

Abstract
New methods for preparation of tailormade fluorine-containing compounds are in extremely high demand in nearly every sctor of chemical industry. The asymmetric construction of quaternary C−F stereogenic centers is the most synthetically challenging and, consequently, the least developed area of research. As a reflection of this apparent methodological deficit, pharmaceutical drugs featuring C−F stereogenic centers constitute less than 1% of all fluorine-containing medicines currently on the market or in clinical development. Here we provide a comprehensive review of current research activity in this area, including such general directions as asymmetric electrophilic fluorination via organocatalytic and transition-metal catalyzed reactions, asymmetric elaboration of fluorine-containing substrates via alkylations, Mannich, Michael, and aldol additions, cross-coupling reactions, and biocatalytic approaches.
Graphical Abstract

1. INTRODUCTION
Fluorine is the 24th most abundant element in the universe and the 13th most common element in the earthjs crust (0.027% by weight).1 For example, the life-forming elements, such as carbon (15th), nitrogen (31st) and sulfur (17th), as well as other halogens [Cl (21th), Br (59th), I (63th)] are significantly less common.2 However, despite its natural abundance, fluorine is virtually completely absent from the biosphere.3 Three major factors prohibiting chemical and biological evolution of fluorine are (1) the three richest natural sources of fluorine, the minerals fluorospar (CaF2), fluorapatite (Ca5(PO4)3F), and cryolite (Na3AlF6) are practically water-insoluble, rendering the corresponding fluoride unavailable for chemical reactions;4 (2) high oxidation potential of fluorine (−3.06 V, greatly higher than the rest of halogens) makes it impossible to form the corresponding hypohalous intermediates necessary for known enzymatic halogenation;5 (3) high hydration energy of fluorine (117 kcal/mol) renders fluoride a very poor nucleophile in an aqueous/biological environment and therefore unsuitable to form organic C−F bonds via typical nucleophilic substitutions.6 Hence, fluorine (fluoride) is virtually xenobiotic except for a handful of monofluoroacetic acid derived compounds.3
Nevertheless, virtually man-made fluoro-organic chemistry is currently one of the most hectic areas of current research, exerting a profound effect on the most vital industries such as energy, food, and healthcare. The first spectacular demonstration of fluorine-enabled technological achievements was made during the Manhattan Project (1942–1946), where fluorinated compounds played an absolutely indispensable role in the separation of fissile U-235 from U-238 via centrifugation as well as development of novel chemically inert, stable, and durable materials. Similarly, stabilizing and electronic effects of fluorination on material properties are currently used in the solar cells industry7–9 and systematic design of functional materials.10 Medicinal applications of fluorinated molecules can be exemplified by positron emission tomography–computed tomography (PET-CT) using radiotracers labeled with 18F nuclei.11,12 Other diagnostic tools are based on high NMR sensitivity of fluorine, rendering it as an ideal marker for biological studies.13 Some particular progress has been made in preparation of various fluorinated amino acids14–19 and their strategic incorporation into peptides and proteins.20,21 Another important medicinal diagnostic technique is 19F magnetic resonance imaging (MRI), a superior alternative to the current diagnostic procedures using harmful ionizing radiation.22,23 This area technology was developed as part of the more general field of fluorous chemistry based on perfluorinated molecules showing omniphobic physicochemical properties.24–26 Even more decisive impact of fluorinated compounds can be seen in modernization of agrochemical industry.27 Thus, about half of newly developed pesticides contain some type of fluorination, 8–30 generally leading to increased environmental and metabolic stability as well as enhanced biological activity. However, the most spectacular impact of fluorine chemistry on modern society is observed in the pharmaceutical industry.31 Thus, according to the recent survey of the new drug candidates currently in phase II–III clinical trials, fluorine is becoming an increasingly common trait, accounting for about 35% of the designed molecules.32,33 Most importantly, fluorine is found in more than half of most-prescribed multibillion-dollar pharmaceuticals.32–34 Furthermore, the beneficial effect of fluorination can be applied in all therapeutic areas for modulation of virtually any type of biological activity. In this regard, it is interesting to note the success of this strategy in the development of small-molecule therapeutics for Ebola virus (EBOV) disease treatment.35
One may agree that a full extent of technological innovations enabled by fluorine chemistry is far from being fully explored, rendering research in this area of great practical potential and socioeconomic impact. Indeed, fostered by numerous practical applications, the current research activity in fluorine chemistry is at an all-time high.36–54 In particular, the development of innovative synthetic methodology, which is providing access to new fluorinated structural motifs with yet unknown physicochemical and biological attributes, is in extremely high demand in nearly every sector of the chemical industry. However, the progress in the development of fluoro-organic methodology was far from balanced. For example, one of the most developed areas is a direct introduction of a trifluoromethyl group and synthetically related processes.55–67 In sharp contrast, the asymmetric construction of carbon-fluorine quaternary stereogenic centers is the most synthetically challenging and, consequently, the least developed area of research. As a reflection of this apparent methodological deficit, pharmaceutical drugs featuring C–F stereogenic centers constitute less than 1% of all fluorine-containing medicines currently on the market or in the clinical development.31–33 Some success has been achieved in the development of enantioselective electrophilic fluorination, and this subject has been intensively reviewed.68–75 On the other hand, the alternative approaches have received much less appreciation in the current literature. Therefore, we trust that a comprehensive review, critically discussing the state-of-the-art of the corresponding methodology, is both strategically timely and scientifically stimulating.
2. MARKETED DRUGS FEATURING QUATERNARY C–F STEREOGENIC CENTERS
Considering the xenobiotic character of fluorine, the idea of modification of bioactive molecules with fluorine atoms, to improve the desired properties, was quite implausible until the early 1950s. Around that time, Fried and Sabo76 were studying a series of hydrocortisones in which the 9α-hydrogen atom was replaced by halogen. They found that iodo-, bromo-, and chloro-derivatives possessed noticeably higher glucocorticoid activity as compared with that of the parent hormones. Most importantly, they discovered that the bioactivity was inversely proportional to the size of the halogen atom, leading them to a logical curiosity to investigate the corresponding fluoro-derivative. 9α-Fluoro hydrocortisone 2 (Scheme 1) was prepared in about 50% yield by treatment of acetate 1 with anhydrous hydrogen fluoride in alcohol/water-free chloroform at 0 °C for 4.5 h.77
Scheme 1.
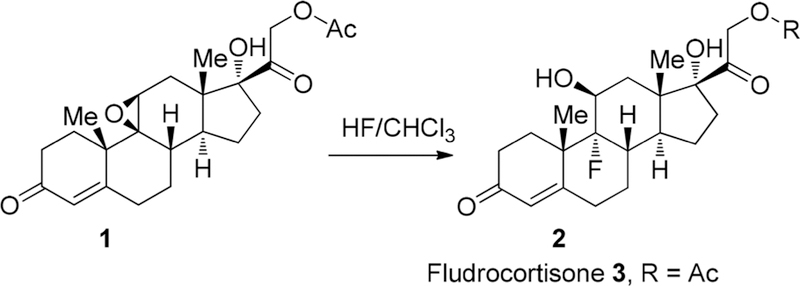
Synthesis and Structure of Fludrocortisone 3
Subsequently, it was shown that 9α-fluoro hydrocortisone acetate 2 possessed astonishing ~10.7 times the activity of nonfluorinated cortisone acetate in the rat liver glycogen assay. Deacetylation of 2 with sodium methylate gave rise to 9α-fluoro hydrocortisone 3, which was patented in 1953 and marketed since August 18, 1955 under the brand names Fludrocortisone, Florinef, and others. Fludrocortisone 3 is still in use for treatment of adrenogenital syndrome, postural hypotension, and adrenal insufficiency and is included in World Health Organization’s list of essential medicines.78 The discovery of fludrocortisone 3 demonstrated that fluorine is a good bioisostere for hydrogen while influencing neighboring functional groups due to its extreme electronegativity. It should also be noted that fludrocortisone 3 was the first fluorine-containing drug approved by the FDA and, at the same time, the first example of pharmaceuticals featuring a quaternary C–F stereogenic center.
Another successful fluorine-containing drug possessing quaternary C–F moiety is synthetic glucocorticoid fluticasone propionate 4 (Figure 1). It is also known in combination with salmeterol 5 under the trade name Advair Diskus, prescribed as an oral inhaler for the treatment of asthma. It is interesting to note that Advair Diskus is in the league of top-performing drugs in terms of prescription and sales rates (>$5.0 billion).79
Figure 1.

Structures of fluticasone propionate 4 and salmeterol 5.
Structurally very similar to fluticasone 4 is difluorinated corticosteroid difluprednate 6 (Scheme 2). This drug possesses a potent clinical efficacy in controlling postoperative inflammation. It was approved by the FDA in June 2008 as the first topical steroid prescribed for inflammation as well as pain associated with ophthalmic procedures.80,81 It was shown that fluorine substitution for hydrogen in the C6 and C9 positions contributes to the potency of the drug, likely due to the increased lipophilicity and corneal penetration.82,83
Scheme 2.
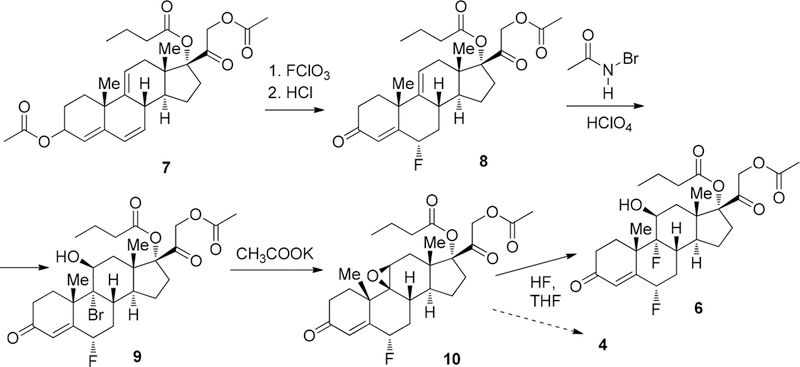
Key Fluorination Steps in the Synthesis of Fluticasone 4 and Difluprednate 6
The first fluorination step in the synthesis of fluticasone 4 and difluprednate 6 involves the selective introduction of the fluorine atom in position 6 of compound 7 using perchloryl fluoride, followed by the removal of secondary acetate moiety to afford intermediate 8. Successive transformation of 8 to bromohydrin 9 was accomplished with bromo acetamide/perchloric acid. The latter was converted into epoxide 10 under very mild basic conditions. The second fluorination step is quite similar to the synthesis of fludrocortisone 3 (Scheme 1) and based on the epoxide ring opening with hydrogen fluoride under dry conditions.84
Another example of drugs possessing quaternary C–F moiety is solithromycin 13 (Scheme 3), the fluoroketolide antibiotic currently under consideration by the U.S. FDA for treatment of moderate to moderately severe community-acquired bacterial pneumonia.85 The drug interrupts bacterial protein synthesis, preventing the growth and reproduction by reversibly binding to the bacterial ribosome.86 It is interesting to note that solithromycin 13 shows noticeably superior inhibition against growth of streptococci carrying a special methyltransferase gene which is thought to be the main cause of the current global macrolide resistance. The effect of the fluorination is difficult to ascertain but can be linked with configurational stability of the fluorinated C2 in solithromycin 13.87
Scheme 3.

Key Fluorination Step in the Synthesis of Solithromycin 13
The key, regioselective electrophilic fluorination step of intermediate 11 was performed using 1-chloromehyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborates) (Selectfluor),88–90 in the presence of KOt-Bu. The final product solithromycin 13 was prepared form 12 in a few steps including copper-catalyzed azide–alkyne cycloaddition with 3-ethynylaniline.91
Hepatitis C is a current pandemic liver disease, affecting 130–150 million people worldwide, with high morbidity and mortality rates. Recent progress in the treatment of this disease was made with the development of a new generation of agents acting directly on the viral protein synthesis.92,93 Among the most effective therapies against HCV infection, is sofosbuvir 14 (Scheme 4), featuring the quaternary stereogenic C–F moiety.94,95
Scheme 4.

Key Fluorination Step in the Synthesis of Sofosbuvir 14
In several of the developed protocols96–99 for preparation of sofosbuvir 14, the key fluorination step involves the treatment of tertiary alcohol 15 with DAST,100,101 proceeding with the inversion of the absolute configuration at C2′. It is proposed that the role of the fluorination in this drug is a stabilization of the 3D geometry around the corresponding stereogenic center. Thus, despite the availability of a 3′ hydroxyl group to act as a nucleophile, sofosbuvir 14 acts as a chain terminator because the 2′ methyl group causes a steric clash with an incoming nucleotide triphosphate.102
The five above-profiled drugs, fludrocortisone 3, fluticasone 4, difluprednate 6, solithromycin 13, and sofosbuvir 14, representing different therapeutic areas, clearly underscore the pharmacophoric importance of the quaternary C–F moiety in the design of modern pharmaceuticals. Quite remarkable is that the C–F quaternary stereogenic centers in these compounds impart rather versatile effects influencing reactivity of the neighboring functional groups, configurational stability of a stereogenic carbon, 3D structure, and overall lipophilicity of the parent biomolecules. Another noticeable trend is that the methodology of fluorine introduction is limited by the reactions discovered 50–70 years ago. One would agree that to realize full pharmaceutical potential of fluorine-containing compounds in general, and these bearing quaternary C–F structural features in particular, there is a critical need for advanced new approaches for chemo- and enantioselective selective construction of quaternary C–F stereogenic centers.
3. MODERN METHODS FOR CONSTRUCTION OF QUATERNARY C–F STEREOGENIC CENTERS
3.1. Introduction of Fluorine
Because of the prevalence of fluorinated pharmaceuticals and pesticides, the asymmetric incorporation of fluorine into organic molecules has attracted considerable attention. Despite significant progress in the development of asymmetric fluorination methodology,71,74 the asymmetric construction of a fluorine-containing quaternary stereogenic center is still synthetically quite challenging. Herein, we aim to comprehensively cover recent advances in the asymmetric fluorination transformations, especially for catalytic electrophilic enantioselective approaches, resulting in formation of a C–F bond of a quaternary stereogenic center.
In 1988, Differding and Lang reported the first electrophilic enantioselective fluorination of enolates using stoichiometric amounts of chiral N-fluoro camphorsultam reagent.103 Subsequently, the development of bench-stable and operationally convenient electrophilic fluorinating reagents such as Selectfluor, N-fluorobenzenesulfonimide (NFSI), and N-fluoro pyridinium salts has marked an important milestone in enantioselective fluorination.104,105 Since 2000, the practical enantioselective methodology leading to construction of a C–F quaternary stereocenter has been blooming due to the development of chiral N-F reagents derived from in situ generated or isolated N-fluoroammonium salts by the combination of equimolar cinchona alkaloids and Selectfluor.106,107 At the same time, the pioneering research related to a catalytic protocol by using TADDOLato/Ti(II) catalyst for enantioselective fluorination of acyclic β-ketone esters with Selectfluor was reported.108 First, we will provide a brief description of a reagent-controlled process, especially the development of chiral N-fluoroammonium salts of cinchona alkaloids, because these studies laid important groundwork for latter methodological advances. Subsequently, comprehensive discussion will be devoted to the catalytic asymmetric scenario including organocatalytic methods (tertiary amine catalysts derived from cinchona alkaloids, primary and secondary amine catalysts via enamine intermediates, cationic and anionic phase-transfer catalyst, etc.) and transition-metal catalyzed transformations. Additionally, F-additions to C=C bonds will be highly emphasized. The fluoro-functionalization of alkenes by electrophilic fluorinating reagent to enantioselective installation of a C–F quaternary stereogenic center is an appealing strategy that converts common alkenes into valuable bioactive fluorinated molecules.
Asymmetric fluorination by using a nucleophilic fluorinating source is much less developed as compared to the electrophilic processes. Here we would like to mention just a handful of known examples. One of them is a stoichiometric, diaster-eoselective fluorination by using a nucleophilic source (Scheme 5). In this case, the anodic fluorination (platinum anode) of the 1,3-oxazolidines 17 derived from l-threonine was performed to afford a-monofluorinated product 18 in 73% yield with 81% de.109 Recently, an iron(II)-catalyzed diastereoselective olefin aminofluorination, which applied a functionalized hydroxylamine 19 as a nitrogen source and Et3N·3HF as a fluorine source, can afford desired fluorinated product 20 bearing a C–F quaternary carbon in 45% yield with >20:1 dr.110
Scheme 5.

Synthesis of α-Monofluorinated Compound 18 (a) and the Target Fluorinated Product 20 (b)
Two special examples related to a catalytic enantioselective approach to construct C–F quaternary carbon by employing nucleophilic fluorinating reagents are presented in Scheme 6. First, the oxidative dearomatization of substituted phenols 21 by PhI(OAc)2 in the presence of HF-pyridine complex was used to generate the fluorinated meso-cyclohexadienones intermediate, which then underwent a enantioselective intramolecular Michael addition sequence catalyzed by chiral secondary amine catalyst C1, leading to enantioenriched fluorinated product 22 in good yield (83%) and good diastereo- and enantioselectivity (>20:1 dr, 99% ee).111 Recently, the combination of ArI/HF-pyridine/mCPBA system has been applied to perform a nucleophilic fluorination of β-dicarbonyl compounds 23 via in situ generation of hypervalent iodine compound ArIF2 by mCPBA, HF, and a catalytic amount of iodoarene (ArI). Subsequently, the catalytic enantioselective scenario was conducted by using substrate with a steric adamantly demanding group and chiral iodoarene, (R)-binaphthyldiiodine, to afford α-fluorinated β-ketoester 24 in moderate yield and moderate enantioselectivity (56% ee).112
Scheme 6.

Intramolecular Michael Addition Sequence Leading to Fluorinated Product 22 (a) and α-Fluorination Leading to Compound 24 (b)
3.1.1. Asymmetric Electrophilic Fluorination.
The diastereoselective electrophilic fluorination to construct C–F quaternary stereogenic centers mainly focused on the α-fluorination of carbonyl compounds. The diastereoselectivity in these reactions is controlled by substrate structures bearing chiral auxiliaries to influence the diastereofacial discrimination of the intermediate enolates. The representative examples of bioactive molecules with various functionalities are provided in Scheme 7. For instance, the electrophilic fluorination of dipeptides 25 bearing quaternary chiral amino acid,113 2′-ketouridine lithium enolates generated from nucleoside analogues 27,114 stabilized sodium enolate generated from azetidinone 30 as antibiotic analogues,115 or malonate 32 bearing a chiral phenylmenthyl auxiliary,116 can afford desired α-fluorinated products 33 with moderate to good diastereoselectivity.
Scheme 7.

Asymmetric Electrophilic Fluorination Affording Compounds 26 (a), 28 (b), 31 (c), and 33 (d)
To achieve the enantioselective fluorination, a wide variety of chiral sulfonamide-type fluorinating reagents had been developed in earlier examples. For instance, Differding and Liang reported enantioselective fluorination of enolates controlled by chiral sulfonamide-type fluorinating reagents, N-fluorocamphorsultam 34, in 1988.103 Then Davis’s reagents 35117,118 and Takeuchi and Shibata’s saccharin-type reagents 36–38119–121 followed (Scheme 8). However, multistep procedures and using toxic or aggressive reagents for their preparation make these chiral N–F reagents unavailable. Meanwhile, unsatisfactory enantioselectivity and narrow substrate scope further limited their application.
Scheme 8.

Chiral N–F Reagents for Enantioselective Fluorination
In 2000, the Cahard and the Shibata groups simultaneously reported the introduction new class of N–F electrophilic reagents 39 and 40 derived from naturally occurring cinchona alkaloids.106,107 In the Cahard’s case, the N-fluoroammonium salts of cinchona alkaloids were isolated and applied in enantioselective fluorination, and the Shibata’s procedure was based on in situ-generated N-fluoroammonium salts. Subsequently, a stoichiometric amount of cinchona alkaloids/Selectfluor combinations, or isolated N-fluoroammoniun salts of cinchona alkaloids, were proven to enable a wide range of substrates, including silyl enol ethers 43,106,122 allylsilanes 46,123 1,3-dicarbonyl compounds (49,51 ),124 lactones,125 enolates 60107,126 oxindoles 54,124 and dipeptides 57,113 to covert to corresponding fluorinated products bearing with C–F quaternary stereogenic centers (44, 47, 50, 52, 55, 58, 61, 63) in good yields and effective enantioselective control (Scheme 9). In 2013, Cahard, Ma and Shibata developed a new chiral fluorinating reagent 42 as analogues of NFSI based on a chiral 1,1′-binaphthyl moiety with axial chirality127 In 2013, the Gouverneur group developed a more reactive chiral N–F reagent 41 based on the structural core of Selectfluor with a chiral environment on the dicationic DABCO core,128 and the application of this reagent in asymmetric fluorocyclization will be discussed in 3.1.1.2. F Additions to C=C Bonds (vide infra).
Scheme 9.

Substrate Types for Enantioselective Fluorination Using N–F Reagents: Silyl Enol Ethers (a), Allylsilanes (b), 1,3- Dicarbonyl Compounds (c), Oxindoles (d), Dipeptides (e), and Enolates (f)
To further verify the synthetic utility for drug development, the enantioelective fluorination of several bioactive molecules was reported (Scheme 10). For instance, when employing the cinchona alkaloids (DHQ)2AQN 66/Selectfluor combinations129 or isolated F-2-NaphtQN-BF4 salts,130 the desired fluorinated oxindole 67, BSM-204352 (MaxiPost), which serves as an effective opener of maxi-K channels, can be prepared with high yields with good enantioselectivity, and the enantioenriched 20-deoxyl-20-fluorocamptothecin 70, which can be capable of mimicking the hydrogen bond acceptor during the inhibition of DNA topoisomerase, can be prepared form asymmetric fluorination of corresponding lactone moiety 68 with good enantioselective control (88% ee).131
Scheme 10.

Enantioselective Fluorination Using Cinchona Alkaloid Derived Reagents: Structural Types 65 (a) and 68 (b)
Additionally, in 2011, the Shibata group reported the preparation of enantiomerically pure 3′-fluorothalidomides 74 by enantiodivergent asymmetric fluorination via the combination of stoichiometric amounts of cinchona alkaloid (dihydroquinine DHQ, 73) and NFSI with ligands and Lewis acids.125 By the combination of DHQ/NFSI with Cu(acac)2 and ligand bipy, the fluorinated R-enantiomer 72 can be synthesized in 81% yield with 77% ee, while with the use of tetramethylethylenediamine (TMEDA) as additive, the corresponding S-enantiomer 72 can be prepared in 88% yield with 78% ee (Scheme 11).
Scheme 11.

Preparation of Enantiomers of 3-Fluorothalidomide 74
3.1.1.1. Organocatalytic Methods
3.1.1.1.1. Tertiary Amine Catalysts Derived from Cinchona Alkaloids and Their Analogues.
Although a stoichiometric amount of cinchona alkaloids/Selectfluor combinations or N-fluoroammoniun salts of cinchona alkaloids enabled a wide range of substrates as mentioned above (Scheme 9 and Scheme 10) to be converted to the corresponding fluorinated products with effective enantioselective control, the organocatalytic approach of the methodology employing catalytic amounts of cinchona alkaloids and electrophilic fluorinating reagents were still highly desirable and attractive, especially for the enantioselective incorporation of fluorine into organic molecules to construct a chiral quaternary stereogenic center.
In 2006, the Shibata group revealed a protocol for the electrophilic fluorination of cyclic acyl enol ethers with five- or six-membered rings 75 to afford α-fluorinated ketones 76 bearing a C–F quaternary carbon center with moderate enantioselectivity (up to 54% ee) by employing a catalytic amount of DHQB or (DhQ)2ANQ(Scheme 12).132 To enable the desired catalytic cycle, the initial transfer fluorination from Selectfluor to cinchona alkaloid catalysts, which was considered to form a temporary electrophilic asymmetric fluorinating N-fluoroammoniun salts to react with substrates followed by enantioselective transfer fluorination and regenerating the catalysts, should suppress the direct electrophilic fluorination of substrates by achiral Selectofluor. Thus, acetyl enol ethers was chosen as preferable substrates instead of more reactive silyl enol ethers and CH2Cl2 was selected as reaction solvent because it can precipitate Selectfluor to further restrain direct fluorination of substrates. Meanwhile, addition of 1.2 equiv inorganic base such as NaOAc was essential to activate the enolates followed by capturing the acetyl cation and counter BF4- in the reaction cycle. Although there are several limitations such as substrates, scope, and enantioselectivity in this research, it has proved that the combination of cinchona alkaloids and Selectfluor can be performed in a catalytic scenario.
Scheme 12.
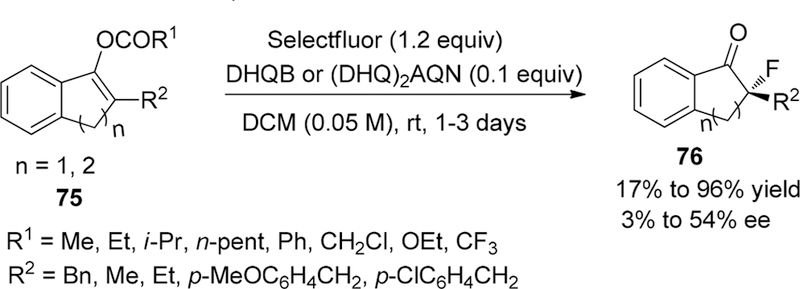
Preparation of α-Fluorinated Ketones 76 Bearing a C–F Quaternary Carbon Center
By 2008, the Shibata group modified their method to further restrain the direct fluorination of more reactive substrates which was supposed to cause inaccessible catalyst regeneration, and they developed the first highly enantioselective catalytic fluorodesilylation reaction of allyl silanes and silyl enol ethers 77 based on the combination of catalytic amount of bis-cinchona alkaloids (C3, C4) and N-fluorobenzenesulfonimide (NFSI) in the presence of excess inorganic base (Scheme 13).133 Then biscinchona alkaloids (DHQ)2PYR or (DHQ)2PHAL (10 mol %)/NFSI (1.2 equiv)/K2CO3 (6.0 equiv) have proven to be an effective catalytic combination for construction of a chiral quaternary carbon center with a fluoro substituent via the fluorodesilylation of allyl silanes (up to 95% ee) and silyl enol ethers (up to 86% ee) with the requirement for bulky substituents on the substrates (when R in C2 position of allyl silanes changed to Me and H, the ee value decrease obviously to 72% and 51%, respectively), and the opposite S-enantiomer of the fluorodesilylation of allyl silanes could be prepared in the presence of the hydroquinidine variant (DHQD)2PYR.
Scheme 13.

Preparation of α-Fluorinated Ketones 78 Bearing a C–F Quaternary Carbon Center
In the plausible catalytic cycle for enantioselective fluorode-silyllation reactions, a stable N-fluoroammonium salt I derived from the combination of NFSI and bis-cinchona alkaloids can react with K2CO3, leading to the formation of corresponding N-fluoroammonium KCO3– salt II, which triggered the fluorodesilylation process followed by enantioselective transfer fluorination from the chiral N-fluoroammonium ion to the substrates (Figure 2). Meanwhile, one dihydroquinine moiety with the open conformation in (DHQ)2PYR confirmed by the X-ray crystal structure analysis was considered to be responsible for the transfer fluorination with high enantioselectivity based on the experimental evidence that N-fluorinated quininium and N-fluorinated dihydoroquinidium salts exist in the open conformations both in solid and solution states.
Figure 2.

