

Conspectus
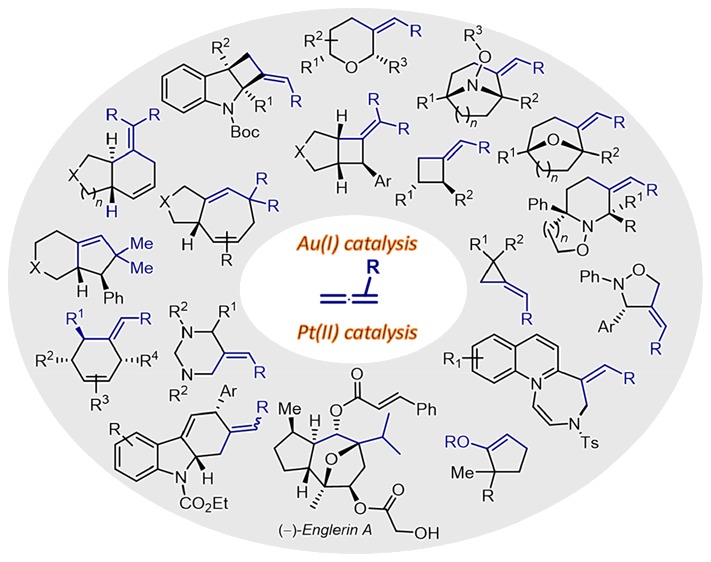
Cycloaddition reactions, by involving the formation of at least two bonds and one cycle in a single operation, represent one of the more practical ways to assemble carbo- and heterocyclic structures from simple acyclic precursors. Especially appealing are formal cycloadditions promoted by transition metals, owing to the ability of these reagents to open mechanisms that are not accessible using classical chemistry. Therefore, along the years, a great variety of annulations based on first-, and particularly second-row transition metals have been discovered. Most of these reactions involve inner sphere mechanisms, with the metal participating via standard oxidative addition or reductive elimination processes. Curiously, metals of the third row like platinum and, especially, gold remained largely unexplored, likely because of the belief that they were inert and expensive. However, from the beginning of this century, many groups realized that these metals can open very interesting mechanistic scenarios and promote novel types of transformations. In particular, the π-acidic, carbophilic behavior of gold(I) complexes, together with the possibility of tuning their reactivity using designed ligands, has triggered important activity in the field. Many gold-catalyzed transformations involved addition or cycloisomerization processes, but during recent years, there have been also important advances in the development of formal cycloaddition reactions. While many of these reactions rely on the activation of alkynes, there has been an increasing number of reports that exploit the peculiar reactivities of allenes and derivatives.
In this Account, we present recent efforts on the development of platinum- and gold-catalyzed formal cycloadditions of allenes. For the sake of simplicity, we only include annulations initiated by a direct metal-promoted activation of the allene moiety. Thus, alternative Pt- or Au-catalyzed reactions wherein the allene does not interact with the metal catalyst are not covered. Upon activation by the metals, allenes generate allyl-cation alkenylmetal species that can behave as 1,2- or 1,3-carbon dipoles in cycloaddition processes. Especially relevant is the reactivity of allenamides. The presence of the amide substituent provides for the generation of gold intermediates with a good balance of reactivity and stability, which can therefore react with the corresponding partners in a controlled manner. Moreover, despite the difficulties associated with the transfer of stereochemical information from chiral linear gold(I) complexes, a variety of enantioselective gold-catalyzed annulations have been discovered.
This Account is organized considering the number of atoms engaged in the annulation process, and when possible, we present the results in a chronological order.
1. Introduction
The last decades have witnessed extraordinary advances in homogeneous transition metal catalysis, mainly using Pd, Ni, Rh, or Ru reagents. Conversely, third row Pt and, especially, Au complexes were largely ignored, in part because they were considered inert.
This situation started to change at the beginning of the century, with the discovery that gold and platinum salts could work as carbophilic catalysts and activate π-bonds of unsaturated substrates.1,2 This type of reactivity is in part associated with relativistic effects, which are particularly pronounced in the case of gold.3 Additional advantages of these metal reagents stem from the possibility of tuning their properties with designed ligands, their low propensity to participate in redox processes, and the simplicity of the associated experimental protocols, which can tolerate air and traces of water.
In 1998, Teles reported that cationic phosphane–gold(I) complexes can catalyze the addition of alcohols to alkynes with high turnover frequencies (Scheme 1A).4 After this seminal contribution, there have been numerous reports dealing with alternative types of inter- and intramolecular additions to alkynes. Mechanistically, it is now well established that coordination of the unsaturated bond of alkynes to these metals triggers their reactivity toward nucleophiles to generate trans-alkenylmetal intermediates (I). These species can be simply trapped with electrophiles to give the corresponding 1,2-trans-functionalized products (Scheme 1B) or, alternatively, can engage in much more complex cycloisomerization and annulation processes.5,6
Scheme 1. Pioneer Work of Teles (A)4 and Typical Reactivity Mode of Alkynes under Pt or Au Catalysis (B).

Alkenes can also coordinate to gold complexes; however, they are kinetically less reactive than alkynes. Even so, during the last years, an important number of gold-catalyzed additions of different types of nucleophiles to nonpolarized alkenes have been developed.7,8
The activation mode of alkynes and alkenes can also be extended to allenes, which can be formally viewed as strained alkenes, featuring two contiguous C–C double bonds. Depending on the substitution pattern of the allene and the type of gold complex used, the intermediate species resulting from the gold-activation of allenes might be either gold-η2-allene complexes (IIa) or related η1-species (IIb–d) in which gold is bound to the former central sp-allenyl carbon. Planar or bent σ-allyl cations of type IIc, as well as zwitterionic carbenes like IId, can be considered as extreme cases, which, nonetheless, help to visualize the dipolar reactivity of allenes under gold catalysis.
Regardless of their precise structure, these gold-intermediate species (II) can then be trapped in a 1,2- or 1,3-manner, depending on the characteristics of the catalyst, nucleophile (Nu), and electrophile (E) (Scheme 2).9−11
Scheme 2. Typical Reactivity Mode of Allenes under Pt or Au Catalysis.

Most Pt- and Au-catalyzed reactions of allenes consist of addition and cycloisomerization processes and usually convey the formation of just one carbon–carbon or carbon–heteroatom bond.12−14 However, the 1,3-dipolar character of η1-metal allyl cation intermediates like IIc or IId suggests the possibility of achieving formal cycloaddition processes in which the allene moiety could work either as a two- or a three-carbon annulation partner.
In this context, in 2007 Toste reported a gold(I)-catalyzed [2 + 2] annulation between allenes and tethered vinyl arenes (Scheme 3A).15 This seminal contribution represented the first use of allenes as two-carbon reaction partners in formal gold-catalyzed cycloadditions. A few months later, our group unveiled a new type of reaction in which allenes behave as a three-carbon partner, namely, an intramolecular [4 + 3] cycloaddition between allenes and 1,3-dienes, in this case promoted by PtCl2 (Scheme 3B).16
Scheme 3. Au(I)-Catalyzed [2 + 2] Cycloadditions of Allenenes,15 and Pt(II)-Catalyzed [4 + 3] Cycloadditions of Allenedienes16.

