

Abstract
Introduction:
Pain relief remains a major public health challenge. The most efficient available painkillers are opioids targeting the mu opioid receptor (MOR). MORs are expressed in the areas of the brain [including pain and respiratory centers] that are important for processing reward and aversion. Thus, MOR activation efficiently alleviates severe pain, but the concomitant reward and respiratory depressant effects pose a threat; patients taking opioids potentially develop opioid addiction and high risk for overdose.
Areas covered:
Ongoing efforts to generate safer opioid analgesics are reviewed here. The design of biased compounds that trigger MOR induced G protein over β-arrestin signaling, peripheral opioids, drugs targeting MORs in heteromers and drugs enhancing endogenous opioid activity are discussed.
Expert opinion:
There is evidence that throttling MOR signaling may lead to an era of opioids that are truly efficient painkillers with lower side effects and risk of overdose. However, few of the drugs derived from the advanced approaches outlined here, are getting approval by regulatory committees for use in clinical settings. Thus, there is an urgent need to (i) better clarify mechanisms underlying the hazardous physiological effects of MOR activation, and (ii) fully validate the safety of these new MOR-based therapies.
Keywords: opioid receptor, biased agonism, allosteric, pain, addiction, opiate, GPCR, drug discovery, heteromer, peripherally limited opioid, pH-dependent agonist, endogenous opioid analogue
1. Introduction
Acute pain is a normal sensation produced by the nervous system to bring attention to a possible injury [1]. Chronic pain is abnormal and may arise from only physiological origins or from a combination of mental and physiological causes requiring patients to seek out pain relief [1]. Chronic pain may be alleviated by acupuncture, nerve stimulation, psychotherapy, relaxation, physical therapy and medication therapies, however the most widely prescribed drugs in the US for pain management are opioids [2, 3]. Opioids are potent painkillers derived from the alkaloid containing opium secretions of the poppy plant (papaver somniferum). Opioid alkaloids stimulate opioid receptors, which belong to the G protein-coupled receptor (GPCR) family and are normally activated by endogenous opioid peptides including endorphins, enkephalins and dynorphins to control pain and stress, and regulate reward and mood [4]. The opioid system is composed of three major subtypes of opioid receptors mu (MOR), delta (DOR), and kappa (KOR). While all three opioid receptors are promising drug targets for different aspects of mood and pain, MORs are the only receptors that can be activated by opioids such as morphine to induce potent analgesia [5, 6]. Evidence from knockout mice determined that this key receptor regulates pain relief and produces reward and is therefore the target of most opioid drug discovery programs [7, 8]. MOR agonists that are currently prescribed for pain management include morphine, oxycodone and fentanyl. Although these MOR targeted opioids are the most potent analgesics clinically available, they are also highly addictive and are at the center of a growing addiction epidemic in North America [9]. In rodent and human brains, MORs are strongly expressed in pain centers (periaqueductal grey, thalamus, cortex and amygdala), but also in respiratory centers of the brainstem and in reward areas (striatum and ventral tegmental area) [10, 11], and thus, these attractive pain targets are also responsible for respiratory complications and the development of addiction [6]. Drug discovery efforts have never been more concentrated on delivering the Holy Grail, i. e. a new opioid that retains the desired MOR-driven analgesic properties but lacks deadly respiratory and unwanted addictive effects.
This review examines current drug discovery strategies towards safer MOR-targeted therapies in pain management. As shown in Figure 1, we summarize drug design approaches targeting MORs based on current literature including biased agonism, peripheral MORs, MORs in heteromers, allostery and exploiting physiology with peptide analogues or modulators.
Figure 1. Drug discovery strategies targeting mu opioid receptors.

This scheme demonstrates current drug design strategies aimed at improving the safety of painkilling drugs by modulating mu opioid receptor (MOR) activity. The approaches discussed here include biased agonists, peripherally limited opioids, bivalent ligands acting at heteromers, allosteric modulators, and various approaches modulating opioid peptides. Colored arrows indicate potential drug induced signaling and physiological response(s) for each drug strategy. Physiological responses associated with signaling pathways are indicated below in matching colors including G protein mediated analgesia and β-arrestin signaling associated to constipation and respiratory depression however the pathway(s) rewarding properties of opioids remain unassociated to either pathway. Abbreviations: plasma membrane (PM), extracellular regulated kinase ½ (ERKs) and G protein-coupled receptor kinases (GRKs), orthosteric site (1), allosteric site (2). References are from the text.
2. Biased agonists
Ligand-receptor interactions have long-been viewed as a two-step process, first the ligand binds to a receptor inducing a conformational change that second, initiates downstream signaling pathways [12]. Over the years pharmacological studies have further demonstrated that ligands may have functional selectivity or “biased agonism” at different receptor-effector (i.e. G proteins, G protein-coupled receptor kinases (GRKs), arrestins) complexes to activate distinct pathways, which together give rise to wanted and unwanted drug effects [13]. In fact, the signaling response depends on the active ligand-receptor-effector complex that is formed and determines the downstream biological consequences [14, 15]. Designing biased ligands to induce a receptor conformation that elicits a distinct and preferred effect on cellular signaling is a current drug discovery strategy that could yield drugs with high potency, low toxicity and few adverse effects [16].
MOR activation involves several effectors and biased agonism at MOR is considered a feasible path to discover new drugs to efficiently kill pain with fewer side effects. Activation of MORs can lead to multiple signaling effects including G protein-dependent (Gi, Go, Gz) and independent signaling, the latter including phosphorylation by kinases such as extracellular regulated kinase ½ (ERKs) and G protein-coupled receptor kinases (GRKs) or recruitment of β-arrestins Figure 1 [12, 17]. Activation of MORs induce G protein-dependent inhibition of cyclic adenosine monophosphate (cAMP) and this G protein pathway is thought to mediate analgesia whereas G protein-independent pathways, internalization/β-arrestin interactions, are proposed to cause adverse effects of opioids such as constipation and respiratory depression [18]. This idea is based on initial findings that mice lacking β-arrestin 2 showed enhanced morphine analgesia with lower constipation and respiratory depression [19]. Additionally, a recent study elegantly addressed the impact of biased agonism at MORs by designing a panel of novel MOR ligands whose signaling properties range from balanced to bias at either G protein or β-arrestin [20]. The ligand’s bias factor for G protein signaling correlated with its’ therapeutic window [20] thus, supporting the notion that MOR signaling targeted to G protein activation would promote analgesia with fewer side effects. Over the last decade therefore, a focus of opioid drug discovery has been to design a biased MOR agonist with a G protein-biased profile [13, 21].
Below, we review several strategies being used to design G protein biased MOR agonists including measuring bias at existing opioids, deriving compounds from natural products (herkinorin, mitragynine and PnPP-19), conventional drug screening (TRV130), and employing MOR structural data (PZM21).
2.1. Bias among existing clinical opioid analgesics
Distinguishing current clinical MOR analgesics by their MOR signaling responses can advance mechanistic understanding underlying drug-induced therapeutic effects. Since the original observation that Met-enkephalin but not morphine promotes receptor internalization [22], many studies have demonstrated biased properties for MOR agonists [23]. A notable recent study by McPherson and colleagues compared the intrinsic efficacies for twenty-two preclinical and clinical MOR agonists in two pathways, G protein activation using GTPγS or β-arrestin recruitment in cells overexpressing MORs [24]. They found a significant positive correlation between the two pathways by analyzing the intrinsic efficacies for the series of MOR agonists [24]. Thus, compounds with low intrinsic efficacy for G protein activation also displayed low intrinsic efficacy for arrestin recruitment while compounds that display high intrinsic efficacy for G protein activation also showed high intrinsic efficacy to recruit arrestin [24]. Although bias factors were not reported, the authors described etorphine, primarily used in veterinary medicine as an animal tranquilizer and fentanyl, a rapid onset surgical analgesic, as having apparent bias toward β-arrestin recruitment [24], the pathway associated with undesirable effects. In contrast, morphine and oxycodone displayed no bias, but rather exhibit low intrinsic efficacy at both G protein and β-arrestin pathways [24]. Finally, among the clinical MOR agonists, buprenorphine was the only drug that did not recruit β-arrestin, thus displaying selectivity for the G protein pathway as do most recently developed biased MOR drugs (see below). Thus, both the intrinsic drug efficacy and the nature of MOR agonist/receptor/effector complexes formed at the membrane contribute to elaborate the signaling response.