A plausible catalytic cycle for cinchona alkaloids catalyzed enantioselective fluorodesilylation.
Subsequently, they investigated the organocatalyzed enantio-selectivre fluorination of oxindoles 79 in order to probe the further synthetic utility of this catalytic strategy (Scheme 14). After screening the reaction conditions, the modified catalyst (DHQD)2AQN (C5, 5 mol %)/NFSI (1.2 equiv)/CsOH-H2O (6.0 equiv) system have proven to be effective to construct the enantioenriched fluorine-substituted quaternary carbon centers (up to 85% ee) in CH3CN/CH2Cl2 (3:4) at low temperature –80 °C.
Scheme 14.
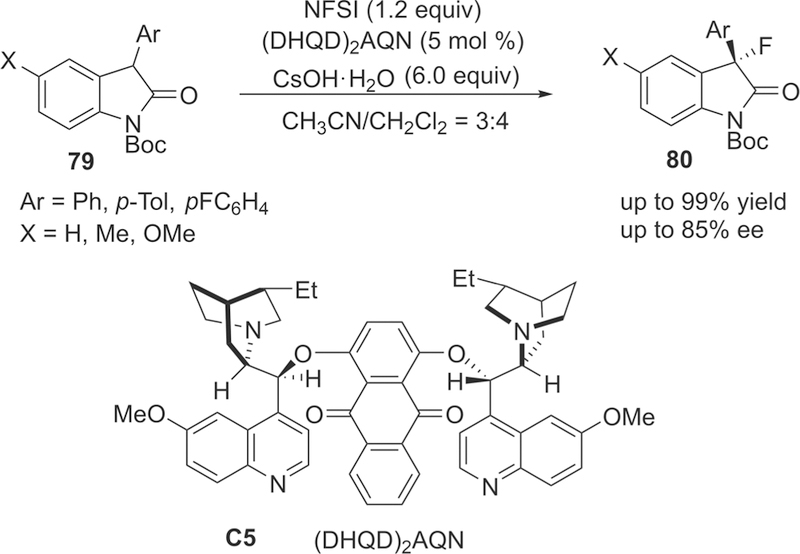
Enantioselective Fluorination of Oxindoles
For the enantioselective fluorodesilylation reactions of silyl enol ethers to construct a C–F quaternary stereogenic center, the major limitation of this protocol was the requirement for a bulky substituent on the substrates to improve the enantioselectivity. Then the Shibata group hypothesized that sterically demanding analogues of NFSI could potentially enhance the enantioselective control in this fluorodesilylation process comparing with NFSI. In 2011, the Shibata group reported the method to improve enantioselectivity of the fluorination products (83, 85) by modifying the electrophilic fluorinating reagents and designed the steric bulky analogues of NFSI, N-fluoro-(3,5-di-tert-butyl-4-methoxy)-benzenesulfonimide 82 (NFBSI) (Scheme 15).134 As mentioned above, the common N-fluorinated ammonium of cinchona alkaloid was presumably to be formed in the initial transfer fluorination reaction in catalytic cycle, the steric hindrance originated from an anion of (3,5-ditert-butyl-4-methoxy) benzenesulfonimide in this N-fluorinated ammonium salt presumably helped to weaken the reactivity of enantioselective fluorination process followed by increasing the enantiomeric excess of the products (the enantioselectivity improved as much as 18% by using NFBSI compared to the use of NFSI).
Scheme 15.

Enantioselective Fluorodesilylation Reactions of Silyl Enol Ethers
In 2013, to explore the influence of different kinds of substituents in NFSI on the fluorinating reactivity and selectivity, the He group reported the enantioselective fluorination of oxindoles 86 to construct a carbon-fluorine quaternary stereogenic center by the combination of bis-cinchona alkaloid (DHQD)2PHAL (69, 5 mol %)/structurally modified N-fluorobenzenesulfonimides 87 (NFSIs) (1.2 equiv)/K2CO3 (6.0 equiv) in CH2Cl2/CH3CN (3:4) at –80 °C with high enantioselectivity (up to 96% ee) (Scheme 16).135 They disclosed that modified NFSI reagents bearing an electron-donating and steric bulky t-butyl group on the para position of the symmetric phenyl ring showed lower electrophilic fluorinating reactivity by cyclic voltammetry and obviously enhanced enantiselectivity compared with using the general NFSI reagent. Furthermore, electron-withdrawing group substituted reagents CF3-NFSI and CF3O-NFSI failed to afford target products due to their instability and decomposition in the presence of K2CO3.
Scheme 16.
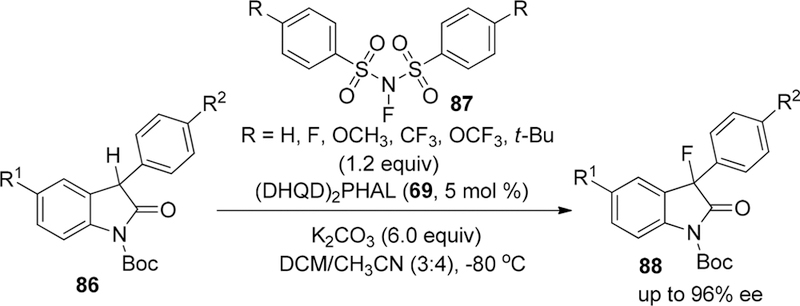
Enantioselective Fluorination of 3-Aryl-oxindoles
In 2015, the Wu group reported electrophilic fluorination of oxindoles 89 via the combination of catalytic amounts of cinchona alkaloid (DHQD)2ANQ(C5, 5 mol %) and modified Selectfluor 90 bearing two (PhSO2)2N-as counterion anions to tune its fluorinating reactivity with low to moderate (up to 55% ee) enantioselectivity (Scheme 17).136 In the construction of a carbon-fluorine quaternary stereogenic centers in oxindoles 91, they provide a protocol to make Selectfluor more compatible with cinchona alkaloid by modifying its corresponding anions.
Scheme 17.

Enantioselective Fluorination of 3-Aryl-oxindoles Using Selectfluor
In 2011, an organocatalyzed asymmetric cascade fluorination–heterocyclization to prepare enantiopure fluorinated hetero-cycles, hexahydropyrrolo[2,3-b]indole or the tetrahydro-2H-furo-[2,3-b]indole skeleton bearing a C–F quaternary benzylic carbon center 93, has been reported by the Gouverneur group. A prochiral indole 92 with a pendant heteronucleophile tethered at the C3 position enables asymmetric fluorocyclization in moderate to good enantioselectivity (52% ee to 84% ee) by the combination of bis-cinchona alkaloid (DHQ)2PHAL (C4, 20 mol %)/NFSI(1.2 equiv)/K2CO3(6.0 equiv) in acetone at −78 °C (Scheme 18). The presence of a substituent at C5 position (R1 ≠ H) led to a markedly improved enantioselective control in this irreversible fluoroquaternization at C3 followed by the intramolecular capture of the transient iminium intermediate by the pendant oxygen or protected nitrogen nucleophile. Under catalytic reaction conditions, only slight decrease of enantiomeric excess and similar yields were observed by comparing with the use of stoichiometric amount of alkaloid and the level of enantioselectivity was found to be dependent on the nature of the nuecleophile (X = O, 66% ee; X = NTs, 64% ee; X = NOMe 80% ee; X = NBoc 78% ee). Additionally, for probing the reaction mechanism, only less than 2% (DHQ)2PHAL+-F canbe detected by 19F-NMR at low temperature −78 °C with or without K2CO3. Thus, they proposed that the enantioselectivity may not be induced by in situ generated transient chiral N-F cinchona species because fluorine transfer from NFSI to (DHQ)2PHAL was proved to be ineffective at low temperature and the associative complexation seemed to take place through the effect of hydrogen bonding138 between the alkaloid catalysts and the indole substrates and/or NFSI.
Scheme 18.

Enantioselective Fluorination–Cyclization of Indoles 92 with a Pendant Heteronucleophile Tethered at C3 Position
In 2015, the Wang group reported the asymmetric electrophilic fluorination of 4-substituted isoxazolinones 94 catalyzed by a bis-cinchona alkaloid (QN)2PYR (C7, 10 mol %) in the presence of NFSI (1.1 equiv) and K3PO4(1.1 equiv) in CHCl3 (0.1 M) at −60 °C. The enantiopure fluorinated heterocycles bearing a fluorine-containing quaternary stereogenic center 95 were prepared in good yields and good enantioselectivities (up to 91% yield, 85% ee).139 Meanwhile, to demonstrate the practical utility of the asymmetric fluorination protocol, the 4-fluoroisoxazolinone derivatives 95a can be provided with high enantioselectivity (>99% ee) after a single recrystallization from gram-scale products which had been generated in 85% yield and 80% ee under optimized reaction conditions (Scheme 19).
Scheme 19.
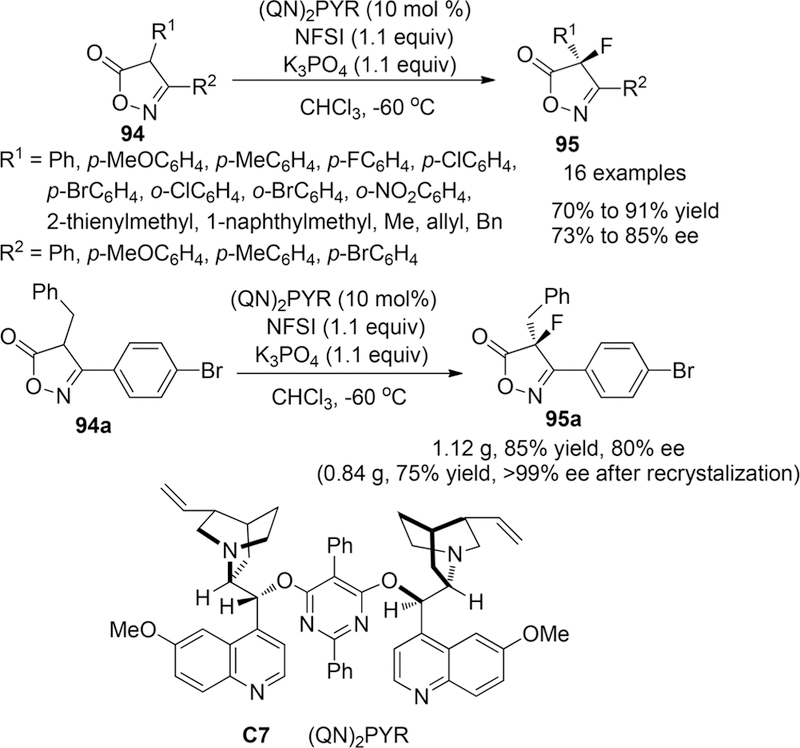
Asymmetric Electrophilic Fluorination of 4-Substituted Isoxazolinones 94
In 2016, the Wang group reported a catalytic asymmetric fluorination process of 4-substituted pyrazolones 96 to provide a series of 4-fluorinated pyrazol-5-ones 97 bearing a C–F quaternary carbon center with good yields and moderate enantioselectivities (from 37% to 81% ee).140 After screening the reaction conditions, the combination of quinine (10 mol %)/NFSI (1.2 equiv)/Cs2CO3 (1.0 equiv)/H2O (2.0 equiv) in CHCl3 (0.05 M) at −60 °C was chosen as the optimized reaction condition (Scheme 20), and they believed that the acceleration effect caused by the addition of water may be due to the enhanced solubility of the inorganic base in the fluorination process.
Scheme 20.
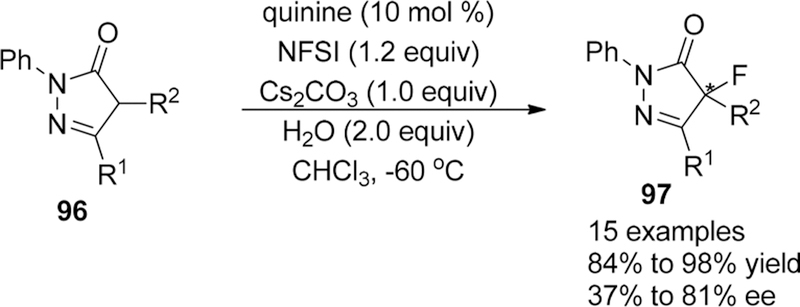
Asymmetric Fluorination of 4-Substituted Pyrazolones 96
3.1.1.1.2. Enamine Catalysis: Chiral Secondary Amine and Primary Amine Catalysis
Chiral aminocatalysis via enamine intermediates has emerged as an appealing strategy for the direct α-fluorination of carbonyl compounds and their analogues, providing access to a fluorinated quaternary stereogenic center in an enantiocontrolled manner.
As for asymmetric electrophilic fluorination of linear aldehydes catalyzed by chiral amino catalysis, the reaction conditions should be screened carefully under the following terms: first, the fluorination process must be faster than directly N-fluorination of the aminocatalyst, and second, difluorinated side products and potential racemization caused by second enamine formation from desired monofluorinated aldehydes, which showed the enhancement in acidity of the α proton due to the introduction of high electronegativity of fluorine, was expected to be rigorously avoided.
Then in 2005, the Jørgensen group reported the asymmetric α-fluorination of linear aldehydes 98 catalyzed by a sterically encumbered chiral pyrrolidine derivative C8 with high enantioselectivity.141 To restrain catalyst degradation caused by N-fluorination of NFSI, lowering the catalysis loading to 1 mol % in specific solvent methyl-tert-butyl ether (MTBE) was found to markedly improve the conversion and enantioselectivity. To explain the configurational stability of the optically active products, the hydrogen atom at the newly formed fluorinated chiral center of the preferable imminium ion intermediaters was expected to be located in a sort of hydrophobic pocket which prevented its abstraction to afford the second enamine intermediate. Then they extended the scope of the reaction to the branched aldehyde (only one case) to afford desired product 99 bearing a C–F quaternary stereocenter in 78% yield with 48% ee by employing a modified sterically less-demanding catalyst C8 (5 mol %) and high temperature (60 °C) (Scheme 21). Although the fluorinated products of branched aldehydes are unable to racemize, as they have no α proton, the enantioselective control for the construction of the C–F quaternary carbon center was still unsatisfactory. Additionally, the α-fluorinated aldehydes were required to derivatize in situ to corresponding optically active β-fluorinated alcohols 100 for subsequent analysis because they could decompose rapidly on silica gel.
Scheme 21.

Asymmetric α-Fluorination of Linear Aldehydes 98
In 2005, the Barbas group revealed the direct asymmetric α-fluorination of branched aldehydes 101 catalyzed by chiral secondary amine catalyst derived from pyrrolidine and its analogues with moderate enantioselectivity (up to 66% ee)142. As α-fluoro aldehydes have been proven to be volatile and thermally unstable because they always decompose upon column purification or distillation conditions, the yields for the synthesis of α-fluoro aldehydes 102 were just measured by 1H NMR spectroscopy and GC analysis of crude reaction mixtures. Subjecting branched aldehydes to the combination of chiral amine catalysts (30 mol %) and the electrophilic fluorinating reagent NFSI (1.2 equiv) in THF (0.25 M), the desired optical active α-fluorinated aldehydes bearing a fluorinated tetrasubstituted stereogenic center can be prepared in 99% NMR yield with 45% ee for acyclic substrate in the presence of a sterically demanding triisopropylsilyl (TIPS) group substituted l-prolinol derivative C9 and in 98% NMR yield with 66% ee for cyclic substrate in the precence of the proline-derived tetrazole catalyst C10 (Scheme 22). Although high enantoselectivity (up to 96% ee) can be provided in the scenario of linear aldehydes to afford fluorinated trisubstituted stereocenters, a stoichiometric amount of the catalyst was required, and commercially available fluorinating reagents such as Selectfluor, 1-fluoro-4-hydroxy-1,4-diazoniabicyclo[2,2,2]octane bis(tetrafluoroborate) (Accufluor), and pyridinium fluorides showed minimal reactivity and afforded the racemic products in very low yield.
Scheme 22.

Asymmetric α-Fluorination of Branched Aldehydes 101
In 2008, the Yamamoto group reported the enantioselective installation a fluorinated chiral quaternary carbon centers in gem-chlorofluoro carbonyls compounds based on organocatalytic asymmetric α-fluorination.143 Various optically active α,α-chlorofluoro aldehydes 104 can be prepared in good yields (62–88%) and with high enantioselectivity (82% to 98% ee) from racemic α-chloroaldehydes 103 (3.0 equiv) catalyzed by Jørgensen catalysts C11 (10 mol %) in the precence of NFSI (1.0 equiv) in MTBE (methyl tert-butyl ether; 0.25 M). Subsequently, assessable enantioenriched α, α-cholorofluoro ketones 106 can be prepared from α, α-chlorofluoro aldehydes 104 via nucleophilic addition of a Grignard reagent followed by oxidation using the Dess-Martin reagent without loss of optical purity (Scheme 23).
Scheme 23.

Asymmetric α-Fluorination of α-Chloroaldehydes 103
Furthermore, α-chloro- α-fluoroaldehydes could be prepared in high enantioselectivities via asymmetric α-fluorination of α-alkyl-α-chloroaldehydes 107 mediated by the Jørgensen–Hayashi catalysts C11 when the starting aldehyde was used in excess over NFSI (Scheme 24). However, when an excess of NFSI with respect to the starting aldehyde was used, the loss in enantiopurity of products were observed. Subsequently, Shibatomi and co-workers insisted on the kinetic resolution process in the asymmetric fluorination mechanism.144 Thus, the asymmetric induction in this transformation required not only the enantiofacial distinction of enamine intermediates for electrophilic fluorination but also a kinetic resolution for the corresponding (S)-α-chloroaldehydes. Moreover, they reported the determination of the absolute configuration of desired α-chloro-α-fluoroaldehydes via X-ray crystallographic analysis of its corresponding α-chloro-α-fluoro-β-keto ester derivatives.
Scheme 24.

Asymmetric α-Fluorination of α-Alkyl-α-chloroaldehydes 107
In 2015, the Brenner-Moyer group extended the asymmetric fluorination of α-chloroaldehydes into a one-pot scenario.145 In other words, they developed a method to install a fluorinated tetrasubstituted stereocenter in gem-chlorofluoro compounds from unfunctionalized aldehydes 109. The starting point for the cascade reactions was to assess the compatibility of the two catalytic reactions, asymmetric chlorination of starting aldehydes, and fluorination of corresponding α-chloroaldehydes. First, N-chlorosuccinimide (NCS) was screened to serve as an electrophilic chlorine source because succinimide, the byproduct of chlorination, had been proven to be harmless for the fluorination step. Subsequently, by employing the l-proline (5 mol %) and NCS (0.95 equiv) in CHCl3 for the chlorination step and the addition of NFSI (0.7 equiv) and Jørgensen-Hayashi catalyst C11 (33 mol %) in methyl tert-butyl ether (MTBE) as cosolvent followed by reduction the unstable gem-chlorofluoro aldehydes in situ, the unfunctionalized aldehydes (1 mmol scale) can be converted to corresponding gem-chlorofluoro alcohols in moderate to good yields (54%−87%) with good enantioselective control (81%−98% ee) (Scheme 25).
Scheme 25.

Enantioselective Preparation of gem-Chlorofluoro Compounds from Unfunctionalized Aldehydes
In 2016, the Juhl group reported a highly diastereoselective access to β-fluoropyrrolidines 111 bearing two adjacent quaternary carbon centers catalyzed by chiral secondary amine catalysts.146 The starting optically pure pyrrolidines were separated by chiral supercritical fluid chromatography (SFC). For achiral pyrrolidine as an enamine catalyst, the anti-products were formed as major products because fluorine approached the least hindered enamine face, and after screening the secondary amine catalysts, they found that imidazolidinone catalysis C12 could enhance the substrate control to increase the ratio of anti-products and jørgensen catalysts C11 can completely reverse the substrate control to afford the syn-β-fluoropyrrolidines (dr >99:1). The observed diastereodivergence was rationalized by the catalyst-induced diastereofacial discrimination. Meanwhile, classicalj0rgensen catalysts C11 enable the kinetic resolution of racemic cyclic α-branched aldehyde bearing a pyrrolidine scaffold followed by reduction in situ to afford a fluorinated β-prolinol analogue 112 with vicinal quaternary stereogenic centers in 31% isolated yield with 95% ee (Scheme 26).
Scheme 26.

Asymmetric Synthesis of Fluorinated β-Prolinol Analogues
Then in 2016, the Juhl group reported a method to enantioselective fluorination of cyclic α-branched aldehydes 113 to afford the desired α-fluorinated aldehydes 114 with a α-fluorinated tetrasubstituted chiral center in high yields and good to high enantioselective control (up to 97% ee).147 It should be noted that the enantioselectivities in the case of “d” (Scheme 27) without branching at the β-positions decreased to 26% ee compared with that of corresponding gem-dimethyl substituted analogue “e” with high enantioselective control in 97% ee. A lower differentiation of the two α-substituets would afford a mixture of E- and Z-enamine intermediates and, steric hindrance demanded gem-dimethyl groups would lead to a better control of the enamine E/Z ratio. The high enantioselective control in this transformation needs a high enamine E/Z equilibrium constant combined with a fast equilibration rate relative to the rate of fluorination process.148
Scheme 27.

Enantioselective Fluorination of Cyclic α-Branched Aldehydes
In the consideration of the steric effect, enabling the pyrrolidine ring of the catalyst to stay farthest away from the oxetane scaffold, the Z-enamine, which gives the Si face exposed to fluorination, is proposed to be dominant for the 4-spirocyclic substituted substrates. In contrast, for 2-spirocyclic substituted substrates, E-enamine intermediates were presumed to be favered and the Si face was shielded by chiral amine catalyst (Figure 3).
Figure 3.
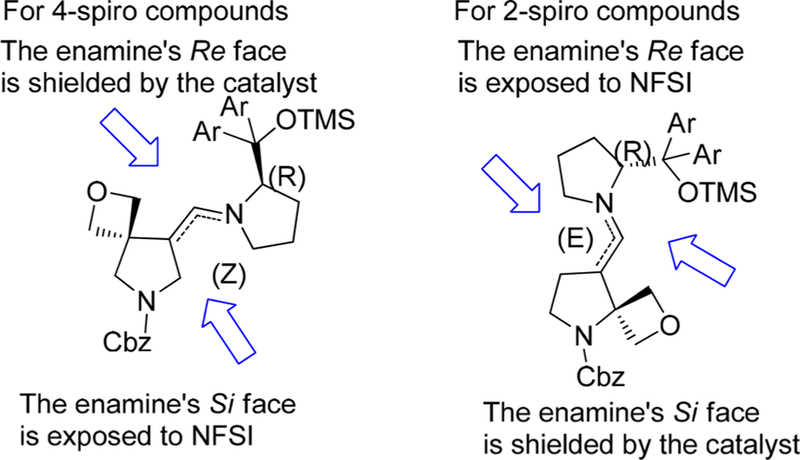
Enamine transition-state geometries to rationalize the enantioselectivity.
In 2016, the Quintavalla group reported a protocol for enantioselective construction of a fluorinated quaternary stereogenic center at α position of α,α-dialkyl aldehydes 115 bearing an enantiomerically pure chiral center in the C β-position catalyzed by chiral secondary amine catalysts (Scheme 28).149 During the catalyst screening, organocatalysts bearing acidic protons were found to clearly improve the reactivity of the electrophilic fluorination process. Thus, the combination of J0rgensen’s diarylprolinols C11 or ent-C11 and trifluoroacetic acid as co-catalyst had been shown to enhance the reaction rate and diastereocontrol. Then treatment of chiral γ-nitroaldehydes and 1.2 equiv of NFSI with the combination of C11 or ent-C11 (15 mol %) and trifluoroacetic acid, in an equimolar amount with respect to the organocatalysts in tert-butyl methyl ether (0.24 M) at room temperature for 17–72 h, afforded the desired fluorinated product 116 with moderate to good yields and with high diastereocontrol (dr up to 97:3) and good enantioselectivity (erup to 99:1).
Scheme 28.

Enantioselective Fluorination of α,α-Dialkyl Aldehydes
Additionally, for the substrate scope, increasing the steric hindrance at the Cα position was found to decrease the reaction yields and diastereocontrol. On the basis of the computational calculations for reaction mechanism, the enantioselectivity was under catalysis control with a very limited role imposed by the stereocenter on Cβ in substrates. Meanwhile, the fluorination process was found to be faster than the E/Z-enamine intermediates equilibration and E-enamines are responsible for the formation of the major products.
In 2006, the Jørgensen group reported the asymmetric α-fluorination of α-branched aldehydes 117 catalyzed by a new type of primary amine catalyst, aminated 8-amino-2-naphthol C13 (Scheme 29), in which the chirality originates from nonbiaryl atropisomerism.150 The primary amine catalyst C13 can be prepared by the asymmetric Friedel-Crafts amination of 8-amino-2-naphthol controlled by aminated 6′-hydroxy cincho-na alkaloids with a nonbiaryl atropisomeric functionalization at the 5′-position of the quinoline core. By utilizing the amine catalyst C13 (5 mol %) and fluorinating reagent NFSI (1.2 equiv) in the solvent mixture of hexane/iPrOH (9:1), the substrates, which have an a-aromatic group without substituent or with electron-withdrawing substituents, can be converted to the corresponding aldehydes 118 bearing a fluorinated quaternary carbon center in moderate yields (up to 60%) and good enantioselective control (up to 90% ee). The chiral induction can be rationally explained by the main E-geometry enamine intermediates, which would only permit the NFSI to attack from the Si-face of the E-enamine. However, for fluorination of aliphatic a-branched aldehydes, the E/Z-isomerism of the enamine intermediates, which can cause undistinguishable faces of the enamine, can be responsible for the poor enantioselectivties (less than 31% ee). Additionally, extrapolating from the X-ray analysis of an acylated analogue of the catalysis, the geometry of the enamine intermediate could be stabilized by the intramolecular hydrogen bonding between the carbonyl oxygen of the Boc group in the N-1 atom in the aminated 8-amino-2-naphthol C13 and the enamine NH (Figure 4).
Scheme 29.

Enantioselective Fluorination of α,α-Dialkyl Aldehydes Followed by the Reduction to the Corresponding Alcohols
Figure 4.

Possible enamine intermediate showing a proposed intramolecular hydrogen bond.
In 2015, the Jacobsen group developed a new primary amine catalyst C14 for the asymmetric α-fluorination of α-branched aldehydes 120 to afford α-fluorinated quaternary stereogenic centers 121 with high yields (from 74% to 99%) and with moderate to high enantioselective control (48–86% ee) on 1.0 mmol scale (Scheme 30).151 Although impressive progress had made in the reaction of chiral secondary amine catalyzed α-fluorination of unbranched aldehydes to provide a-trisubstituted products, secondary amines are inappropriate for the reactions of a-branched aldehydes due to the steric demands of the reacting partners.152 Meanwhile, unfavorable tautomer equilibration and poorer control of E/Z selectivity of enamine intermediates are the problems inherent to primary amine catalysts, which can induce the formation of less steric hindered enamines.
Scheme 30.

Enantioselective Fluorination of α,α-Dialkyl Aldehydes via Enamine Intermediates
On the basis of the bifunctional primary aminothioureas designed to activate the hindered carbonyls via formation the nucleophilic enamines and simultaneously activate the electrophiles via hydrogen bonding interaction, the benzamide analogue C14 was designed and screened as the most effective catalyst (20 mol %), while the dual H-bond donor in aminothioureas and its urea analogue had proved unnecessary. Meanwhile, the combination of achiral acids such as trifluoroacetic acid (TFA, 20 mol %) and inorganic base additives NaHCO3 (1.0 equiv) can enhance both reaction rate and enantioselective control. For probing the scope of substrates, α-aryl-α-methyl substituted aldehydes (12 examples) afforded α-fluorinated products with good enantioselective control (from 69% to 86% ee) and enhanced enantiomeric purity after recrystallization (up to 99% ee), while α-ethyl-substituted and α, α-dialkyl branched aldehydes afforded desired products with significantly lower enantioselectivity.
Additionally, a one gram scale of starting branched aldehydes, 2-phenylpropionaldehyde, can be converted to the corresponding fluorinated products in 99% yield and with 80% ee under optimized reaction conditions. On the basis of the computational analysis (lowest energy calculated structures on B3LYP/6–31G(d)), a plausible stereoinduction model was supported. The intramolecular H-bond between enamine NH and the benzamide carbonyl serves to rigidify the catalyst backbone, and one aryl ring of the terphenyl moiety locating in the one face of the enamine could block accessing to incoming fluorinated electrophile. The E-enamines leading to the formation of R-products was calculated to lie 1.28 kcal-mol–1 lower than corresponding Z-enamine which could induce the minor S-enantiomers. Thus, the enantioselective control may be determined and limited by the E/Z ratio of the enamine intermediates (Scheme 30).
By 2016, the Shibatomi group reported the enantioselective fluorination of a-branched aldehydes 123 to introduce a fluorine atom onto a tertiary carbon center catalyzed by the chiral primary amine C15 (Scheme 31).153 Although similar catalyst structure had already been designed in 1996,154 there was no research related to its applications. Additionally, the steric hindrance of aryl substituent (Ar = 3,5-tBuC6H3) in 3,3′-positions on the binaphthyl backbone of the primary amine catalysis exerted impact on the asymmetric induction because employing catalysts without aryl substituents (replacement Ar group by H) in 3,3′-positions provided nearly racemic products. Treatment of rac-aldehydes (1.5 equiv) and NFSI (1.0 equiv) in the presence of primary amine catalysis C15 (10 mol %) and 3,5-dinitrobenzoic acid (10 mol %) as co-catalysis in toluene at 0 °C for 20–48 h, the fluorinated products were isolated after reduction to corresponding primary alcohols 125 with good enantioselectivity (up to 99% ee). The substrate scope was found to be limited to α-alkyl-α-aryl aldehydes as low yields, or disappointingly low enantioselectivity was observed for α,α-dialkyl aldehydes. Subsequently, the resulting α-fluoroaldehydes bearing a quaternary stereogenic center can transform to corresponding α-hydroxyacetals via C–F bond cleavage without obviously loss of enantiomeric purity.
Scheme 31.