These two reports stimulated further research to exploit the reactivity of allenes and derivatives in gold- and platinum-catalyzed annulations. In the following sections, we will summarize recent advances in this topic, with special emphasis on the work developed in our laboratory.
2. Intramolecular Reactions
2.1. [2 + 2] Cycloadditions
As indicated, in 2007 Toste demonstrated that the cationic gold complex prepared from Ph3PAuCl and AgBF4 catalyzes the intramolecular [2 + 2] annulation of N- and C-tethered allenenes (1), to give cyclobutene-containing bicycles (2, Scheme 4A).15 The same authors also developed an asymmetric variant, albeit enantioselectivities were quite dependent on the nature of the tether connecting the allene and alkene. Thus, allenenes bearing geminal diesters in the tether underwent highly enantioselective annulations using a DTBM-Segphos-based gold catalyst (Au1/AgBF4),15 whereas allenenes bearing N-tosyl connecting groups provided good ee values only when phosphoramidite–gold catalysts like (R,R,R)-Au2/AgBF4 were employed.17 Additionally, Fürstner showed that a Taddol-based phosphoramidite–gold complex such as (R,R,R,R)-Au3/AgBF4 is also effective for promoting this type of [2 + 2] cycloaddition (Scheme 4B).18
Scheme 4. Gold(I)-Catalyzed [2 + 2] Cycloaddition of Allenenes.

A stepwise process involving the activation of the allene to give a gold-linked allylcation, followed by regioselective addition of the alkene, was proposed. The resulting carbocationic intermediate (III, Scheme 4C) is then trapped by the alkenyl-gold moiety to give the corresponding cyclobutane. DFT calculations carried out by the same group17 revealed that intermediate III is better described as a metallacyclic species, IV, wherein the gold atom establishes an electrostatic interaction with the benzylic carbocation, resulting in its stabilization by the filled d-shell of the electron rich gold. This pseudo-metallacyclic intermediate enables a better rationalization of the observed stereoselectivities and also explains the stereochemistry of the products obtained when the reaction is carried out in the presence of external nucleophiles.17
2.2. [4 + 3] Cycloadditions
Early in 2008, our group demonstrated that is possible to induce intramolecular [4 + 3] annulations between dienes and allenes, using PtCl2 as catalyst.16 The reaction, which represented the first use of allenes as three-carbon components in metal-catalyzed cycloadditions, allowed a wide variety of functionalized bicyclo[5.3.0]decanes to be built from easily accessible allenedienes, in a fully diastereoselective manner. One year later, we demonstrated that these reactions could also be promoted, under milder conditions, by gold(I) catalysts like IPrAuCl (Au4)/AgSbF6.19 With terminal disubstituted allenes (R2, R3 = alkyl), the reaction generates products of type 4 as single isomers; however, with monosubstituted analogs (R3 = H), it affords isomeric adducts 5 or mixtures of 4 and 5. Also in 2009, Toste reported the same annulations, in this case promoted by the gold catalyst Au5/AgSbF6, featuring the JohnPhos ligand (Scheme 5A).20
Scheme 5. Gold- and Platinum-Catalyzed [4 + 3] Cycloadditions of Allenedienes.

DFT calculations supported a mechanism starting with the activation of the allene by the carbophilic catalyst, followed by a concerted [4C(4π) + 3C(2π)] diene-allylcation cycloaddition to produce a metal–carbene intermediate (VI). Then a 1,2-hydrogen or a 1,2-alkyl shift, with concomitant elimination of the metal, generates the observed bicyclic products (4 or 5). The observed stereochemistry can be explained assuming an exo-like transition state (Scheme 5B).
Importantly, in 2011 we reported an enantioselective version of this [4 + 3] cycloaddition, using the chiral phosphoramidite–gold complex (R,R,R)-Au6.21 The transformation occurs with high levels of enantioselectivity provided that monosubstituted allenes are employed (R3 = H). Importantly, the reaction tolerates longer tethers (n = 1 or 2) as well as the presence of alkyl substituents at the allene internal position; thus, enantioenriched 5,7- and 6,7-bicyclic systems featuring quaternary stereocenters at their ring fusions can be obtained with excellent enantioselectivities (Scheme 5A).
Our group has also reported an important application of this [4 + 3] allenediene cycloaddition in the asymmetric total synthesis of (−)-englerin A, a guaiane sesquiterpene that has raised notable interest because of its antitumoral potential.22 In this case, the platinum catalyst generated from equimolar amounts of PtCl2 and P(C6F5)3 (5%) provided the best results in the cycloaddition of the designed allenediene 7, so the desired cycloadduct 8, featuring the trans-fused guaiane skeleton, was obtained in good yield and complete diastereoselectivity. This adduct was readily transformed into englerin A by using highly selective oxygenation processes and designed functional group transformations (Scheme 6).
Scheme 6. Synthesis of Englerin A Relying on a Pt-Catalyzed [4 + 3] Allenediene Cycloaddition.

2.3. [4 + 2] Cycloadditions
During the study of the above [4 + 3] cycloadditions, we observed that substrates bearing terminally disubstituted allenes provided cyclohexanes (9), instead of cycloheptane products (4 or 5), when a phosphite–gold precatalysts such as Au7 or chiral phosphoramidites like Au6 were used (Scheme 7).23 Similar observations were reported, almost simultaneously, by Toste and co-workers.20 Experimental data and theoretical calculations suggested that both seven- and six-membered ring cycloadducts, arise from the same cycloheptenyl–gold carbene intermediate of type VI (Scheme 7A). Strong σ-donor ligands at gold (such as IPr or JohnPhos) favor its evolution through a 1,2-H or alkyl migration, to give the seven-membered rings. However, with the bulky π-acceptor phosphite ligand, the carbene undergoes a ring contraction (1,2-alkyl internal migration), to give cyclohexyl adducts 9.23 Subsequent work by Fürstner further confirmed that the divergence mainly depends on the electronic characteristics of the ligand. They built gold complexes bearing NHC ligands with similar σ-donor abilities but different π-acceptor characteristics and found that, for the same allenediene, the proportion of the formal [4 + 2] cycloadducts increases with the π-acceptor character of the ligand.24
Scheme 7. Gold(I)-Catalyzed [4 + 2] Cycloaddition of Allenedienes.

