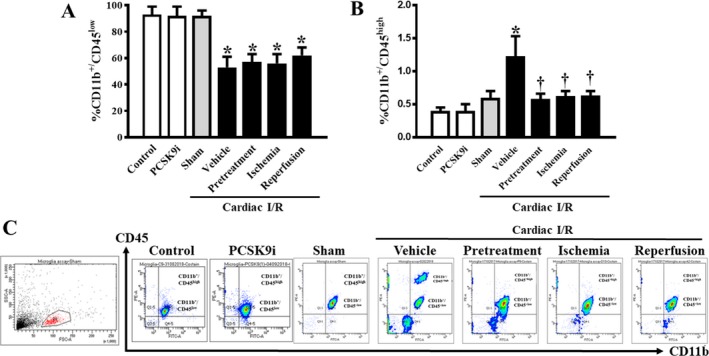
Effects of PCSK9 inhibition on microglial count after cardiac I/R injury. A, Percentage of CD11b+/CD45+ microglia of the brains. B, Percentage of CD11b+/CD45+high microglia of the brains. C, Representative flow cytometry images of microglial count. I/R, ischemia/reperfusion; CD, cluster of differentiation; PCSK9i, proprotein convertase subtilisin/kexin type 9 inhibitor; Control, rat group with no surgical intervention; PCSK9i, rats group with no intervention and treated with PCSK9i for 180 minutes; Sham, rat group with open chest surgery; Vehicle, rats treated with vehicle at 15 minutes before cardiac I/R; Pretreatment, rats treated with PCSK9i at 15 minutes before cardiac I/R; Ischemia, rats treated with PCSK9i at 15 minutes during ischemic period; Reperfusion, rats treated with PCSK9i at the onset of reperfusion period (n=4–10 per group). *P<0.05 vs control, PCSK9i, and sham, † P<0.05 vs vehicle.
In the rats without cardiac I/R, PCSK9i administration did not affect the levels of CD11b+/CD45+low and CD11b+/CD45+high, compared with control rats (Figure 2A and 2B). The representative pictures of CD11b+ and CD45+ cells by flow cytometry are shown in Figure 2C. These findings suggested that cardiac I/R increased microglia activity, as indicated by a decrease in CD11b+/CD45+low microglia as well as an increase in CD11b+/CD45high microglia. It was also found that PCSK9i administration before cardiac I/R, during ischemia, and at the onset of reperfusion led to similarly reduced microglia hyperactivity.
The microglia morphology was determined using confocal microscopy. The phenotype of microglia was established using Iba1 staining, which allowed the measurement of the dendrite volume and length, the number of Iba‐1‐positive cells, and the complexity of the processes, by using the area under the curve of Sholl analysis. Microglia morphology was determined from 3 areas from the CA1 region per brain slice, with 3 brain slices per animal. Our data demonstrated that, in rats with cardiac I/R injury and vehicle treatment, the number of Iba‐1 positive (P<0.001) cells and microglial dendrite volume (P<0.05) were significantly increased, while filament length (P=0.0327) and dendrite complexity significantly decreased (P<0.01), when compared with those of the sham group (Figure 3B through 3F). Interestingly, pretreatment with PCSK9i restored all aforementioned parameters to levels comparable to the sham group (Figure 3B through 3F). However, PCSK9i treatment during cardiac ischemia and at the onset of reperfusion did not exert any effects on microglial activity or morphology (Figure 3B through 3F). In the rats without cardiac I/R, PCSK9i administration did not alter the number of Iba‐1 positive cells, microglial dendrite volume, and filament length, compared with control rats (Figure 3B through 3F). These findings suggest that cardiac I/R injury could induce microglia activation, and only pretreatment with PCSK9i restored microglia activity to the basal levels of the sham group.
Figure 3.
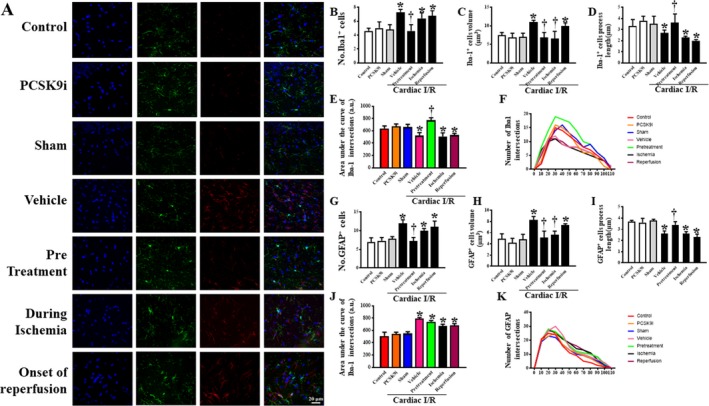
Effects of PCSK9 inhibition on microglial and astrocytic morphology after cardiac I/R injury. A, Representative confocal microscopy images of microglia (Iba1), astrocytes (GFAP), neuronal nuclei (DAPI), and all merged. Statistical analysis for the microglia fraction including: B, number of Iba1‐positive cells (units); C, dendrite volume (μm3); D, Iba1 filament dendrite length (μm); E, dendritic complexity as area under the curve of the Sholl analysis; F, dendritic complexity as slope of Sholl analysis; G, number of GFAP–positive cells (units); H, dendrite volume (μm3); I, GFAP filament dendrite length (μm); J, dendritic complexity as area under the curve of the Sholl analysis; K, dendritic complexity as slope of Sholl analysis. I/R, ischemia/reperfusion; Iba1, ionized calcium‐binding adapter molecule 1; GFAP, glial fibrillary acidic protein; PCSK9i, proprotein convertase subtilisin/kexin type 9 inhibitor; Control, rat group with no surgical intervention; PCSK9i, rats group with no intervention and treated with PCSK9i for 180 minutes; Sham, rat group with no surgical intervention/treatment; Vehicle, rats treated with vehicle at 15 minutes before cardiac I/R; Pretreatment, rats treated with PCSK9i at 15 minutes before cardiac I/R; Ischemia, rats treated with PCSK9i at 15 minutes during ischemic period; Reperfusion, rats treated with PCSK9i at the onset of reperfusion period (n=4–10 per group). *P<0.05 vs sham, † P<0.05 vs vehicle.