Enantioselective Fluorination of α-Aryl-α-alkyl aldehydes
Recently, the Luo group reported a reagent-controlled enantioselectivity switch for organocatalytic asymmetric fluorination of acyclic and cyclic β-ketoesters and β -ketoamides or 1,3-diketones to construct fluorinated quaternary stereogenic centers by a single chiral primary amine catalysis C16 (Scheme 32).155,156 By employing two commercial available electrophilic fluorination reagents NFSI and N-fluoro-pyridinium salt (NFPy), the two R- and S- enantiomers can be prepared with good enantioselectivity, in both cases tuning by one single chiral primary-secondary diamine catalysis C16.
Scheme 32.
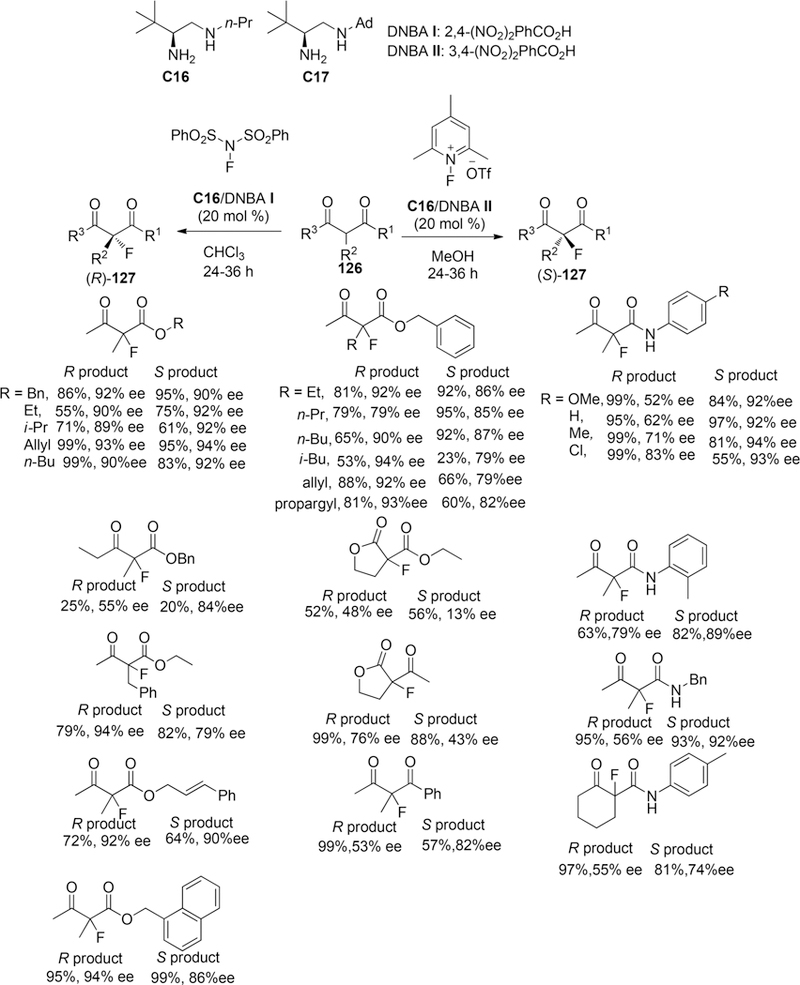
Enantioselective Fluorination of β-Ketoesters 126
For the R-selective process, the reaction of β-ketoesters 126 were performed under the combination of catalysis C16 (20 mol %) and dinitrobenzoic acid DNBA-I (20 mol %) with NFSI in chloroform at room temperature for 24–36 h, and R-products 127 (up to 93% ee) can be prepared through an enamine based intermolecular F-attack transition state model I tuning by hydrogen bonding interaction between the sulfonyl moiety in NFSI and an the protonated ammonium N–H in amine catalysis to favor the Re-facial fluorination. On the other hand, with the combination of catalysis C16 (20 mol %) and dinitrobenzoic acid DNBA-II (20 mol %) with N-fluoro-pyridinium salts as the fluorination reagent in methanol at room temperature, the desired S-configuration products 127 can be obtained in good enantioselectivity (up to 99% ee) via transition state model II, which controlled by the electrostatic repulsion between the cationic charged ammonium in amine catalysis and cationic charged pyridinium species (Figure 5).
Figure 5.
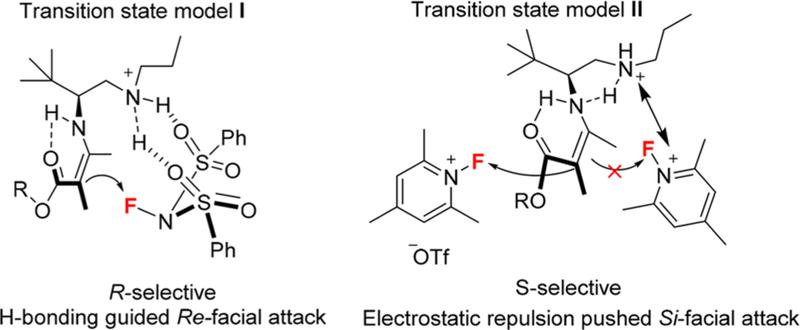
Proposed transition states (I and II) for the two enantioselectivity switch fluorination reactions.
Additionally, improvement in enantioselectivity can be observed by alternation of the acidic additive from TfOH to dinitrobenzoic acids which can also simplify the reaction manipulation as the resulting salts were bench-stable crystal solids, and primary amine C16 performed equally as well as its more bulky counterparts such as adamantyl primary amine C17. Thus, transition state model II would be mainly dominated by electrostatic repulsion of two positively charged species not by steric effect, which also had been demonstrated by DFT calculation at B3LYP/6–31G* level of approximation.
To probe the scope of substrates, for the R-selective process which was controlled by the H-bonding transition state model I, under optimized reaction conditions, a variety β-keto esters including alkyl, benzyl, allyl, and cinnamyl esters afforded the desired products with high ee value (89%−93% ee) in good yields. Then a variety of β-ketoamides including N-aliphatic and N-aryl amides, 1,3-diketone, and lactone-type substrates provided moderated to good enantioselectivity (from 43% to 83% ee). For S-selective reactions which controlled by electrostatic mode II, the yields and enantioselectivity can be comparable to the corresponding R-selective process with the exception of reactions for β-ketoamides, wherein the S-selective process afforded high enantioselectivity (84–94% ee). Additionally, a gram-scale reaction also was conducted to probe the utility of the fluorination reaction of benzyl 2-methyl-3-oxobutanoate and the desired R-selective product was prepared in good yield (0.952 g, 85% yield) with 94% ee.
Recently, the Xu group reported the enantioselective fluorination of β-ketoesters 128 catalyzed by the combination of cinchona alkaloid-derived chiral primary amines QN-NH2 C18 and l-leucine C19 as dual organocatalysts in good yields and only moderate enantioselectivity (up to 55% ee) (Scheme 33).157 Furthermore, only racemic α-fluorinated ketones 129 can be obtained in the absence of cocatalyst l-leucine, and the function of each catalyst is still unclear.
Scheme 33.

Asymmetric Fluorination of β-Ketoesters 128
3.1.1.1.3. Phase-Transfer Catalysis
The interaction of charged intermediates and reagents in organic transformations with a charged, chiral catalyst has emerged as a powerful strategy for enantioselective synthesis,158 and the quaternary ammonium cation in a cinchona-derived phase-transfer catalyst could form a nucleophilic ionic complex with an anion of the nucleophile, which will induce the approaching of the electrophiles from the least sterically hindered face of the ionic complex.159
In 2002, the Kim group reported the catalytic enantioselective electrophilic fluorination of β-keto esters 130 to construct a fluorinated quaternary stereogenic carbon center promoted by quaternary ammonium salts derived from cinchona alkaloids as phase-transfer catalysts (Scheme 34).160 To enhance the enantioselective control, the introduction of a bulky subunit, the (3,5-di-tert-butyl-4-methoxy)benzyl group, into the position of the bridgehead nitrogen of cinchona alkaloids was taken into consideration in catalyst design. Treatment of cyclic β-keto esters with NFSI in the presence of a catalytic amount of cationic phase-transfer catalyst C20 (10 mol %) and inorganic base K2CO3 or Cs2CO3 in toluene afforded the desired α-fluoro β-keto esters 131 in good yields and moderate enantiomeric excess (from 48% ee to 69% ee). Then for acyclic substrate 132, NaH was required in the fluorination process and only 40% ee can be observed.
Scheme 34.

Enantioselective Electrophilic Fluorination of β-Keto Esters 130
Then in 2004, they extended the asymmetric fluorination reactions catalyzed by chiral quaternary ammonium salts to other substrates such as acyclic α-cyano acetate derivatives (Scheme 35).161 Treatment aromatic groups substituted α-cyano acetates 134 with the cinchona alkaloid derived catalyst C20 (10 mol %) and fluorinating reagent NFSI in the presence of Cs2CO3 in toluene gave the desired α-fluorinated products 135 in good yields and good enantioselectivity (from 73% ee to 76% ee).
Scheme 35.

Asymmetric Fluorination of α-Cyano Acetates 134
In 2013, the Lu group revealed the asymmetric fluorination of indane carboxylates 136 bearing a sterically hindered t-butyl ester group catalyzed by quaternary ammonium salts derived from cinchona alkaloid to install a fluorine containing quaternary carbon centers (Scheme 36).162 When employing catalyst C22 in which the C–9 hydroxy function in cinchonine was protected by sterically bulky adamantoyl group (catalyst C22 was developed by the Jørgensen group in the year of 2006163), the desired α-fluorinated products 137 were prepared with satisfactory enantioselectivity (up to 94% ee).
Scheme 36.

Asymmetric Fluorination of Indane Carboxylates 136
Additionally, they insisted that the negatively charged enolate intermediate, paired with ammonium cation in catalyst C22 via ionic interaction, was proposed to place into the groove between the quinoline and quinuclidine in transition state (Figure 6). Thus, the sterically hindered adamantoyl group blocked effectively the bottom face of the plane in enolate intermediate, leading the approaching of electrophile from another face with high enantioselectivity.
Figure 6.

Proposed transition model for asymmetric fluorination.
The C9 position functions in cinchona alkaloids serving as a molecular hinge because four low energy conformers (anti-open, anti-closed, syn-open, sjn-closed) can be generated by the internal rotations around the C8-C9 and C9-C4′ bonds (Figure 7).164,165 Thus, governing the internal rotations in cinchonium based catalysts to modulate the conformations and reactivity was highly desirable. In 2012, the Gilmour group reported a class of chiral, fluorinated cinchonium salts for enantio-induction in electrophilic fluorination of β-ketoesters 138 (Scheme 37).166 They provided a strategy by using fluorine stereoelectronic and electrostatic effects (a fluorine-ammoniumion gauche effect σC−H → σ*C−F; Fδ− … N+) for conformational dynamics control (restricting the rotation about C8–C9 bond in catalysis). The antiperiplanar alignment of the C–F bond positioned in C9 of cinchona alkaloid and C–N+ bonds was stereoelectronically disfavored (mismatch between donor and acceptor orbitals), thus ruling out the possible conformers anti-closed and open-closed. Meanwhile, they found that the installation of the N-benzyl group clearly impacted the rotation about C9–C4′ bonds in catalysis, leading to the anti-open conformation (X-ray crystal analysis) as majority conformers in the solid state.
Figure 7.

Fluorine–ammonium ion gauche effect controlling the conformation of 9-fluoro-cinchonine.
Scheme 37.
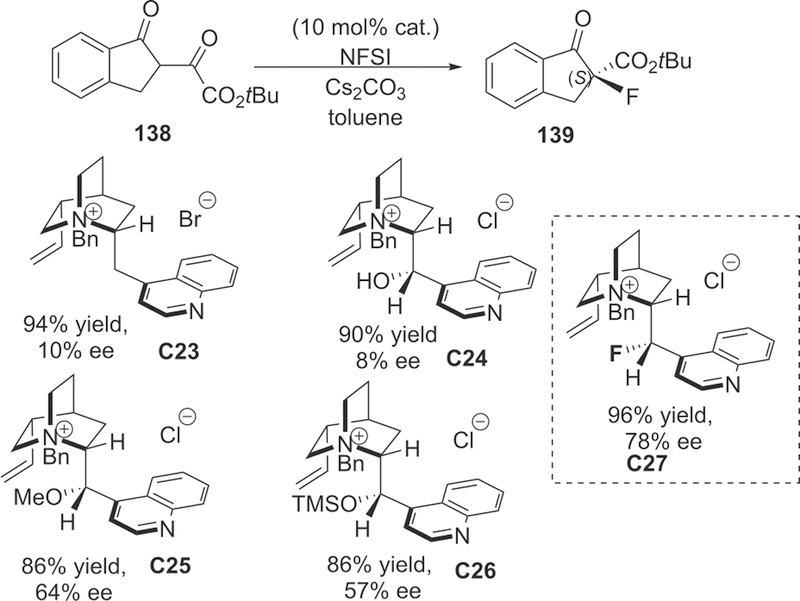
Asymmetric Electrophilic Fluorination of β-Ketoesters 138
For the asymmetric fluorination reaction, as the principle governing catalyst–substrate recognition is electrostatic interaction in nature (ion pairing), the effect of counterion (such as Cl–, F–, BF4–, PF6–, SbF6–) was investigated and no appreciable variation in enantioselectivity was observed, and for the fluorination of tert butyl-1-indanone-2-carboxylate, catalysts with H, OH, OMe, and OTMS group located in C9 position, showed lower enantioselective control than fluorinated cinchonium salts C27 (78% ee). Additionally, the 1H NMR spectra for the combination of catalysis/Cs2CO3/substrate were investigated to probe the structure of the catalyst–substrate complex.
In 2010, the Maruoka group developed a chiral bifunctional phase-transfer catalyst introducing bis(diarylhydroxmethyl) substituents at 3,3′-positions of the chiral binaphthyl core and incorporating the scaffold of thiomorpholine-derived quaternary ammonium salts, which can be applied to asymmetric α-fluorination of t-butyl Indane carboxylates and their analogues 140 in high yields and high enantioselective control (8 examples, from 88% ee to 98% ee) (Scheme 38).167 In the catalysis design, the free hydroxyl group in the moiety of bis(diarylhydroxmethyl) substituent in catalyst C28 had been proven to be crucial role to obtain high enantioselective control because corresponding methyl-protected catalysts could only afford racemic mixtures.
Scheme 38.
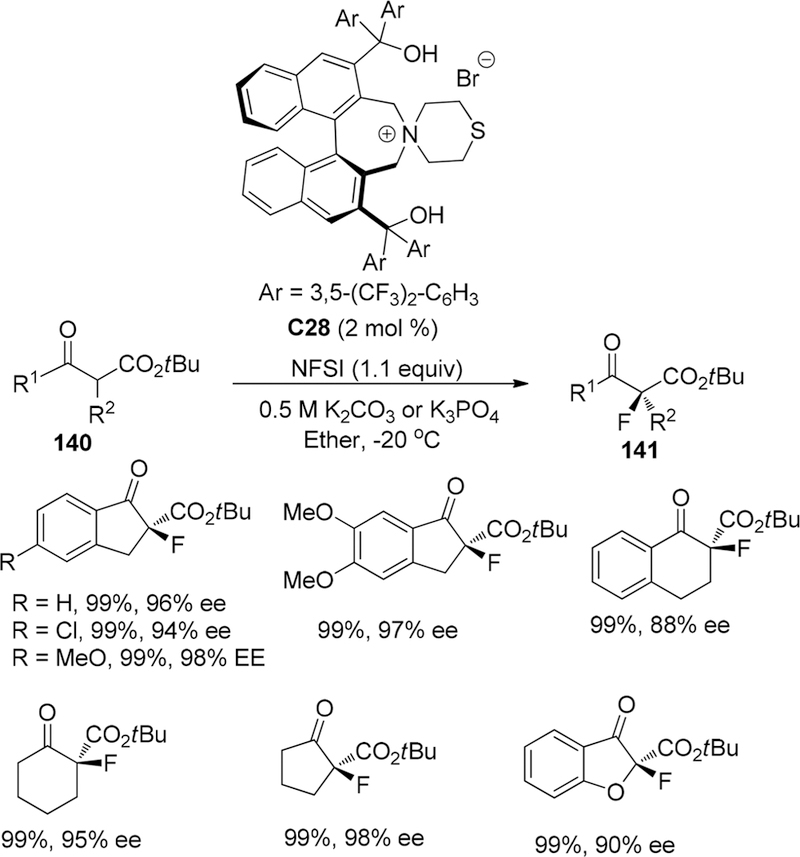
Asymmetric α-Fluorination of t-Butyl Indane Carboxylates and Their Analogues 140
On the basis of the X-ray analysis of a morpholine-derived bifunctional catalyst, a proposed transition state mode was rationalized by forming an ammonium Z-enolate, which could stabilized by the hydrogen bonding between the enolate oxygen and one hydroxyl group in bis(diarylhydroxmethyl) substituents and ionic interaction between ammonium salt and enolate anion (Figure 8). However, for acyclic β-keto esters, only low enantioselectivity (5–20% ee) can be observed.
Figure 8.

Proposed transition state structure for asymmetric fluorination of β-keto esters.
Although many chiral ammonium salts derived catalysts had been reported in a catalytic electrophilic fluorination, further design and development of new chiral phase-transfer catalysts are still attractive research subjects.168 In 2013, Ma, Cahard, and co-authors reported the asymmetric electrophilic fluorination of 3-substituted benzofuran-2(3H)-ones 142 via phase-transfer catalyst C29 (2 mol %) based on chiral P-spiro phosphonium scaffold to afford the desired products 143 bearing a fluorinated quaternary stereogenic center in high yields and only moderate enantioselectivity (up to 56% ee) (Scheme 39).169 The chiral phosphonium salt C29 was first developed for asymmetric electrophilic amination of benzofuran-2(3H)-ones in the year of 2011.170 Tailoring of the inorganic base (K2HPO4) and initial concentration of the substrate was considered to be crucial for this liquid-liquid phase transfer transformation. Additionally, for the substrates benzofuran-2(3H)-ones, the phosphonium salts would serve as preferred catalysts because various quaternary ammonium salts derived from cinchona alkaloids afford poor enantioselectivity (less than 8% ee).
Scheme 39.
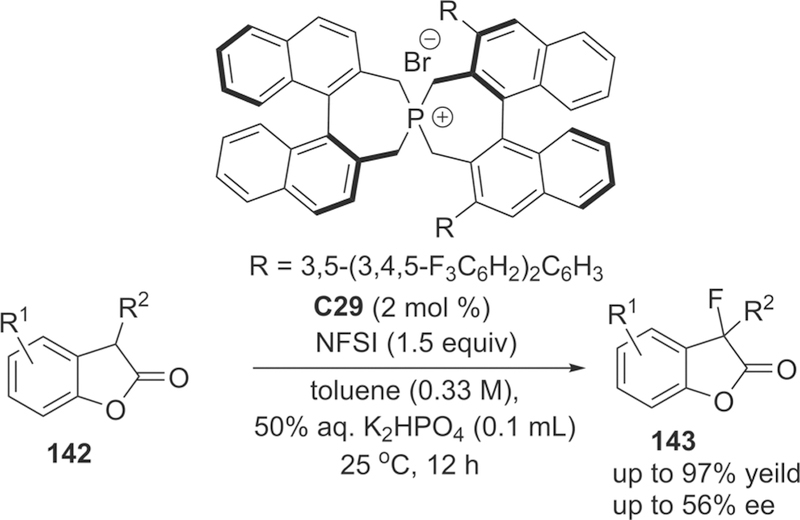
Asymmetric Electrophilic Fluorination of 3-Substituted Benzofuran-2(3H)-ones 142
While catalytic electrophilic fluorination via anionic intermediates through ionic interaction (ion pairing) with chiral cationic catalyst, such as quaternary ammonium and phosphonium salt, is well precedented, reports of analogues charge-inverted processes have been rather less explored. The chiral anion phase-transfer catalysis,158,171 which can bring an insoluble cationic promoter into solution, provides a platform for asymmetric fluorination that proceed via cationic intermediates or that utilize cationic reagents. While the first example of chiral anion PTC was reported by the Toste group,172 they expanded their methodology to prove the versatility of chiral phosphoric acids for a broad range of substrate classes by using Selectflor, which is normally insoluble in nonpolar media. They envisioned that a lipophilic chiral anionic catalyst, bulky chiral phosphate anions, could extract an insoluble cationic reagent such as Selecfluor from insoluble phase into the organic phase. Ion-pairing of the cationic reagent with the chiral phosphate anions would then provide a chiral environment for the desired enantioselective fluorination. Subsequently, the Toste group developed many attractive catalytic methods to construct a C–F quaternary carbon center with high enantioselectivity by employing the lipophilic, bulky phosphate anions as phase-transfer catalyst. Detailed discussion of asymmetric fluorination of alkenes such as fluoro-cyclization or fluorinative dearomatization process will be presented in 3.1.1.2 F-Additions to C=C Bonds (vide infra).
In 2014, the Toste group developed a dual catalysis method for asymmetric fluorination of α-branched cyclohexanones 144 to generate quaternary fluorine-containing stereocenters involving the merge of two separate catalytic cycles: a chiral lipophilic BINOL derived phosphate anion as phase-transfer catalyst C30 to active Selectfluor and enamine catalyst employing protected amino acids.173 First, they hypothesized that the incorporation of amine catalysis would form a transient enamine intermediate as a hydrogen bond donor which can attach to the soluble chiral electrophilic fluorinating reagent generated by the ion exchange between the lipophilic chiral phospahate anion and achiral tetrafluoroborate counteranions of insoluble Selectfluor (Figure 9). Then they also demonstrated the necessity of the chiral controlling elements on both catalysts, anionic phase-transfer catalyst, and chiral primary amine catalyst, in order to obtain high enantioselective control, because in the absence of either, both yield and enantiomeric excess are poor (less than 10% in each case). Additionally, they also found that the small amount of water was critical to achieve high enantioselectivity because the inconsistent levels of moisture in dry inorganic base Na2CO3, which had been replaced by Na2CO3H2O, can cause some unpredictable outcomes.
Figure 9.