Nonetheless, steric properties of the ancillary ligands also affect the selectivity of the cycloadditions.25 Along these lines, Sigman and Toste have recently shown that the Au–Cl bond distance in the gold complexes, which is significantly affected by both the electronic and remote steric influence of the ancillary ligand, is an excellent indicator of the net σ-donation of the ligand at gold, and it can be easily used to explain and even predict the [4 + 2]/[4 + 3] selectivity.26
In consonance with this mechanistic scenario, in which the carbene intermediate VI already includes all the stereochemical information of the cycloaddition, chiral phosphoramidite–gold complexes like (R,R,R)-Au6 are also able promote highly enantioselective [4 + 2] cycloadditions of allenedienes bearing terminally disubstituted allenes. Similar observations were also published by Toste, who developed the related phosphoramidite–gold complex (S,S,S)-Au8, as well as the phosphite–gold precatalyst Au9.27 On the other hand, Fürstner demonstrated that the Taddol-based phosphoramidite–gold complex (R,R,R,R)-Au3/AgBF4 also provides good selectivities in the [4 + 2] cycloaddition of allenedienes (Scheme 7B).28
2.4. [2 + 3] Cycloadditions
As shown above, allenes can undergo gold-promoted intramolecular [2 + 2] cycloadditions with alkenes or behave as three-carbon partners in cycloadditions with dienes. One could then wonder whether they could also work as 3C components in annulations with alkenes.20,24 Toste’s group has shown that while the reaction of allenene 1a(n = 0) with Ph3PAuCl/AgBF4 provides the alkylidenecyclobutane 2a, the use of the σ-donor JohnPhos ligand at gold, with a very related precursor (1b, n = 1), led to the majority formation of the cyclopentene 10b (Scheme 8). The formation of this [3 + 2] adduct was explained in terms of the preferred formation of a cyclopentyl-gold carbene intermediate (VII), mostly due to the steric properties of the ligand at gold.26
Scheme 8. Gold(I)-Catalyzed [3 + 2] and [2 + 2] Cycloadditions of Allenenes.

3. Intermolecular Reactions
In 2009, Iwasawa demonstrated that allenyl silylethers (11) can engage in intermolecular Pt-catalyzed formal annulations with alkenyl ethers (Scheme 9).29 The authors proposed a mechanism that involves the addition of the enol ether to the platinum-containing 1,3-dipole IX. The resulting intermediate X might evolve either to the cyclopentyl Pt-carbene XI or to the cyclobutyl species XII, respective precursors of cyclopentene or cyclobutane adducts (12 and 13). Different factors such as the type of ligand and solvent or the bulkiness of the silyl group at the allene moiety influence the [3 + 2]/[2 + 2] ratio (Scheme 9). Thus, while the use of [PtCl2(C2H4)2]/P(o-tol)3 favors the [3 + 2] pathway, the analog Pt-complex bearing a bulky trialkynylphosphine instead of P(o-tol)3, promotes the [2 + 2] cycloaddition.30 In any case, the use of an allenylether is critical, likely because its electron rich character stabilizes putative cationic intermediates. This methodology constitutes another illustrative example of the versatility of allenes as 2C or 3C components in metal-catalyzed cycloadditions.
Scheme 9. Platinum(II)-Catalyzed Cycloaddition of Allenyl Silylethers and Alkenyl Ethers.

3.1. [4 + 2] Cycloadditions
In addition to intramolecular reactions, our group has also pursued the development of intermolecular cycloadditions between allenes and dienes. Eventually, in 2011, we discovered that N-allenamides like 14, can efficiently participate as 2C partners in gold-catalyzed formal [4 + 2] cycloadditions with neutral or electron rich 1,3-dienes. These reactions represented the first examples of intermolecular formal cycloadditions of allenes catalyzed by gold (Scheme 10A).31 The annulation was better promoted by AuCl or by IPrAuCl (Au4)/AgSbF6, which led to cyclohexenes of type 16 with good yields and excellent regio- and diastereoselectivities.32 It is important to note that carbon-substituted allenes or allenyl ethers like 11 generated complex mixtures of products. Therefore, the use of allenamides seems key to warrant an appropriate balance between reactivity and stability of the different reaction intermediates.
Scheme 10. Gold(I)-Catalyzed Intermolecular [4 + 2] Cycloadditions between Allenamides and 1,3-Dienes and Mechanistic Scenario.

Experimental data and DFT calculations suggest a complex mechanism with several possible pathways that depend on the electronic properties of both the gold catalyst and the diene unit (Scheme 10B).33 A common feature for all of them is the easy formation of initial gold–zwitterionic species of type XIII. Then, when neutral dienes such as isoprene are used, this gold-intermediate reacts with the diene via concerted pathways. Thus, with IPr as gold ligand, a direct [4 + 2] cycloaddition between the diene and the terminal double bond of XIII is favored (route b); however, with AuCl, calculations suggest that a concerted [4 + 3] cycloaddition to give the cycloheptenyl carbene XIV, followed by ring contraction, could be operative (route a). Both pathways explain the formation of the same [4 + 2] cycloadduct 16a, but the latter is slightly favored (by ∼2 kcal mol–l). On the other hand, with electron rich dienes such as 1-methoxydiene 15b, a stepwise process involving the formation of an acyclic intermediate like XV is preferred (route c, Scheme 10B).
A related stepwise pathway also explains the eventual formation of cyclobutanes of type 17, which were observed as side products with neutral dienes, particularly when using IPrAuCl/AgSbF6 (Scheme 11). Thus, after formation of the gold(I)-activated allenamide derivative XIII, a regioselective addition of the diene (e.g., isoprene) would lead to the carbocationic species XVI. This intermediate might conformationally evolve to facilitate an interaction of the gold atom with the internal carbon of the allylic carbocation (XVII). An eventual ring closure provides either cyclobutane or cyclohexene adducts, with the overall activation barrier to the cyclobutane being favored by over 5 kcal·mol–1. Thus, this pathway would be reponsible for the generation of cyclobutanes like 17a, whereas the formation of the major Diels–Alder adducts (e.g., 16a) is better explained in terms of a concerted annulation from XIII, which has a significantly lower overall energy barrier.
Scheme 11. Explanation for the Formation of Minor [2 + 2] Adducts.

In 2012, we reported an enantioselective variant of the [4 + 2] cycloaddition reaction between dienes and allenamides (Scheme 12A).34 Specifically, we found that the Lassaletta’s gold complex Au10, featuring a triazole unit embedded in a highly crowded axially chiral environment, was able to promote these annulations between allenamides and aryl- or alkyl-substituted 1,3-dienes with excellent enantioselectivities and with complete regio- and diastereoselectivities.
Scheme 12. Gold(I)-catalyzed [4 + 2] cycloadditions between allenamides and 1,3-dienes.