Buprenorphine (Subutex) is worth a specific comment. This drug has long been endorsed for opioid use disorder therapy, but has been less exploited for its potent analgesia [25]. Buprenorphine binds to all opioid receptors, producing partial agonism at MOR, inverse agonism at KOR, antagonism at DOR and weak agonism at nociception/orphaninFQ peptide receptor (NOP) [26, 27, 28]. As an analgesic, buprenorphine is as efficient as morphine and is also approved for chronic pain prescription with low risk of side effects (hyperalgesia, abuse, respiratory depression and overdose) [26, 27, 28]. Furthermore transdermal buprenorphine is an available efficient method of pain management that causes less constipation and sedation compared to oral opioids [29]. Thus, there is a growing consensus that buprenorphine is an already available safer MOR agonist for patient pain management [27, 30]. However, careful consideration about patient dosing is advised as buprenorphine is not always suitable for acute pain and may produce mild side effects such as constipation, dependence and withdrawal symptoms [31].
2.2. Biased MOR agonists derived from natural products
Here, we discuss three drugs derived from natural sources that activate MORs in a Gi-biased manner. Salvia divinorum, is from the Sage family and has been used for mystical purposes due to its hallucinogenic properties [32]. Herkinorin, was synthesized from salvinorin A, the active component of Salvia divinorum, through addition of an aromatic group, and is the first MOR biased agonist derived from a natural product [13, 33, 34]. Although herkinorin is selective for MOR, lower binding also occurs at KOR and even lower at DOR [35]. Herkinorin has been shown, in rodents to produce potent analgesia [36] and in cells to be G protein biased, possessing poor β-arrestin 2 recruitment [37] however its utility is not yet realized due to poor permeability crossing the blood brain barrier.
Kratom is a plant that has been used for stimulant, pain-killing and sedative effects and its main alkaloid is mitragynine [38]. Mitragynine pseudoindoxyl is an oxidative rearrangement product of the corynanthe alkaloid mitragynine and activates MOR signaling at G proteins but does not recruit β-arrestin 2 and strongly produces analgesia in rodents [39]. Importantly, mitragynine specifically activates MORs and antagonizes DOR suggesting that the mechanism of action may involve both opioid receptors [39]. However, in the U.S. the drug enforcement agency (DEA) has categorized the active component in kratom as a schedule I drug potentially limiting the therapeutic reach of this drug.
Finally, an antinociceptive synthetic peptide, PnPP-19, that contains nineteen amino acid residues of the toxin δ-CNTX-Pn1c (PnTx2–6) isolated from the venom of the spider, Phoneutria nigriventer [40]. PnPP-19 was demonstrated to induce both peripheral and central nociception [41] with low potency but selective activation of MORs inducing G protein stimulated indirect inhibition of calcium channels without β-arrestin recruitment [42] and by modulating calcium channels [42] and cannabinoid receptors [41]. It is interesting to note that the non-opioid calcium channel inhibitors like gabapentin, pregabalin and ziconotide are effective safer clinical analgesics for some pain patient populations [43] and thus, targeting MORs to inhibit calcium channels albeit as an indirect outcome of G protein signaling, is another promising avenue to develop safer MOR analgesics [44]. Thus, PnPP-19 may potentially offer improved design strategies for MOR opioid analgesics.
2.3. Conventional drug screening methods
While the study of naturally sourced MOR agonists may lead to defining novel modes of MOR activation, conventional drug screening efforts also lead to novel compounds with the desired G protein-biased signaling profile. One biased MOR agonist that has been identified from existing chemical library screening is Trevena’s TRV130 [45]. Compared to morphine, TRV130 activates Gi/o protein signaling without β-arrestin recruitment [45], and shows rapid analgesia in mice and rats using the hot plate test [45] and in mice using tail immersion test [46]. Note however that another preclinical study reported TRV130-induced side effects including constipation and potential for abuse in rodents [46, 47]. Remarkably, TRV130 is the first G protein-biased MOR agonist that has gone to clinical trials as a treatment focusing on post-surgery pain patients [48]. Clinical studies are expected to evaluate this compound in the clinic.
2.4. MOR structure-based drug design
An innovative drug discovery strategy is to use the solved crystal structural data of the drug target. Recent drug discovery efforts have used crystal structures of aminergic family GPCRs to computationally dock molecules, determining ligands with new scaffolds and nanomolar range potencies [50]. The inactive mouse MOR 2.8 Å crystal structure was determined in 2012 by X-Ray crystallography [51]. The binding pocket permits the ligand to bind deeply within a solvent-exposed space, distinct from the more commonly observed deeply buried binding pockets in other GPCRs. In a first structure-based MOR targeted campaign, over 3 million compounds were computationally docked against the inactive MOR structure to identify a novel chemical entity entirely distinct from known MOR agonists, which was further optimized to produce PZM21 [52]. This novel MOR opioid has unmatched selectivity, potency and biased G protein signaling that produces long-lasting analgesia with less unfavorable behavioral properties (respiratory depression, locomotor and reward) [52]. Additionally, PZM21 activation leads to low MOR internalization and undetectable β-arrestin 2 recruitment even in the presence of overexpressed GRK2 [52]. PZM21 encompasses a potential to be a safer pain killer however, a recent study has demonstrated that in their experimental conditions PZM21 could produce respiratory depression at the same level as morphine [53] suggesting that further research will be needed to uphold the high promise of this new opioid.
3. Other strategies to design safer compounds targeting MORs
Although much of the focus in the last decade has been on developing biased compounds, several other promising strategies are actively pursued including, targeting peripheral MORs, heteromers, allosteric sites on MORs or modulating endogenous MOR peptides. In fact, these approaches existed before the concept of biased agonism even existed.
3.1. Peripherally limited opioids
The concept of peripherally limited ligands arose from the desire to limit addictive central effects of opioids such as reward [54, 55, 56]. Peripherally limited compounds are generally designed to be hydrophilic to lower their ability to cross the blood brain barrier. Loperamide (Imodium) was the first commercial peripherally limited MOR agonist. Loperamide was also shown to reduce heat and mechanical hyperalgesia in nerve-injured rats [58] yet, in human it is not used as an analgesic. Since then, new designs have been used to generate peripherally limited MOR agonists including arylacetamide triazaspiro morphinan based (DiPOA), and peptidic compounds (DALDA) [54]. However, these new modifications have a disadvantage of reducing the ligand binding affinity at MORs and some of the compounds can still cross the blood brain barrier at high doses [54]. Another strategy has been to use morphine covalently attached to a hyperbranched polyglycerol (PG-M) by a cleavable linker to relegate morphine to peripheral release in selective inflamed tissue without crossing the blood-brain barrier [59]. This strategy offers safer opioid analgesics by peripherally restricting and surpassing central and intestinal side effects, however the efficacy of this approach still needs to be determined in humans.