Proposed dual catalytic cycle for the enantioselective fluorination of ketones.
Under the optimized reaction condition, by the combinations of chiral phosphoric acid C31 (5 mol %)/protected amino acids A (20 mol %)/ketone (2.0 equiv)/Selectfluor (1.0 equiv)/Na2CO3·H2O (2.0 equiv) in toluene at room temperature for 40 h, 2-aryl group substituted cyclohexanones 146 exhibited compatibility to match the two chiral catalysts, leading to high enantioselectivity (from 83% to 94% ee) (Scheme 40). However, no fluorination was observed for 2-alkylcyclohexanones and closely related acyclic ketones.
Scheme 40.
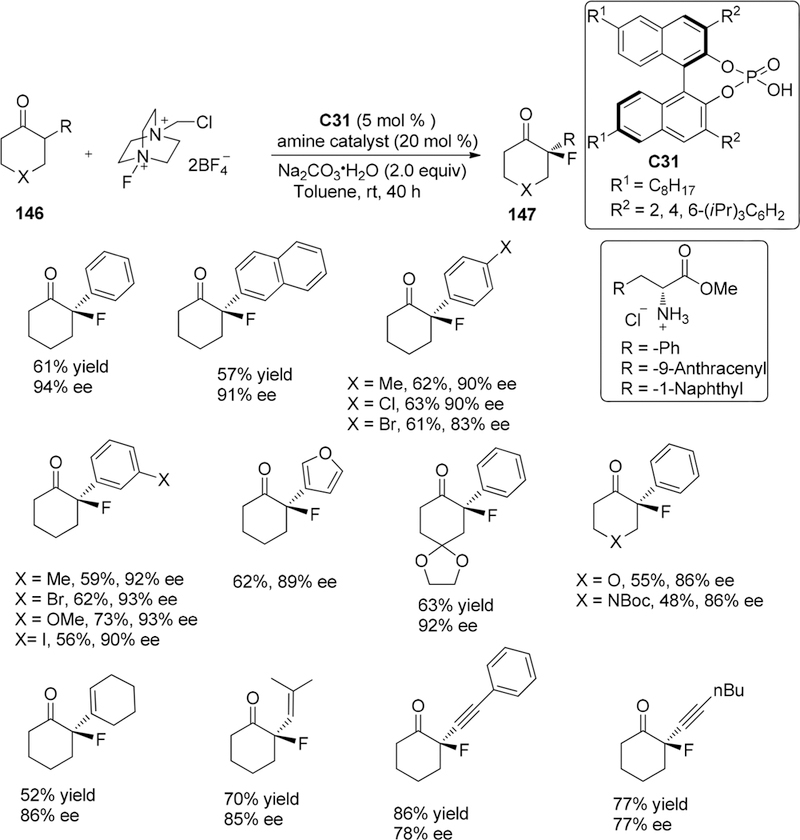
Enantioselective Fluorination of Cyclohexanones 146
3.1.1.1.4. Bifunctional Organocatalysts Based on (Thio)urea Backbone.
(Thio)ureas were commonly employed as hydrogen-bonding donors in catalyst design. The combination of (thio)urea scaffolds to activate and control the reactivity of electrophiles with other catalytically active motifs, such as quaternary ammonium salts, to activate nucleophiles, can be expected to form a remarkable bifunctional catalytic system for introduction fluorine into bioactive molecules.
In 2012, the Niu group reported the thiourea–tertiary amine C32 catalyzed enantioselective fluorination of β-keto esters 148 to construct a C–F quaternary carbon center with high enantioselectivity (up to 99% ee) (Scheme 41).174 After screening the thiourea catalysts, they found that the more steric hindered catalysts produced almost racemic products and they demonstrated the necessity for both thiourea scaffold and tertiary amine motif in order to achieve high enantioselectivity by comparing various catalysts bearing similar core structures. Additionally, the catalyst loading also played a critical role in stereochemical control because the ee value was observed to increase with the increasing catalyst loading (50 mol % catalyst C32,99% ee). Then the catalyst loading can be tailored to 10 mol % when employing DMAP (10 mol %) as base in MeOH.
Scheme 41.
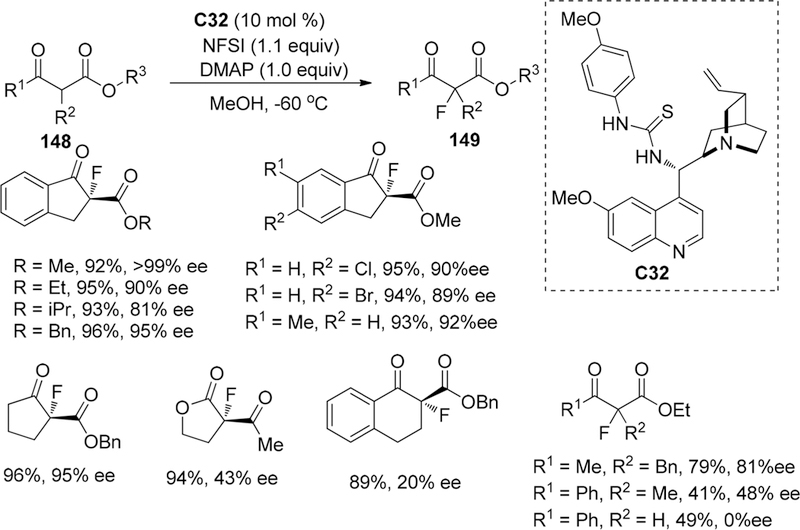
Enantioselective Fluorination of β-Keto Esters 148
Additionally, The alkoxy group in indanone carboxylate derivatives had shown a great influence on the enantiomeric excess of products because the bulky tertiary butyl group substituted β-keto esters can only afford racemic products (Scheme 41), and the acyclic β-keto esters and tetralone derivatives generally provided poor enantioselectivity. Subsequently, the authors hypothesized that the thiourea group serving as a hydrogen donor, which can bond to NFSI and the 1,3-dicarbonyl group, could interacted with the tertiary amine group (basic center) in the bifunctional catalyst (Figure 10).
Figure 10.
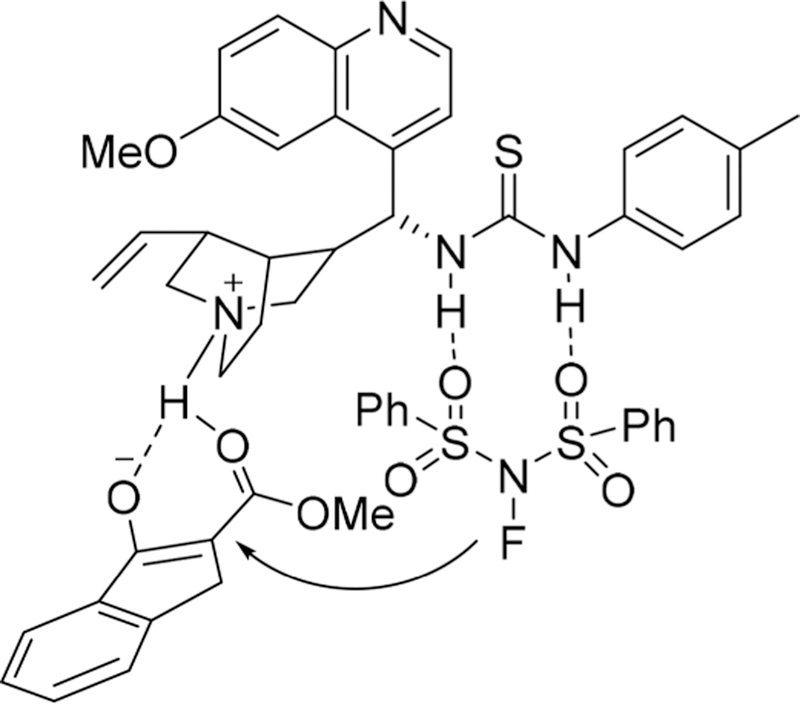
Proposed transition state for asymmetric fluorination catalyzed by thiourea–tertiary amine.
In 2014, the Waser group reported the synthesis of a new class of systematically modified (thio)urea-containing quaternary ammonium salts catalysts based on 1,2-trans-cyclohexanedi-amine chiral backbones and evaluation of its catalytic potential in asymmetric α-fluorination of β-keto esters.175 Meanwhile, the importance of the bifunctional nature of these catalysts, including the (thio)urea motif, serving as hydrogen bonds donor, and the quaternary ammonium scaffold as phase-transfer catalyst, was demonstrated by control experiments as only racemic mixture can be obtained when employing simplified monofunctional analogues. For the preparation of catalyst C33, they provided an alternative synthesis sequence proceeding through an early quaternization, followed by the late-stage introduction of (thio)urea scaffold.
After screening the catalysts, for the electrophilic fluorination reactions, they found that the urea-containing catalysts with an ester-containing aryl group on the urea scaffold performed better than corresponding thioureas. Additionally, increasing the steric hindrance in the moiety of quaternary ammonium was beneficial, leading to some enhancement in enantioselectivity. Thus, by employing catalyst C33 (2 mol %) and NFSI (1.1 equiv) in the presence of K3PO4 (0.5 M aqueous, 2.0 equiv) in m-xylene at −10 °C, the β-keto esters especially for adamantly esters can be converted to the corresponding fluorinated products 151 with good enantioselectivity (up to 86% ee) (Scheme 42).
Scheme 42.
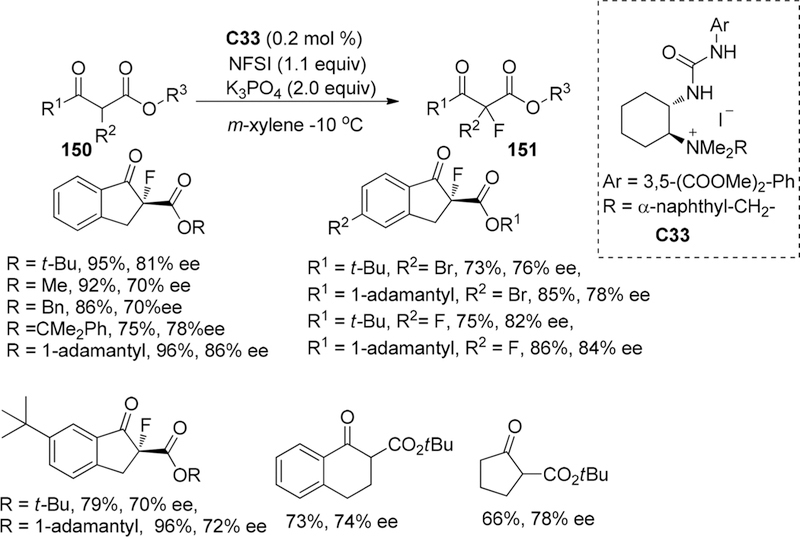
Asymmetric Fluorination of β-Keto Esters 150
The proposed bifunctional activation transition state model is shown in Figure 11. While the ammonium moiety was considered to activate the enolate anion of keto-esters via ionic interaction, the urea would attach to NFSI proceeding through hydrogen bonding effect, leading to Re facial recognition by the fluorinating reagent.
Figure 11.

Proposed activation model of the urea-containing chiral quaternary ammonium salt.
In 2012, the Ma group reported a diastereo- and enantioselective one-pot multistep 1,4-addition and fluorination sequence between pyrazolone derivatives 152 and nitro-olefins 153 catalyzed by the chiral tertiary-amine-thiourea catalyst and achiral benzoic acid as co-catalyst, leading to enantioselective formation of adjacent stereogenic centers, including a C–F quaternary carbon (Scheme 43).176 To achieve this desired asymmetric sequential transformation, various bifunctional chiral tertiary-amine–thiourea catalysts were initially screened to match the tandem process. Eventually, they found that the saccharide motif derived from l-glucopyranose in catalyst C34 was critical for controlling the stereochemistry of the two adjacent stereogenic centers in fluorinated products, comparing with the dramatic decrease in diastereo- and enantioselectivity when using similar bifunctional catalysts without a saccharide moiety. In addition, the combination of saccharide-derived catalyst C34 with external weak acids, such as benzoic acid, was found to improve the enantioselectivity. The access to the keto tautomer of pyrazolin-5-one, which can be activated by protonated tertiary-amine-thiourea catalysts, was proven to be critical for achieving the transformation.
Scheme 43.

Asymmetric Fluorination of Pyrazolone Derivatives 152
With the aim to explore the scope of substrates, various nitro-styrene derivtives with different substitution patterns on the aromatic ring and alkyl-substituted nitro-alkenes were studied and found to afford the desired products with high level of stereoselectivity (Scheme 43). The 1,4-conjugated addition products can be isolated in quantitative yields with high enantioselectivity by suppressing the fluorination step. However, subjecting Michael addition product to triethylamine and NFSI, the decrease in diastereoselectivity was observed compared with the unchanged stereoselectivity when employing chiral catalyst C34 for the second dearomatization–fluorination transformation. Thus, the authors insisted that the chiral bifunctional catalyst not only controls the stereochemistry in the first Michael addition step but also plays important role in diastereoselective formation of the C–F bond in the second step.
Proposed mechanism for this one-pot sequential transformation involving two catalytic cycles is presented in Figure 12. In the Michael addition sequence, the nitro-olefin is assumed to attach the thiourea group via hydrogen bonding, while the enol forms of the pyrazolone substrates could coordinate to the multiple hydrogen bonding interaction help the 1,4-addition ammonium center through hydrogen bonding to achieve high products to dissociate from the catalyst and form a highly level of enantioselectivity. In the subsequent fluorination step, organized structure for interaction with NFSI.
Figure 12.

Proposed mechanism for the 1,4-addition and fluorination sequence.
In 2013, the Ma group extended their methodology to the synthesis of chiral fluorinated isoxazol-5(4H)-ones 156 bearing a C–F quaternary carbon center via similar sequential conjugate addition/dearomatizative fluorination transformations in high yields with good stereoselectivity (Scheme 44).177 After optimization of reaction conditions, inorganic base NaCO3 (1.2 equiv) was found to be essential for dearomatizative fluorination process. In addition, the combination of the (S,S)-1,2-diphenylethane-1,2-diamine moiety with d-glucopyranose in catalyst design was found to enhance the stereoselectivity because the erosion of the enantiomeric purity was observed when using a thiourea–tertiary amine catalyst without the saccharide motif. Furthermore, catalyst C35 bearing a cyclic tertiary amine substituents was particularly good for the transformation of various aryl group substituted isoxazol-5(4H)-ones 155 and nitroalkenes 153 to the corresponding fluorinated isoxazol-5(4H)-ones 156 in good stereoselectivies (97:3 to 99:1 dr, 62% to 92% ee). On the other hand, alkyl substituted nitro-olefins were found to be unsuitable for this tandem process and no desired products were obtained. Finally, step-by-step control experiments were performed to probe the determining factors in controlling the stereoselectivity of the fluorination step. Similar results had been obtained, as shown above (Figure 12), for the bifunctional catalyst based on thiourea backbone, which was supposed to control the stereochemistry of both Michael addition and subsequent fluorination via hydrogen bonding.
Scheme 44.
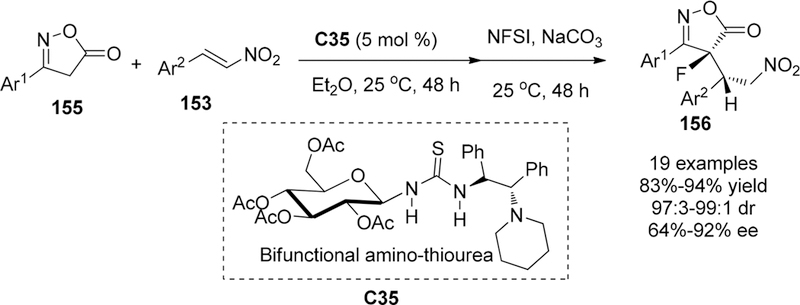
Conjugate Addition/Dearomatizative Fluorination of Isoxazol-5(4H)-ones 155
3.1.1.1.5. Miscellaneous Catalysis.
An organocatalytic one-pot and tandem intramolecular oxa-Michael addition/electrophilic fluorination transformations for the preparation of chiral monofluorinated flavanones bearing a C–F quaternary stereo-center has been revealed by the Zhao group in 2009.178 After the evaluation of different bifunctional catalysts with structural modifications based on quinine and quinidine, the sterically demanding catalysts were proven to be inferior and the trifluoromethyl group containing catalyst C36 was chosen as the best candidate. Solvent effect was detected in reaction optimization, the decrease of enantioselectivity was observed in chlorine-containing solvents, and a slight increase in the ee values was shown in Et2O or methyl tert-butyl ether at the expense of marked erosion in the yields due to a poor solubility of NFSI in these two solvents. Thus, the desired fluorinated flavanones 158 can be prepared in high yields with good enantioselectivity (up to 96% ee) in the presence of quinidine-derived bifunctional catalyst C36 (15 mol %), NFSI (1.5 equiv), and Na2CO3 (1.2 equiv) in toluene at room temperature (Scheme 45).
Scheme 45.

oxa-Michael Addition/Electrophilic Fluorination Transformations
Although the organocatalyst seems to play a role in the electrophilic fluorination step, the oxa-Michael addition was assumed as an enantio-discriminating step, which was considered to be induced by the bifunctional catalyst with a free hydroxyl group through hydrogen bonding to activate both nucleophiles and electrophilic acceptors. Subsequently, the oxygen nucleo-phile was supposed to attack the Re face of C=C bond in α,β-unsatuated ketones, leading to the R-configured intermediate, which would further induce subsequent electrophilic fluorination step to form a C–F quaternary stereocenter in an enantioselective manner.
In 2015, the Wang group reported an organocatalytic asymmetric one-pot and tandem Friedel−Crafts/fluorination process to enable the construction of two vicinal tetrasubstituted stereocenters containing a C−F quaternary carbon in high diastereo- and enantioselectivity (>20:1 dr and up to >99:1 ee).179 The tandem transformations delivered a class of fluorinated oxindole−pyrazolone adducts via the enantioselective Friedel−Crafts addition of 4-nonsubstituented pyrazolones 160 to isatin-derived ketimines 159 followed by subsequent diastereoselective electrophilic fluorination of the pyrazolone scaffold. In addition, by employing quinine squaramide catalyst C37, which can be used in low loading (0.5 mol %) without obvious impact on the yield and enantioselectivity, the gram scale experiments was efficiently achieved with fully maintained stereoselectivity, demonstrating the practical utility of this tandem transformations (Scheme 46). To probe the stereochemical determining factors in each step of the one-pot sequential process, control experiment via step-by-step procedure had been performed. The stereoselectivity of the electrophilic fluorination of pyrazolone moiety in oxindole−pyrazolone adducts was proven to be a pure substrate-controlled process, which is independent of the chiral catalyst C37.
Scheme 46.
Tandem Friedel–Crafts/Fluorination Process: Structural Generality (1), Large-Scale Synthesis (2), and Elucidation of the Stereochemical Outcome (3)
In 2012, the Sun group reported the enantioselective synthesis of β,γ-unsaturated α-fluoro-esters catalyzed by N-heterocyclic carbene C38.180 They hypothesized that enals with a leaving group in the γ position could activate byNHC catalyst to form an NHC-bound dienolate, which was expected to react with electrophilic fluorinating reagents to afford α-fluorinated carbonyl compounds. Although the NHC-catalyzed α C−F bonds formation process proceeded in good yield with high enantioselective control (up to 95% ee) in a broad range of enals, a substituent in the α-position significantly slowed down the reaction rate; the desired α-fluoro-ester 163 (only one case) bearing a C−F quaternary carbon center was obtain in poor yield (5%) with moderate enantioselectivity (55% ee) (Scheme 47).
Scheme 47.

Preparation of α-Fluoro-ester 163
Despite the significant progress in asymmetric electrophilic fluorination catalyzed by organocatalysts based on the skeleton of cinchona alkaloids or (thio)urea, the development of chiral guanidines, which combine the strong basicity of the guanidine moiety and hydrogen-bond donor capacity of its conjugate acid, was still less reported because the lack of a general chiral scaffold that is both easily available and readily tunable in steric and electronic properties.
In 2014, the Wang group developed a series of chiral guanidines based on the tartaric acid skeleton for asymmetric fluorination by using NFSI. Guanidine catalyst C39 containing 2,6-diisopropylaniline fragment was recognized as a superior promoter for enantioselective fluorination of 1,3-dicarbonyl and α-cyano carbonyl compounds 164 with high yield and moderate to good ee value (up to 84% ee).181 Notably, β-keto esters bearing a six-membered ring also gave the desired α-fluorinated products with good enantioselectivity (80% ee), and fivemembered cyclic α-cyano carbonyl compounds gave inferior enantioselectivity (39% ee), as compared with the six-membered cyclic substrate (82% ee). Furthermore, cyclic β-diketones afforded the desired products in high yields with poor enantioselectivity. This method also had proven to be unsuitable for acyclic substrate such as acyclic β-keto esters (Scheme 48).
Scheme 48.

Enantioselective Fluorination of 1,3-Dicarbonyl and α-Cyano Carbonyl Compounds 164
In 2014, the Akiyama group reported the enantioselective fluorination of β-keto esters 166 catalyzed by chiral sodium phosphate C40.182 Two active intermediates, sodium enolate and sodium phosphate, were supposed to form simultaneously in the presence of a slight excess (1.1 equiv) of an inorganic base NaCO3, and the corresponding fluorinated products 167 for indanone and benzofuranone derivatives were obtained in good yield (up to 98%) with good enantioselectivity (up to 92% ee). An investigation of the ester group in substrates showed that the sterically demanding group such as tert-butyl ester (decreasing to 60% ee) was unsuitable for this asymmetric process (Scheme 49). Also, a decreased selectivity was observed in reaction of six-membered ring containing substrate due to the loss of structural rigidity.
Scheme 49.
Enantioselective Fluorination of β-Keto Esters 166
For probing the mechanism for the origin of enantiomeric control, the authors argued that the sodium enolate and sodium phosphate species were essential for achieving the selectivity (Figure 13). To rule out the anionic phase-transfer process, under optimized reaction conditions, low yield and very low enantioselectivity (14%) were observed by using Selectfluor instead of NFSI and no reaction can be detected by using N-fluoropyridinium triflate salt as a fluorinating reagent. Thus, a 12-membered ring transition state mode combining the Lewis base activation of sodium enolate by oxygen in the phosphate moiety sodium of phosphate moiety was proposed to rationalize the with the Lewis acidic activation between the oxygen in NFSI and origin of enantioselectivity.
Figure 13.
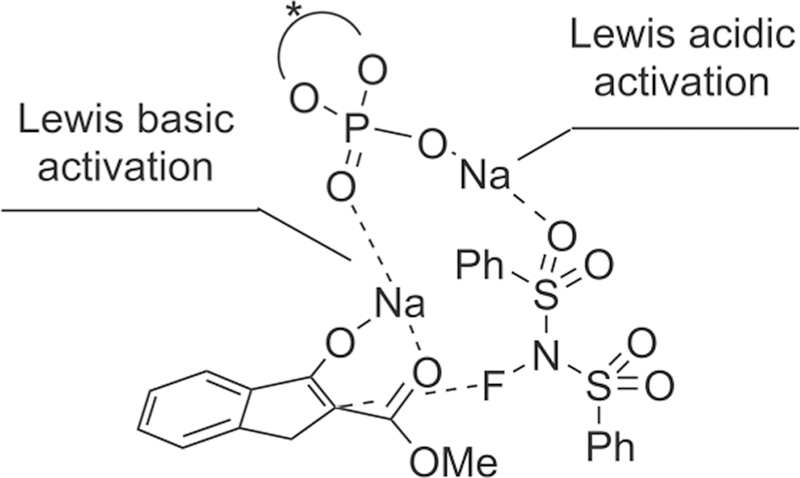
Proposed transition state mode for fluorination catalyzed by chiral sodium phosphate.
In 2014, the Fu group reported the catalytic enantioselective coupling of aryl alkyl ketenes 168 with NFSI and C6F5ONa to afford fluorinated products 169 bearing a C–F quaternary carbon center in the α-position to the carbonyl group in high yield with high enantioselectivity (up to 98% ee) (Scheme 50).183 First, the generation of tertiary alkyl fluorides was observed in very low conversion (<5% yield) by the combination of a planar-chiral nucleophilic catalyst (C41) (10 mol %)/NFSI (1.0 equiv)/ketenes (1.0 equiv) without addition of external nucleophiles. Then they hypothesized that the inaccessible catalyst regeneration may be caused by the stability of the proposed N-acylpyridinium intermediate toward (SO2Ph)2N– anion. After screening different type of nucleophiles, sodium pentafluorophenoxide was found to be essential for turnover, releasing the catalyst (C41) from stable N-acylpyridinium intermediate.
Scheme 50.
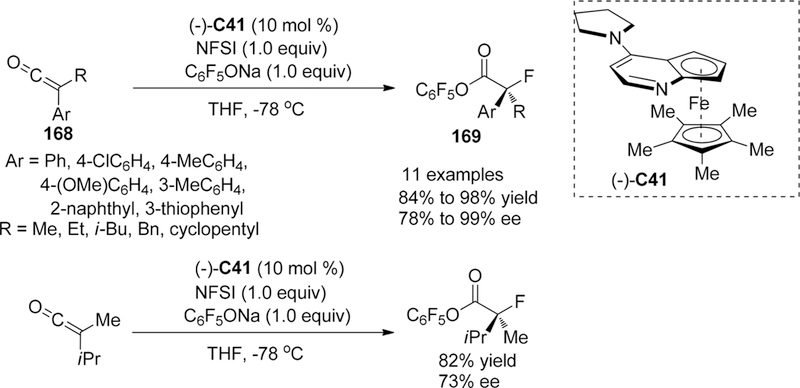
Enantioselective Coupling of Aryl Alkyl Ketenes 168
The lower ee value was observed with large alkyl groups in aryl alkyl ketenes (for instance, the ee value decreasing from 98% to 80% when changing methyl to cyclo-pentyl), and the gram scale experiment was proven to proceed without the loss of yield and enantioselectivity. Meanwhile, a special case about a dialkyl ketene substrate, methyl, and iPr group substituted ketene gave a promising level of enantioselectivity (73% ee) for the asymmetric fluorination.
For probing the reaction mechanism, two catalytic cycles involving a C41-derived chiral enolate as key intermediate (Figure 14) and C41-derived chiral N–F reagent were provided to rationalize the enantio-induction. To gain insight into the operative pathway, the ketene was proven to be involved in the rate-determining step via kinetic study to rule out the pathway related to the formation of C41-derived chiral N–F reagent, and the enantioselectivity was expected to be determined by the fluorine transfer from NFSI to the enolate intermediate generated by the nucleophilic addition of C41 to ketene. Additionally, the enantioenriched N-acylpyridinium salt intermediate can be isolated in the absence of an added nucleophile and confirmed by X-ray analysis of its derivative.
Figure 14.
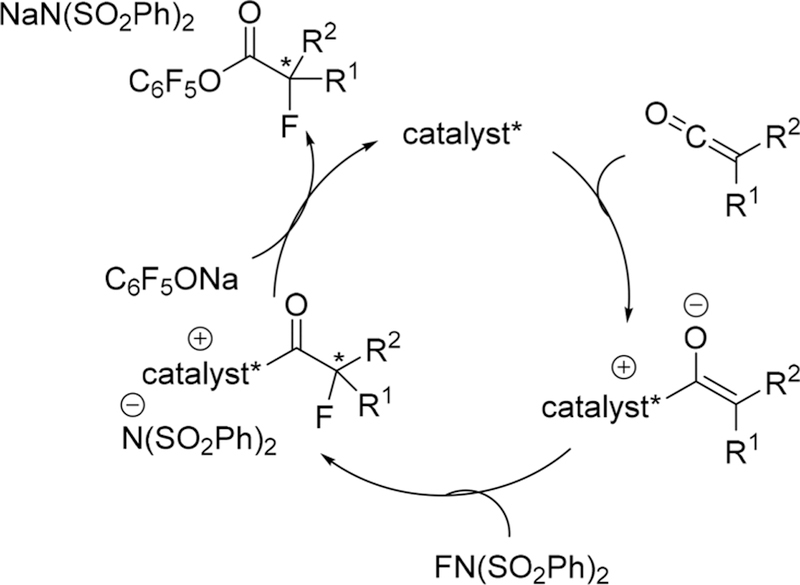
Possible mechanism for the C41-catalyzed enantioselective fluorination of ketenes.
3.1.1.2. F-Additions to C=C Bonds
The fluoro-functionalization of alkenes by electrophilic fluorinating reagent, as an enantioselective installation of a C–F quaternary stereogenic center, is an appealing strategy that converts feedstock alkenes into valuable fluorinated molecules for further application. The asymmetric fluoro-cyclizations of active C=C bonds, such as acyl enols,132 allyl silanes, and silyl enol ethers133 or prochiral indoles with a pendant heteronucleophile tethered at C3 position,137 catalyzed by cinchona alkaloids in the presence of Selectfluor or NFSI, has been already discussed in this work (3.1.1.1.1. Tertiary Amine Catalysts Derived from Cinchona Alkaloids and Their Analogues). Thus, in this section, we will focus on other asymmetric protocols for fluorination of C=C bonds leading to the construction of fluorine containing quaternary stereocenters. The anionic phase-transfer strategy by using catalysts derived from chiral phosphoric acids highlights the development in asymmetric fluorination of alkenes, including fluoro-cyclization, 1,4-aminofluorination of conjugated dienes, and fluorinative dearomatization of phenols, etc. (vide infra). Additionally, some one-pot and tandem processes involving fluorination postcyclization or nucleophilic addition, the initial nucleophilic sequence followed by fluorination, will also be discussed in this section because it is indirect or formal fluorination of alkenes. Although the preceding review related to asymmetric fluoro-cyclizations of alkenes was provided by the Gouverneur group in 2014,184 we will further cover advances in the construction of a C–F quaternary carbon center via electrophilic addition to C=C.
3.1.1.2.1. Substrates or Reagents Controlled Asymmetric Fluoro-Functionalization of Alkenes.
In 2013, the Li group reported the silver-catalyzed phosphono-fluorination of unactivated alkenes 170, the condensation of various alkenes with diethyl phophite and Selectfluor, via a radical process to afford the desired β-fluorinated alkylphosphonates 171 in high yield.185 Then silver-catalyzed oxidative generation of phosphonyl radicals and fluorine atom transfer were proposed to rationalize the reaction mechanism, and two cases were provided to show the diastereoselective control (substrate control) in this transformation (Scheme 51).
Scheme 51.

Phosphono-fluorination of Unactivated Alkenes 170
In 2013, the Gouverneur group reported the asymmetric electrophilic fluoro-cyclization of indenes 172 bearing a 2-phenylethyl substituent at the C2 position as nucleophiles. First, the nonenantioselective version of this transformation, under the optimized reaction condition (N-fluoro-2,6-dichloropyridinium triflate (1.1 equiv) in the presence of inorganic base NaHCO3 (3.0 equiv) in nitromethane {(0.05 M) at 40 °C for 1 h}, the desired fluoro-cyclization products 173 were predominantly formed as the corresponding sjn-diastereomers (>20:1 dr) (Scheme 52)
Scheme 52.