In addition to these examples, in 2013 Rossi and Vicente,35 as well as Zhang,36 reported [4 + 2] annulations between N-allenamides and C2- or C3-styryl indoles (Scheme 12B). The cycloadditions of 3-styryl indoles (20) were also described in an asymmetric fashion using phosphoramidite–gold catalysts so that tetrahydrocarbazoles 22 could be obtained with excellent enantioselectivities (Scheme 12C).
3.2. [2 + 2] Cycloadditions
The observation that cyclobutanes, derived from a formal [2 + 2] annulation between the allenamide and one of the double bonds of the diene, could be obtained as side products (Scheme 11)31 led us to investigate the viability of a [2 + 2] cycloaddition between allenamides and alkenes.37 We eventually found that the phosphite–gold(I) complex Au7/AgSbF6 was very efficient to promote these cycloadditions between carbonyl-allenamides and styrenes or enamides, in most cases with excellent or even complete diasteroselectivities (Scheme 13A). Chen38 and González39 also reported closely related [2 + 2] cycloadditions with electron-rich alkenes, but using N-sulfonyl allenylamides as reaction partners and Au7′ or Au5/AgSbF6 as catalysts (Scheme 13B,C). The reactions were proposed to take place through a stepwise process involving the initial attack of the alkene to the gold-activated allenamide to give a carbocationic intermediate that evolves to the product by ring closure (Scheme 13D). The observation of the same stereoisomeric product 25 from both the E- and Z-enamide precursor 24, is highly consistent with the formation of acyclic carbocationic intermediates like XVIII.37 However, the retention of the stereochemistry of cis-β-deuterated styrene (26) observed by Chen38 indicates that, at least for this type of alkene and gold(I) catalyst, the reaction might involve gold-stabilized intermediates of type XIX (Scheme 13E).
Scheme 13. Gold(I)-Catalyzed [2 + 2] Cycloadditions between Allenamides and Alkenes.

Importantly, González also developed an enantioselective version of the intermolecular [2 + 2] cycloaddition with styrenes, using phosphoramidite–gold complexes like (R,R,R)-Au12 and (S,R,R)-Au2 (Scheme 13F).40,41
In 2014, we demonstrated that the cationic complex [(JohnPhos)Au(NCMe)]SbF6 (Au5″) was a very efficient catalyst for promoting [2 + 2] annulations between allenamides and a wide variety α,β-unsaturated N,N-dialkyl hydrazones (28, Scheme 14A).42 The reaction was proposed to proceed via a stepwise mechanism involving the regioselective addition of the unsaturated hydrazone, which behaves as a vinylogous aza-enamine, to the gold-activated allenamide to afford the acyclic intermediate XX. A stereoselective cyclization would lead to the observed cyclobutanes with cis stereochemistry. In consonance with the formation of intermediates XX, both Z- and E-isomers of a particular α,β-unsaturated hydrazone (R1 = Ph, R2 = H) provided the same adduct as a single isomer, with both phenyl and hydrogen in a cis disposition.
Scheme 14. Recent Examples of Gold-Promoted [2 + 2] Cycloadditions with Electron-Rich Allenes.

More recently, Bandini43 and Zhang36 reported additional examples of [2 + 2] cycloadditions between electron rich allenes and indoles (Scheme 14B,C). In particular, the cycloaddition of 2,3-disubstituted N-Boc indoles (30) with several allenamides or allenyl arylethers was effectively promoted by Au5″, to give densely functionalized cyclobutanindolines (31). The presence of an electron-withdrawing group at the indolic nitrogen was critical, presumably because of the increased electrophilicity of the dearomatized intermediate XXI. The use of the chiral gold catalyst derived from DTBM-Segphos allowed them to obtain cyclobutanindolines of type 31, featuring two carbon quaternary stereocenters, with excellent enantioselectivities (Scheme 14B). On the other hand, Zhang introduced chiral gold complexes bearing sulfonamide–phosphane ligands (e.g., Au13) in the asymmetric annulations of C3-styryl indoles (Scheme 14C).44
3.3. [3 + 2], [5 + 2], and [1 + 2] Dipolar Cycloadditions
In the above annulations, the allenamide behaves as a two-carbon annulation partner. We are not aware of reports on gold- or platinum-based catalysis in which allenamides participate as a three-carbon unit. This is quite surprising, owing to the precedent of Iwasawa on the platinum-promoted reaction of allenyl silylethers (Scheme 9). Nonetheless, it is possible to achieve alternative formal [3 + 2] cycloadditions, confronting the allenamides with 1,3-dipoles that behave as 3-atom partners. Therefore, Chen demonstrated that azomethine ylides of types 34 and 35 can react with N-carbonyl and N-sulfonyl allenamides to give the corresponding [3 + 2] cycloadducts in good yields and complete regioselectivity (Scheme 15A).45,46 Interestingly, the analogous reaction with nitrones (38) was also developed in asymmetric fashion using the phosphoramidite–gold complexes (R,R,R)-Au14 and (R,S,S)-Au15 (Scheme 15B).
Scheme 15. Gold(I)-Catalyzed Cycloadditions between N-Allenamides and 1,n-Dipoles.

Alternative types of 1,n-dipoles might also engage in gold-catalyzed formal cycloadditions with N-allenamides. Thus, Yoo recently reported the feasibility of [5 + 2] cycloadditions between N-allenamides and quinolinium zwitterions (1,5-dipoles, 40) to yield a variety of fused 1,4-diazepine derivatives (41) in a stereospecific manner (Scheme 15C).47 On the other hand, Maulide reported formal gold-catalyzed [2 + 1] cycloadditions between allenamides and stabilized sulfonium ylides (1,2-dipoles, 42) to give alkylidenecyclopropanes of type 43 (Scheme 15D).48
3.4. [2 + 2 + 2] Cycloadditions
A particularly challenging extension of the gold-promoted chemistry of allenes is related to the development of tandem annulations involving three different components. Relying on the stepwise mechanisms proposed for the [2 + 2] cycloadditions, we demonstrated that it is possible to intercept the carbocation generated after the addition of the alkene to the gold-activated allenamide by tethered carbonyl groups. The resulting oxonium is then trapped in a Prins-like cyclization to give oxa-bridged medium sized carbocycles (Scheme 16).49 This formal [2 + 2 + 2] annulation between allenamides and oxo-alkenes is best promoted by the phosphite–gold complex Au7″. A wide range of carbonyl-tethered alkenes (44) participated in the process providing their respective oxa-bridged seven-, eight-, and even nine-membered ring cycloadducts (45) in good yields. An enantioselective version of this process was also disclosed, using (R)-DTBM-Segphos(AuCl)2 (Au1)/AgNTf2, for the assembly of eight-membered rings, and the phosphoramidite complex (S,R,R)-Au12/AgNTf2, for the synthesis of cycloheptenes.
Scheme 16. Gold(I)-Catalyzed Formal [2 + 2+2] Cycloaddition between Allenamides and Carbonyl-Tethered Alkenes.