Recently, integration of the active MOR structural data [60] with computational simulation approaches brought on a key advance in producing a nontoxic opioid painkiller that selectively activates peripheral MORs in inflamed tissues [60, 61]. An accepted hallmark of injured tissue is lowered pH, the study designed a ligand that is protonated and thus able to activate MOR only in inflamed tissue. This pH-sensitive ligand yielded inflammatory-restricted analgesia in rats without central or intestinal adverse effects. This new compound (NFEPP) produces an analgesic effect during neuropathic and abdominal pain [61]. An interesting extension of this work comes from another closely related compound, FF3, that revealed an important criteria for the development of pH-sensitive ligands to possess selective peripheral analgesia without side effects, is to have a pKa that corresponds to the pH of injured tissue [62]. Taken together, the targeting an “acidic” version of MOR represents a pioneering approach to produce safer opioids.
3.2. MORs in heteromers for pain relief
There is considerable biochemical and functional evidence for the existence of MOR and DOR heteromers with distinct pharmacology [63] [64]. Furthermore, knock-in mice expressing MOR-mCherry and DOR-eGFP showed MOR/DOR co-localization in neurons from regions regulating pain, such as periaqueductal gray and pons [10]. Targeting MORs in heteromers to design safer painkillers, may thus be an approach that would take advantage of the restricted tissue distribution of heteromers and unique signaling properties of such complexes [65]. Several compounds selectively targeting MOR-DOR heteromers have been developed and produce analgesia with less tolerance [64]. Among them are, (i) CYM51010, a small molecule identified by ligand screening that produces an analgesic effect similar to morphine in mice [64], (ii) MDAN-21 a bivalent ligand in which a MOR agonist (oxymorphone) was linked to DOR antagonist (naltrindole) inducing thermal analgesia with lower dependence in rhesus monkeys [66] and eluxadoline (viberzi), a clinical orally active MOR agonist and DOR antagonist for the treatment of diarrhea-predominant irritable bowel syndrome [67, 68]. However, recent in-vivo evidence also shows that, in fact, MOR and DOR co-expressed in the same neurons internalize and function independently [69] questioning the development of novel analgesics based on the concept of MOR-DOR heteromerization. Another well-documented heteromer consists of MOR in complex with metabotropic glutamate receptor 5 (mGluR5). A bivalent ligand, MMG22, has been developed to activate MOR-mGluR5 heteromers and produces potent reversal of tactile hypersensitivity [70] and reduces neuropathic pain [71] in rodents. Altogether, preclinical studies support the notion that MOR targeting in heteromers in-vivo is an option, but further mechanistic studies are required and clinical evaluation remains a far-reaching goal.
Another approach is to take advantage of buprenorphine’s mixed pharmacology, which has been proposed to exert agonism at mu, with also weak agonism at the, inverse agonism at kappa and antagonism at delta opioid receptors [26]. Recently, a buprenorphine analogue was developed with bifunctionality, at MOR and the NOP receptor, as a potential non-addictive analgesic [72]. A translational study in primates, demonstrates that BU08028 induces robust analgesia with less reinforcing effects as compared to cocaine, remifentanil or buprenorphine and no respiratory depression was observed at 30-fold higher than effective analgesic doses [73, 74]. The combination of an exceptional pharmacological and behavioral profile for buprenorphine should inspire future studies to identify the structural components, cellular spatiotemporal signaling dynamics and physiological responses of buprenorphine’s effects. With these mechanistic insights in hand, perhaps, modifications on buprenorphine may produce an innovative opioid with excellent therapeutic profile for pain management.
3.3. Allosteric MOR drugs
Another approach to developing new opioid painkillers with less side effects is to design allosteric modulators [75, 76]. Endogenous and classical clinical MOR drugs bind to an orthosteric site on the receptor to activate MOR through a change in structural conformation [75, 76]. An appealing strategy is to take advantage of another ligand binding site on the MOR, the allosteric site, to modulate orthosteric activation [75, 76]. These allosteric ligands could either facilitate MOR signaling and are called positively modulators (PAMs), or either inhibit MOR signaling and are named negative allosteric modulators (NAMs) [75, 76]. Based on this concept, PAMs could be used in combination with classical orthosteric binding opioid analgesics such as morphine, fentanyl or oxycodone to reduce the dose of these opioid painkillers, and thus making them safer [5]. Another possible use of the allosteric ligand is to enhance the signaling of endogenous opioids which are naturally released during pain or stress [77]. This option is advantageous because it should in principle induce a temporary and local analgesic response at the exact site where the pain is occurring in the body, limiting the risk of developing side effects. BMS-986122 was recently identified as a PAM from a high-throughput campaign [78]. BMS-986122 enhances endomorphin signaling by binding to the Na (+) modulatory site of MOR [78]. Additional compounds of the MOR-PAM family such as BMS-986187, are now proposed as new therapeutics with potentially fewer side effects [75, 79]. All these allosteric compounds are currently tested preclinically and will need to be evaluated clinically. Furthermore, cellular studies have suggested salvinorin A, THC and cannabidiol are potential NAMs at MORs but in vivo studies are required to evaluate the physiological effects of MOR NAMs and whether they may be useful clinical compounds [75].
3.4. Endogenous opioid peptide analogues and modulators
Another effort has been made to generate endogenous opioid peptide analogues [80]. This approach is appealing as it will use the naturally produced analgesic properties of endogenous opioids and improve at the same time their stability and bioavailability. Several methods have been used during the last few decades to make peptides druggable such as glycosylation, cyclization, modification of the C- and N-terminal and packaging of opioid peptides and are reviewed in detail in [80]. These strategies are still in their early stages, requiring a significant need of development due to the challenges in stabilizing peptides with short in-vivo half-life, and increasing their bioavailability making them capable of crossing the blood brain barrier. Another interesting approach consists of increasing peptide levels by inhibiting the enzyme(s) responsible for endogenous opioid peptide degradation [81]. The enzymes that degrade endogenous enkephalin opioid peptides are known as enkephalinases. The designed inhibitors of enkephalinases such as aminopeptidase N and neutral endopeptidase neprilysn (NEP), are called racecadotril, ubenimex (bestatin), and RB-101. Remarkably, preclinical observations support that these enkephalinase inhibitors are capable of producing efficient analgesia without respiratory depression, reward, tolerance and constipation [82]. This strategy is very appealing because it circumvents exogenous opioid use thereby avoiding dependency issues and lethal complications like respiratory depression, however until now enkephelinase inhibitors have not made it into the clinic to treat pain.
4. Conclusion
Today’s potent opioid analgesics are unmatched in pain relief, but their highly addictive rewarding euphoric effects and lethal respiratory complications have made their use a danger for over 3000 years. We have reviewed recent MOR targeting efforts to evaluate current attempts to design an opioid with efficient pain relief and minimal side effects, a major goal in biomedicine. Clinicians and researchers alike are arduously working towards producing a safer MOR targeted therapeutic that will hopefully soon come to fruition. In fact, the current opioid epidemic is inducing a revival of MOR drug discovery campaigns with innovative strategies that hold real promise to deliver the Holy Grail, i. e. safer opioid analgesics. Last but not least, efforts to develop more efficient non-opioid analgesics represent another entire research field to address the social challenges of opioid pain relief [1, 2, 3, 5].
5. Expert opinion
As summarized in Table 1, we compared how current opioids available to clinicians match up to opioids under development in terms of structure, origin, DEA classification of the United States, signaling and therapeutic relevance. Going forward there is a critical need to elucidate how to distinguish physiological effects of opioids in terms of receptor activities and signaling responses to aid drug discovery efforts. There are several considerations about physiological contexts that may impact the outcome of ligand-receptor interactions. First, interrogations as to how distinct drugs impose dissimilar conformational changes in the receptor, how much intrinsic efficacy a ligand has [83] and the duration that a ligand occupies a receptor may offer key distinguishing criteria. Second, is that ligands with “location-bias”, a property of alkaloid opioids but not peptides, can activate MORs to signal at subcellular membrane compartments such as golgi membranes to induce different signaling consequences [84]. A third aspect, is that the signaling context of MOR may be determined by environment as was shown for dopamine receptors which exhibit functional selectivity based on the availability of transducers known as “system bias” [85, 86]. Therefore, each opioid has the potential to activate MORs and induce diverse responses depending on how they bind to MORs, where they bind to MORs and the biological context in which they act.