Electrophilic Fluoro-cyclization of Indenes 172
Later, they tried to develop an enantioselective variant of this fluoro-cyclizative transformation. The chiral N-F reagents derived from the combination of cinchona alkaloids and Selectfluor failed to proceed due to the decreased nucleophilic ability of carbon nucleophiles. Subsequently, anionic phase-transfer catalysts were also proven to be inactive because of the requirement for a polar solvent. Thus, a more reactive chiral N–F reagent 175 based on the structural core of Selectfluor with chiral environment on the dicationic DABCO core was prepared. The reagent 175, bearing the para-electron-withdrawing CF3 substituents on the aryl rings, was found to be the most reactive. Thus, the tuning of the reactivity and solubility profile of this new N–F reagent can be achieved by tailoring the steric and electronic properties of the aryl group on the DABCO core. Subsequently, by using chiral N–F reagent 175 (1.5 equiv) in 1,4-dioxane, several fluorotetrahydro-5H-indeno-[2,1-c]-quinolones 176 can be prepared in good yield (up to 99%) with good enantioselectivity (ee values averaging 71%). Additionally, the structural variation of the substrates can exert a dramatic impact on the enantioselectivity because the corresponding hexahydrobenzo[k]phenanthridine was formed with low enantioselectivity (19% ee) (Scheme 53).
Scheme 53.
Preparation of Fluorotetrahydro-5H-indeno-[2,1-c]quinolones 176
3.1.1.2.2. One-Pot and Tandem Process for Fluoro-functionalization of Alkenes.
In 2007, the Ma group reported enantioselective tandem Nazarov cyclization/electrophilic fluorination sequence catalyzed by Lewis acid, Cu(OTf)2/(R)-Ph-bis(oxazoline) C42, leading to enantioenriched 1-indanone derivatives 178 with adjacent carbon- and fluorine-substituted quaternary and tertiary stereocenters with moderate to good stereoselective control (up to >49:1 dr, 95.5% ee) (Scheme 54).186 In their initial exploration, they envisioned that the electrophilic reagents can capture the metal-bound enolate intermediate generated by the first 4n-electrocyclization sequence, and the trans-fluorinated indanone derivatives were predominantly formed (dr >49:1) in the presence of achiral Lewis acid catalysts. A similar asymmetric intramolecular oxa-Michael addition/electrophilic fluorination tandem process, catalyzed by an organocatalyst, has already been discussed (3.1.1.1.5 Miscellaneous Catalysis).
Scheme 54.
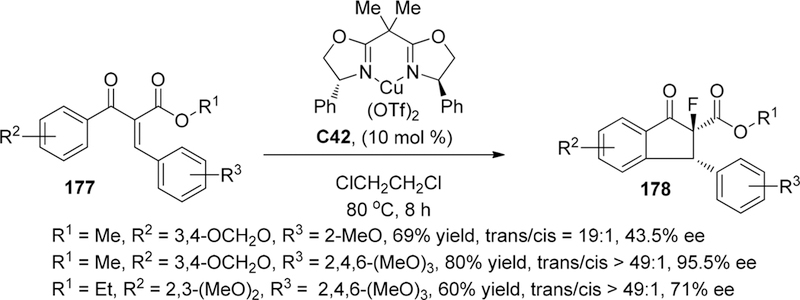
Enantioselective Tandem Nazarov Cyclization/Electrophilic Fluorination Sequence
In 2011, the Ma group reported the copper-catalyzed tandem 1,4-addition/fluorination sequence with a range of acyclic alkylidene β-keto esters 179 and dialkyl-zinc reagents to install a fluorine-substituted quaternary stereocenter adjacent to carbon tertiary stereocenter in good yields and high diastereo- and enantioselective control (up to >99:1 dr, 98% ee) (Scheme 55).18 To further narrow the space around the P-ligated copper center to improve the stereoselectivity, the sterically bulky substituents were incorporated onto the 3- and 3′-positions of the binaphthol moiety of the axial chirality. Latter, after evaluation of a series of well-defined modular modification on the biphenyl scaffold at 3- and 3′-positions of the binaphthol, chiral monodentate phosphoramidite ligand L2 was found to perform better in this tandem transformation.
Scheme 55.

Tandem 1,4-Addition/Fluorination Sequence of Acyclic Alkylidene β-Keto Esters 179
Subsequently, a broad range of alkylidene β-keto esters bearing aryl and heteroaromatic rings can be converted to the corresponding fluorinated products in good yields (72–91%) and satisfactory stereoselectivity (82–97% ee). A gram scale experiment, performed without erosion of the enantiomeric purity of the product, was conducted in order to verify the synthetic utility of this asymmetric tandem transformation (Scheme 55). In addition, the one-pot tandem process was demonstrated to be crucial for controlling the diastereoselectivity in the second electrophilic fluorination because the obvious decrease in diastereoselectivity was observed when the reaction was conducted in a stepwise manner (Scheme 56).
Scheme 56.

Asymmetric Fluorination of Alkylidene β-Keto Esters
3.1.1.2.3. F-Additions to C=C Bonds Catalyzed by Anionic Phase-Transfer Catalyst
In 2011, the Toste group reported the highly enantioselective fluoro-cyclization of alkenes 181, including electronically disadvantaged alkenes, with a cationic fluorinating agent and chiral binaphthol-derived phosphates via a phase-transfer process in which chiral anionic catalyst brings an insoluble positively charged reagents or cationic reaction intermediates into the organic phase (Scheme 57).188 First, they envisioned that the lipophilic, bulky chiral phosphate anions bearing the hydrophobic alkyl chains attached to the chiral binaphthol backbone such as catalyst C43 could exchange one or both tetrafluoroborate anions associated with Selectfluor, which normally is insoluble in nonpolar media, to bring the versatile cationic fluorinating agent into organic phase to form a chiral ion pair, bringing the chiral environment for subsequent fluoro-cyclization wherein a pendant nucleophile can attack a π-bond activated by an electrophilic fluorine source. To verify their hypothesis toward the anionic phase-transfer process, enantioenriched spiro-fused oxazolines were prepared from the sequence of fluorination of the enol ether moiety followed by attacking the oxocarbenium ion generated on the initial step by the amide carbonyl moiety in dihydropyran substrates. It should be pointed out that a good yield and only modest decrease in enantioselectivity can be observed for employing less electro-rich alkenes such as an unactivated olefin with only alkyl substituents.
Scheme 57.
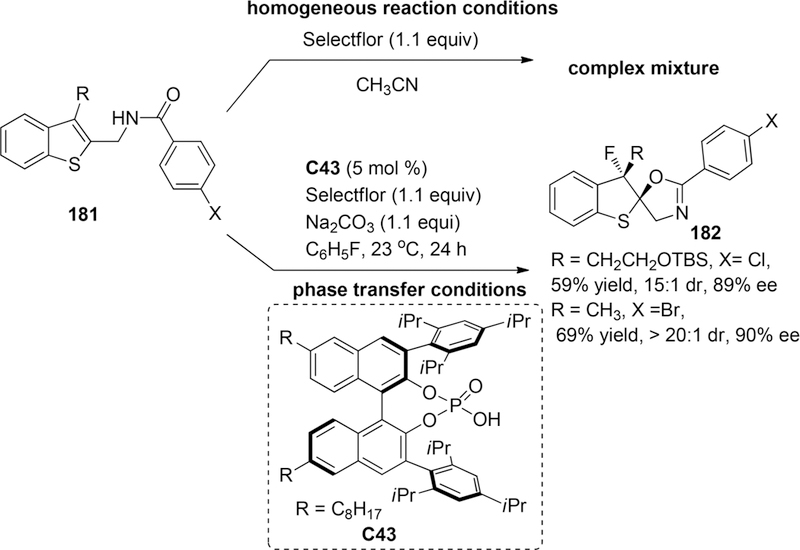
Enantioselective Fluoro-cyclization of Alkenes 181
Benzothiophene substrates (two examples) can convert to corresponding spiro-fused oxazolines bearing a C–F quaternary carbon center in moderate yield with good diastereoselectivity (up to >20:1) and enantioselectivity (up to 90% ee). Additionally, the results of control experiments showed that the anionic phase-transfer protocol indeed improved the tolerance toward sensitive functionality, as only a complex mixture can be detected when using Selectfluor under homogeneous conditions, and the slow introduction of the fluorinating agent into solution via forming a chiral ion pair and a reduction in fluorinating reactivity in nonpolar solvent, were considered to be responsible for the improved chemoselectivity.
To explore the mechanism for this phase-transfer protocol, the nonlinear effect between the enantiomeric purity of the catalyst and products was observed by employing a catalyst with six different levels of enantiomeric enrichment, suggesting that both tetrafluoroborate anions in Selectlor are exchanged for chiral phosphate before the reaction with substrates. Then a catalytic cycle was proposed (Figure 15) where two equivalents of phosphate undergo salt metathesis with dicationic Selectfluor to provide chiral soluble ion pairs, which would induce the asymmetric fluoro-cyclization of alkene substrates.
Figure 15.
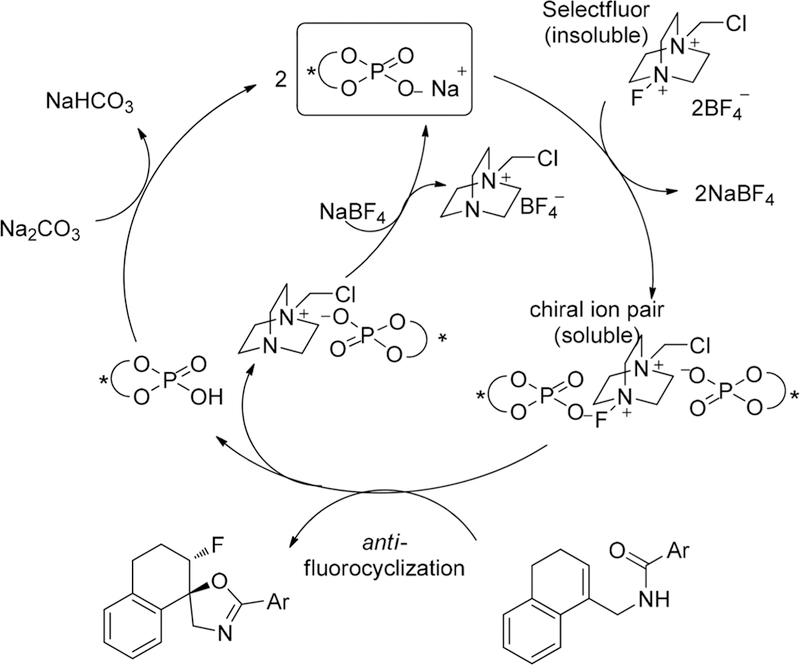
Proposed catalytic cycle for chiral anion phase-transfer catalyst.
Then in 2012, the Toste group had extended their anionic phase-transfer catalysis strategy by using BINOL-derived phosphate to encompass the enantioselective fluorination of cyclic enamides.189 After evaluation of the catalysts and optimization of reaction parameters, a combination of C43 and apolar hexane was chosen as a superior promoter for obtaining high enantioselective control. The inorganic base Na2CO3 was proven to be essential to activate phosphoric acids because low conversion with poor stereochemical selectivity was observed in the absence of Na2CO3.
The enantioenriched α-fluorinated benzoyl-imines 184 bearing a quaternary fluorinated stereocenters can be prepared from corresponding six-membered ring and five-membered ring enamides 183 with the toleration of various functionality such as methyl, allyl, phenyl, and benzyl groups in good yields (up to 94%) with high enantioselectivity (up to 98% ee). In some cases, the addition of 3-hexanol (5.0 equiv) as an additive was needed to improve the enantioselectivity. Additionally, as for 2-phenyl-cyclohexanone-derived enamide, a slightly reduced enantiose-lectivity (87% ee) and moderate yield (58%) was detected. To further probe the scope of cyclic enamides, 2-methylcyclohex-anone-derived enamide showed sluggish conversion under the optimized reaction conditions. Meanwhile, enantioenriched geminal chlorofluoro and bromofluoro benzoy-limines can be prepared from chloro- and bromo-substituted enamides in good yields with high enantioselectivity (Scheme 58).
Scheme 58.
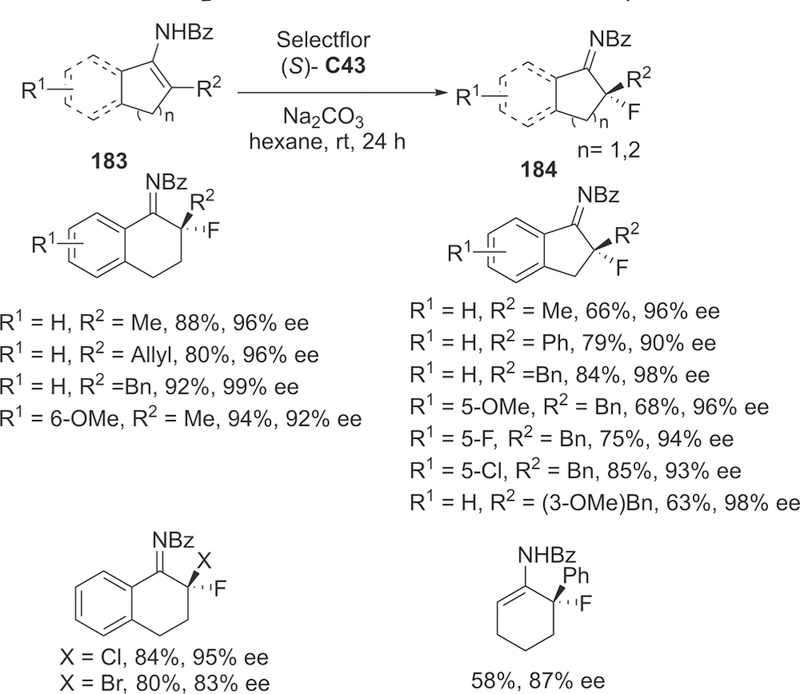
Preparation of α-Fluorinated Benzoyl-imines 184
Ahypothesis as to the origin of the observed enantioselectivity was proposed to rationalize the BINOL-derived phosphoric acid catalyzed asymmetric fluorination. Because of the bifunctional nature of chiral sodium phosphate, one oxygen atom in a phosphate anion would attach to Selectfluor via ion pairing interaction while simultaneously activating the enamide via hydrogen bonding between another oxygen atom in a phosphate anion and a NH proton in enamide. Thus, the steric bulk of the tetralone moiety in enamide was supposed to locate in an “open” quadrant, with the relatively small amide group positioning as a “closed” quadrant (Figure 16). The proposed transition state model was further demonstrated by the experimental results that the high tolerance toward substitutions on enamides. The tetralone moiety in enamide was considered to stay away from the catalyst center in a favored transition state model and thus had no effect on catalyst–substrate binding.
Figure 16.

Proposed transition state model for asymmetric fluorination of cyclic enamides catalyzed by anionic phase-transfer catalyst.
In 2012, the Toste group extended the methodology for asymmetric fluorination of cyclic enamides to tandem oxyfluorination transformations of acyclic aldehyde-derived enamides.190 The N-acyliminium ions, which generated from the fluorination of enamides, could allow the catalyst-controlled addition of an external oxygen nucleophiles via hydrogen bonding interaction, leading to enantioenriched α-fluoro N,O-aminals. The challenge in this tandem process was enabling the sequential enantioselective transformations via utilizing a single catalyst, which could control the formation of second stereo-center to match or mismatch the inherent diastereo-control derived from the initially installed chiral center. Finally, phenyl-substituted doubly axially chiral phosphate C44 was proven to produce a clear enhancement in enantioselectivity due to a more rigid and constrained pocket for the acyclic (Z)-configured enmides. Both aromatic and aliphatic substituted enamides were compatible in these one-pot tandem oxy-fluorination transformations. Moreover, the enantioenriched C–F quaternary carbon center can be constructed from the hydroxy-fluorination reaction of the E and Z benzoyl enamide 185 derived from 2-phenylpropionaldehyde (Scheme 59). The (E)-configured substrate reacted with poor diastereoselectivity and very high enantioselectivity (>99% ee), giving rise to anti-products 186 due to the results of double stereodifferentiation. For the (Z)-configured substrate, the syn-isomer was obtained as the major diastereomer (4:1 dr) with reduced enantioselectivity (83% ee) without obvious double stereodifferentiation effect.
Scheme 59.
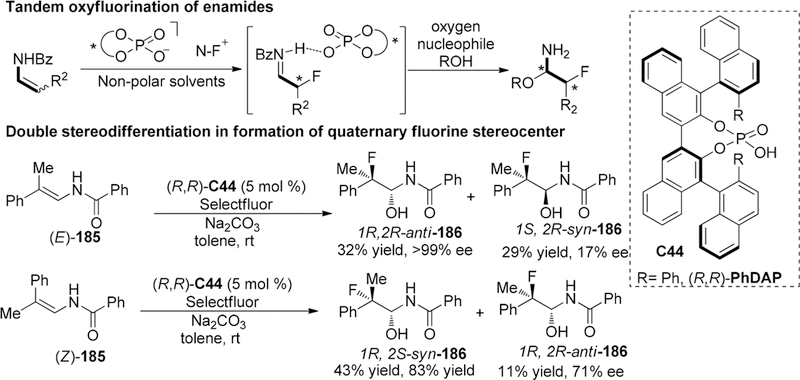
Tandem Oxyfluorination Transformations of Acyclic Aldehyde-Derived Enamides
In 2013, the Toste group reported the enantioselective 1,4-aminofluorocyclization of conjugated 1,3-dienes 179 catalyzed by lipophilic chiral phosphate as an anionic phase-transfer catalyst, leading to the 6-endo-trig cyclization to provide allylic fluorides 188 bearing a C–F quaternary stereocenter (Scheme 60).191 For evaluation of the BINOL-derived phosphate catalysts, with the aim of enhancing the solubility and selectivity of Selectflor without compromising reactivity, (R)-C45 was chosen as the superior candidate to produce higher enantiomeric excess. Additionally, the inorganic base was found to be critical to control the reactivity and selectivity in this tandem transformation. Thus, the high conversion and enantioselectivity for the desired fluorinated products generated by 6-endo-trig cyclization was observed by using the combination of (R)-C45 (10 mol %)/Na3PO4 (1.2 equiv)/Selectfluor (1.5 equiv) in solvent PhCF3 (0.1 M) at room temperature for 36 h.
Scheme 60.

Enantioselective 1,4-Aminofluorocyclization of Conjugated 1,3-Dienes
To account for the origin of the observed stereochemistry of the fluorine containing quaternary carbon center, the authors provided a plausible transition state (Figure 17). Two probable pathways can rationalize the observed diastereoselectivity, selective 1,4-anti-addition to the diene via concerted process or through a stepwise process that produces an equilibrating allyl cation intermediate in which one isomer can react preferentially. Subsequently, no reaction was detected for the Z-configured substrate, supporting the concerted reaction pathway that would be blocked by increasing a 1,3 strain in the transition state of the Z-configured substrate. For probing the substrate scope, under phase-transfer conditions, satisfactory conversion and stereo-selectivity can be observed for electron-rich substrates, which generally decompose in a homogeneous acetonitrile solution of Selectfluor. Although far from the reactive center, substitutions on benzamide arene showed impact on the selectivity and 4-tert-butylbenzamide as the nucleophile gave the best results.
Figure 17.

Suggested origin of diastereo- and regioselectivity for fluoro-amination.
When further expanding the substrate scope, the less-reactive diene derived from cyclohexene reacted sluggishly under optimized conditions with Selectfluor as the fluorinating reagent. Thus, to increase the electrophilicity of the fluorine source, structurally modified Selectfluor derivatives were prepared by replaceing the chlorine atom with electron-deficient aryl group. The new Selectfluor type derivatives were proven to be more powerful than Selectfluor in the preparation of octahydro-isoquinoline compounds with a C–F quaternary carbon center (Scheme 61).
Scheme 61.
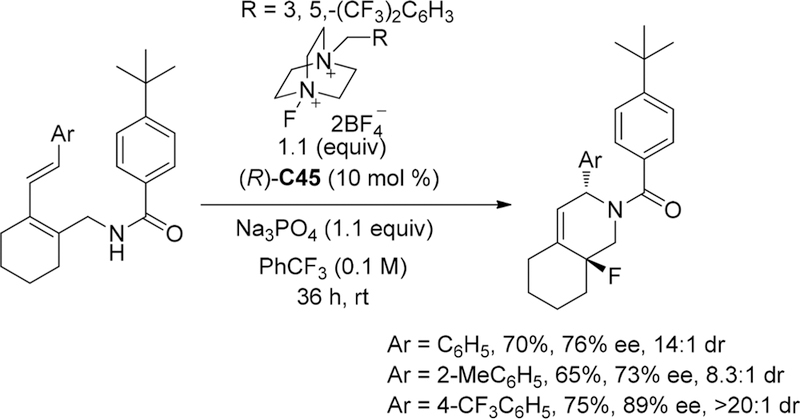
Preparation of Octahydro-isoquinoline Compounds with a C–F Quaternary Carbon Center
Directing groups (DG), which enable the functionalization of nonpolar bonds, to maintain the beneficial polar interactions between the substrate and chiral catalyst, have been proven to be powerful tool for the achievement of selectivity in asymmetric transformations. In 2013, the Toste group revealed a delicate strategy for electrophilic fluorination of alkenes 189 by the combination of directing groups and chiral phosphate anion C46 as the phase-transfer catalyst, leading to the enantioselective construction of quaternary C–F bonds in β-amino and phenolic allylic fluorides 190.192 The anionic conjugate bases of chiral phosphoric acids can abstract the positively charged electrophilic intermediate from insoluble phase to the reaction solution via ion pairing interaction. Meanwhile, the hydrogen bonding interaction, which can play a critical role in the enhancement of stereoselectivity, can help substrate to attach to chiral phosphate anion. Thus, they envisioned that an allylic hydrogen-bonding DG would help to direct an ion-paired chiral phosphate species via transition state organization for asym-metric fluorination of alkenes (Scheme 62).
Scheme 62.

Enantioselective Electrophilic Fluorination of Alkenes 189
In consideration of previous successful examples with amides as pendant nucleophiles, the allylic amides were chosen as DGs to verify their hypothesis. After evaluation of the phosphoric acids, (R)-C46 can gave improved stereoselectivity due to a tighter binding pocket, and very low enantiomeric excess for fluorination to alkenes was observed in the absence of DG. Additionally, the distance requirement for the directing group was proven to be essential for obtaining high stereoselectivity compared with homoallylic amide analogous with very low enantioselectivity. Subsequently, investigation of the influence of steric and electronic properties on enantioselectivity was performed to gain better understanding of the requirement for an effective DG. Steric parameters of DG seem to be more important because the increased enantioselectivity was found with the increased steric bulk. Then the substrate scope was explored; substrates with different ring size and benzamide substitution patterns, electron-donating groups substituted fused benzene ring on the bicyclic core, and heterocyclic substrates, proceeded in good yields with high enantioselectivity (up to 97% ee). Additionally, 2-hydroxyphenyl was found to be suitable DG as hydrogen-bond donor in the asymmetric electrophilic fluorination to alkenes, directing the enantioselective construction of β-phenolic tertiary and quaternary fluorides with aryl or alkyl substituents. Then they further expended this DG strategy to asymmetric fluorination of allylic alcohols via in situ generation boronic acid monoester as a directing group.75,193
In 2013, the Toste group expended the anionic phase-transfer strategy to the direct and highly enantioselective intermolecular fluorinative dearomatization of simple phenols to incorporate a quaternary fluorine stereocenter catalyzed by a BINOL-derived phosphate (S)-C45.194 The direct asymmetric dearomatization was considered as a challenging issue because it requires discrimination between enantiotopic faces of the arene during the dearomatization process. They hypothesized that the hydrogen bonding interaction between the donor Phen-OH and the acceptor, phosphoryl oxygen moiety in the soluble chiral phosphate-Selectfluor ion pair, might help the discrimination of the two enantiotopic faces of the phenols, leading to high levels of enantioselectivity. In addition, the reaction of Selectfluor with phenols under homogeneous conditions can afford multiple products depending on substrate structure and reaction conditions. Subjecting 5,6,7,8-tetrahydro-1-naphthol to optimized reaction conditions, the combination of chiral phosphoric acid (S)-C45 (5 mol %) in the presence of Selectfluor and inorganic base Na2CO3 in toluene at room temperature, the desired ortho-fluorinated product 192 can be prepared in 75% yield with 96% ee. The advantage of employing 2,3-disubstituted phenols 191 as substrates are as follows: First, the ortho-selective fluorinative dearomatization would afford the major products. Second, the greater steric differentiation between two sides of the substrates close to the hydrogen-bonding donor position of binding to the catalyst would enhance the enantioselectivity. Then various 2,3-di- and 2,3,4-trisubstituted phenols can be converted to corresponding ortho-fluorinated product with high enantioselectivity control (87–96% ee) (Scheme 63).
Scheme 63.

Asymmetric Fluorination of 2,3-Disubstituted Phenols 191
In the case of o-cresol without substitutions occupying the C–3 position, the [4 + 2] cycloaddition of the chiral 2,4-cyclo-hexadienone intermediates 194 generated from the initial fluorination was observed with good enantioselectivity (Scheme 64), and increasing steric demand of the ortho substituent by using o-benzylphenol, higher eantioselectivity (97% ee) can be detected. Substrates with benzyl, phenyl, isopropyl, and cyclohexyl group at 2-, 2,4-positions in phenol can afford the [4 + 2] dimer products 195 with good enantioselectivity, and a decreased stereochemical selectivity can be observed by using 2,5-substituted substrates due to the decreasing in the steric differentiation of two sides of phenols (Scheme 64). Subsequently, the [4 + 2] dimer products can undergo retro-[4 + 2]/[4 + 2] sequence with N-phenylmaleimide and cyclopentadiene dimer to deliver a single diastereomer without erosion of enantiomeric purity.
Scheme 64.

Tandem Fluorination-[4 + 2] Cycloaddition
Additionally, para-fluorinated products can be prepared in low to moderate yields with good enantioselectivity (86% ee) by incorporating a germinal 8,8′ disubstitution moiety without the possibility of ortho-selective fluorinative dearomatization (Scheme 65). The para-fluorinated substrate 197 was designed in accordance with the hypothesis that a clear steric distinction between two sides of phenol was essential for improving enantioselectivity. Subsequently, the fluorinated analogue of natural product (–)-grandifloracin was prepared from silylox-ymethylphenol to verify the utility of this method.
Scheme 65.

Preparation of Fluorinated Analogue of Natural Product (–)-Grandifloracin
In 2017, the You group reported the asymmetric fluorinative dearomatization of tryptamide derivatives via cascade fluorocyclization catalyzed by chiral phosphate anion derived from BINOL backbone as anionic phase-transfer catalyst C47, leading to construction of two consecutive quaternary stereogenic centers in fluorinated pyrroloindolines 199 with good enantioselectivity (up to 97% ee).195 Positive results can be observed when screening polar solvents under homogeneous conditions, the combination of catalyst C47 (5 or 10 mol %) and proton sponge (1.1 equiv) and Selectfluor (1.1 equiv) in mixed solvents of fluoro-benzene and acetonitrile (1:1) was optimized to be the best conditions. For exploration substrates scope, N-Boc protected tryptamines with varied electron-donating (5-Me, 5-MeO, 5-t-Bu) or electron-withdrawing group (5-F, 5-Cl, 5-Br, 5-CF3, 5-CO2Et) substituted at the C5 position of the indole moiety were well tolerated (82%−90% ee) (Scheme 66), and 6-Br, 4,6-dihalo-, and 2-alkyl group substituted substrates were also evaluated to provided desired fluorocylization products in moderate yields with good enantioselectivity (85%–97% ee). Additionally, the gram-scale experiment proceeded well without the obvious erosion in enantiomeric purity of the fluorinated products.
Scheme 66.
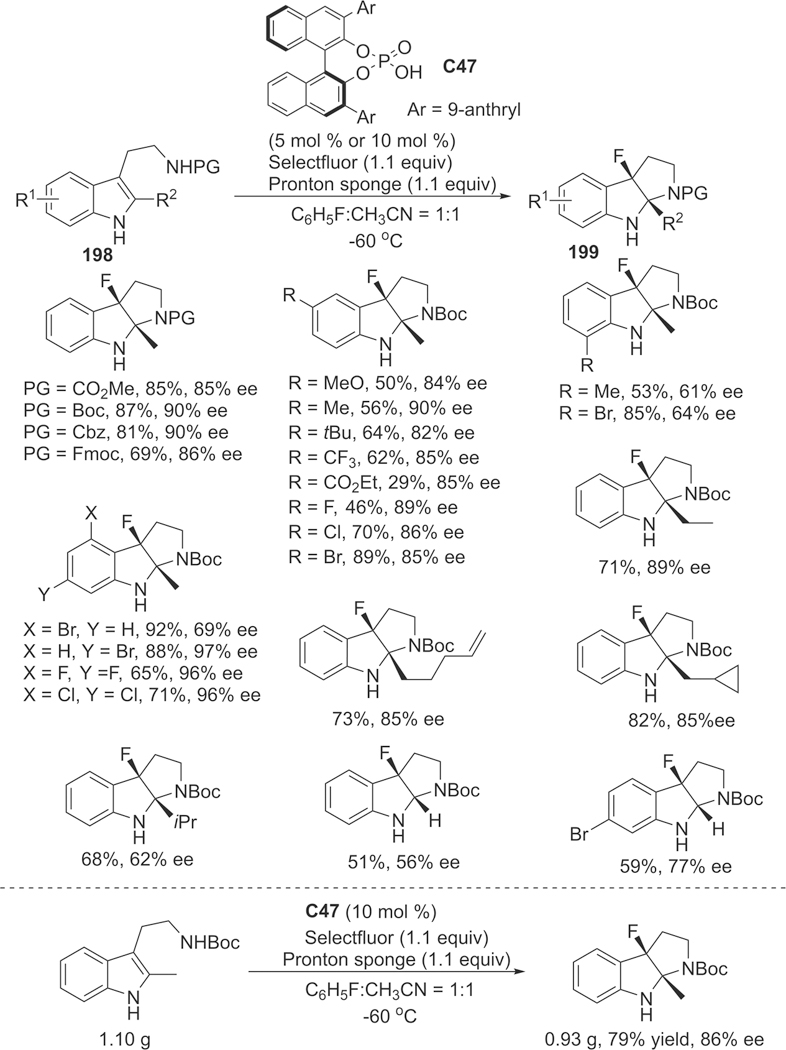
Asymmetric Fluorinative Dearomatization of Tryptamide Derivatives
The reaction of 3,5-dimethyl substituted substrate gave the hydroxyl group substituted products in 72% yield with 93% ee (Scheme 67). The authors insisted that the fluorinated dearomatized product was thermally unstable due to the steric hindrance on the indole ring, leading to cleavage of the C–F bond to form a stable benzyl carbenium intermediate. To probe the reaction mechanism, the effects of acid and base additives were examined. HBF4 released from Selectfluor would accelerate the reaction because of the strong background reaction without the addition of chiral phosphoric acids and proton sponge. The hypothesis about the strong background reaction was further supported by the evidence that faster reaction can be detected when external HBF4 (1.0 equiv) was added and sluggish performance can be observed when proton sponge (1.2 equiv) was employed. Thus, the function of proton sponge in this reaction system is to neutralize HBF4 in situ and further restrains the racemic transformation.
Scheme 67.
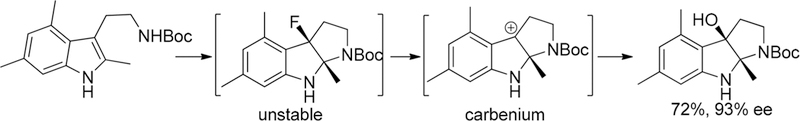
Asymmetric Fluorinative Dearomatization of 3,5-Dimethyl Substituted Substrate
3.1.1.3. Transition-Metal Catalyzed Reactions
The first example of the catalytic enantioselective α-fluorination of β-keto esters using chiral [TiCl2(TADDOLato)L2] complex as a catalyst was reported by the Togni group in 2000.108 Two years later, the Sodeoka group reported Pd(II)-BINAP complex-catalyzed enantioselective α-fluorination of β-keto esters.196 These papers triggered extensive further work on enantiose-lective electrophilic fluorination of a variety of carbonyl compounds catalyzed by various chiral transition metal complexes, involving not only Ti and Pd but also other late transition metals such as Ni, Cu, Ru, and Zn. This strategy has been shown to have a broad scope. The key chiral intermediates of these reactions are transition metal bidentate enolate complexes generated under acidic or neutral conditions (Scheme 68). This section deals with these transition-metal-catalyzed fluorination reactions.
Scheme 68.