Our group has also developed a fully intermolecular version of this annulation, using allenamides, electron rich alkenes, and aldehydes as reaction partners (Scheme 17).50 Again, the phosphite–gold complex Au7″ was very efficient for a broad range of both cyclic and acyclic alkenes, as well as diverse types of aldehydes. The corresponding tetrahydropyrans were obtained in good to excellent yields, and with moderate to complete diastereoselectivities in favor of the 2,6-cis isomer (46). In the case of acyclic alkenes, the best results were obtained with 1,1-disubstituted styrene derivatives or enolethers. Likely, the higher stabilization of the carbocationic intermediate resulting from the initial addition of the alkene to the allenamide (XXIV) retards the ring closure to the cyclobutene product, which is eventually observed as minor side product. Stereochemical studies with α,β-disubstituted alkenes and β-deuterium labeled styrenes revealed that, when α-monosubstituted alkenes are used, the reaction takes place through an intermediate of type XXIV, which preserves the stereochemical information of the parent alkene. This species evolves through a nucleophilic anti attack of the carbonyl moiety, leading to oxonium XXV and to the product as a single isomer at the C5–C6 centers (Scheme 17B). On the other hand, with α,α-disubstituted alkenes, the additional stabilization of the tertiary cation labilizes the interaction with the gold atom and therefore facilitates the formation of acyclic carbocationic species like XXVI or of the epimeric intermediate XXIV′, with the consequent loss of the initial alkene geometry.
Scheme 17. Gold(I)-Catalyzed formal [2 + 2 + 2] Cycloaddition between Allenamides, Alkenes, and Aldehydes.

More recently, we developed an enantioselective version of this intermolecular [2 + 2 + 2] cycloaddition (Scheme 18).51 Specifically, we found that the triazole-based gold complex Au10/AgNTf2 affords the major 2,6-cis-tetrahydropyrans (46) with good to excellent enantioselectivities. Although the ee values of the minor 2,6-trans adducts (46′) were typically low, these isomers could be obtained in higher amounts and with remarkable enantioselectivities, of up to 90%, with the Vanol-derived catalyst (S,R,R)-Au12/AgNTf2.
Scheme 18. Enantioselective [2 + 2 + 2] Cycloaddition between Allenamides, Alkenes, and Aldehydes.

Importantly, very recently, we expanded the scope of these formal cycloadditions to nitrogenated compounds by developing a gold(I)-catalyzed [2 + 2 + 2] annulation process between allenamides and alkenyl-oxime ethers (Scheme 19A).52 This methodology allowed us to obtain both piperidine (49)and tropane (50) skeletons, depending on whether the alkene is O- or C-tethered to the oxime moiety. The gold complex Au7″ was again the most effective catalyst, allowing the azacyclic products to be obtained with moderate to excellent yields and with complete selectivity. Moreover, we found that the efficiency and the diastereoselectivity is completely determined by the E/Z-stereochemistry of the parent oxime ether. Additionally, we also developed an enantioselective version of the two manifolds using (S,R,R)-Au12/AgNTf2. Thus, a wide range of O- and C-tethered alkenyl-oximes gave their corresponding azabicyclic adducts with enantiomeric excesses from moderate to excellent.
Scheme 19. Gold(I)-Catalyzed Cycloaddition between Allenamides and Alkenyl Oximes.

Importantly, we found that using oximes instead of imines is critical for obtaining these results. Indeed, mechanistic studies confirmed that related imines like 51 do not participate in the annulations due to the strong coordination to the gold, which results in catalyst deactivation. The oxygen atom of the oxime ethers makes their adjacent nitrogen less nucleophilic, thereby decreasing their ability to coordinate the gold(I) complex (Scheme 19B).
Recently, Sun also contributed to the synthesis of N-heterocycles by developing a formal [2 + 2 + 2] cycloaddition of allenamides and 1,3,5-triazines, which behave as aldimine reservoirs through an in situ retro-cyclotrimerization process (Scheme 20).53 Interestingly, Ph3PAuCl/NaBArF efficiently catalyzed the reaction of a range of 1,3,5-triazines with both electron rich allenamides and electron-deficient allenoates to provide two types of products, 54 and 55, respectively. Different pathways account for this divergence. In the case of allenamides, two independent molecules of the aldimine, generated in the reaction media, are sequentially added to the gold-activated allenamide to yield 54 after a cyclization/deauration process (Scheme 20A). However, for allenoates, a coordination to the carbonyl group to generate a gold-activated Michael acceptor was proposed, so that sequential addition of two aldimines, followed by a ring closing and alkene isomerization, provided the adducts 55 (Scheme 20B).54,55
Scheme 20. Gold(I)-Catalyzed Cycloaddition between 1,3,5-Triazines and Allenamides or Allenoates.