Astrocyte morphology and activity were also analyzed by confocal microscopy. Data showed that rats with cardiac I/R injury and vehicle treatment had increased numbers of glial fibrillary acidic protein–positive cells (P<0.05), astrocytic dendrite volume (P<0.05), and complexity (P<0.01), and decreased filament length (P<0.05) (Figure 3G through 3K), when compared with those of the sham group. Pretreatment with PCSK9i led to the restoration of cell number (P<0.05), astrocytic cell volume (P<0.01), and an increase in filament length (P<0.01), when compared with those of rats with cardiac I/R injury and vehicle treatment (Figure 3G through 3K). PCSK9i treatment during ischemia led to restored dendrite volume (P<0.01) and increased filament length, when compared with those of rats with cardiac I/R injury and vehicle treatment (Figure 3H and 3I). PCSK9i treatment at the onset of reperfusion only restored the length of processes (P<0.01), when compared with those of rats with cardiac I/R injury and vehicle treatment (Figure 3I). However, none of the treatments affected dendrite complexity following cardiac I/R injury (Figure 3J). Similar to the microglial morphology, PCSK9i administration did not alter the number of astrocytic‐positive cells, astrocytic dendrite volume, filament length, and complexity compared with control rats (Figure 3G through 3K). The representative pictures from confocal microscopy of microglia and astrocytes are shown in Figure 3A. These findings suggested that cardiac I/R injury increased astrocytic activation, and pretreatment with PCSK9i provided greater efficacy in reducing astrocytic activation following cardiac I/R than the treatment in the other groups.
PCSK9 Inhibition Reduced the Expression of Aβ and Ameliorated the Reduction of Dendritic Spine Density in Response to Cardiac I/R Injury
One phenomenon of neurodegeneration can be characterized by amyloid plaque formation, which was assessed in these experiments as the expression of Aβ and APP protein levels. In the brains of rats with cardiac I/R injury and vehicle treatment, Aβ was found to be significantly increased by ≈2.4‐fold (P<0.001), when compared with that of the sham group (Figure 4A). Interestingly, treatment with PCSK9i at all time points of cardiac I/R injury attenuated the protein levels of Aβ to the basal level of the sham group (P<0.05). However, the level of APP, the precursor protein of Aβ, was found to be at the same level across all groups (Figure 4B). We also calculated the ratio of Aβ to the APP, and our data showed that the ratio of Aβ/APP was increased in rats with cardiac I/R injury and vehicle treatment, when compared with that of the sham group (Figure 4C). Treatment with PCSK9i at all time points of cardiac I/R reduced the ratio of Aβ/APP, when compared with that of rats with cardiac I/R injury and vehicle treatment (Figure 4C). However, the administration of PCSK9i without cardiac I/R did not alter the levels of Aβ, APP, and Aβ/APP ratio, compared with control rats (Figure 4A through 4C). These findings suggested that cardiac I/R injury induced Aβ aggregation, and PCSK9i effectively reduced brain Aβ levels in rats with cardiac I/R injury.
Figure 4.
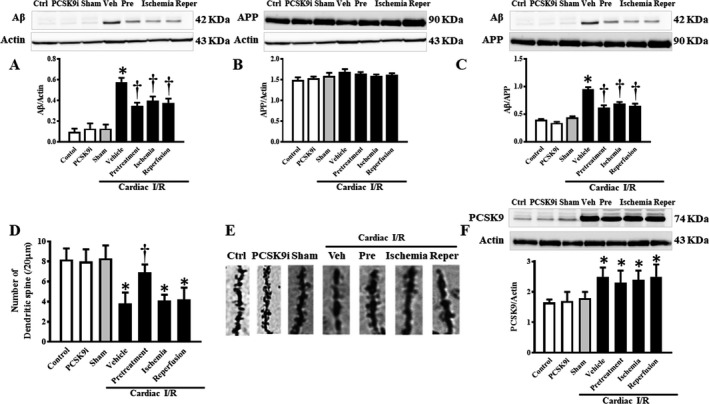
Effects of PCSK9 inhibition on Aβ and APP protein expression levels and dendritic spine density after cardiac I/R injury. A, Protein levels of Aβ established by Western blotting, normalized to actin. B, Protein levels of APP by Western blotting, normalized to actin. C, Protein levels of Aβ by Western blotting, normalized to APP. D, Dendritic spine density per 20 μm apical tertiary dendrite; E, Representative images of dendritic spines. F, PCSK9 levels by Western blotting, normalized to actin. Aβ, amyloid beta; I/R, ischemia/reperfusion; Veh, vehicle; Pre, pretreatment; Reper, reperfusion; PCSK9, Proprotein convertase subtilisin/kexin type 9; PCSK9i, proprotein convertase subtilisin/kexin type 9 inhibitor; Control, rat group with no surgical intervention; PCSK9i, rats group with no intervention and treated with PCSK9i for 180 minutes; Sham, rat group with no surgical intervention/treatment; Vehicle, rats treated with vehicle at 15 minutes before cardiac I/R; Pretreatment, rats treated with PCSK9i at 15 minutes before cardiac I/R; Ischemia, rats treated with PCSK9i at 15 minutes during ischemic period; Reperfusion, rats treated with PCSK9i at the onset of reperfusion period (n=4–10 per group). *P<0.05 vs sham, † P<0.05 vs vehicle.