Intermediate Transition Metal Bidentate Enolate Complexes
3.1.1.3.1. Titanium Catalysis.
In the Togni’s first paper,108 they showed that the isolated and well-defi ned [TiCl2(TADDOLato)L2] complex C48 acted as an excellent catalyst for enantioselective fluorination reaction with Selectfluor. The α-fluorinated products 201 were obtained in good to excellent yields (up to 90% ee) (Scheme 69a). They examined naphthyl and phenyl complexes (C48a: R = 1-naphthyl, L = acetonitrile, and C48b: R = phenyl, L2 = DME) and found that better enantioselectivity was consistently obtained with the naphthyl complex C48a. Later, they applied their catalysts to various β-keto esters, β-keto thioesters, β-keto amides, and 1,3-diketones.197 Although moderate to high asymmetric induction was observed for the acyclic substrates tested (55–90% ee), poor asymmetric induction was observed for some cyclic β-keto esters and 1,3-diketones. They speculated that noncatalyzed background fluorination of the enol form might account for the decrease of ee of the products because poorer enantioselectivity was observed for substrates that exist mainly in enol form rather than keto form (Scheme 69b). When milder NFSI or NFPY-BF4 was used as the fluorinating reagent instead of highly reactive Selectfluor, increased enantioselectivity was observed for such highly enolized substrates but not for nonenolized substrates, but the reactions using these reagents were very slow.197–199
Scheme 69.

Asymmetric Fluorination of Acyclic (a) and Cyclic β-Keto Esters (b)
For these reactions, care is necessary to exclude moisture. Addition of a small amount of water deactivated the catalyst, and the reaction slowed down dramatically.198 To clarify the mechanism of their [TiCl2(TADDOLato)L2] complex-catalyzed fluorination reaction, they performed computational and experimental studies.200,201 The absolute stereochemistry of the major enantiomer from the reaction catalyzed by [TiCl2(R,R-TADDOLato)L2] complex was determined to be (S). To understand the origin of the enantioselectivity and to elucidate in detail the mechanism of the fluorination step, DFT calculations were performed for the [TiCl(R,R-TADDOLato)(enolate)-(NCMe)] complex. The results suggested that in the lowest energy complex, the Re-face of the enolate is completely shielded and the fluorine atom can only be delivered from the opposite side (Scheme 70a).200 This model well explained the absolute stereochemistry. Next, they tried to prepare and isolate the Ti–enolate 1:1 complex. However, only the 1:2 complex was obtained. The solution structure of the [Ti(R,R-TADDOLato)-(enolate)2] complex was analyzed in detail by NMR. Six diastereomeric forms are possible for complexes of the type [Ti(R,R-TADDOLato)(enolate)2]. In the case of the complex containing the enolate of 2-methyl-3-oxopentanoic acid benzyl ester, several different species were observed, but two major isomers were observed in 7:3 ratio, and the predominant isomer was identified as the C2 symmetric Face-on-Re/Face-on Re diastereomer, while the other major isomer was the C1 symmetric Face-on-Re/Face-on Si diastereomer (Scheme 70b). Finally, they determined the crystal structure of the 1:2 complex (for this experiment, they used [Ti(S,S-TADDOLato)(enolate)2] having opposite chirality to that used for the catalytic reaction). The C2-symmetric Face-on-Si/Face-on-Si structure corresponds to the major isomer observed in the NMR experiment, in which the Si face of both enolate planes is completely shielded. Similar NMR experiments for the 1:2 complexes prepared from other keto esters showed that the most abundant diastereomer was always the C2 symmetric Face-on-Re/Face-on Re diastereomer, but the abundance of the other diastereomers varied depending on the structure of the keto esters. The existence of isomeric forms of complexes containing either one or two carbonyl-enolato units, differing by the enantioface being shielded, explains why the enantioselectivity obtained with this system rarely exceeds ca. 90%. The existence of many different configurational and diastereomeric isomers of the octahedral Ti complex with a chiral bidentate ligand makes it difficult to predict the reaction outcome.201
Scheme 70.

Enolate Re-Face (a) and Si-Face (b) Shielding Considerations
The Togni group also applied their Ti catalyst C48a to the enantioselective sequential fluoro-chlorination and chloro-fluorination of β-keto esters with active methylene 202 instead of active methine (Scheme 71).202 The opposite enantiomer was obtained simply by changing the sequence of addition of Selectfluor and NCS, and the α-chloro-α-fluoro-β-ketoesters were obtained with up to 65% ee.
Scheme 71.
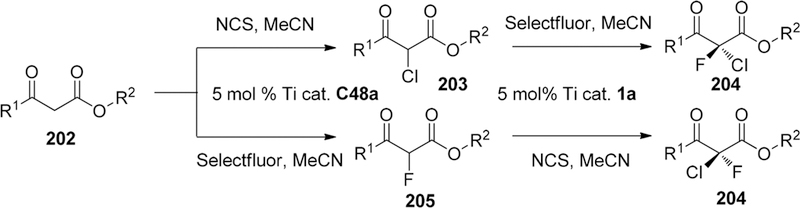
Enantioselective Sequential Fluoro-chlorination and Chloro-fluorination of β-Keto Esters
3.1.1.3.2. Palladium Catalysis.
In 2002, the Sodeoka group reported the first example of enantioselective fluorination reactions catalyzed by palladium complex.196 They found that their original Pd aqua and μ-hydroxo complexes with BINAP-type bisphosphine ligands C49 and C50 (Scheme 72a) both worked well as catalysts when NFSI was used as a fluorinating reagent. The fluorination of β-keto esters 206 proceeded smoothly at 20 °C or even lower temperature with excellent enantioselectivity. More than 90% ee was achieved for both acyclic and cyclic substrates (Scheme 72b). In 2007, the Kim group reported asymmetric fluorination reaction of α-chloro-β-keto ester using the Pd monoaqua complex C51 (Scheme 72b).203
Scheme 72.

Enantioselective Fluorination Reactions Catalyzed by Palladium Complex (a) and Its Application for Acyclic and Cyclic Substrates (b)
This Pd-catalyzed reaction was tolerant ofair and moisture and could be carried out even in an open flask. The reaction proceeded readily in various solvents such as THF, acetone, iPrOH, EtOH, and even in pure H2O without significant loss of enantioselectivity,204 which is different from the case of the water-sensitive Ti complex (Scheme 73). The reaction catalyzed by C50b (X = TfO) also proceeded in ionic liquid [himim][BF4], affording the fluorinated product with comparable ee to that obtained from the reaction in EtOH, although a longer reaction time was required. After simple extraction of the reaction mixture with ether, the fluorination product, remaining NFSI, and coproduct benzenesulfone-imide were efficiently extracted into the ether phase, whereas the cationic Pd complex remained in the ionic liquid. Therefore, the recovered ionic liquid solution of the catalyst could be directly used for the next reaction. Catalyst immobilized in the ionic liquid was recycled no less than 10 times, maintaining excellent enantioselectivity (91% ee) (Figure 18).205
Scheme 73.
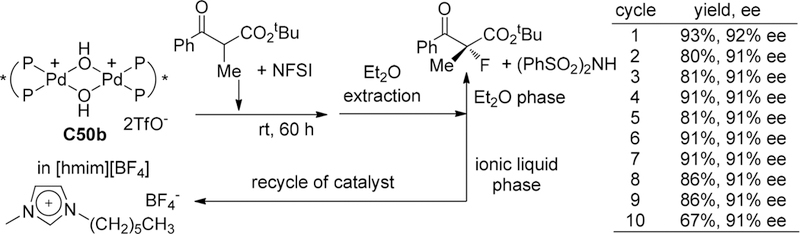
Pd-Catalyzed Fluorination of β-Keto Esters
Figure 18.

The Sodeoka group observed clean formation of the stable bidentate Pd enolate upon simple mixing of Pd complex C50 with 1,3-diketone and β-keto ester in NMR experiments.206–208 Recently, they succeeded in solving the crystal structure of the (R,R)-BINAP-Pd-enolate complex (Figure 18).208 As expected, the complex has typical square-planar geometry with bidentate coordination of diketone. The bulky tert-butyl group of the substrate is oriented toward the Si face to avoid steric repulsion with the ligand phenyl group. Therefore, NFSI should approach from the less crowded Re-face to give the (R)-product. Different from the octahedral titanium complex, the Pd (II) complex strongly favors square-planar geometry, and therefore, in the case of the complex with a C2-symmetric bidentate chiral ligand, only one enolate complex is generated in the reaction mixture. This is presumably the reason for the robustness and the wide substrate scope of this Pd-catalyzed system.
Interestingly they observed rapid formation of the Pd enolate of the less acidic β-keto amide 208a, and the fluorination reaction proceeded in a highly enantioselective manner (Scheme 74a). They also solved the crystal structure of the Pd enolate complex of β-keto amide, in which the tert-butyl group lies in the plane of the amide backbone, and the square-planar structure is distorted. As a result, the ligand phenyl group on the Si-face side is located closer to the α-carbon of the enolate.208 Diesters 208b and ester–amides 208c were also good substrates. In the case of less acidic ester–amide type substrates, addition of a catalytic amount of amine such as 2,6-lutidine was effective to accelerate the reaction without decreasing the enantioselectivity (Scheme 74b).209,210 Recently, the Kim group reported reactions of several other keto amides 208d using Pd monoaqua complex C51b as a catalyst and 2,6-di-tert-butyl-4-methylpyridine (DTBMP) as a base (Scheme 74c).211
Scheme 74.

Pd-Catalyzed Fluorination of β-Keto Amides (a) and Their Heterocyclic Analogues (b)
It is noteworthy that this Pd-catalyzed asymmetric fluorination chemistry has been successfully applied to the multikilogram-scale synthesis of a spleen tyrosine kinase (SYL) inhibitor, which has potential applications in a number of therapeutic areas, including rheumatoid arthritis, B-cell lymphoma, and asthma/rhinitis.212 The GlaxoSmithKline group performed a thorough optimization of the reaction processes to support preclinical and clinical evaluation of this inhibitor. They established a process using menthyl ester as a substrate for the fluorination reaction. Asymmetric fluorination of menthyl ester catalyzed by (S)-BINAP-Pd complex worked well on a large scale, and 43.7 kg of the desired fluorinated product was obtained as crystalline solid with perfect selectivity and in 68% yield in two steps from the inexpensive ethyl ester (Scheme 75). This process provided 8 kg of SYK inhibitor in a single batch. This example illustrates the robustness of this Pd-catalyzed chemistry and its potential for industrial application.
Scheme 75.

Large-Scale Preparation of Commercial SYK Inhibitor
The Sodeoka group reported that the Pd catalysts C49 and C50 also worked well for not only 1,3-dicarbonyl compounds but also other types of carbonyl compounds. Fluorination reaction of β-keto phosphonate esters 210 afforded fluorophosphonate 211 with excellent enantioselectivity and yield (Scheme 76a).213,214 The excellent enantioselectivity and absolute stereochemistry of the product suggest that keto phosphonate esters also form a bidentate Pd enolate. Independently, the Kim group reported a similar reaction using the monoaqua Pd complex C51.214–217 The diaqua complex C49 and the monoaqua complex C51 showed basically similar reactivity and selectivity. The Sodeoka and the Kim groups also reported asymmetric fluorination of α-aryl-α-cyanophosphate esters, α-aryl-α-cyano-acetate esters, and α-aryl-α-cyanosulfones 212 catalyzed by Pd μ-hydroxo complex C50 and monoaqua complex C51 (Scheme 76b).218–222 The Pd complex with SPANphos ligand L3 also worked well for the asymmetric fluorination of α-phenyl-α-cyano-acetate.223
Scheme 76.
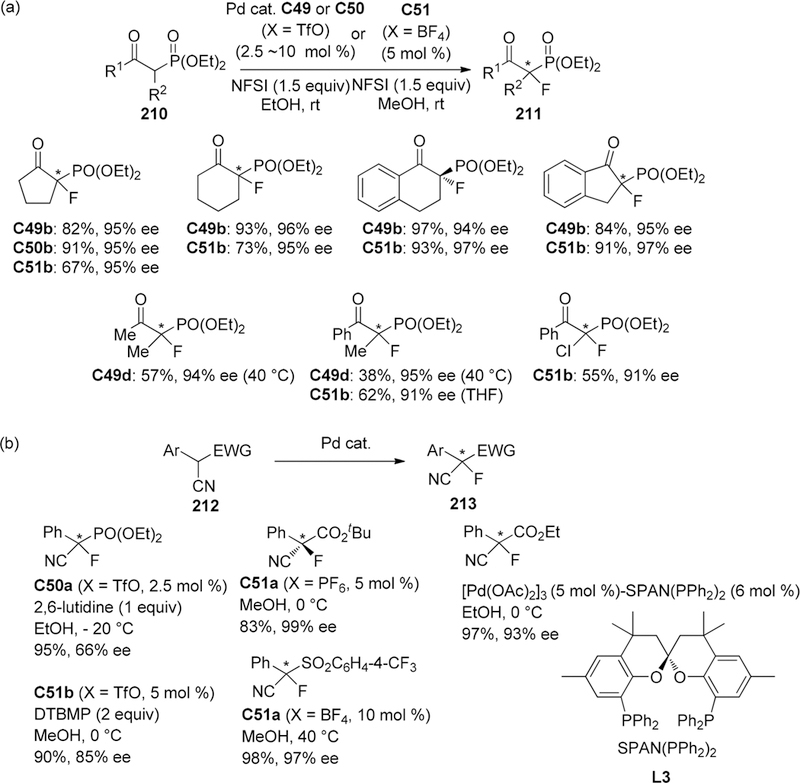
Asymmetric Fluorination of β-Keto Phosphonate Esters (a) and α-Aryl-α-cyanophosphate Esters (b)
In 2005, the Sodeoka group reported enantioselective fluorination of oxindole derivatives.224 The fluorination reaction of unprotected oxindole derivatives was not successful; the reaction was very slow and the enantioselectivity was very low, suggesting that the acidity of the oxindole is not high enough and the monodentate enolate would be conformationally too flexible. Therefore, the effects of electron-withdrawing and coordinating protecting groups were investigated. Introduction of a carbamate-type protecting group dramatically improved the reactivity, as well as the enantioselectivity, although an acyl group was far less potent.210 The Boc group was the best, and various oxindole derivatives 215 were obtained with excellent enantioselectivity (Scheme 77a). This result indicated that this Pd enolate chemistry is potentially available not only for carbonyl compounds having an electron-withdrawing/coordinating functional group at the α-position but also for imide-type compounds. This reaction was applied to the enantioselective synthesis of MaxiPost (BMS 204352) 67, which is a potent potassium channel modulator.224 Monofluorination of a α-methylene substrate is normally very difficult. When mono-fluorination of tert-butyl 3-oxo-3-phenyl propionate was examined, the difluorinated product (4%) was produced even when only 1 equiv of NFSI was used, and the obtained monofluorinated product (54% yield) was completely racemic, indicating rapid enolization of the monofluoro β-keto ester.196,204 In the case of the 3-unsubstituted N-Boc oxindole, the reaction in THF afforded a 29% yield of monofluorinated oxindole with 21% ee. Interestingly, when the solvent was changed to a 1:1 mixture of 1,2-dichloroethane and methanol, selective ring-opening proceeded in situ, and the phenyl acetic acid derivative was obtained in 53% yield with 93% ee (Scheme 77b). Normally 2.5 mol % catalyst C49 or C50 was used for experiments, but in 2014, Yang and Wu published an interesting paper.225 They prepared a series of substituted NFSI derivatives and examined the potency of these compounds as fluorinating reagents in this Pd-catalyzed fluorination of N-Boc-protected oxindole derivatives. They succeeded in reducing the amount of catalyst C50a to 0.5 mol % by using (4-F-C6H4SO2)2NF in diethyl ether (Scheme 77c). Recently, asymmetric fluorination of oxindoles using chiral NHC carbene Pd complex was also examined, but the enantioselectivity was only moderate (up to 59%).226
Scheme 77.
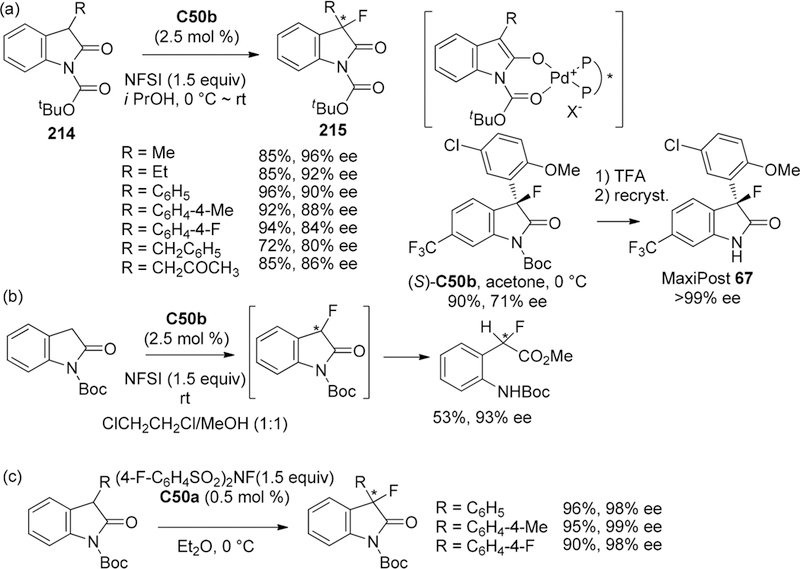
Enantioselective Fluorination of Oxindole Derivatives; 3-Substituted N-Boc Oxindoles (a), 3-Unsubstituted N-Boc Oxindoles (b), and Conditions Using only 0.5 mol % of the Catalysts (c)
The Sodeoka group also reported asymmetric fluorination of α-ketoester 216 catalyzed by the μ-hydroxo complex C50.227 Interestingly, the monofluorinated product 217 was obtained with high optical purity for this substrate (Scheme 78).
Scheme 78.
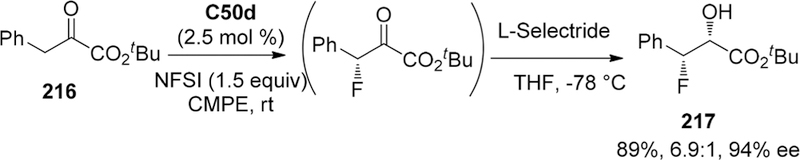
Asymmetric Fluorination of α-Ketoester 216
Pd complexes also catalyze many other types of reactions, such as allylic substitution and reactions via C–H activation, and recently, several interesting enantioselective fluorination reactions based on such chemistries have been reported.228–231 These reactions are not discussed here because they do not involve asymmetric construction of a C–F quaternary stereogenic center.
3.1.1.3.3. Nickel Catalysis.
In 2004, the Shibata group reported that Ni- and Cu-bis(oxazoline) complexes also worked well for the enantioselective fluorination reaction of β-keto esters.232 The reaction of 2-tert-butoxycarbonyl-1-indanone 218a catalyzed by the Ni complex prepared from Ni(ClO4)2 and (S,S)-Box-Ph ligand L4 using NFSI as fluorinating reagent gave the (R)-fluorinated product 219a with 93% ee in 87% yield. Interestingly, the opposite enantiomer was obtained when Cu(OTf)2 was used instead of Ni(ClO4)2 (Scheme 79a). In these reactions, the enantioselectivity seems to be solvent-dependent. Multiple coordination geometries such as square-planar, tetrahedral, square-pyramidal, and octahedral are known for the Ni(II) and Cu (II) complexes, and this may account for the observed variations of the ee and the enantio-switching. This problem was solved by using a C2-symmetric tridentate ligand, which is expected to form an octahedral bidentate Ni enolate complex. The fluorination reaction of various β-keto esters 218 catalyzed by 10 mol % of the Ni complex prepared from Ni(ClO4)2 and (R,R)-DBFOX-Ph ligand L5 proceeded smoothly to afford the fluorinated product 219 with excellent enantioselectivity (up to 99% ee). The catalyst loading could be reduced to 2 mol % without loss of enantioselectivity. Asymmetric fluorination of oxindole derivatives was also successfully performed and applied to the synthesis of MaxiPost 67. In the case of oxindole, Ni(OAc) 2–4H2O was used as a catalyst precursor (Scheme 79b).233
Scheme 79.

Ni- and Cu-Catalyzed Fluorination Using (S,S)-Box-Ph Ligand (a) and (R,R)-DBFOX-Ph Ligand (b)
Similar Ni-catalyzed fluorination reactions of β-keto esters using various other chiral ligands L6-L9 have also been reported by several groups (Scheme 80).234–239 Among them, Shibatomi and Iwasa reported a unique N,N,N-tridentate pyridine ligand L6 with both chiral binaphthyl and oxazoline groups. Fluorination of β-keto esters proceeded in a highly enantioselective manner (up to 99% ee) in the presence of Ni(ClO4)2 salt. It is noteworthy that their ligand also worked well with Mg(ClO4)2 to afford highly optically active fluorinated products (up to 99% ee).234,235
Scheme 80.
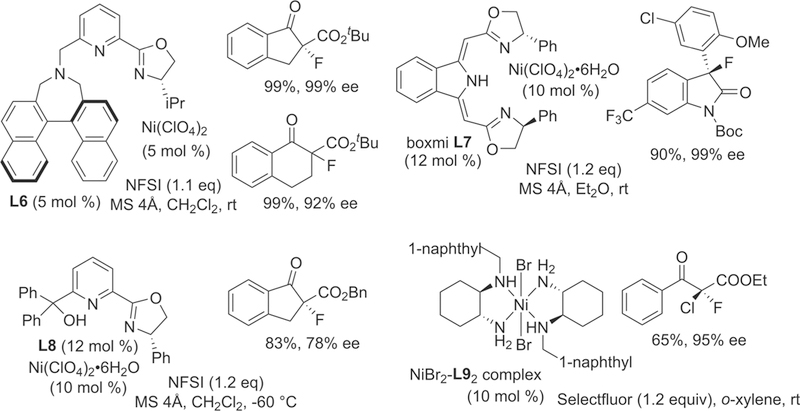
Asymmetric Ni-Catalyzed Fluorination of β-Keto Esters
Ni-Catalyzed asymmetric fluorination of 3-(2-arylacetyl)-2-thiazolidinones 220 was also achieved.240 In 2007, the Sodeoka group reported the first catalytic asymmetric monofluorination reaction for this type of substrate, catalyzed by (R)-BINAP-NiCl2 complex C52 in the presence of triethyl silyl triflate and 2,6-lutidine (up to 88% ee) (Scheme 81a). In 2008, the Shibata group also applied their (R,R)-DBFOX-Ni catalyst to this type of substrate (up to 78% ee).241 In 2009, they found that addition of HFIP accelerated the reaction, and the reaction at −60 °C afforded α-aryl-α-fluoroacetic acid derivatives 221 with excellent enantioselectivity (up to 99% ee) (Scheme 81b).242
Scheme 81.
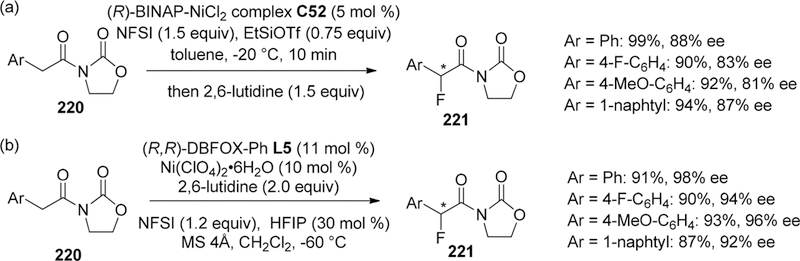
Ni-Catalyzed Asymmetric Fluorination Using (R)-BINAP (a) and (R,R)-DBFOX (b) Ligands
3.1.1.3.4. Copper Catalysis.
In 2004, the first Cu-catalyzed enantioselective fluorination of β-keto esters was reported by Ma and Cahard.243 The Cu complex prepared from the bisoxazoline ligand (R,R)-Box-Ph L4 and Cu(OTf)various other chiral ligands were also reported.th various other chiral ligands were also reported. The Bolm group reported asymmetric fluorination catalyzed by the chiral sulfoximine L10-Cu complex (Scheme 82b).244 The Kesavan group reported the reaction using (S,S)-Nap-(R,R)-Box ligand L11. Relatively high enantioselectivity was obtained for less bulky ethyl ester substrates.245 The Shibatomi and Iwasa group reported a fluorination reaction using a unique chiral spiro oxazoline ligand L12, affording products with excellent enantioselectivity (up to 99% ee). 246 The Du group reported a reaction catalyzed by diphenylamine-linked bis(thiazoline) L13-Cu(OTf)2 complex; although the substrate scope was very narrow, some products were obtained with up to 99% ee.247,248 Just recently, a reaction using this ligand under solvent-free conditions in a ball mill was also reported.249 The Xu group examined Ph-BINMOL-derived salan L14-Cu complex and found that fluorination reaction of 1-indanone-2-carboxylate derivatives proceeded with excellent enantioselectivity (up to 99% ee).250 Queneau examined Cu-catalyzed fluorination reaction using their sugar-modified bipyridine ligands, but only low asymmetric induction was observed (up to 32% ee).251
Scheme 82.