Table 1 |.
Opioids under development: structure, signaling and therapeutic relevance
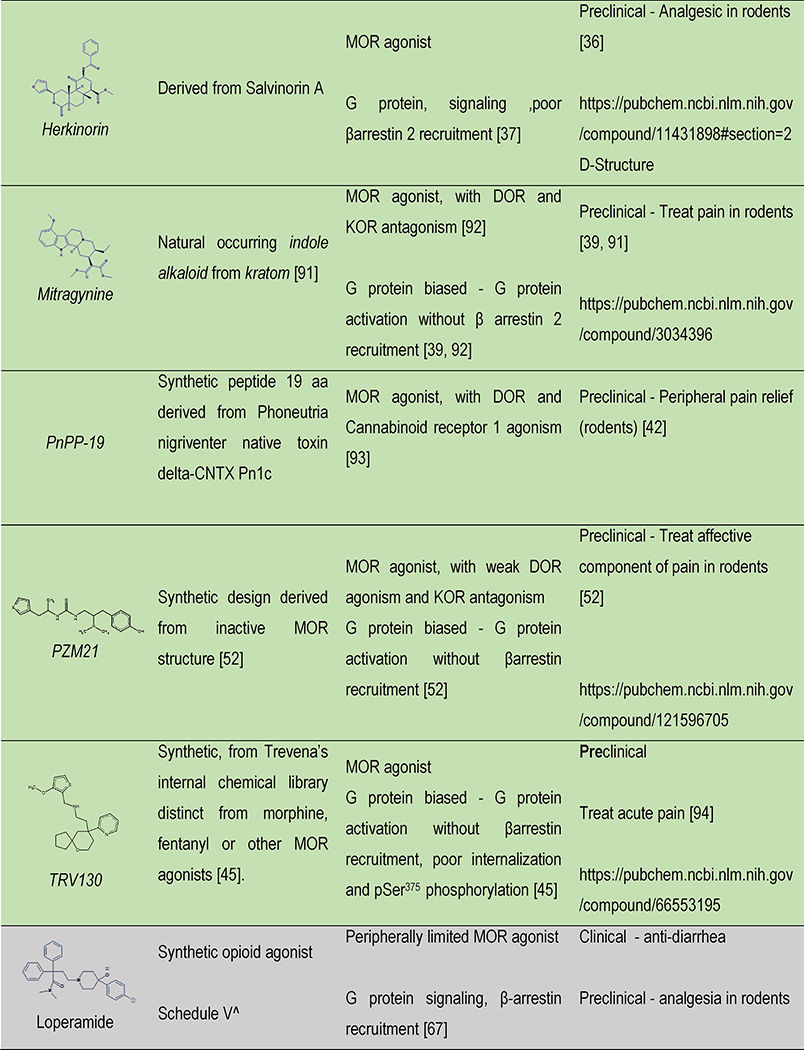 |
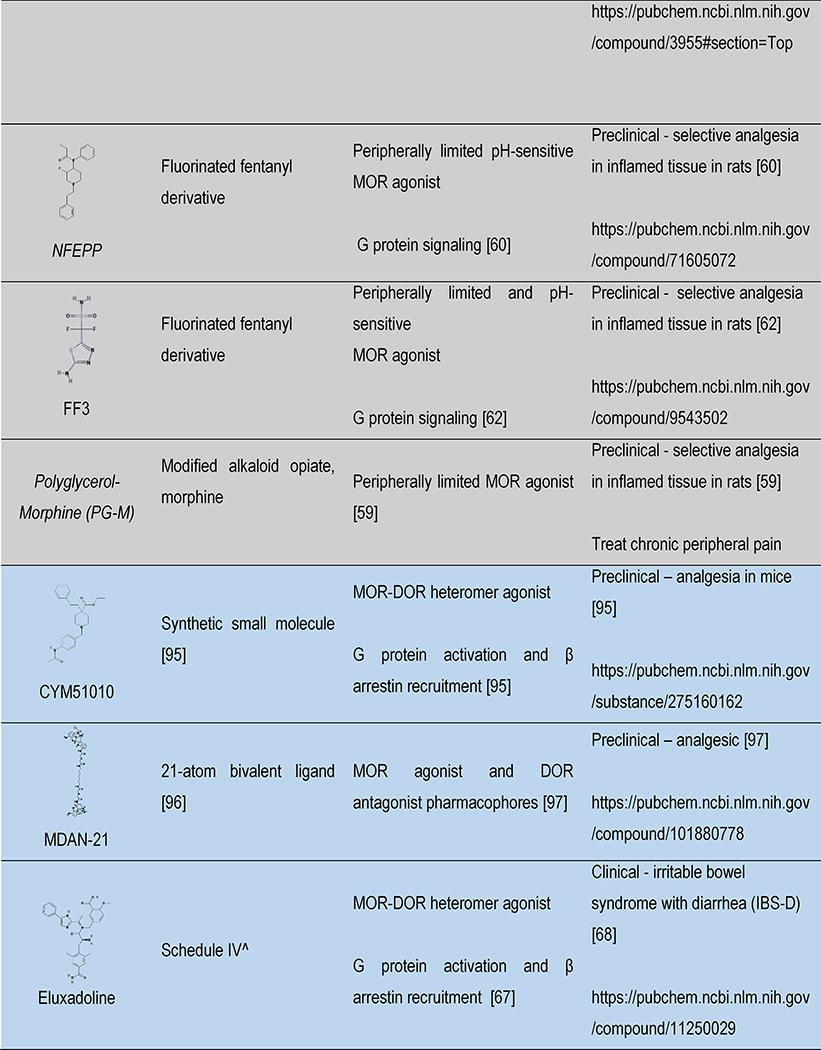 |
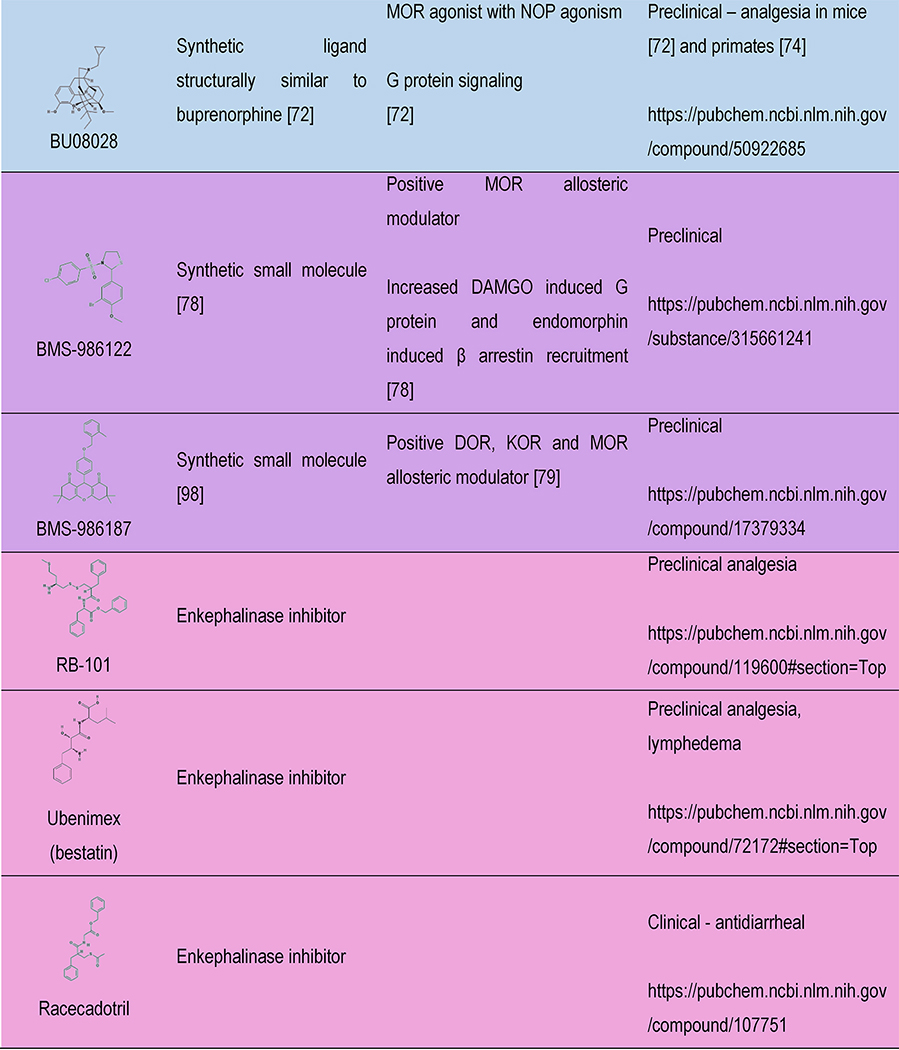 |
Color indicates the drug discovery strategy and matches with the colors in Figure 1. ^ indicates schedule of controlled substances and are up to date as of 01-Oct-18 according to https://www.deadiversion.usdoj.gov/schedules/
A common point among all the strategies that we have reviewed here, is the desire to produce efficient analgesic opioids with less side effects. The side effects that present the most risk to the patient are lethal respiratory depression and addictive euphoria [3]. Peripherally-limited opioids lack these unwanted effects but analgesic reach may depend on the type of pain [87]. Strategies like biased agonism, aim to reduce risk of developing lethal respiratory depression by designing ligands with lower β-arrestin signaling. At the cellular level, measurement of MOR induced β-arrestin signaling is a distinguishing criterion frequently used to predict whether a drug will produce side effects like constipation and respiratory depression [20]. However, addressing the highly-addictive euphoric and reinforcing effects of MOR agonists is more complex [17]. Examining rewarding effects of G protein-biased agonists like TRV130 and PZM21 using conditioned place preference (CPP) showed that MOR agonists which lack β-arrestin signaling and potently activate G proteins are devoid of reinforcing effects [45, 52]. These results may suggest that G protein pathways are not mediating reward properties of opioids however, there is also evidence that the rewarding effects of morphine are not mediated through β-arrestin signaling as β-arrestin knockout mice showed enhanced morphine CPP [88]. Furthermore, the current understanding on the neurobiology of opioid reward is that MORs are expressed on GABAergic interneurons of the ventral tegmental area (VTA) and mediate, presumably via G protein signaling, the disinhibition of VTA-dopaminergic projection to nucleus accumbens (Nac) pathway producing dopamine release in the Nac leading to euphoria [6, 55, 89, 90]. These observations provoke the question as to whether it is possible to treat pain without inducing reward, as pain relief is a process that is inherently rewarding. Furthermore, an important physiological aspect to clarify is to understand how biased ligands are producing different effects in preclinical analgesia tests. For example, PZM21 produced an analgesic effect in the affective but not in reflexive pain tests [52], whereas TRV130 produced an analgesic effect in both tests [45]. A better understanding, identification and characterization of MOR activities through the G protein pathway with respect to dissecting rewarding and analgesic properties will be critical for the discovery and designing of safer MOR agonists.