Dendritic spine density was assessed on 3 tertiary branches of randomly chosen CA1 hippocampal neurons. The apical 20‐nm section of the dendrites was chosen. Our results showed that the dendritic spine count of rats with cardiac I/R injury and vehicle treatment was decreased by ≈2‐fold (P<0.01), when compared with that of the sham group (Figure 4D and 4E). Only pretreatment with PCSK9i increased dendritic spine density by ≈1.6‐fold (P<0.01) with respect to vehicle treatment (Figure 4D and 4E). However, treatment with PCSK9i during cardiac ischemia and at the onset of reperfusion did not lead to an increase in the number of dendritic spines. In rats without cardiac I/R, PCSK9i did not affect the levels of dendritic spine density, compared with control rats (Figure 4D and 4E). These findings suggested that although treatment with PCSK9i at all experimental time points of cardiac I/R injury reduced Aβ aggregation, only pretreatment with PCSK9i increased dendritic spine density in rats with cardiac I/R injury.
The levels of PCSK9 in the brain were determined using an immunoblot to determine the direct effects of PCSK9i on the brain with and without cardiac I/R injury. PCSK9i did not decrease brain PCSK9 protein levels in rats without cardiac I/R. The levels of PCSK9 were increased in rats with cardiac I/R injury and vehicle treatment, when compared with that of the sham group (Figure 4F). However, we found some unexpected results in that treatment with PCSK9i at any time point could not reduce PCSK9 levels in the brain (Figure 4F).
PCSK9 Inhibition Reduced Brain Inflammation, But Did Not Reduce Apoptosis in Response to Cardiac I/R Injury
Data from the Western blot analysis showed that the levels of p‐NFκB/NFκB were increased following cardiac I/R, and PCSK9i effectively reduced the levels of p‐NFκB/NFκB (P<0.05). In rats without cardiac I/R, PCSK9i administration did not alter the levels of p‐NFκB/NFκB (Figure 5A).
Figure 5.
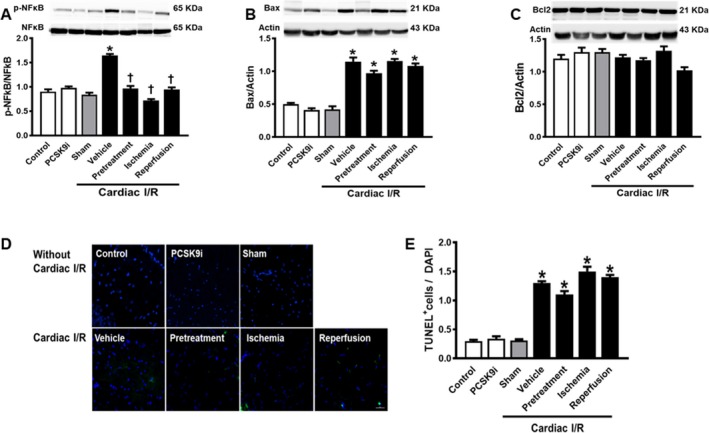
Effects of PCSK9 inhibition on brain inflammation and apoptosis. A, p‐NFκB/NFκB established by Western blotting. B, Protein levels of Bax by Western blotting, normalized to actin. C, Protein levels of Bax by Western blotting, normalized to actin. D, Representative picture of TUNEL. E, TUNEL + cells/DAPI. I/R, ischemia/reperfusion; Veh, vehicle; Pre, pretreatment; Reper, reperfusion; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling; PCSK9i, Proprotein convertase subtilisin/kexin type 9 inhibitor; Control, rat group with no surgical intervention; PCSK9i, rats group with no intervention and treated with PCSK9i for 180 minutes; Sham, rat group with no surgical intervention/treatment; Vehicle, rats treated with vehicle at 15 minutes before cardiac I/R; Pretreatment, rats treated with PCSK9i at 15 minutes before cardiac I/R; Ischemia, rats treated with PCSK9i at 15 minutes during ischemic period; Reperfusion, rats treated with PCSK9i at the onset of reperfusion period (*P<0.05 vs. sham, † P<0.05 vs. vehicle. n=4–10 per group).