Cu-Catalyzed Enantioselective Fluorination of β-Keto Esters Using (R,R)-Box-Ph (a), Chiral Sulfoximine, Nap-(R,R)-Box, Chiral Spiro Oxazoline, Diphenylamine-Linked Bis(thiazoline), Ph-BINMOL-Derived Salan Ligands (b), and DNA as a Catalyst (c)
The Shibata group reported a unique approach for asymmetric fluorination. The combination of achiral Cu catalyst C53 and salmon testis DNA as the chiral source afforded fluorinated indanone derivatives with up to 74% ee (Scheme 82c).252 By using a chiral Cu catalyst, simultaneous C–C and C–F bond formation was also achieved (See Schemes 54–56).
3.1.1.3.5. Ruthenium Catalysis.
The Togni and Mezzetti group reported asymmetric fluorination of 1,3-dicarbonyl compounds 206 catalyzed by chiral dicationic ruthenium PNNP complex C54 prepared in situ from [RuCl2]PNNP)] complex and (Et3O)PF6 in 2004 (Scheme 83).253–255 The reactivity and selectivity were solvent-sensitive, and higher reaction rate and ee value were observed in CH2Cl2/Et2O mixed solvent compared with CH2Cl2. They also isolated the Ru enolate complex and solved its crystal structure, revealing a tetradentate apical–equatorial coordination of PNNP ligand similar to that of C54; they discussed the absolute stereochemistry of the product based on the enolate structure. The same catalyst C54 was used for oxidative fluorination of aldehyde with AgHF2 as a fluorine source. Although the eantioselectivity was low (up to 27% ee), they proposed an interesting reaction mechanism, in which Ru(lV) species is involved.256
Scheme 83.
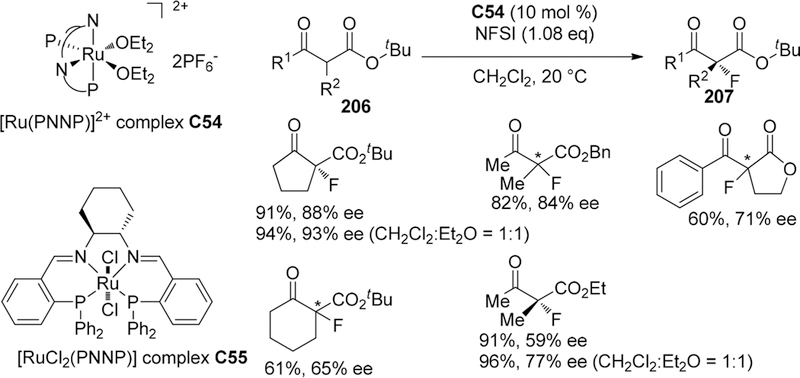
Asymmetric Fluorination of 1,3-Dicarbonyl Compounds 206 Catalyzed by Chiral Dicationic Ruthenium PNNP Complex
3.1.1.3.6. Other Metal Catalysis.
In addition to Ti, Pd, Ni, Cu, and Ru, many other transition metal catalysts have also been examined for asymmetric fluorination. In 2004, the Cahard group reported that (R,R)-BOX-Ph complexes of not only Cu(OTf)2 (Scheme 82a) but also various other metal salts such as Mg(ClO4)2, Zn(OTf)2, Sc(OTf)3, and La(OTf)3 can catalyze the fluorination reaction of β-keto estere.243,257 Although only low enantioselectivity (up to 17% ee) was observed for the reaction using Mg and lanthanoid complexes, reasonably high asymmetric induction was observed when the (R,R)-BOX-Ph-Zn(OTf) 2 complex was used as a catalyst (up to 74% ee). They also tested a heterobimetallic complex, Al–Li–BINOL, and obtained moderate asymmetric induction (up to 67% ee) when NFPY-BF4 was used as fluorinating reagent. In 2005, J0rgesen reported that (R,R)-DBFOX-Zn(ClO4) 2 catalyzed asymmetric fluorination of β-keto phosphonates 210 (Scheme 84a).258 Several other groups also tried asymmetric fluorination catalyzed by a zinc salt with chiral ligands, but in most cases, the enantioselectivity was inferior to that obtained with Cu or Ni atalysts having the same ligands.236,239,244,245,247–249,251 In 2008, the Shibata and Toru group reported enantioselective fluorination of methyl tert-butyl malonate esters 222.259 They first applied their conditions for the asymmetric fluorination reactions of β-keto esters using Ni catalyst233 to the malonate esters and obtained high asymmetric induction (up to 89% ee). But much higher enantioselectivities were achieved by using the (R,R)-DBFOX-Ph L5-Zn(OAc)2 complex, and the desired fluoromalonate esters 223 were obtained with excellent enantioselectivity (up to 99% ee). It is noteworthy that this reaction is applicable for the preparation of α-fluoro-α-heteroatom-substituted malonate esters (Scheme 84b).
Scheme 84.
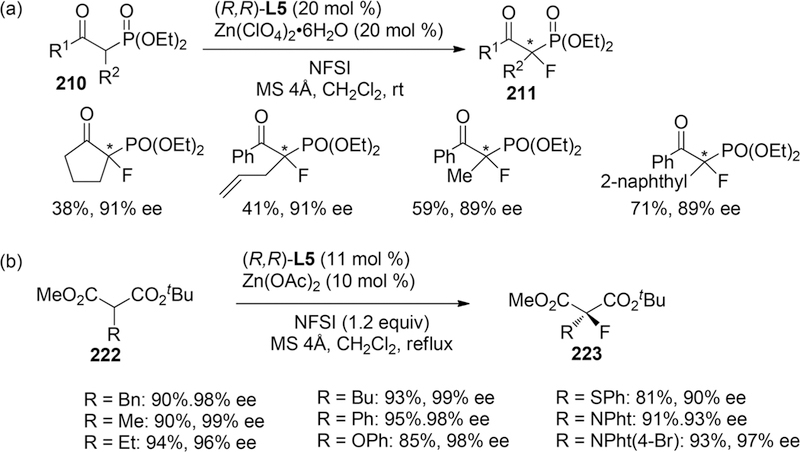
Asymmetric Fluorination of β-Keto Phosphonates (a) and Alkyl tert-Butyl Malonate Esters (b)
In 2006, the Inanaga group reported the first successful examples of rare earth metal complex-catalyzed asymmetric fluorination reaction (Scheme 85a).260 They found that the scandium complex of F8-BINOL C56 worked well as a catalyst for asymmetric fluorination of β-keto esters 218 (up to 88% ee). In 2012, the Feng group achieved highly enantioselective fluorination reaction of oxindole by using their original chiral N-oxide ligand L15 (Scheme 85b).261 In contrast to other transition metal catalysts, this catalyst can fluorinate unprotected oxindole derivatives to give the fluorooxindoles with excellent enantioselectivity. By using this reaction, they synthesized MaxiPost 67 with 96% ee directly.
Scheme 85.

Rare Earth Metal Complex-Catalyzed Asymmetric Fluorination
In 2010, the Kawatsura and Itoh group reported that cobalt salen complex was an efficient catalyst for the enantioselective fluorination of β-keto esters (Scheme 86a).262 Furthermore, in 2014, the Xu and Che group reported that iron(III)-salan complex C57 catalyzed fluorination reaction quite efficiently to give highly optically active β-ketoesters and oxindoles (Scheme 86b).263
Scheme 86.

Enantioselective Fluorination of β-Keto Esters Using Chiral Cobalt–Salen Complex (a) and Iron(III)–Salan Complex (b)
3.2. Asymmetric Elaboration of F-Containing Substrates
3.2.1. Alkylations.
3.2.1.1. Alkylation Reaction of Fluorinated Carbonyl Compounds.
In 1999, Arai, Shioiri, and co-authors developed an asymmetric catalytic alkylation reaction between α-fluoro cyclic ketone 226 and benzyl bromide with cinchonine based quaternary ammonium bromide as chiral catalyst C58 (Scheme 87).264 The chiral catalyst was optimized via variation of substitution on the N-benzyl group, and the results showed that the permethyl-substituted phenyl one was the best choice affording the corresponding α-alkyl, α-fluoro cyclic ketone 227 with C–F quaternary stereogenic center in up to 91% ee. Several benzyl bromide and allyl bromide could react smoothly with α-fluoro cyclic ketone with moderate chemical yields. The further transformation of the obtained product 227 has also been carried out to afford corresponding ester compounds 228 via a one-pot Ru-catalyzed oxidation process.
Scheme 87.

Asymmetric Catalytic Alkylation of Cyclic α-Fluoro Ketones
In 2009, Maruoka also developed a phase-transfer catalyzed approach for the synthesis of α-alkyl-β-keto esters 230 with α-fluorinated quaternary center from fluorinated keto-esters 229 and alkyl halides (Scheme 88).265 The asymmetric organocatalytic reaction used N-spiro quaternary ammonium salt C59 as chiral phase-transfer catalyst and CsOH as base, affording the product in 68–89% chemical yields and 65–88% ee. Several types of alkyl halides, such as Bn, aryl, allyl, propargyl, and methyl halides, were well tolerated.
Scheme 88.
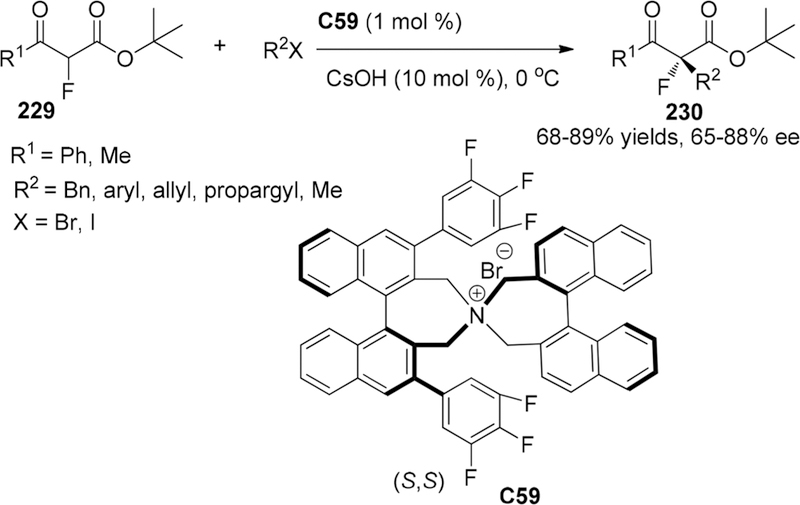
Phase-Transfer Catalyzed Asymmetric Alkylations
In 2016, Zhou, Hartwig and co-authors reported a Pd-catalyzed asymmetric arylation of α-fluoroketones 231 with aryl bromides as coupling electrophiles (Scheme 89).266 The reaction used the combination of palladium complex and BINOL-derived monophosphine as catalyst, resulting in α-fluoro-α-aryl carbonyl compounds 232 in 63–91% yields and 80–94% ee. In this work, aryl triflates also have been developed as aryl precursors to react with α-fluoroketones, and the similar level of yields and enantioselectivities was obtained. Besides α-fluoroketones, the in situ generated fluorinated enolates from gem-diols also were used as the coupling partners. Comparing to the results from α-fluoroketones, this detrifluoroacetylative arylation reaction afforded better chemical yield and enantioselectivity.
Scheme 89.

Pd-Catalyzed Asymmetric Arylation of α-Fluoroketones
Shortly after that, the Hartwig group used α-fluorooxindoles 233 as coupling partners for the asymmetric Pd-catalyzed arylation reaction with aryl triflates (Scheme 90).267 The combination of Pd(dba)2/(R)-Segphos L18 as a catalyst was employed for this reaction, which catalyzed this transformation to form the α-aryl-α-fluorooxindoles 234 with good yields and high enantioselectivities.
Scheme 90.
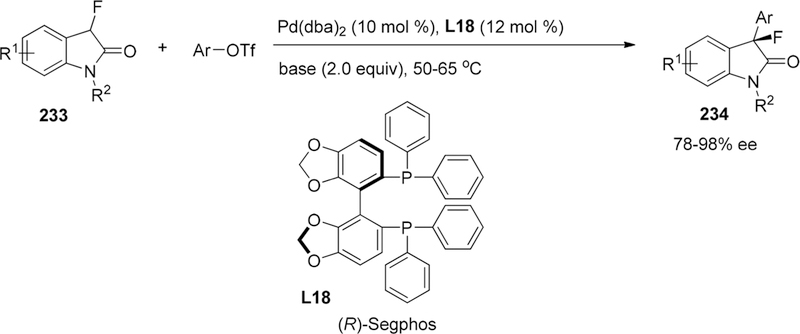
Asymmetric Pd-Catalyzed Arylation Reaction with Aryl Triflates
In 2007, Paquin and co-authors reported a Pd-catalyzed asymmetric allylation reaction of silyl protected enolates 235 by using oxazoline derived (S)-t-Bu-PHOX as chiral ligand L19 and tetrabutylammonium triphenyldifluorosilicate (TBAT) as additive (Scheme 91).268 This methodology used allyl ethyl carbonate 236 as allyl precursor, allowing preparation of the α-allyl, α-fluoro cyclic ketone 237 with 52–93% yields and 83–95% enantioselectivities. The five-, six- and seven-membered silyl enolates were all tolerated in this reaction.
Scheme 91.

Pd-Catalyzed Asymmetric Allylation Reaction of Silyl Protected Enolates
Asymmetric allylic alkylation represents another efficient strategy for the preparation of chiral α-allyl-α-fluoro carbonyl compounds bearing a quaternary stereogenic C–F center (Scheme 92).269 Tan, Jiang, and co-authors reported in 2013 an asymmetric allylic alkylation reaction of linear α-fluoro-β-keto-esters 238 by using (DHQD)2PHAL 69 as organocatalyst. The reaction used Morita-Baylis-Hillman carbonates 239 as electrophilic reagents and afforded the corresponding allylic alkylated product 240 in good chemical yields, good enantilselectivities, and moderate diastereoselectivities.
Scheme 92.
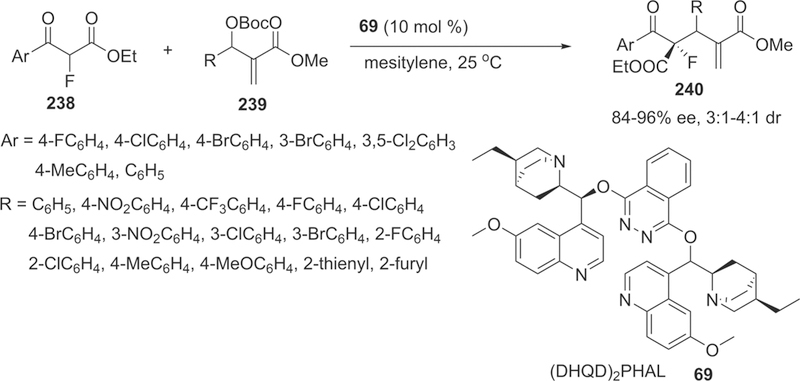
Asymmetric Allylic Alkylation Reaction of Linear α-Fluoro-β-keto Esters
In 2014, Chen, Guo, and co-authors reported a one-pot Pd-catalyzed asymmetric allylic alkylation reaction for the preparation of α-allyl-α-fluoro ketones 242 with the use of phosphinooxazoline (S)-t-Bu-PHOX L19 as chiral ligand (Scheme 93).270 In the presence of strong base (LiHDMS), acyclic α-fluoro ketones 241 were converted into the corresponding tertiary fluorinated enolates, which then reacted with allyl enol carbonates to give the final product in 30–91% chemical yields and 60–90% ee. In the case of allyl enol carbonates substrates, the steric hindrance showed almost no effect on the reaction outcome, and the same level of yield and enantioselectivity was found.
Scheme 93.
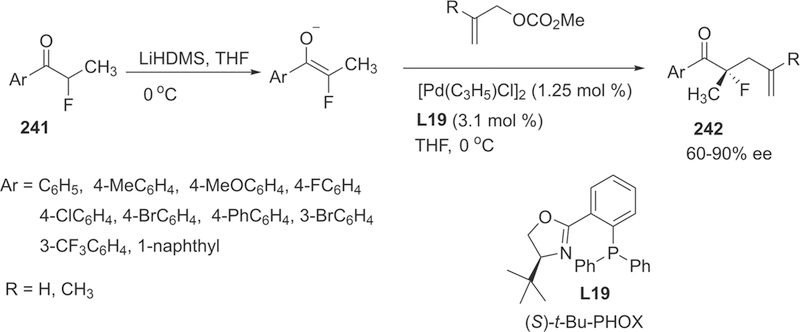
Pd-Catalyzed Asymmetric Allylic Alkylation α-Fluoro Ketones
Recently, the Wolf group developed an asymmetric allylic alkylation reaction between tertiary fluorinated enolates generated from C3-fluorinated oxindoles 233 and allylic acetates/carbonates 243 (Scheme 94).271 The optimization of chiral ligand disclosed that (S)-t-Bu-PHOX L19 was the most efficient one, and the combination with [η3-C3H5ClPd]2 catalyzed the allylic alkylation reaction to give the 3-fluorinated carbon quaternary oxindoles 244 with excellent enantioselectivities (>99% ee) and diastereoselectivities (92:8 → 99:1). The regioselectivity of this asymmetric alkylation reaction has also been examined by using nonsymmetrically substituted allylic acetates, and also excellent regioselectivity was found.
Scheme 94.

Asymmetric Allylic Alkylation of Tertiary Fluorinated Enolates Generated from C3-Fluorinated Oxindoles
3.2.1.2. Alkylation Reaction of Fluorinated Allyl Enol Carbonates.
In 2005, the Nakamura group reported a Pd-catalyzed enantioselective decarboxylative reaction of α-fluorinated ketoester 245 under room temperature by using chiral phosphines as ligands (Scheme 95).272 The optimization studies on chiral ligand showed that the substituent on the oxazoline dramatically affects the enantioselectivity, and (S)-t-Bu-PHOX L19 gave the best enantioselectivity (up to 99% ee). The reactions of cyclic substrates, including five-, six-, and seven-membered ketoesters, gave good chemical yields and excellent enantioselectivities. However, the obviously lower ee value was obtained from the reactions of linear substrates. This decarboxylative reaction provides an alternative way for chiral α-allyl-α-fluoro ketones 246 containing quaternary C–F center.
Scheme 95.
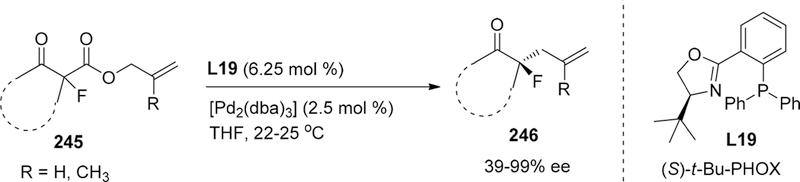
Pd-Catalyzed Enantioselective Decarboxylative Allylation of α-Fluorinated Ketoesters
The Paquin group developed another decarboxylative method for the preparation of chiral α-allyl-α-fluoro ketones 248 bearing a quaternary stereogenic C–F center by using fluorinated cyclic allyl enol carbonates 247 as starting materials (Scheme 96).273 This asymmetric Pd-catalyzed allylation reaction also used (S)-t-Bu-PHOX L19 as chiral ligand, which converted several fluorinated allyl enol carbonates into corresponding products in 58−97% yields and 30−94% enantioselectivity. The enantioselectivity of this reaction relied on the ratio of ligand to metal catalyst. Lower ratio of ligand to metal catalyst gave the better ee value, and less than 1:1.67 of this ratio is necessary for affording good stereo outcomes. The nonfluorinated cyclic allyl enol carbonates also have been examined, and no such phenomenon was found. The ratios of 1:1.67 and 1.25:1 gave almost the same enantioselectivity.
Scheme 96.

Pd-Catalyzed Allylation Reaction Using (S)-t-Bu-PHOX Ligand
3.2.1.3. Alkylation Reaction of α-Halo-α-fluoroketones.
In 2014, the Fu group developed a Ni/bis(oxazoline)-catalyzed asymmetric method for the synthesis of α-keto tertiary alkyl fluorides 250 via Negishi reactions (Scheme 97).274 They used racemic α-halo-α-fluoroketones 249 as starting materials to react with organozinc reagents, resulting in the corresponding fluorinated carbon quaternary keteones with moderate chemical yields and excellent enantioselectivities. The most challenge task of this work is the use of gem-dihalides as electrophiles, and only the C–Br bond was selectively broken in this reaction. This reaction provides a new strategy for the construction of fluorine-containing ketones with C–F quaternary stereogenic center.
Scheme 97.
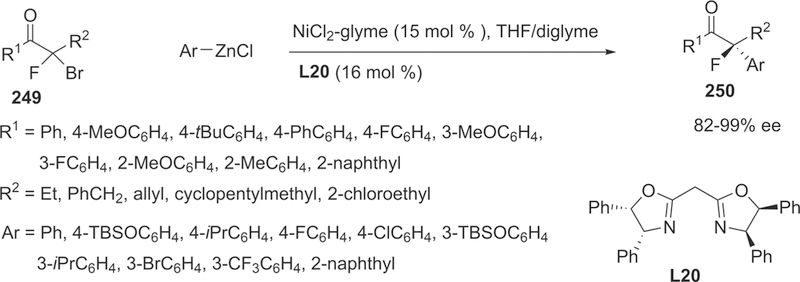
Ni/bis(oxazoline)-Catalyzed Asymmetric Alkylation of Racemic α-Halo-α-fluoroketones
3.2.2. Mannich Addition Reactions.
3.2.2.1. Mannich Addition Reactions of Chiral Imines.
In 2014, the Han group used detrifluoroacetylatively in situ generated fluorinated enolates as new nucleophiles for the Mannich reaction with CF3-sulfinylimine without the use of any transition-metal catalysts, affording the corresponding product in excellent chemical yields and high diastereoselectivities.275 This reaction provides a generalized method for the preparation of α, α-difluoro-β-trifluoromethyl ketones. After that, the Han group designed a cyclic keto-hydrate 251 as the precursor for the tertiary fluorinated enolate, which was used for the construction of quaternary α-fluoro-β-keto-amines 252 (Scheme 98).276 This cyclic keto-hydrate could work well in the detrifluoroacetylative Mannich reaction and react with chiral N-sulfinyl-imines 253 very smoothly via C–C bond cleavage. The reaction was conducted under simple conditions and could complete within 5 min, affording the expected products with excellent chemical yields and >98:2 diastereoselectivities. Besides the usual chiral CF3-imine, several other chiral imines bearing fluoro-containing groups also were examined in this reaction, which also were well tolerated.
Scheme 98.
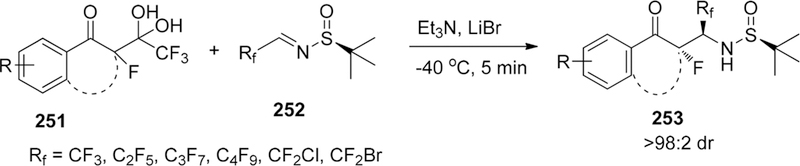
Mannich Reactions of Detrifluoroacetylatively in Situ Generated Fluorinated Enolates
In 2016, the Han group further extended the substrate scope from cyclic keto-hydrates to cyclic amido-hydrates 254 for the asymmetric detrifluoroacetylative277,278 Mannich reaction with fluorinated imines (Scheme 99).279 The cyclic amido-hydrates were synthesized from 3-fluoroindolin-2-ones and generated a new type of fluorinated amide enolates in the presence of DIPEA and LiBr. This is the first example of fluorinated amide enolate and could react well with several types of fluorinated imines to give the corresponding α-fluoroalkyl-β-amino-indolin-2-ones 255 bearing C–F quaternary stereogenic centers. The reaction tolerated a wide range of hydrates, and even the substrates containing free N-H group also worked very well in the reaction. The reactions also showed high diastereoselectivities, and only one diastereomer was found for all the cases (>98:2 dr).
Scheme 99.
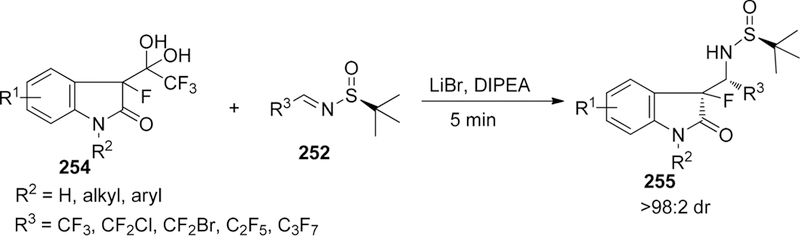
Mannich Reactions of Detrifluoroacetylatively in Situ Generated Enolates Derived from 3-Fluoroindolin-2-ones
Following this work, the Han group continued their work on the asymmetric detrifluoroacetylative Mannich reaction of 3-fluoroindolin-2-one-derived tertiary enolates with fluorinated imines.280,281 The trifluoromethyl group of (S)-N-tert-butane-sulfinylaldimines was further extended to CHF2, CIF2, C4F9, C4HF8, C5F11, and C6HF12. Their reactivities were investigated by reaction with detrifluoroacetylatively in situ generated 3-fluoroindolin-2-one-derived enolates. The results showed that also good yields and excellent diastereoselectivities were obtained.
In 2017, the Han group designed and synthesized a N-tert-butylsulfinyl-(perfluoro)benzaldimine 256 and used this imine as an electrophile in the asymmetric detrifluoroacetylative Mannich reactions (Scheme 100).282 Pentafluorophenyl (perfluorophenyl) is an electronic antipode of the common phenyl group,283–285 which also has been widely used in the chemical and biological area. The Mannich addition reactions of in situ generated enolates proceeded smoothly to give the corresponding quaternary β-perfluorophenyl-amino-indolin-2-ones 257 with moderate yields and diastereoselectivities. Comparing with the results from the reaction of CF3-imine, the reaction of this imine resulted in obviously lower chemical yields and stereochemical outcomes. It should also be mentioned that free N-H 3-fluoroindolin-2-one-derived hydrates were tolerated in this reaction.
Scheme 100.

Asymmetric Detrifluoroacetylative Mannich Reactions Using N-tert-Butylsulfinyl-(perfluoro)benzaldimine
The detrifluoroacetylative Mannich reactions with indolin-2-ones derived gem-diols have been reported by the Han group to react with several types of fluorinated aldimines. Then, in 2017, the Han group examined their reactivities in the reactions with nonfluorinated (Ss)-sulfinylimines 258 (Scheme 101).286 The optimization of reaction conditions disclosed that the reaction could be completed within 10 min at 0 °C. Several types of regular imines with aryl, heteroaryl, alkyl, alkenyl, and alkynyl groups have been examined in the reaction and gave the expected quaternary α-fluoro-β-amino-indolin-2-ones 259 in good yields and excellent diastereoselectivities were obtained.
Scheme 101.

Detrifluoroacetylative Mannich Reactions of Indolin-2-ones with Fluorinated Aldimines
In 2016, the Li group reported an asymmetric Mannich reaction between α-fluoro esters 260 and N-tert-butylsulfinyl imines 258 in the presence of strong base LiHDMS at −70 °C (Scheme 102).287 Three examples of α-fluoro-α-branched ester substrates, including benzyl, 2-naphthylmethyl, and BnOCH2CH2-, have been investigated in this reaction, affording the corresponding quaternary α-fluorinated β-amino acids 261 in good chemical yields. The diastereoselectivities were moderate, and three diastereomers were detected in most cases.
Scheme 102.