Article Highlights.
Opioid analgesics are unsurpassed painkillers but are hampered by severe adverse effects
Emerging biased agonists may soon be available as safer opioids
There are new approaches to the development of peripherally limited opioids as safer painkillers
Strategies to develop safer opioid pain killers include targeting MORs in heteromers, MOR allostery and the development of endogenous opioid peptide analogues
There is an urgent need to clarify the mechanisms underlying the unwanted effects of MOR activation; this may pave safer ways to target MORs
Acknowledgments
Funding
This work was funded by National Institute of Health (National Institute of Drug Abuse Grant number. 05010 and National Institute on Alcohol Abuse and Alcoholism, Grant number 16658 awarded to BL Kieffer), the Canada Fund for Innovation and the Canada Research Chairs awarded to E Darcq and BL Kieffer.
Footnotes
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers
- 1.Basbaum AI, Bautista DM, Scherrer G, et al. Cellular and molecular mechanisms of pain. Cell. 2009. October 16;139(2):267–84. doi: 10.1016/j.cell.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reuben DB, Alvanzo AA, Ashikaga T, et al. National Institutes of Health Pathways to Prevention Workshop: the role of opioids in the treatment of chronic pain. Ann Intern Med. 2015. February 17;162(4):295–300. doi: 10.7326/M14-2775. [DOI] [PubMed] [Google Scholar]
- 3.Volkow N, Benveniste H, McLellan AT. Use and Misuse of Opioids in Chronic Pain. Annu Rev Med. 2018. January 29;69:451–465. doi: 10.1146/annurev-med-011817-044739. [DOI] [PubMed] [Google Scholar]
- 4.Bodnar RJ. Endogenous Opiates and Behavior: 2016. Peptides. 2018. March;101:167–212. doi: 10.1016/j.peptides.2018.01.011. [DOI] [PubMed] [Google Scholar]
- 5.Valentino RJ, Volkow ND. Untangling the complexity of opioid receptor function. Neuropsychopharmacology. 2018. September 24. doi: 10.1038/s41386-018-0225-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Darcq E, Kieffer BL. Opioid receptors: drivers to addiction? Nat Rev Neurosci. 2018. August;19(8):499–514. doi: 10.1038/s41583-018-0028-x. [DOI] [PubMed] [Google Scholar]
- 7.Kieffer BL. Opioids: first lessons from knockout mice. Trends Pharmacol Sci. 1999. January;20(1):19–26. [DOI] [PubMed] [Google Scholar]
- 8.Matthes HW, Maldonado R, Simonin F, et al. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature. 1996. October 31;383(6603):819–23. doi: 10.1038/383819a0.** Employing genetically modified mice, this report demonstrated for the first time that morphine induced responses including, analgesic and side effects, are only mediated by MORs.