In brain of rats with cardiac I/R, Bax expression was increased, along with an increased %TUNEL‐positive cells (P<0.05) (Figure 5B and 5D). However, PCSK9i administration did not alter the levels of Bax and %TUNEL‐positive cells following cardiac I/R (Figure 5B, 5D, and 5E). In brain of rats without cardiac I/R, PCSK9i administration did not alter Bax and %TUNEL‐positive cells (Figure 5B, 5D, and 5E). For Bcl2 protein expression, the level of Bcl2 was not affected by both cardiac I/R and PCSK9i administration (Figure 5C). These data suggested that a single dose of PCSK9i administration did not do enough to reduce brain apoptosis following cardiac I/R.
Discussion
In this study, our results demonstrated an association between PCSK9 inhibition and the brain of rats with cardiac I/R injury. Our findings showed that cardiac I/R caused a decreased number of CD11b+/CD45+low and increased CD11b+/CD45+high microglia, increased the activity of microglia and astrocytes, increased Aβ production, and decreased dendritic spine density. Pretreatment with PCSK9i was the most effective regimen to protect the brain following cardiac I/R injury because it reduced CD11b+/CD45+high cell numbers, microglial and astrocytic activation, Aβ production, and attenuated the reduction of dendritic spine density. Under physiological condition, PCSK9i did not exert favorable outcomes on microglial and astrocytic activity, Aβ levels, and dendritic spine density. It is interesting that these favorable effects of PCSK9i were found to be independent of the PCSK9 levels.
Recent research has shown that PCSK9 is involved in inflammation and apoptosis. This is in addition to its previously well‐established role in lipid homeostasis.10, 11, 12 A previous study has shown that PCSK9 expression is stimulated in response to inflammation and stress23 and PCSK9 siRNA is able to reduce inflammation by inhibiting NF‐kB activation.10 In the brain, the primary responders to neuro‐inflammation are microglia,24 which are now believed to be involved in the secondary injury of the brain following ischemic insults.25 Physiologically, microglia are adapted for scanning the neuronal environment in long and complex processes. Upon the detection of injury, they undergo progressive morphological and functional changes that allow them to acquire the inflammatory harmful M1 phenotype, or beneficial M2 phenotype.24, 26, 27 Our results are substantiated by other literature, as microglial Iba1+ processes became shorter, less complex, and thicker in response to cardiac I/R injury, suggesting that microglia tending towards an ameboid shape or M1 phenotype indicate the activation and the acquisition of phagocytic properties.27 The number of Iba1+ cells was also significantly increased, representing a hallmark of inflammation in the brain.28, 29 Moreover, microglia are the cells in the brain that express CD11b, and CD11b is the most frequently used marker of microglia, and can be detected by flow cytometry.30 CD45 is a surface marker of monocytes and is used together with CD11b to determine the function of microglia. Previous studies have shown that microglia expressed CD11b+ with CD45+low during their steady state,31, 32 and CD45 is expressed at a high intensity during brain injury and inflammation.32 There are studies suggesting that CD11b+/CD45high are found to be associated with the M1 phenotype of microglia.30, 32 According to the role of CD45high in an inflammatory response, CD45high is reported to relate to macrophages infiltrating from the periphery.33, 34 Pertinent to this, our flow cytometry analysis revealed that the numbers of CD11b+/CD45+high were increased, and the numbers of CD11b+/CD45+low were decreased following cardiac I/R. Interestingly, the effect of PCSK9i on microglia has never been studied. Our findings have shown that pretreatment with PCSK9i reduced all aforementioned parameters of microglial activation. Therefore, this may suggest a role for the PCSK9 inhibitor in reducing the pro‐inflammatory wave by preventing CD11b+/CD45+high cell infiltration, which is associated with the M1 phenotype of microglia.
The other important cell type intimately linked to neuronal injury is the astrocyte.35 Similarly to microglia, astrocytes may exist in 2 very different phenotypes, differentiated by their pro‐survival or destructive properties.36 Reactive astrocytosis begins immediately after the ischemic insult and is characterized by intense proliferation, increased glial fibrillary acidic proteins expression, hypertrophy, and secretion of pro‐inflammatory factors.35, 37, 38 In accordance with this, our results show thicker, longer, and more complex astrocytic processes, supporting the hypertrophic phenotype of astrocytes. In addition, intense proliferation was observed. The effect of PCSK9i on astrocytes following cardiac I/R injury or any other type of brain insult has not yet been investigated. Our study showed that PCSK9i managed to attenuate both the proliferation and hypertrophy of astrocytes, but not the complexity of process. The effect of PCSK9i on astrocytes in this study suggested that PCSK9i might inhibit the destructive effect of astrocytes following cardiac I/R injury. However, only a few studies have investigated the protective/destructive effects of reactive astrocytosis and it is not thoroughly understood which morphology of astrocytes is likely to be present during the inactivated stages.35, 36