4. Conclusions and Outlook
Allenes and allenamides offer an unusual potential to participate in different types of platinum- and especially gold-catalyzed annulation reactions. Upon activation, they generate allyl-cation alkenylmetal species that can behave as 1,2- or 1,3-carbon dipoles in cycloaddition processes. While alkyl substituted allenes have shown great utility in intramolecular reactions, intermolecular annulations require the use of allenamides or related derivatives. Likely, the presence of the amide substituent provides a good balance of reactivity and stability in the gold activated derivatives, so that they can react with the corresponding partners in a controlled manner. The range of reactivities discovered include formal [4 + 3], [4 + 2], [2 + 2], [2 + 3], [3 + 2], [2 + 1], [5 + 2], and [2 + 2 + 2] annulations. While in the intramolecular cases, allenes can participate as 2C or 3C components, there is only one intermolecular example of reactions in which they work as 3C partners. On the other hand, despite the difficulties associated with the transfer of stereochemical information from chiral linear gold(I) complexes to the newly created stereocenters, a variety of enantioselective gold-catalyzed annulations have been discovered.
Current and future work in the field might be focused on further developing the 3C reactivity of allenes and on the discovery of annulations that allow one to build relevant heterocyclic and specially azaheterocyclic products from simple materials. Tandem or synergistic processes that combine different types of reactions in a single operation are also appealing. Finally, further studies on enantioselective variants and to gain deeper mechanistic insights are also warranted. With all this knowledge at hand, we will also witness the development of new synthetic applications.
Acknowledgments
Financial support from the Spanish MINECO (SAF2016-76689-R, CTQ2017-84767-P, FPU fellowship to I.V), the Xunta de Galicia (ED431C 2017/19, 2015-CP082, Centro Singular de Investigación de Galicia accreditation 20162019 ED431G/09), the ERDF, and ERC (Adv. Grant No. 340055) is acknowledged. We are extremely indebted to all our co-workers who enthusiastically contributed to this research line over the past decade (their names are indicated in the references).
Biographies
José Luis Mascareñas completed his Ph.D. at the University of Santiago in 1988. He was a postdoctoral fellow at Stanford University under the supervision of Prof. Paul Wender (1989–1990) and became permanent professor in 1993 and full professor in 2005 at the University of Santiago. He has been visiting scholar at Harvard University (1992 and 1995) and visiting scientist at the University of Cambridge (2009) and MIT (2013). He received the Organic Chemistry (2009) and Gold medal (2015) awards of the Spanish Royal Society of Chemistry. In 2014, he received an ERC-Advanced Grant and in 2016 was selected as member of EURASC.
Iván Varela graduated in Chemistry at the University of Santiago in 2013. In 2014, he obtained his master degree, and one year later he was awarded with a FPU fellowship from the Spanish Ministry of Education to carry out his Ph.D. studies, which are focused on the development of new gold-catalyzed cycloadditions.
Fernando López obtained his Ph.D. in 2003 at the University of Santiago de Compostela (USC). He carried out two predoctoral stays with Profs. Erick M. Carreira and John F. Hartwig and a postdoctoral stay with Prof. Ben Feringa (2004−2006). In 2006, he returned to the USC as a Ramón y Cajal Fellow and, in 2008, he was granted a Tenured Scientist position at the Spanish National Research Council (CSIC), joining the IQOG. In 2009, he received the Young Investigator Award of the Spanish Royal Society of Chemistry. Since 2012, he has been assigned at the CiQUS (USC), and in 2018, he was granted a Senior Scientist Researcher position at CSIC.
The authors declare no competing financial interest.
References
- Hashmi A. S. K., Toste F. D., Eds. Modern Gold Catalyzed Synthesis; Wiley VCH: Weinheim, Germany, 2012. [Google Scholar]
- Fürstner A.; Davies P. W. Catalytic Carbophilic Activation: Catalysis by Platinum and Gold π Acids. Angew. Chem., Int. Ed. 2007, 46, 3410–49. 10.1002/anie.200604335. [DOI] [PubMed] [Google Scholar]
- Gorin D. J.; Toste F. D. Relativistic Effects in Homogeneous Gold Catalysis. Nature 2007, 446, 395–403. 10.1038/nature05592. [DOI] [PubMed] [Google Scholar]
- Teles J. H.; Brode S.; Chabanas M. Cationic Gold(I) Complexes: Highly Efficient Catalysts for the Addition of Alcohols to Alkynes. Angew. Chem., Int. Ed. 1998, 37, 1415–1418. . [DOI] [PubMed] [Google Scholar]
- Dorel R.; Echavarren A. M. Gold(I)-Catalyzed Activation of Alkynes for the Construction of Molecular Complexity. Chem. Rev. 2015, 115, 9028–9072. 10.1021/cr500691k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L. M. A Non-Diazo Approach to α-Oxo Gold Carbenes via Gold-Catalyzed Alkyne Oxidation. Acc. Chem. Res. 2014, 47, 877–888. 10.1021/ar400181x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarucci M.; Bandini M. New Developments in Gold-Catalyzed Manipulation of Inactivated Alkenes. Beilstein J. Org. Chem. 2013, 9, 2586–2614. 10.3762/bjoc.9.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chianese A. R.; Lee S. J.; Gagné M. R. Electrophilic Activation of Alkenes by Platinum(II): So Much More Than a Slow Version of Palladium(II). Angew. Chem., Int. Ed. 2007, 46, 4042–4059. 10.1002/anie.200603954. [DOI] [PubMed] [Google Scholar]
- Malacria M.; Fensterbank L.; Gandon V.. Activation of Allenes by Gold Complexes: A Theoretical Standpoint. In Computational Mechanisms of Au and Pt Catalyzed Reactions; Alcaide B., Soriano E., Eds.; Springer, 2011; Vol. 302, pp 157–182. [DOI] [PubMed] [Google Scholar]
- Yang W.; Hashmi A. S. K. Mechanistic Insights into the Gold Chemistry of Allenes. Chem. Soc. Rev. 2014, 43, 2941–2955. 10.1039/c3cs60441a. [DOI] [PubMed] [Google Scholar]