- 9.Curbing prescription opioid dependency. Bull World Health Organ. 2017. May 1;95(5):318–319. doi: 10.2471/BLT.17.020517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Erbs E, Faget L, Scherrer G, et al. A mu-delta opioid receptor brain atlas reveals neuronal co-occurrence in subcortical networks. Brain structure & function. 2015. March;220(2):677–702. doi: 10.1007/s00429-014-0717-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pecina M, Karp JF, Mathew S, et al. Endogenous opioid system dysregulation in depression: implications for new therapeutic approaches. Mol Psychiatry. 2018. June 28. doi: 10.1038/s41380-018-0117-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Williams JT, Ingram SL, Henderson G, et al. Regulation of mu-opioid receptors: desensitization, phosphorylation, internalization, and tolerance. Pharmacol Rev. 2013. January;65(1):223–54. doi: 10.1124/pr.112.005942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Madariaga-Mazon A, Marmolejo-Valencia AF, Li Y, et al. Mu-Opioid receptor biased ligands: A safer and painless discovery of analgesics? Drug Discov Today. 2017. November;22(11):1719–1729. doi: 10.1016/j.drudis.2017.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kenakin T The Effective Application of Biased Signaling to New Drug Discovery. Mol Pharmacol. 2015. December;88(6):1055–61. doi: 10.1124/mol.115.099770. [DOI] [PubMed] [Google Scholar]
- 15.Costa-Neto CM, Parreiras ESLT, Bouvier MA Pluridimensional View of Biased Agonism. Mol Pharmacol. 2016. November;90(5):587–595. doi: 10.1124/mol.116.105940. [DOI] [PubMed] [Google Scholar]
- 16.Thompson GL, Lane JR, Coudrat T, et al. Systematic analysis of factors influencing observations of biased agonism at the mu-opioid receptor. Biochem Pharmacol. 2016. August 1;113:70–87. doi: 10.1016/j.bcp.2016.05.014. [DOI] [PubMed] [Google Scholar]
- 17.Al-Hasani R, Bruchas MR. Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology. 2011. December;115(6):1363–81. doi: 10.1097/ALN.0b013e318238bba6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Raehal KM, Schmid CL, Groer CE, et al. Functional selectivity at the mu-opioid receptor: implications for understanding opioid analgesia and tolerance. Pharmacol Rev. 2011. December;63(4):1001–19. doi: 10.1124/pr.111.004598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bohn LM, Lefkowitz RJ, Gainetdinov RR, et al. Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science. 1999. December 24;286(5449):2495–8. [DOI] [PubMed] [Google Scholar]
- 20.Schmid CL, Kennedy NM, Ross NC, et al. Bias Factor and Therapeutic Window Correlate to Predict Safer Opioid Analgesics. Cell. 2017. November 16;171(5):1165–1175 e13. doi: 10.1016/j.cell.2017.10.035.** This study compares bias factors with the analgesia–respiratory depression therapeutic window, and predicts safer opioid pain killers.
- 21.Smith JS, Lefkowitz RJ, Rajagopal S. Biased signalling: from simple switches to allosteric microprocessors. Nat Rev Drug Discov. 2018. January 5. doi: 10.1038/nrd.2017.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keith DE, Murray SR, Zaki PA, et al. Morphine activates opioid receptors without causing their rapid internalization. J Biol Chem. 1996. August 9;271(32):19021–4. [DOI] [PubMed] [Google Scholar]
- 23.Pradhan AA, Smith ML, Kieffer BL, et al. Ligand-directed signalling within the opioid receptor family. Br J Pharmacol. 2012. November;167(5):960–9. doi: 10.1111/j.1476-5381.2012.02075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McPherson J, Rivero G, Baptist M, et al. mu-opioid receptors: correlation of agonist efficacy for signalling with ability to activate internalization. Mol Pharmacol. 2010. October;78(4):756–66. doi: 10.1124/mol.110.066613.* This is the first work to thoroughly determine the intrinsic efficacies of 22 MOR agonists to activate Gi and/or β arrestin pathways.
- 25.Butler S Buprenorphine-Clinically useful but often misunderstood. Scand J Pain. 2013. July 1;4(3):148–152. doi: 10.1016/j.sjpain.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 26.Davis MP, Pasternak G, Behm B. Treating Chronic Pain: An Overview of Clinical Studies Centered on the Buprenorphine Option. Drugs. 2018. July 26. doi: 10.1007/s40265-018-0953-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fishman MA, Kim PS. Buprenorphine for Chronic Pain: a Systemic Review. Curr Pain Headache Rep. 2018. October 5;22(12):83. doi: 10.1007/s11916-018-0732-2. [DOI] [PubMed] [Google Scholar]
- 28.Hale M, Urdaneta V, Kirby MT, et al. Long-term safety and analgesic efficacy of buprenorphine buccal film in patients with moderate-to-severe chronic pain requiring around-the-clock opioids. J Pain Res. 2017;10:233–240. doi: 10.2147/JPR.S120170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Skaer TL. Dosing considerations with transdermal formulations of fentanyl and buprenorphine for the treatment of cancer pain. J Pain Res. 2014;7:495–503. doi: 10.2147/JPR.S36446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ehrlich AT, Darcq E. Recommending buprenorphine for pain management. Pain Manag. 2019. January 1;9(1):13–16. doi: 10.2217/pmt-2018-0069. [DOI] [PubMed] [Google Scholar]
- 31.Vlok R, An GH, Binks M, et al. Sublingual buprenorphine versus intravenous or intramuscular morphine in acute pain: A systematic review and meta-analysis of randomized control trials. Am J Emerg Med. 2018. May 24. doi: 10.1016/j.ajem.2018.05.052. [DOI] [PubMed] [Google Scholar]
- 32.Valdes LJ 3rd, Diaz JL, Paul AG. Ethnopharmacology of ska Maria Pastora (Salvia divinorum, Epling and Jativa-M.). J Ethnopharmacol. 1983. May;7(3):287–312. [DOI] [PubMed] [Google Scholar]
- 33.Marmolejo-Valencia AF, Martinez-Mayorga K. Allosteric modulation model of the mu opioid receptor by herkinorin, a potent not alkaloidal agonist. J Comput Aided Mol Des. 2017. May;31(5):467–482. doi: 10.1007/s10822-017-0016-7. [DOI] [PubMed] [Google Scholar]
- 34.Harding WW, Tidgewell K, Byrd N, et al. Neoclerodane diterpenes as a novel scaffold for mu opioid receptor ligands. J Med Chem. 2005. July 28;48(15):4765–71. doi: 10.1021/jm048963m. [DOI] [PubMed] [Google Scholar]
- 35.Butelman ER, Rus S, Simpson DS, et al. The effects of herkinorin, the first mu-selective ligand from a salvinorin A-derived scaffold, in a neuroendocrine biomarker assay in nonhuman primates. J Pharmacol Exp Ther. 2008. October;327(1):154–60. doi: 10.1124/jpet.108.140079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lamb K, Tidgewell K, Simpson DS, et al. Antinociceptive effects of herkinorin, a MOP receptor agonist derived from salvinorin A in the formalin test in rats: new concepts in mu opioid receptor pharmacology: from a symposium on new concepts in mu-opioid pharmacology. Drug Alcohol Depend. 2012. March 1;121(3):181–8. doi: 10.1016/j.drugalcdep.2011.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Groer CE, Tidgewell K, Moyer RA, et al. An opioid agonist that does not induce mu-opioid receptor--arrestin interactions or receptor internalization. Mol Pharmacol. 2007. February;71(2):549–57. doi: 10.1124/mol.106.028258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jansen KL, Prast CJ. Ethnopharmacology of kratom and the Mitragyna alkaloids. J Ethnopharmacol. 1988. May-Jun;23(1):115–9. [DOI] [PubMed] [Google Scholar]
- 39.Varadi A, Marrone GF, Palmer TC, et al. Mitragynine/Corynantheidine Pseudoindoxyls As Opioid Analgesics with Mu Agonism and Delta Antagonism, Which Do Not Recruit beta-Arrestin-2. J Med Chem. 2016. September 22;59(18):8381–97. doi: 10.1021/acs.jmedchem.6b00748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Silva CN, Nunes KP, Torres FS, et al. PnPP-19, a Synthetic and Nontoxic Peptide Designed from a Phoneutria nigriventer Toxin, Potentiates Erectile Function via NO/cGMP. J Urol. 2015. November;194(5):1481–90. doi: 10.1016/j.juro.2015.06.081. [DOI] [PubMed] [Google Scholar]
- 41.Freitas AC, Pacheco DF, Machado MF, et al. PnPP-19, a spider toxin peptide, induces peripheral antinociception through opioid and cannabinoid receptors and inhibition of neutral endopeptidase. Br J Pharmacol. 2016. May;173(9):1491–501. doi: 10.1111/bph.13448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Freitas ACN, Peigneur S, Macedo FHP, et al. The Peptide PnPP-19, a Spider Toxin Derivative, Activates mu-Opioid Receptors and Modulates Calcium Channels. Toxins (Basel). 2018. January 15;10(1). doi: 10.3390/toxins10010043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Durkin B, Page C, Glass P. Pregabalin for the treatment of postsurgical pain. Expert Opin Pharmacother. 2010. November;11(16):2751–8. doi: 10.1517/14656566.2010.526106. [DOI] [PubMed] [Google Scholar]
- 44.Chan HCS, McCarthy D, Li J, et al. Designing Safer Analgesics via mu-Opioid Receptor Pathways. Trends Pharmacol Sci. 2017. November;38(11):1016–1037. doi: 10.1016/j.tips.2017.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.DeWire SM, Yamashita DS, Rominger DH, et al. A G protein-biased ligand at the mu-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J Pharmacol Exp Ther. 2013. March;344(3):708–17. doi: 10.1124/jpet.112.201616.** This report identified a novel MOR biased agonist, TRV130, with promising properties to become a safer opioid pain killer.