Neuronal axons and dendrites are surrounded by structures such as the myelin sheath with a high cholesterol composition.39 Studies have shown that PCSK9 monoclonal antibodies reduce circulating cholesterol by ≈60%, in which high cholesterol level has been the major risk factor for the development of neurodegenerative diseases such as Alzheimer disease.16, 40 Thus, the reduction of cholesterol should be beneficial for reducing the impact of Alzheimer disease. However, there has been concern that lowering circulating cholesterol levels has been implicated in Parkinson disease as well as other forms of dementia.41 Previous studies have shown that Aβ level is prone to increase in response to cardiac I/R injury.4, 6 Although there was a significant reduction in Aβ with the onset of all PCSK9i treatments, these findings could not imply a direct effect of PCSK9 on the levels of Aβ. The reasons were that contradictory findings of the association of PCSK9 and Aβ production have been found. Courtemanche and colleagues proposed a positive relationship between PCSK9 and Aβ production42; however, Liu and colleagues did not support any correlations between PCSK9 and APP or Aβ.12
PCSK9 has also been implicated in the potential healthy development of the nervous system as PCSK9 knockout in zebrafish resulted in lethality of the embryos.43 Moreover, PCSK9 levels were found to be very elevated during the developmental stages of mouse embryos.44 Those findings suggest that PCSK9 might be involved in neuronal development and plasticity. However, it is pertinent to this investigation that dendritic spines, which are protrusions involved in neuroplasticity, have been shown to be destabilized by ischemic insults.45 This was confirmed by the significant decrease in dendritic spine density following cardiac I/R in our findings. The dendritic spines decreased as a response to the reduction of synaptic activity following an inadequate energy supply.46 Nonetheless, microglia are normally involved in dendritic spine density maintenance through pruning or growth stimulation by brain‐derived neurotrophic factor secretion. During reactive gliosis, microglia secrete IL‐1β,47, 48 which possibly led to a decline in spine density in the present study. Moreover, increased Aβ levels are able to induce microglial activation and loss of dendritic spines.49 Although the effect of PCSK9i on dendritic spines following ischemic insults has not been studied, we could speculate that PCSK9i might attenuate the reduction of dendritic spine density by decreasing microglial activation and/or Aβ production.
It is interesting that PCSK9i did not reduce PCSK9 levels in the brain following cardiac I/R injury; however, pretreatment with PCSK9i treatment effectively enhanced dendritic spine density, possibly via decreasing neuro‐inflammation and Aβ aggregation in the brain. Therefore, these favorable effects of PCSK9i might be driven as a result of the modulation of systemic inflammation. Our finding was supported by a previous recent study, which showed that PCSK9 inhibition reduced the activation of immune cells in human peripheral blood monocytes, leading to a decrease in pro‐inflammatory cytokine production including tumor necrosis factor‐α, IL‐1β, and IL‐6. Also, this anti‐inflammatory effect is independent of the lipid‐lowering effect.50 Therefore, our findings suggest that PCSK9i mainly reduced the activity of brain innate immunity, and acted neuroprotectively. We propose that PCSK9i acts via reducing systemic inflammation. The proposed underlying mechanism of PCSKi on the brain following cardiac I/R injury is shown in Figure 6. In addition, PCSK9i acute injection during cardiac ischemia and at the onset of reperfusion is ineffective, invalidating the idea that postischemic intervention with PCSK9i would be clinically interesting.
Figure 6.
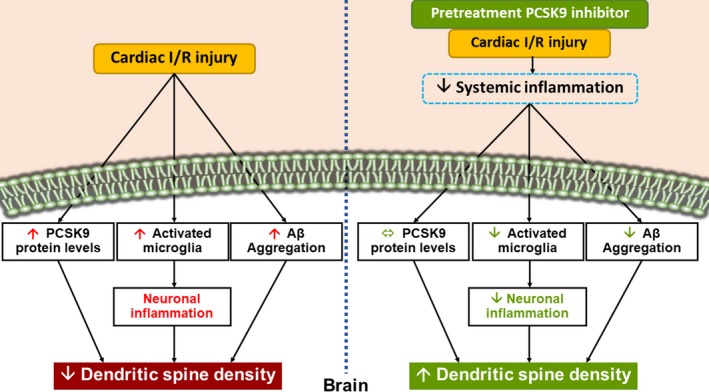
A summary diagram showing the possible effects of PCSK9 inhibition on the brain following cardiac I/R injury. During cardiac I/R injury, the brain PCSK9 levels had increased, along with neuronal inflammation, and Aβ aggregation, leading to a reduction in dendritic spine density. Treatment with PCSK9i increased dendritic spine density by reducing neuronal inflammation and Aβ aggregation, but it did not affect the brain PCSK9 levels. Thus, we proposed that the beneficial effects of PCSK9i on the brain were possibly because of a reduction of systemic inflammation during cardiac I/R injury. Aβ indicates amyloid beta; I/R, ischemia/reperfusion; PCSK9i, proprotein convertase subtilisin/kexin type 9 inhibitor.
Study Limitations
Regarding the lack of benefit when acute PCSK9i administration was done during ischemia and reperfusion, these findings may not be clinically useful. However, there are at least 2 points obtained from this study that could encourage the potential benefits of using PCSK9i in cardiac I/R condition. First, PCSK9i has been used as a antihyperlipidemic drug. Therefore, our results provided some good news that those who use PCSK9i for their treatment could have some degree of neuroprotective benefits when acute MI occurs. However, the present study used only a single dose of PCSK9i. It is possible that during ischemia and at onset of reperfusion, a higher dose is required for its potential neuroprotection. Future studies are needed to elucidate this hypothesis.
Conclusion
The PCSK9i reduced brain damage following cardiac I/R injury as indicated by inhibition of microglial/astrocyte hyperactivity, a decrease in Aβ aggregation, and an increase in dendritic spine density. This study suggests the possible therapeutic advantages of the use of PCSK9i for hyperlipidemic individuals and a potential new strategy for the improvement of neurological outcomes following cardiovascular events.