- Brooner R. E. M.; Widenhoefer R. A. Cationic, Two-Coordinate Gold π Complexes. Angew. Chem., Int. Ed. 2013, 52, 11714–11724. 10.1002/anie.201303468. [DOI] [PubMed] [Google Scholar]
- Barreiro E. M.; Adrio L. A.; Mimi Hii K. K.; Brazier J. B. Coinage Metal Catalysts for the Addition of O-H to C = C Bonds. Eur. J. Org. Chem. 2013, 2013, 1027–1039. 10.1002/ejoc.201201441. [DOI] [Google Scholar]
- Xiong H.; Wang H.; He L.; Zhang Y.; Zeng Z. Application of Gold Catalyst in Nucleophilic Addition of Allene. Chin. J. Org. Chem. 2011, 31, 466–479. [Google Scholar]
- Belmont P.; Parker E. Silver and Gold Catalysis for Cycloisomerization Reactions. Eur. J. Org. Chem. 2009, 2009, 6075–6089. 10.1002/ejoc.200900790. [DOI] [Google Scholar]
- Luzung M. R.; Mauleón P.; Toste F. D. Gold(I)-Catalyzed [2 + 2]-Cycloaddition of Allenenes. J. Am. Chem. Soc. 2007, 129, 12402–12403. 10.1021/ja075412n. [DOI] [PubMed] [Google Scholar]
- Trillo B.; López F.; Gulías M.; Castedo L.; Mascareñas J. L. Platinum-Catalyzed Intramolecular [4C + 3C] Cycloaddition Between Dienes and Allenes. Angew. Chem., Int. Ed. 2008, 47, 951–954. 10.1002/anie.200704566. [DOI] [PubMed] [Google Scholar]
- González A. Z.; Benitez D.; Tkatchouk E.; Goddard W. A.; Toste F. D. Phosphoramidite Gold(I)-Catalyzed Diastereo- and Enantioselective Synthesis of 3,4-Substituted Pyrrolidines. J. Am. Chem. Soc. 2011, 133, 5500–5507. 10.1021/ja200084a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teller H.; Flügge S.; Goddard R.; Fürstner A. Enantioselective Gold Catalysis: Opportunities Provided by Monodentate Phosphoramidite Ligands with an Acyclic TADDOL Backbone. Angew. Chem., Int. Ed. 2010, 49, 1949–1953. 10.1002/anie.200906550. [DOI] [PubMed] [Google Scholar]
- Trillo B.; López F.; Montserrat S.; Ujaque G.; Castedo L.; Lledós A.; Mascareñas J. L. Gold-Catalyzed [4C+3C] Intramolecular Cycloaddition of Allenedienes: Synthetic Potential and Mechanistic Implications. Chem. - Eur. J. 2009, 15, 3336–3339. 10.1002/chem.200900164. [DOI] [PubMed] [Google Scholar]
- Mauleón P.; Zeldin R. M.; González A. Z.; Toste F. D. Ligand-Controlled Access to [4 + 2] and [4 + 3] Cycloadditions in Gold-Catalyzed Reactions of Allene-Dienes. J. Am. Chem. Soc. 2009, 131, 6348–6349. 10.1021/ja901649s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso I.; Faustino H.; López F.; Mascareñas J. L. Enantioselective Gold(I)-Catalyzed Intramolecular (4 + 3) Cycloadditions of Allenedienes. Angew. Chem., Int. Ed. 2011, 50, 11496–11500. 10.1002/anie.201105815. [DOI] [PubMed] [Google Scholar]
- Nelson R.; Gulías M.; Mascareñas J. L.; López F. Concise, Enantioselective, and Versatile Synthesis of (−)-Englerin A Based on a Platinum-Catalyzed [4C+3C] Cycloaddition of Allenedienes. Angew. Chem., Int. Ed. 2016, 55, 14359–14363. 10.1002/anie.201607348. [DOI] [PubMed] [Google Scholar]
- Alonso I.; Trillo B.; López F.; Montserrat S.; Ujaque G.; Castedo L.; Lledós A.; Mascareñas J. L. Gold-Catalyzed [4C+2C] Cycloadditions of Allenedienes, including an Enantioselective Version with New Phosphoramidite-Based Catalysts: Mechanistic Aspects of the Divergence between [4C+3C] and [4C+2C] Pathways. J. Am. Chem. Soc. 2009, 131, 13020–13030. 10.1021/ja905415r. [DOI] [PubMed] [Google Scholar]
- Alcarazo M.; Stork T.; Anoop A.; Thiel W.; Fürstner A. Steering the Surprisingly Modular π-Acceptor Properties of N-heterocyclic Carbenes: Implications for Gold Catalysis. Angew. Chem., Int. Ed. 2010, 49, 2542–2546. 10.1002/anie.200907194. [DOI] [PubMed] [Google Scholar]
- Benitez D.; Tkatchouk E.; Gonzalez A. Z.; Goddard W. A.; Toste F. D. On the Impact of Steric and Electronic Properties of Ligands on Gold(I)-Catalyzed Cycloaddition Reactions. Org. Lett. 2009, 11, 4798–4801. 10.1021/ol9018002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian A. H.; Niemeyer Z. L.; Sigman M. S.; Toste F. D. Uncovering Subtle Ligand Effects of Phosphines Using Gold(I) Catalysis. ACS Catal. 2017, 7, 3973–3978. 10.1021/acscatal.7b00757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez A. Z.; Toste F. D. Gold(I)-catalyzed Enantioselective [4 + 2]-Cycloaddition of Allene-dienes. Org. Lett. 2010, 12, 200–203. 10.1021/ol902622b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teller H.; Corbet M.; Mantilli L.; Gopakumar G.; Goddard R.; Thiel W.; Furstner A. One-point Binding Ligands for Asymmetric Gold Catalysis: Phosphoramidites with a TADDOL-Related but Acyclic Backbone. J. Am. Chem. Soc. 2012, 134, 15331–15342. 10.1021/ja303641p. [DOI] [PubMed] [Google Scholar]
- Kusama H.; Ebisawa M.; Funami H.; Iwasawa N. Platinum(II)-Catalyzed Intermolecular [3 + 2] Cycloaddition of Propadienyl Silyl Ethers and Alkenyl Ethers. J. Am. Chem. Soc. 2009, 131, 16352–16353. 10.1021/ja907633b. [DOI] [PubMed] [Google Scholar]
- Ebisawa M.; Kusama H.; Iwasawa N. Selective Intermolecular [2 + 2] Cycloaddition Reaction Using Platinum(II) Catalyst with Hollow-shaped Triethynylphosphine. Chem. Lett. 2012, 41, 786–788. 10.1246/cl.2012.786. [DOI] [Google Scholar]
- Faustino H.; López F.; Castedo L.; Mascareñas J. L. Gold(I)-catalyzed Intermolecular (4 + 2) Cycloaddition of Allenamides and Acyclic Dienes. Chem. Sci. 2011, 2, 633–637. 10.1039/c0sc00630k. [DOI] [Google Scholar]
- For a related [4 + 2] cycloaddition using allenyl-ethers, instead of allenamides, see:; Wang G.; Zou Y.; Li Z.; Wang Q.; Goeke A. Cationic Gold(I)-Catalyzed Intermolecular [4 + 2] Cycloaddition between Dienes and Allenyl Ethers. Adv. Synth. Catal. 2011, 353, 550–556. 10.1002/adsc.201000597. [DOI] [Google Scholar]
- Montserrat S.; Faustino H.; Lledós A.; Mascareñas J. L.; López F.; Ujaque G. Mechanistic Intricacies of Gold-Catalyzed Intermolecular Cycloadditions between Allenamides and Dienes. Chem. - Eur. J. 2013, 19, 15248–15260. 10.1002/chem.201302330. [DOI] [PubMed] [Google Scholar]