- 46.Altarifi AA, David B, Muchhala KH, et al. Effects of acute and repeated treatment with the biased mu opioid receptor agonist TRV130 (oliceridine) on measures of antinociception, gastrointestinal function, and abuse liability in rodents. J Psychopharmacol. 2017. June;31(6):730–739. doi: 10.1177/0269881116689257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Negus SS, Freeman KB. Abuse Potential of Biased Mu Opioid Receptor Agonists. Trends Pharmacol Sci. 2018. November;39(11):916–919. doi: 10.1016/j.tips.2018.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Drug and Device News. P T 2017. April;42(4):223–265. [PMC free article] [PubMed] [Google Scholar]
- 49.Singla N, Minkowitz HS, Soergel DG, et al. A randomized, Phase IIb study investigating oliceridine (TRV130), a novel micro-receptor G-protein pathway selective (mu-GPS) modulator, for the management of moderate to severe acute pain following abdominoplasty. J Pain Res. 2017;10:2413–2424. doi: 10.2147/JPR.S137952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kieffer BL. Drug discovery: Designing the ideal opioid. Nature. 2016. September 08;537(7619):170–171. doi: 10.1038/nature19424. [DOI] [PubMed] [Google Scholar]
- 51.Manglik A, Kruse AC, Kobilka TS, et al. Crystal structure of the micro-opioid receptor bound to a morphinan antagonist. Nature. 2012. March 21;485(7398):321–6. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Manglik A, Lin H, Aryal DK, et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature. 2016. September 8;537(7619):185–190. doi: 10.1038/nature19112.** This study is the first to employ computational screening based on the MOR structure to discover a novel MOR Gi- biased agonist with promising biological properties towards safer analgesics.
- 53.Hill R, Disney A, Conibear A, et al. The novel mu-opioid receptor agonist PZM21 depresses respiration and induces tolerance to antinociception. Br J Pharmacol. 2018. July;175(13):2653–2661. doi: 10.1111/bph.14224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stein C, Machelska H. Modulation of peripheral sensory neurons by the immune system: implications for pain therapy. Pharmacol Rev. 2011. December;63(4):860–81. doi: 10.1124/pr.110.003145. [DOI] [PubMed] [Google Scholar]
- 55.Badiani A, Belin D, Epstein D, et al. Opiate versus psychostimulant addiction: the differences do matter. Nat Rev Neurosci. 2011. October 5;12(11):685–700. doi: 10.1038/nrn3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stein C Opioid Receptors. Annu Rev Med. 2016;67:433–51. doi: 10.1146/annurev-med-062613-093100. [DOI] [PubMed] [Google Scholar]
- 57.Lacy BE. Review article: an analysis of safety profiles of treatments for diarrhoea-predominant irritable bowel syndrome. Aliment Pharmacol Ther. 2018. October;48(8):817–830. doi: 10.1111/apt.14948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chung C, Carteret AF, McKelvy AD, et al. Analgesic properties of loperamide differ following systemic and local administration to rats after spinal nerve injury. Eur J Pain. 2012. August;16(7):1021–32. doi: 10.1002/j.1532-2149.2012.00148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gonzalez-Rodriguez S, Quadir MA, Gupta S, et al. Polyglycerol-opioid conjugate produces analgesia devoid of side effects. Elife. 2017. July 4;6. doi: 10.7554/eLife.27081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Spahn V, Del Vecchio G, Labuz D, et al. A nontoxic pain killer designed by modeling of pathological receptor conformations. Science. 2017. March 3;355(6328):966–969. doi: 10.1126/science.aai8636.** The authors designed a novel MOR agonist that acts only under conditions of inflammation and targets a ‘pathological’ (acidic) receptor structure, opening a new approach in the design of safer opioid pain killers.
- 61.Rodriguez-Gaztelumendi A, Spahn V, Labuz D, et al. Analgesic effects of a novel pH-dependent mu-opioid receptor agonist in models of neuropathic and abdominal pain. Pain. 2018. November;159(11):2277–2284. doi: 10.1097/j.pain.0000000000001328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Spahn V, Del Vecchio G, Rodriguez-Gaztelumendi A, et al. Opioid receptor signaling, analgesic and side effects induced by a computationally designed pH-dependent agonist. Sci Rep. 2018. June 12;8(1):8965. doi: 10.1038/s41598-018-27313-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fujita W, Gomes I, Devi LA. Heteromers of mu-delta opioid receptors: new pharmacology and novel therapeutic possibilities. Br J Pharmacol. 2015. January;172(2):375–87. doi: 10.1111/bph.12663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gomes I, Ayoub MA, Fujita W, et al. G Protein-Coupled Receptor Heteromers. Annu Rev Pharmacol Toxicol. 2016;56:403–25. doi: 10.1146/annurev-pharmtox-011613-135952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Massotte D In vivo opioid receptor heteromerization: where do we stand? Br J Pharmacol. 2015. January;172(2):420–34. doi: 10.1111/bph.12702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Aceto MD, Harris LS, Negus SS, et al. MDAN-21: A Bivalent Opioid Ligand Containing mu-Agonist and Delta-Antagonist Pharmacophores and Its Effects in Rhesus Monkeys. Int J Med Chem. 2012;2012:327257. doi: 10.1155/2012/327257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fujita W, Gomes I, Dove LS, et al. Molecular characterization of eluxadoline as a potential ligand targeting mu-delta opioid receptor heteromers. Biochem Pharmacol. 2014. December 1;92(3):448–56. doi: 10.1016/j.bcp.2014.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Scarpellini E, Laterza L, Ianiro G, et al. Eluxadoline for the treatment of diarrhoea-predominant irritable bowel syndrome. Expert Opin Pharmacother. 2016. July;17(10):1395–402. doi: 10.1080/14656566.2016.1182982. [DOI] [PubMed] [Google Scholar]
- 69.Wang D, Tawfik VL, Corder G, et al. Functional Divergence of Delta and Mu Opioid Receptor Organization in CNS Pain Circuits. Neuron. 2018. April 4;98(1):90–108 e5. doi: 10.1016/j.neuron.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Akgun E, Javed MI, Lunzer MM, et al. Ligands that interact with putative MOR-mGluR5 heteromer in mice with inflammatory pain produce potent antinociception. Proc Natl Acad Sci U S A. 2013. July 9;110(28):11595–9. doi: 10.1073/pnas.1305461110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Peterson CD, Kitto KF, Akgun E, et al. Bivalent ligand that activates mu opioid receptor and antagonizes mGluR5 receptor reduces neuropathic pain in mice. Pain. 2017. December;158(12):2431–2441. doi: 10.1097/j.pain.0000000000001050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Khroyan TV, Polgar WE, Cami-Kobeci G, et al. The first universal opioid ligand, (2S)-2-[(5R,6R,7R,14S)-N-cyclopropylmethyl-4,5-epoxy-6,14-ethano-3-hydroxy-6-meth oxymorphinan-7-yl]-3,3-dimethylpentan-2-ol (BU08028): characterization of the in vitro profile and in vivo behavioral effects in mouse models of acute pain and cocaine-induced reward. J Pharmacol Exp Ther. 2011. March;336(3):952–61. doi: 10.1124/jpet.110.175620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li JX. Buprenorphine analogue BU08028 is one step closer to the Holy Grail of opioid research. Proc Natl Acad Sci U S A. 2016. September 13;113(37):10225–7. doi: 10.1073/pnas.1612752113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ding H, Czoty PW, Kiguchi N, et al. A novel orvinol analog, BU08028, as a safe opioid analgesic without abuse liability in primates. Proc Natl Acad Sci U S A. 2016. September 13;113(37):E5511–8. doi: 10.1073/pnas.1605295113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Livingston KE, Traynor JR. Allostery at opioid receptors: modulation with small molecule ligands. Br J Pharmacol. 