Sources of Funding
This work was supported by the Thailand Research Fund grants: RTA6080003 (S.C. Chattipakorn), RSA6080056 (Palee), TRG6080005 (Apaijai), the Royal Golden Jubilee Program PHD/0111/2559 (Saiyasit and S.C. Chattipakorn), PHD/144/2558 (Maneechote and N. Chattipakorn), the NSTDA Research Chair grant from the National Science and Technology Development Agency Thailand (N. Chattipakorn), and the Chiang Mai University Center of Excellence Award (N. Chattipakorn).
Disclosures
None.
(J Am Heart Assoc. 2019;8:e010838 DOI: 10.1161/JAHA.118.010838)
References
- 1. Palmerini T, Tomasi L, Barozzi C, Della Riva D, Mariani A, Taglieri N, Leone O, Ceccarelli C, De Servi S, Branzi A, Genereux P, Stone GW, Ahamed J. Detection of tissue factor antigen and coagulation activity in coronary artery thrombi isolated from patients with ST‐segment elevation acute myocardial infarction. PLoS One. 2013;8:e81501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ramirez‐Sanchez J, Pires ENS, Meneghetti A, Hansel G, Nunez‐Figueredo Y, Pardo‐Andreu GL, Ochoa‐Rodríguez E, Verdecia‐Reyes Y, Delgado‐Hernández R, Salbego C, Souza DO. JM‐20 treatment after MCAO reduced astrocyte reactivity and neuronal death on peri‐infarct regions of the rat brain. Mol Neurobiol. 2018. Available at: https://link.springer.com/article/10.1007%2Fs12035-018-1087-8. Accessed January 3, 2019. [DOI] [PubMed] [Google Scholar]
- 3. Ibanez B, James S, Agewall S, Antunes MJ, Bucciarelli‐Ducci C, Bueno H, Caforio ALP, Crea F, Goudevenos JA, Halvorsen S, Hindricks G, Kastrati A, Lenzen MJ, Prescott E, Roffi M, Valgimigli M, Varenhorst C, Vranckx P, Widimský P. 2017 ESC guidelines for the management of acute myocardial infarction in patients presenting with ST‐segment elevation. Rev Esp Cardiol (Engl Ed). 2017;70:1082. [DOI] [PubMed] [Google Scholar]
- 4. Kumfu S, Charununtakorn ST, Jaiwongkam T, Chattipakorn N, Chattipakorn SC. Humanin prevents brain mitochondrial dysfunction in a cardiac ischaemia‐reperfusion injury model. Exp Physiol. 2016;101:697–707. [DOI] [PubMed] [Google Scholar]
- 5. Nour M, Scalzo F, Liebeskind DS. Ischemia‐reperfusion injury in stroke. Interv Neurol. 2013;1:185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pratchayasakul W, Sivasinprasasn S, Sa‐Nguanmoo P, Proctor C, Kerdphoo S, Chattipakorn N, Chattipakorn SC. Estrogen and DPP‐4 inhibitor share similar efficacy in reducing brain pathology caused by cardiac ischemia‐reperfusion injury in both lean and obese estrogen‐deprived rats. Menopause. 2017;24:850–858. [DOI] [PubMed] [Google Scholar]
- 7. Evonuk KS, Prabhu SD, Young ME, DeSilva TM. Myocardial ischemia/reperfusion impairs neurogenesis and hippocampal‐dependent learning and memory. Brain Behav Immun. 2017;61:266–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhao XS, Wu Q, Peng J, Pan LH, Ren Z, Liu HT, Jiang ZS, Wang GX, Tang ZH, Liu LS. Hyperlipidemia‐induced apoptosis of hippocampal neurons in apoE(‐/‐) mice may be associated with increased PCSK9 expression. Mol Med Rep. 2017;15:712–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bingham B, Shen R, Kotnis S, Lo CF, Ozenberger BA, Ghosh N, Kennedy JD, Jacobsen JS, Grenier JM, DiStefano PS, Chiang LW, Wood A. Proapoptotic effects of NARC 1 (= PCSK9), the gene encoding a novel serine proteinase. Cytometry. 2006;69:1123–1131. [DOI] [PubMed] [Google Scholar]
- 10. Tang Z, Jiang L, Peng J, Ren Z, Wei D, Wu C, Pan L, Jiang Z, Liu L. PCSK9 siRNA suppresses the inflammatory response induced by oxLDL through inhibition of NF‐kappaB activation in THP‐1‐derived macrophages. Int J Mol Med. 2012;30:931–938. [DOI] [PubMed] [Google Scholar]
- 11. Kysenius K, Muggalla P, Matlik K, Arumae U, Huttunen HJ. PCSK9 regulates neuronal apoptosis by adjusting ApoER2 levels and signaling. Cell Mol Life Sci. 2012;69:1903–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu LS, Bai XQ, Gao Y, Wu Q, Ren Z, Li Q, Pan LH, He NY, Peng J, Tang ZH. PCSK9 promotes oxLDL‐induced PC12 cell apoptosis through the Bcl‐2/Bax‐caspase 9/3 signaling pathway. J Alzheimers Dis. 2017;57:723–734. [DOI] [PubMed] [Google Scholar]
- 13. Wu Q, Tang ZH, Peng J, Liao L, Pan LH, Wu CY, Jiang ZS, Wang GX, Liu LS. The dual behavior of PCSK9 in the regulation of apoptosis is crucial in Alzheimer's disease progression (review). Biomed Rep. 2014;2:167–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Piao MX, Bai JW, Zhang PF, Zhang YZ. PCSK9 regulates apoptosis in human neuroglioma u251 cells via mitochondrial signaling pathways. Int J Clin Exp Pathol. 2015;8:2787–2794. [PMC free article] [PubMed] [Google Scholar]