- Francos J.; Grande-Carmona F.; Faustino H.; Iglesias-Sigüenza J.; Díez E.; Alonso I.; Fernández R.; Lassaletta J. M.; López F.; Mascareñas J. L. Axially Chiral Triazoloisoquinolin-3-ylidene Ligands in Gold(I)-Catalyzed Asymmetric Intermolecular (4 + 2) Cycloadditions of Allenamides and Dienes. J. Am. Chem. Soc. 2012, 134, 14322–14325. 10.1021/ja3065446. [DOI] [PubMed] [Google Scholar]
- Pirovano V.; Decataldo L.; Rossi E.; Vicente R. Gold-catalyzed Synthesis of Tetrahydrocarbazole Derivatives through an Intermolecular Cycloaddition of Vinyl Indoles and N-Allenamides. Chem. Commun. 2013, 49, 3594–3596. 10.1039/c3cc41514g. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Zhang P.; Liu Y.; Xia F.; Zhang J. Enantioselective Gold-Catalyzed Intermolecular [2 + 2] versus [4 + 2]-Cycloadditions of 3-Styrylindoles with N-Allenamides: Observation of Interesting Substituent Effects. Chem. Sci. 2015, 6, 5564–5570. 10.1039/C5SC01827G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faustino H.; Bernal P.; Castedo L.; López F.; Mascareñas J. L. Gold(I)-Catalyzed Intermolecular [2 + 2] Cycloadditions between Allenamides and Alkenes. Adv. Synth. Catal. 2012, 354, 1658–1664. 10.1002/adsc.201200047. [DOI] [Google Scholar]
- Li X.-X.; Zhu L.-L.; Zhou W.; Chen Z. Formal Intermolecular [2 + 2] Cycloaddition Reaction of Alleneamides with Alkenes via Gold Catalysis. Org. Lett. 2012, 14, 436–439. 10.1021/ol202703a. [DOI] [PubMed] [Google Scholar]
- Suárez-Pantiga S.; Hernández-Díaz C.; Piedrafita M.; Rubio E.; González J. M. Phosphite-Gold(I)-Catalyzed [2 + 2] Intermolecular Cycloaddition of Enol Ethers with N-Allenylsulfonamides. Adv. Synth. Catal. 2012, 354, 1651–1657. 10.1002/adsc.201200043. [DOI] [Google Scholar]
- Suárez-Pantiga S.; Hernández-Díaz C.; Rubio E.; González J. M. Intermolecular [2 + 2] Reaction of N-Allenylsulfonamides with Vinylarenes: Enantioselective Gold(I)-Catalyzed Synthesis of Cyclobutane Derivatives. Angew. Chem., Int. Ed. 2012, 51, 11552–11555. 10.1002/anie.201206461. [DOI] [PubMed] [Google Scholar]
- Huang W.; Zhang Y.-C.; Jin R.; Chen B.-L.; Chen Z. Synthesis of Axially Chiral 1,2,3-Triazol-5-ylidene–Au(I) Complex and Its Application in Enantioselective [2 + 2] Cycloaddition of Alleneamides with Alkenes. Organometallics 2018, 37, 3196–3209. 10.1021/acs.organomet.8b00524. [DOI] [Google Scholar]
- Bernal-Albert P.; Faustino H.; Gimeno A.; Asensio G.; Mascareñas J. L.; López F. Gold(I)-Catalyzed Intermolecular Cycloaddition of Allenamides with α,β-Unsaturated Hydrazones: Efficient Access to Highly Substituted Cyclobutanes. Org. Lett. 2014, 16, 6196–6199. 10.1021/ol503121q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia M.; Monari M.; Yang Q.-Q.; Bandini M. Enantioselective Gold Catalyzed Dearomative [2 + 2]-Cycloaddition between Indoles and Allenamides. Chem. Commun. 2015, 51, 2320–2323. 10.1039/C4CC08736D. [DOI] [PubMed] [Google Scholar]
- Hu H.; Wang Y.; Qian D.; Zhang Z.-M.; Liu L.; Zhang J. Enantioselective Gold-Catalyzed Intermolecular [2 + 2]-Cycloadditions of 3-Styrylindoles with N-Allenyl Oxazolidinone. Org. Chem. Front. 2016, 3, 759–763. 10.1039/C6QO00087H. [DOI] [Google Scholar]
- Zhou W.; Li X.-X.; Li G.-H.; Wu Y.; Chen Z. Gold Catalyzed [3 + 2] Cycloaddition of N-Allenyl amides with Azomethine Imines. Chem. Commun. 2013, 49, 3552–3554. 10.1039/c3cc41258j. [DOI] [PubMed] [Google Scholar]
- Li G.-H.; Zhou W.; Li X.-X.; Bi Q.-W.; Wang Z.; Zhao Z.-G.; Hu W.-X.; Chen Z. Gold Catalyzed Enantioselective Intermolecular [3 + 2] Dipolar Cycloaddition of N-Allenyl Amides with Nitrones. Chem. Commun. 2013, 49, 4770–4772. 10.1039/c3cc41769g. [DOI] [PubMed] [Google Scholar]
- De N.; Song C. E.; Ryu D. H.; Yoo E. J. Gold-Catalyzed [5 + 2] Cycloaddition of Quinolinium Zwitterions and Allenamides as an Efficient Route to Fused 1,4-Diazepines. Chem. Commun. 2018, 54, 6911–6914. 10.1039/C8CC02570C. [DOI] [PubMed] [Google Scholar]
- Sabbatani J.; Huang X.; Veiros L. F.; Maulide N. Gold-Catalyzed Intermolecular Synthesis of Alkylidenecyclopropanes through Catalytic Allene Activation. Chem. - Eur. J. 2014, 20, 10636–10639. 10.1002/chem.201402935. [DOI] [PubMed] [Google Scholar]
- Faustino H.; Alonso I.; Mascareñas J. L.; López F. Gold(I)-Catalyzed Cascade Cycloadditions between Allenamides and Carbonyl-Tethered Alkenes: An Enantioselective Approach to Oxa-Bridged Medium-Sized Carbocycles. Angew. Chem., Int. Ed. 2013, 52, 6526–6530. 10.1002/anie.201302713. [DOI] [PubMed] [Google Scholar]
- Faustino H.; Varela I.; Mascareñas J. L.; López F. Gold(I)-Catalyzed [2 + 2 + 2] Cycloaddition of Allenamides, Alkenes and Aldehydes: A Straightforward Approach to Tetrahydropyrans. Chem. Sci. 2015, 6, 2903–2908. 10.1039/C5SC00295H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela I.; Faustino H.; Díez E.; Iglesias-Sigüenza J.; Grande-Carmona F.; Fernández R.; Lassaletta J. M.; Mascareñas J. L.; López F. Gold(I)-Catalyzed Enantioselective [2 + 2 + 2] Cycloadditions: An Expedient Entry to Enantioenriched Tetrahydropyran Scaffolds. ACS Catal. 2017, 7, 2397–2402. 10.1021/acscatal.6b03651. [DOI] [Google Scholar]
- Marcote D. C.; Varela I.; Fernández-Casado J.; Mascareñas J. L.; López F. Gold(I)–Catalyzed Enantioselective Annulations between Allenes and Alkene-tethered Oxime Ethers: A Straight Entry to Highly Substituted Piperidines and Aza-bridged Medium-sized Carbocycles. J. Am. Chem. Soc. 2018, 140, 16821–16833. 10.1021/jacs.8b10388. [DOI] [PubMed] [Google Scholar]
- Peng S.; Cao S.; Sun J. Gold-Catalyzed Regiodivergent [2 + 2+2]-Cycloadditions of Allenes with Triazines. Org. Lett. 2017, 19, 524–527. 10.1021/acs.orglett.6b03691. [DOI] [PubMed] [Google Scholar]
- See also:; Peng S.; Ji D.; Sun J. Gold-Catalyzed [2 + 2 + 2 + 2]-Annulation of 1,3,5-Hexahydro-1,3,5-Triazines with Alkoxyallenes. Chem. Commun. 2017, 53, 12770–12773. 10.1039/C7CC07554E. [DOI] [PubMed] [Google Scholar]
- For a gold-catalyzed allenamide cyclotrimerization, see; Hernández-Díaz C.; Rubio E.; González J. M. Gold-Catalyzed Allenamide [2 + 2 + 2]-Cyclotrimerization. Eur. J. Org. Chem. 2016, 2016, 265–269. 10.1002/ejoc.201501364. [DOI] [Google Scholar]