2018. July;175(14):2846–2856. doi: 10.1111/bph.13823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Remesic M, Hruby VJ, Porreca F, et al. Recent Advances in the Realm of Allosteric Modulators for Opioid Receptors for Future Therapeutics. ACS Chem Neurosci. 2017. June 21;8(6):1147–1158. doi: 10.1021/acschemneuro.7b00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stein C New concepts in opioid analgesia. Expert Opin Investig Drugs. 2018. October;27(10):765–775. doi: 10.1080/13543784.2018.1516204. [DOI] [PubMed] [Google Scholar]
- 78.Burford NT, Clark MJ, Wehrman TS, et al. Discovery of positive allosteric modulators and silent allosteric modulators of the mu-opioid receptor. Proc Natl Acad Sci U S A. 2013. June 25;110(26):10830–5. doi: 10.1073/pnas.1300393110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Livingston KE, Stanczyk MA, Burford NT, et al. Pharmacologic Evidence for a Putative Conserved Allosteric Site on Opioid Receptors. Mol Pharmacol. 2018. February;93(2):157–167. doi: 10.1124/mol.117.109561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Olson KM, Lei W, Keresztes A, et al. Novel Molecular Strategies and Targets for Opioid Drug Discovery for the Treatment of Chronic Pain. Yale J Biol Med. 2017. March;90(1):97–110. [PMC free article] [PubMed] [Google Scholar]
- 81.Noble F, Roques BP. Protection of endogenous enkephalin catabolism as natural approach to novel analgesic and antidepressant drugs. Expert Opin Ther Targets. 2007. February;11(2):145–59. doi: 10.1517/14728222.11.2.145. [DOI] [PubMed] [Google Scholar]
- 82.Roques BP, Fournie-Zaluski MC, Wurm M. Inhibiting the breakdown of endogenous opioids and cannabinoids to alleviate pain. Nat Rev Drug Discov. 2012. April;11(4):292–310. doi: 10.1038/nrd3673. [DOI] [PubMed] [Google Scholar]
- 83.Livingston KE, Mahoney JP, Manglik A, et al. Measuring ligand efficacy at the mu-opioid receptor using a conformational biosensor. Elife. 2018. June 22;7. doi: 10.7554/eLife.32499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Stoeber M, Jullie D, Lobingier BT, et al. A Genetically Encoded Biosensor Reveals Location Bias of Opioid Drug Action. Neuron. 2018. June 6;98(5):963–976 e5. doi: 10.1016/j.neuron.2018.04.021. PubMed PMID: 29754753.** The authors employed a real-time biosensor that recognizes the active conformation of MORs and reveals that peptidic opioids act at plasma membrane and endosomes whereas non-peptide opioids uniquely act at MORs in golgi membranes.
- 85.Urs NM, Gee SM, Pack TF, et al. Distinct cortical and striatal actions of a beta-arrestin-biased dopamine D2 receptor ligand reveal unique antipsychotic-like properties. Proc Natl Acad Sci U S A. 2016. December 13;113(50):E8178–E8186. doi: 10.1073/pnas.1614347113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Marcott PF, Gong S, Donthamsetti P, et al. Regional Heterogeneity of D2-Receptor Signaling in the Dorsal Striatum and Nucleus Accumbens. Neuron. 2018. May 2;98(3):575–587 e4. doi: 10.1016/j.neuron.2018.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stein C Targeting pain and inflammation by peripherally acting opioids. Front Pharmacol. 2013;4:123. doi: 10.3389/fphar.2013.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bohn LM, Gainetdinov RR, Sotnikova TD, et al. Enhanced rewarding properties of morphine, but not cocaine, in beta(arrestin)-2 knock-out mice. J Neurosci. 2003. November 12;23(32):10265–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Johnson SW, North RA. Opioids excite dopamine neurons by hyperpolarization of local interneurons. J Neurosci. 1992. February;12(2):483–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wise RA, Rompre PP. Brain dopamine and reward. Annu Rev Psychol. 1989;40:191–225. doi: 10.1146/annurev.ps.40.020189.001203. [DOI] [PubMed] [Google Scholar]
- 91.Kruegel AC, Grundmann O. The medicinal chemistry and neuropharmacology of kratom: A preliminary discussion of a promising medicinal plant and analysis of its potential for abuse. Neuropharmacology. 2018. May 15;134(Pt A):108–120. doi: 10.1016/j.neuropharm.2017.08.026. [DOI] [PubMed] [Google Scholar]
- 92.Kruegel AC, Gassaway MM, Kapoor A, et al. Synthetic and Receptor Signaling Explorations of the Mitragyna Alkaloids: Mitragynine as an Atypical Molecular Framework for Opioid Receptor Modulators. J Am Chem Soc. 2016. June 1;138(21):6754–64. doi: 10.1021/jacs.6b00360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.da Fonseca Pacheco D, Freitas AC, Pimenta AM, et al. A spider derived peptide, PnPP-19, induces central antinociception mediated by opioid and cannabinoid systems. J Venom Anim Toxins Incl Trop Dis. 2016;22:34. doi: 10.1186/s40409-016-0091-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liang DY, Li WW, Nwaneshiudu C, et al. Pharmacological Characters of Oliceridine, a mu-Opioid Receptor G-Protein[FIGURE DASH]Biased Ligand in Mice. Anesth Analg. 2018. July 21. doi: 10.1213/ANE.0000000000003662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gomes I, Fujita W, Gupta A, et al. Identification of a mu-delta opioid receptor heteromer-biased agonist with antinociceptive activity. Proc Natl Acad Sci U S A. 2013. July 16;110(29):12072–7. doi: 10.1073/pnas.1222044110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Portoghese PS, Akgun E, Lunzer MM. Heteromer Induction: An Approach to Unique Pharmacology? ACS Chem Neurosci. 2017. March 15;8(3):426–428. doi: 10.1021/acschemneuro.7b00002. [DOI] [PubMed] [Google Scholar]
- 97.Daniels DJ, Lenard NR, Etienne CL, et al. Opioid-induced tolerance and dependence in mice is modulated by the distance between pharmacophores in a bivalent ligand series. Proc Natl Acad Sci U S A. 2005. December 27;102(52):19208–13. doi: 10.1073/pnas.0506627102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Burford NT, Livingston KE, Canals M, et al. Discovery, synthesis, and molecular pharmacology of selective positive allosteric modulators of the delta-opioid receptor. J Med Chem. 2015. May 28;58(10):4220–9. doi: 10.1021/acs.jmedchem.5b00007. [DOI] [PMC free article] [PubMed] [Google Scholar]