- 15. Stoekenbroek RM, Kastelein JJP. Proprotein convertase subtilisin/kexin type 9: from genetics to clinical trials. Curr Opin Cardiol. 2018;33:269–275. [DOI] [PubMed] [Google Scholar]
- 16. Koren MJ, Lundqvist P, Bolognese M, Neutel JM, Monsalvo ML, Yang J, Kim JB, Scott R, Wasserman SM, Bays H; MENDEL‐2 Investigators . Anti‐PCSK9 monotherapy for hypercholesterolemia: the MENDEL‐2 randomized, controlled phase III clinical trial of evolocumab. J Am Coll Cardiol. 2014;63:2531–2540. [DOI] [PubMed] [Google Scholar]
- 17. Giugliano RP, Pedersen TR, Park JG, De Ferrari GM, Gaciong ZA, Ceska R, Toth K, Gouni‐Berthold I, Lopez‐Miranda J, Schiele F, Mach F, Ott BR, Kanevsky E, Pineda AL, Somaratne R, Wasserman SM, Keech AC, Sever PS, Sabatine MS; FOURIER Investigators . Clinical efficacy and safety of achieving very low LDL‐cholesterol concentrations with the PCSK9 inhibitor evolocumab: a prespecified secondary analysis of the Fourier trial. Lancet. 2017;390:1962–1971. [DOI] [PubMed] [Google Scholar]
- 18. Kereiakes DJ, Robinson JG, Cannon CP, Lorenzato C, Pordy R, Chaudhari U, Colhoun HM. Efficacy and safety of the proprotein convertase subtilisin/kexin type 9 inhibitor alirocumab among high cardiovascular risk patients on maximally tolerated statin therapy: the ODYSSEY COMBO I study. Am Heart J. 2015;169:906–915.e913. [DOI] [PubMed] [Google Scholar]
- 19. Robinson JG, Farnier M, Krempf M, Bergeron J, Luc G, Averna M, Stroes ES, Langslet G, Raal FJ, El Shahawy M, Koren MJ, Lepor NE, Lorenzato C, Pordy R, Chaudhari U, Kastelein JJ; ODYSSEY LONG TERM Investigators . Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372:1489–1499. [DOI] [PubMed] [Google Scholar]
- 20. Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, Kuder JF, Wang H, Liu T, Wasserman SM, Sever PS, Pedersen TR; FOURIER Steering Committee and Investigators . Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376:1713–1722. [DOI] [PubMed] [Google Scholar]
- 21. Ninomiya JK, L'Italien G, Criqui MH, Whyte JL, Gamst A, Chen RS. Association of the metabolic syndrome with history of myocardial infarction and stroke in the third National Health and Nutrition Examination Survey. Circulation. 2004;109:42–46. [DOI] [PubMed] [Google Scholar]
- 22. Palee S, Weerateerangkul P, Chinda K, Chattipakorn SC, Chattipakorn N. Mechanisms responsible for beneficial and adverse effects of rosiglitazone in a rat model of acute cardiac ischaemia‐reperfusion. Exp Physiol. 2013;98:1028–1037. [DOI] [PubMed] [Google Scholar]
- 23. Feingold KR, Moser AH, Shigenaga JK, Patzek SM, Grunfeld C. Inflammation stimulates the expression of PCSK9. Biochem Biophys Res Commun. 2008;374:341–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Morrison HW, Filosa JA. A quantitative spatiotemporal analysis of microglia morphology during ischemic stroke and reperfusion. J Neuroinflammation. 2013;10:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ritzel RM, Patel AR, Grenier JM, Crapser J, Verma R, Jellison ER, McCullough LD. Functional differences between microglia and monocytes after ischemic stroke. J Neuroinflammation. 2015;12:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Taylor RA, Sansing LH. Microglial responses after ischemic stroke and intracerebral hemorrhage. Clin Dev Immunol. 2013;2013:746068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Benakis C, Garcia‐Bonilla L, Iadecola C, Anrather J. The role of microglia and myeloid immune cells in acute cerebral ischemia. Front Cell Neurosci. 2014;8:461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li W, Viengkhou B, Denyer G, West PK, Campbell IL, Hofer MJ. Microglia have a more extensive and divergent response to interferon‐alpha compared with astrocytes. Glia. 2018;66:2058–2078. [DOI] [PubMed] [Google Scholar]
- 29. Frick T, Springe D, Grandgirard D, Leib SL, Haenggi M. An improved simple rat model for global cerebral ischaemia by induced cardiac arrest. Neurol Res. 2016;38:373–380. [DOI] [PubMed] [Google Scholar]
- 30. Jeong HK, Ji K, Min K, Joe EH. Brain inflammation and microglia: facts and misconceptions. Exp Neurobiol. 2013;22:59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang GX, Li J, Ventura E, Rostami A. Parenchymal microglia of naive adult C57BL/6J mice express high levels of B7.1, B7.2, and MHC class II. Exp Mol Pathol. 2002;73:35–45. [DOI] [PubMed] [Google Scholar]
- 32. Greter M, Lelios I, Croxford AL. Microglia versus myeloid cell nomenclature during brain inflammation. Front Immunol. 2015;6:249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fumagalli S, Perego C, Pischiutta F, Zanier ER, De Simoni MG. The ischemic environment drives microglia and macrophage function. Front Neurol. 2015;6:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gabrusiewicz K, Ellert‐Miklaszewska A, Lipko M, Sielska M, Frankowska M, Kaminska B. Characteristics of the alternative phenotype of microglia/macrophages and its modulation in experimental gliomas. PLoS One. 2011;6:e23902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gleichman AJ, Carmichael ST. Astrocytic therapies for neuronal repair in stroke. Neurosci Lett. 2014;565:47–52. [DOI] [PubMed] [Google Scholar]
- 36. Abeysinghe HC, Bokhari L, Quigley A, Choolani M, Chan J, Dusting GJ, Crook JM, Kobayashi NR, Roulston CL. Pre‐differentiation of human neural stem cells into gabaergic neurons prior to transplant results in greater repopulation of the damaged brain and accelerates functional recovery after transient ischemic stroke. Stem Cell Res Ther. 2015;6:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gliem M, Krammes K, Liaw L, van Rooijen N, Hartung HP, Jander S. Macrophage‐derived osteopontin induces reactive astrocyte polarization and promotes re‐establishment of the blood brain barrier after ischemic stroke. Glia. 2015;63:2198–2207. [DOI] [PubMed] [Google Scholar]
- 38. Feng D, Guo B, Liu G, Wang B, Wang W, Gao G, Qin H, Wu S. FGF2 alleviates PTSD symptoms in rats by restoring GLAST function in astrocytes via the JAK/STAT pathway. Eur Neuropsychopharmacol. 2015;25:1287–1299. [DOI] [PubMed] [Google Scholar]
- 39. Saher G, Quintes S, Nave KA. Cholesterol: a novel regulatory role in myelin formation. Neuroscientist. 2011;17:79–93. [DOI] [PubMed] [Google Scholar]
- 40. Benn M, Nordestgaard BG, Frikke‐Schmidt R, Tybjaerg‐Hansen A. Low LDL cholesterol, PCSK9 and HMGCR genetic variation, and risk of Alzheimer's disease and Parkinson's disease: Mendelian randomisation study. BMJ. 2017;357:j1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Garcia‐Sanz P, Orgaz L, Fuentes JM, Vicario C, Moratalla R. Cholesterol and multilamellar bodies: lysosomal dysfunction in GBA‐Parkinson disease. Autophagy. 2018;14:717–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Courtemanche H, Bigot E, Pichelin M, Guyomarch B, Boutoleau‐Bretonniere C, Le May C, Derkinderen P, Cariou B. PCSK9 concentrations in cerebrospinal fluid are not specifically increased in Alzheimer's disease. J Alzheimers Dis. 2018;62:1519–1525. [DOI] [PubMed] [Google Scholar]
- 43. Poirier S, Prat A, Marcinkiewicz E, Paquin J, Chitramuthu BP, Baranowski D, Cadieux B, Bennett HP, Seidah NG. Implication of the proprotein convertase NARC‐1/PCSK9 in the development of the nervous system. J Neurochem. 2006;98:838–850. [DOI] [PubMed] [Google Scholar]
- 44. Seidah NG, Benjannet S, Wickham L, Marcinkiewicz J, Jasmin SB, Stifani S, Basak A, Prat A, Chretien M. The secretory proprotein convertase neural apoptosis‐regulated convertase 1 (NARC‐1): liver regeneration and neuronal differentiation. Proc Natl Acad Sci USA. 2003;100:928–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Koleske AJ. Molecular mechanisms of dendrite stability. Nat Rev Neurosci. 2013;14:536–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Khatri N, Man HY. Synaptic activity and bioenergy homeostasis: implications in brain trauma and neurodegenerative diseases. Front Neurol. 2013;4:199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chiang CS, Stalder A, Samimi A, Campbell IL. Reactive gliosis as a consequence of interleukin‐6 expression in the brain: studies in transgenic mice. Dev Neurosci. 1994;16:212–221. [DOI] [PubMed] [Google Scholar]
- 48. Dharmarajan S, Fisk DL, Sorenson CM, Sheibani N, Belecky‐Adams TL. Microglia activation is essential for BMP7‐mediated retinal reactive gliosis. J Neuroinflammation. 2017;14:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dorostkar MM, Zou C, Blazquez‐Llorca L, Herms J. Analyzing dendritic spine pathology in Alzheimer's disease: problems and opportunities. Acta Neuropathol. 2015;130:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Liu A, Frostegard J. PCSK9 plays a novel immunological role in oxidized LDL‐induced dendritic cell maturation and activation of T cells from human blood and atherosclerotic plaque. J Intern Med. 2018. Available at: https://onlinelibrary.wiley.com/doi/abs/10.1111/joim.12758. Accessed January 3, 2019. [DOI] [PubMed] [Google Scholar]