Mannich Reactions between α-Fluoro Esters and N-tert-Butylsulfinyl imines
In 2016, the Li group reported a similar asymmetric Mannich reaction betweenα-fluoro-α-branched esters 260 and N-tert-butylsulfinyl imines 258 with the use of strong base LiHDMS at −70 °C (Scheme 103).288 In this work, they extended the fluoro esters scope, and the esters bearing α-aryl, alkyl, and allyl were well tolerated, affording good chemical yields. The moderate diastereoselectivities were obtained, and four diastereomers were detected in most cases. The stereoselectivity of this asymmetric Mannich reaction mostly relied on the steric hindrance of the substitutions on both esters and imines. Substrates with high steric hindrance usually gave excellent results.
Scheme 103.
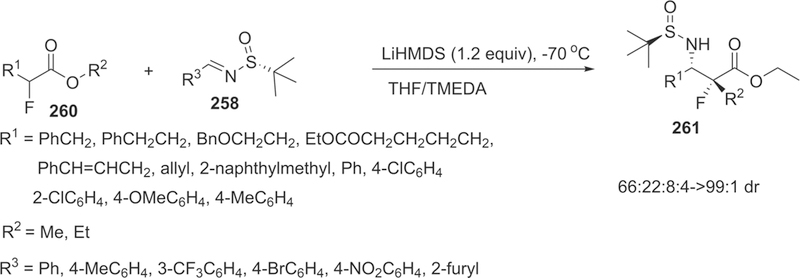
Substrate Generality of the Mannich Reactions between α-Fluoro Esters and N-tert-Butylsulfinyl Imines
Later, the Li group reported α-fluoro ketones 231 as nucleophiles for the asymmetric Mannich reaction with N-tert-butylsulfinylimines. Comparing with the reactions on α-fluoro esters,287,288 this reaction used NaHDMS as a strong base and ether as asolvent (Scheme 104).289 Cyclic α-fluoro ketones could react with a wide range of sulfinylimines bearing varieties of substituents, including aryl, alkyl, and vinyl groups. Also, obviously improved diastereoselectivities were obtained, and usually two diastereomers were detected for most cases.
Scheme 104.

Mannich Reactions of α-Fluoro Ketones with N-tert-Butylsulfinylimines
3.2.2.2. Asymmetric Catalytic Mannich Addition Reactions of Imines.
Besides asymmetric methods based on chiral auxiliary developed for the synthesis of quaternary C–F compounds, asymmetric catalytic Mannich reaction was an alternative way to such structural units. In 2009, Huang, Lu and co-authors reported an organocatalytic Mannich reaction between α-fluorinated ketoesters 263 and Boc-imines 264 with trypto-phan-based bifunctional thiourea C60 as the chiral catalyst (Scheme 105).290 The reaction was conducted at −50 °C, resulting in the corresponding α-fluoro- β-ketoesters 265 bearing fluorinated quaternary stereocenters with excellent chemical yields, high diastereoselectivities, and enantioselectivities. Both aliphatic and aromatic ketones reacted with aliphatic/aromatic aldimines very well. The authors also did some derivatization experiments on the products by converting them into α-fluoro-β-amino acids and α-fluoro-β-lactams via cascade reduction, hydrolysis, and cyclization.
Scheme 105.

Organocatalytic Mannich Reaction between α-Fluorinated Ketoesters and N-Boc-Protected Imines
Then in 2011, the Kim group reported a related Pd-catalyzed asymmetric Mannich reaction. They used the similar substrates, α-fluoro- β-ketoesters 263 and N-Boc-aldimines 264, by using palladium complexes and BINAP L21 as the chiral catalyst (Scheme 106).291 The reaction could be conducted under room temperature, resulting in the expected α-aminated α-fluoro-β-keto esters 265 featuring fluorinated tetrasubstituted carbon centers in excellent enantioselectivities (94−98% ee). The only limitation of this methodology is the diastereoselectivity, and poor diastereoselectivities were obtained for most cases.
Scheme 106.
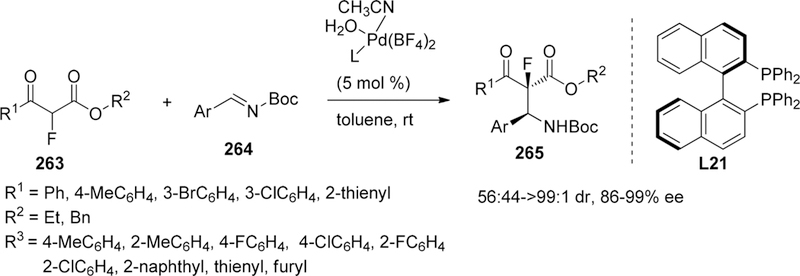
Pd-Catalyzed Asymmetric Mannich Reactions of α-Fluoro-β-ketoesters with N-Boc-aldimines
In 2016, Wennemers and co-authors reported an asymmetric organocatalyzed Mannich addition reaction of imines 267 with α-fluorinated monothiomalonates 266 as nucleophiles (Scheme 107).292 The re action used only 1 mol % of bifunctional cinchona alkaloid–thiourea C60 as a chiral organocatalyst, which catalyzed the reaction very well, affording the corresponding α-fluorinated β-amino thioesters 268 in excellent yields (80−99%), high diastereo (4:1 → 20:1 dr) and enantioselectivities (91−99.9% ee). The protecting group on the amino group showed obvious effect on the chemical yields, and usually the Cbz-protect imines affords obviously higher yields. Furthermore, a wide range scope of imines was tolerated in this system, and even aliphatic aldehyde-derived imines worked very well, resulting in good chemical yields.
Scheme 107.

Asymmetric Organocatalyzed Mannich Addition Reactions of α-Fluorinated Monothiomalonates with Imines
In 2010, Jiang, Tan and co-authors reported an asymmetric organocatalytic Mannich reaction of imines 270 by using β-keto acetyloxazolidinone 269 as a new nucleophile, which provided another method for the synthesis of α-fluoro- β-amino acid derivatives 271 bearing quaternary fluorinated carbon centers (Scheme 108).293 This asymmetric Mannich reaction used (S,S)-bicyclic, guanidine C62 as organocatalysis, and was carried out at room temperature, converting varieties of β-keto acetyloxazoli-dinone into the corresponding product in excellent yields and high enantio- and diastereoselectivities. The substitutions on the two substrates showed an obvious effect on the stereoselectivity of this reaction. The bulky groups substituted imine and β-keto acetyloxazolidinone gave almost completely controlled stereo-chemical outcome.
Scheme 108.

Asymmetric Mannich Reactions of Imines with β-Keto Acetyloxazolidinone Protected β-Keto-α-fluoro Esters
In 2011, the Jiang group reported α-fluorinated aromatic cyclic ketones 231 as nucleophiles for the Mannich reaction with imines 272 by using (S,S)-bicyclic, guanidine C62 as chiral organocatalysis (Scheme 109). The reaction was conducted under lower temperature (−20 °C) with 1,2-dichloroethane as solvent, provided the corresponding β-amino ketone 273 in moderate diastereoselectivities and excellent enantioselectivites. Azodicarboxylates 274 were also tried to be reacted with α-fluorinated aromatic cyclic ketones under the similar catalytic asymmetric system. The expected amination products 275 were obtained with high enantioselectivities.
Scheme 109.

Mannich Reactions of α-Fluorinated Aromatic Cyclic Ketones with Imines Catalyzed by (S,S)-Bicyclic Guanidine
The Trost group also developed an asymmetric catalytic Mannich reaction for the synthesis of β-amino ketone 276 bearing α-fluorinated quaternary stereogenic centers (Scheme 110).295 α-Fluorinated aromatic ketones 231 were used as nucleophiles to react with Boc-protected aldimines with the combination of ZnEt2/(R,R)-prophenol L22 as chiral catalyst. Optimization ofreaction conditions disclosed that increasing the reaction temperature could give the higher chemical yield, and showed no effect on the diastereo- and enantioselectivity. The highest yield (98%) was found when the reaction was conducted 80 °C. The gram-scale study was also carried out, and excellent chemical yield (98%) and stereochemical outcome (>20:1 dr, 99% ee) were obtained.
Scheme 110.

Asymmetric Catalytic Mannich Reactions Catalyzed by ZnEt2/(R,R)-Prophenol
The efficient organocatalytic Mannich reaction of α-fluoro cyclic ketones can also be accomplished by using α-amidosulfones as starting materials. Recently, Yan, Song, and co-authors developed an asymmetric Mannich reaction between α-fluoro cyclic ketones 277 and α-amidosulfones 278 with the use of Song’s chiral oligoEGs C63 as a catalyst (Scheme 111).296 In the presence of catalyst and KF, the in situ generation of imine from α-amidosulfones and generation of enolate from cyclic ketone was performed, which reacted with each other with the aid of a catalyst, affording the corresponding α-fluorinated amino ketones 279 in good chemical yields, good diastereose-lectivities, and high enantioselectivities. It should be mentioned that increasing the temperature to 70 °C gave the best yield and has no effect on the diastereo- and enantioselectivities.
Scheme 111.

Mannich Reactions of α-Fluoro Cyclic Ketones Using Song’s Chiral oligoEGs C63 Catalyst
The Wang group revealed a Cu-catalyzed asymmetric detrifluoroacetylative Mannich reaction for synthesis of compounds 281 bearing fluorinated quaternary stereogenic centers (Scheme 112).297 They employed 2-fluoro-1,3-diketones/hydrates 251 as the precursors of fluorinated enolates to react with isatin derived ketimines 280 in the presence of chiral anthracenyl-substituted cyclohexane-1,2-diamine L23. The reaction was conducted under low temperature (−45 or −30 °C), affording the corresponding 3-substituted 3-amino-2-oxindoles 281 with vicinal tetrasubstituted carbon centers in excellent yields (67–99%), good diastereoselectivities (4:1 → 20:1 dr) and enantioselectivities (66–94% ee).
Scheme 112.
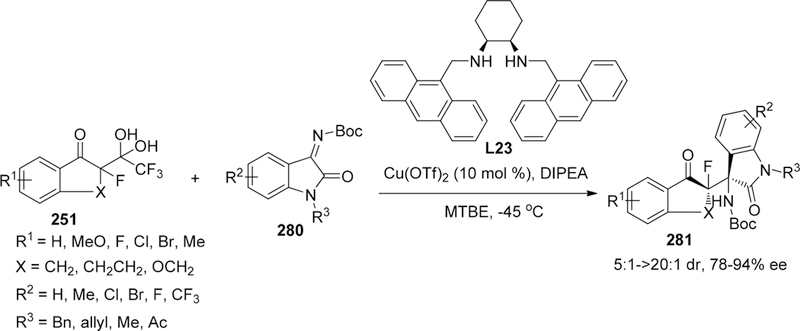
Asymmetric Detrifluoroacetylative Mannich Reactions Using Chiral Anthracenyl-Substituted Cyclohexane-1,2-diamine as Organocatalyst
3.2.3. Aldol Addition Reactions.
3.2.3.1. Aldol Addition Reactions of Detrifluoroacetylatively Generated Fluorinated Enolates.
In 2010, Colby298 and co-authors developed a new 1,1,1,3,3-pentafluoro-2,4-dione system for the preparation of unprotected fluorinated enolates via C–C bond cleavage with the release of trifluoroacetic acid.91 This detrifluoroacetylative reaction could happen under mild conditions, and the resulted fluorinated enolates are the active intermediates, which have been used as the nucleophiles in the aldol reactions. The Colby group has applied these in situ generated enolates in the aldol reaction with varieties of aldehydes to afford the fluorinated hydroxy ketone within about 3 min.298 Then, the Wolf group used this new detrifluoroacetylative approach in aldol reaction with trifluoromethyl ketone pentafluorinated β-hydroxy ketone.299 In 2013, the Wolf group explored the first example on the asymmetric catalytic cascade detrifluoroacetylation and aldol reaction by using copper(II) triflate and chiral bisoxazoline ligand as a catalyst.300 Such 2,2-difluoro-1,3-diketones also have been used as the precursors of fluorinated enolates for asymmetric aldol reaction with N-benzyl isatins.301
In 2013, the Wu group reported a new type of linear detrifluoroacetylative precursors, trifluoromethyl α-fluorinated β-keto gem-diols 282, which were used as the substrates for the organocatalytic asymmetric aldol reactions (Scheme 113).302 The C–C bond cleavage of this monofluorinated gem-diols proceeded smoothly to generate the fluorinated enolates, which reacted very well with varieties of N-benzyl isatins 283 in the presence of cinchona alkaloid derived thiourea C64. The reaction was conducted at room temperature, resulting in the corresponding product 284 with excellent yields and high diastereo- and enantioselectivities. Such gem-diols showed lower reactivity to aliphatic aldehydes, dramatically lower chemical yields, and enantioselectivities were observed.
Scheme 113.

Detrifluoroacetylative Aldol Reactions Using Cinchona Alkaloid Derived Thiourea Catalyst
Then, in 2015, the Han group reported a new cyclic α-fluorinated β-keto gem-diols 251, which could be prepared conveniently from cyclic ketones via a two-step process (Scheme 114).303 These cyclic gem-diols were successfully applied as the substrates for asymmetric detrifluoroacetylative aldol reaction. The reaction was carried out under mild conditions by using copper(II)/chiral bisoxazoline L24 as a catalyst, affording the expected α-fluoro-β-hydroxy ketones 285 in 84–96% yields and up to 99:1 dr and 98% ee. Only the aromatic aldehydes, phenylacetaldehyde, and α,β-unsaturated aldehyde were examined in this system. This asymmetric detrifluoroacetylative aldol reaction provides a new access to the α-fluoro-β-hydroxy ketones bearing C–F quaternary stereogenic centers.
Scheme 114.
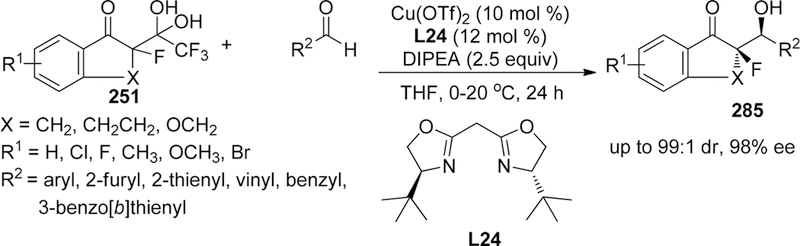
Detrifluoroacetylative Aldol Reactions Using Copper(II)/Chiral Bisoxazoline as a Catalyst
As it should be routinely done in a work on catalytic asymmetric synthesis,304 the authors conducted SDE (self-disproportionation of enantiomers) tests by achiral chromatography305 and sublimation306,307 of some randomly selected aldol products. While the sublimation SDE test was negative, the SDE by achiral chromatography showed noticeable magnitude of about 11% Δee.308 For example, a routine isolation of a product of 84% ee via achiral chromatography resulted in an enantiomerically enriched (88% ee) first fraction and enantiodepleted (77% ee) last fraction, suggesting that extra care should be taken in the avaluation of the stereochemical outcome. Furthermore, taking into account that fluorine and fluorine-containing groups are SDE enabling substituents,309–311 such SDE tests should be a mandatory part of high-quality research in this area.
Using the similar catalytic conditions, Han, Soloshonok, and co-authors tried to use aliphatic aldehydes as substrates for this detrifluoroacetylative aldol reaction (Scheme 115).312 A wide range of aliphatic aldehydes have been used as substrates, which could be successfully transferred into the corresponding α-fluoro-β-hydroxy ketones 285 with moderate chemical yields. This reaction was conducted at room temperature, affording the aldol adducts as well as the byproduct 286 due to the low reactivity of aldehydes. Especially, almost no desired α-fluoro-β-hydroxy ketone product was obtained at all in the cases of the aldehydes with high steric hindrance.
Scheme 115.

Detrifluoroacetylative Aldol Reactions of Alkyl Aldehydes in the Presence of Copper(II)/Chiral Bisoxazoline as a Catalyst
Recently, Han, Soloshonok, and co-authors developed a Cucatalyzed asymmetric detrifluoroacetylative aldol reaction of aldehydes with in situ generated tertiary enolates derived from fluoro-indolinones (Scheme 116).313 CuI was optimized as the best catalyst, which catalyzed the transformation efficiently in the presence of bisoxazoline chiral ligand L25, resulting in α-fluoro-β-hydroxy-indolin-2-ones 287 containing C–F quaternary stereogenic centers in good yields and enantioselectivities. The solvent showed an obvious effect on the reaction outcome, and iPrOH/THF was found to be the best reaction media with the ratio of 1:1. The reaction showed broad substrate generality, and even the aliphatic aldehydes and unprotected N-H fluoro-indolinone gem-diols could be well tolerated to give good yields and enantioselecitivities. The SDE tests via achiral chromatography,314 conducted on these aldol addition products, gave moderate magnitude of about 4% Δee.
Scheme 116.

Detrifluoroacetylative Aldol Reactions of Aldehydes with Tertiary Enolates Derived from Fluoro-indolinones
Recently, Han, Soloshonok, Pan, and co-authors designed a one-pot reaction for the asymmetric synthesis of bicyclic ketoesters containing C–F quaternary stereogenic center, with cyclic α-fluorinated β-keto gem-diols and ortho-phthalaldehyde as starting materials.315 The reaction proceeded smoothly via a complex four-step process, including detrifluoroacetylation, aldol addition, intramolecular cyclization, and oxidation. The combination of trimethylamine and LiBr was used for the detrifluoroacetylative-aldol step and then using PCC to oxidize the intermediate to the final product. This Cu-catalyzed reaction gave bicyclic keto-esters in good chemical yields but poor diastereoselectivity and enantioselectivity.
Shortly after that, the Han group develop a new asymmetric catalytic system for the synthesis of bicyclic keto-esters 290 containing a C–F quaternary stereogenic center (Scheme 117).316 The ester-aldehyde 288, instead of ortho-phthalalde-hyde, was used as the electrophile, which was found to react very well with detrifluoroacetyl derivatives in situ generated enolates via aldol addition and intramolecular substitution. This Cu-(OTf)2/chiral bisoxazoline-catalyzed asymmetric reaction was conducted at room temperature, affording fluorinated bicyclic keto-esters with moderate yields (49−54%) and good diastereo-(80:20−97:3) and enantioselectivity (59−96%).
Scheme 117.
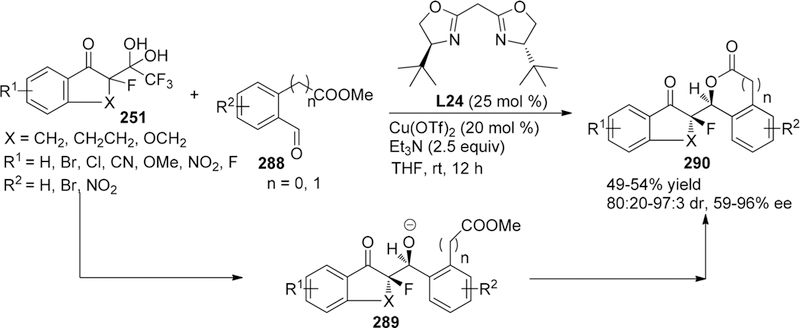
Tandem Detrifluoroacetylative Aldol-Cyclization Reactions
3.2.3.2. Aldol Addition Reactions of Fluorinated Silyl Enol Ethers.
In 2014, the Zhou group developed an asymmetric organocatalytic aldol reaction between fluorinated silyl enol ethers 291 and isatins 292, which used quinine or cinchonine alkaloid urea derivatives as chiral catalysts (C65 and C66) (Scheme 118).317 In the cases of 5-halo groups substituted isatins, the reactions used C66 organocatalyst, affording the expected product 293 in 81−86% ee. The reaction was conducted at −20 °C resulting in up to 98% chemical yields, however, the reaction needed long time for completion (1−5 days). The reaction used fluorinated silyl protected keto-enolates as substrates, generating two adjacent new quaternary stereogenic center, with one of them featuring a C–F quaternary stereogenic center.
Scheme 118.
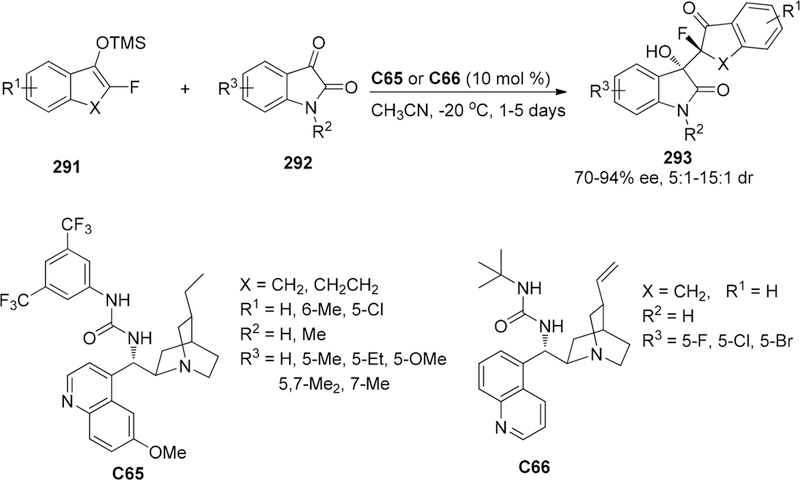
Asymmetric Organocatalytic Aldol Reactions between Fluorinated Silyl Enol Ethers and Isatins
3.2.3.3. Aldol Addition Reactions of Decarboxylative Generated Fluorinated Enolates.
In 2016, the Wennemers group used cinchonine alkaloid urea derivatives C67 as organocatalysts for the asymmetric aldol reactions between in situ generated fluorinated enolates from fluoromalonic acid half-thioesters 294 and aldehydes with THF as solvent under 0 or 10 °C (Scheme 119).318 They used fluoromalonic acid half-thioesters as masked fluoro enolates, which were transferred to the functionalized fluorinated β-hydroxyl thioesters 295 under mild conditions in moderate to good chemical yields (45−87%). The results showed that aromatic aldehydes and half-thioester bearing electron-donating groups usually worked better, resulting in higher enantioselectivities. They applied this synthetic method for the synthesis of fluorinated atorvastatin, which gave excellent enantioselectivity and diastereoselectivity (>99% ee, > 20:1 dr).
Scheme 119.
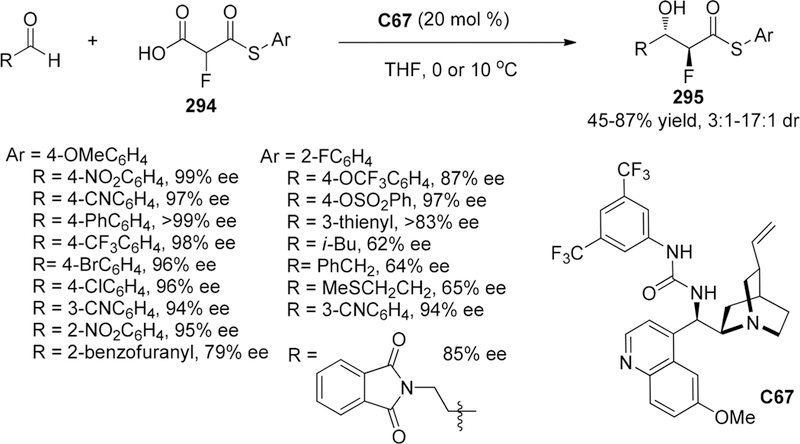
Organocatalytic Asymmetric Aldol Reactions between Fluorinated Enolates of Fluoromalonic Acid Half-Thioesters and Aldehydes
3.2.4. Michael Addition Reactions.
Asymmetric Michael reaction is the other method for the construction of quaternary stereocenter bearing a fluorine atom. A number ofprocedures for the asymmetric addition reaction of fluorinated nucleophiles bearing a carbonyl group to Michael acceptors have been reported.
3.2.4.1. Michael Addition Reactions with Nitro Styrenes as Acceptors.
Asymmetric organocatalytic Michael reactions between nitroolefins and fluorinated β-ketoesters have been well developed for the construction of Michael products with tetrasubstituted C–F centers. In 2009, the Lu group used cinchona alkaloid derived thiourea C67 as the catalyst for this transformation (Scheme 120).319 Optimization of reaction conditions showed that the trifluoromethyl substituted quinidine was the best choice and could catalyze the reaction smoothly to give the corresponding α-substituted α-fluoro-β-ketoester 296 in almost quantitative yields and high diastereo-(2.5:1−19:1) and enantioselectivities (95−99% ee). The reaction also showed a wide substrate generality, and varieties of aryl and alkyl nitroolefins worked well with fluorinated nucleophiles. It should be mentioned that the bulky t-butyl ester group resulted in a dramatically lower chemical yield and decreased diatereoselectivity.
Scheme 120.

Asymmetric Organocatalytic Michael Reactions between Nitroolefins and Fluorinated β-Ketoesters
In 2009, the Wang group also developed an organocatalytic asymmetric Michael addition reaction between α-fluoroketoesters 263 to nitroolefins (Scheme 121).320 The reaction used cinchona alkaloid-derived compound C68 bearing a bulky 9-phenanthrenyl substitution as the chiral catalyst and conducted the reaction under room temperature. Only one example of α-fluoroketoester was used to react with several nitroolefins to give the product 296 in excellent chemical yields and high enantioselectivity. The limitation of this method is the moderate diastereoselectivity (1.7−4:1 dr). One of the products was further converted into the cyclic compound, pyrrolidine, via reduction in the presence of Raney Ni.
Scheme 121.

Organocatalytic Asymmetric Michael Addition Reactions between α-Fluoroketoesters to Nitroolefins
In 2009, the Kim group successfully used chiral bifunctional thiourea containing a tertiary amine C69 as an organocatalyst for the Michael reaction of β-ketoesters bearing an α-fluorine atom to nitroalkenes (Scheme 122).321 Under the optimized reaction conditions, varieties of α-fluorinated-β-ketoesters 263 could add smoothly with nitroalkenes to give the corresponding chiral Michael adducts 296 bearing a fluorinated quaternary carbon center in moderate diastereoselectivities and excellent enantio-selectivities (83 → 99% ee). A bulky alkyl group on the ester moiety was required for the high diastereoselectivity. Changing the ester group from isopropyl ester to ethyl ester led to a noticeable reduction in diastereoselectivity.
Scheme 122.

Bifunctional Thiourea–Tertiary Amine Organocatalytic Michael Reactions of β-Ketoesters
In 2009, Zou, Zhao, and co-authors applied a primary-secondary diamine C70 as an organocatalyst for the asymmetric Michael reactions of α-fluoro-β-ketoesters 263 with nitroolefins (Scheme 123).322 The optimization of reaction conditions showed that the diamine bearing steric bulky groups gave the best stereochemical outcome. On the other hand, the addition of acids as additives obviously improved the reaction efficiency, and the combination of TfOH/p-NBA (p-nitrobenzoic acid) could promote the reaction with better yields. In this reaction, both alkyl and aryl ketoesters reacted well with nitro styrenes, affording the corresponding γ-nitro-α-fluoro-β-ketoesters 296 bearing a C–F quaternary carbon center in excellent diastereoselectivities (6:1 → 20:1) and enantioselectivities (89−99% ee). The 19F NMR experiments tracing the process of the reaction showed that the catalytic cycle contains the enamine activation.
Scheme 123.
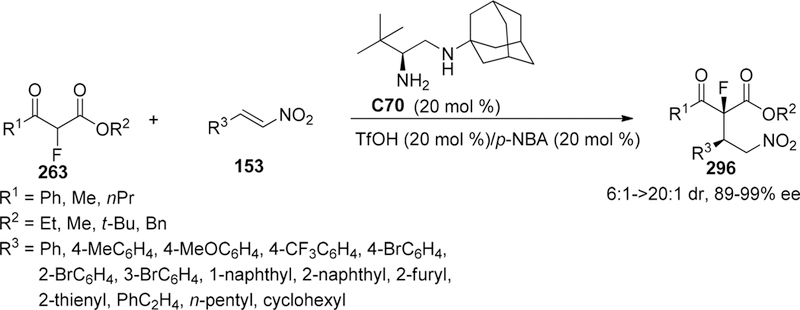
Asymmetric Michael Reactions of α-Fluoro-β-ketoesters with Nitroolefins
α-Fluorinated α-sulfonyl ketones are an efficient type of nucleophile due to their convertibility into varieties of functionalized monofluoromethylated compounds, which are crucial structural unit in many biological compounds. In 2012, Zhao and co-authors applied the α-fluorinated α-sulfonyl ketones 297 in an asymmetric organocatalytic Michael addition reaction to nitro-olefins (Scheme 124).323 They used tertiary amine based thiourea C71 as the organocatalyst for this reaction, which afforded the γ-nitro-α-sulfonyl ketones 298 in excellent yields (70−93%) and high diastereo-(5:1−20:1) and enantioselectivities (86−96%). The alkyl group substituted nitroolefins also worked very well in this reaction, and no reduction on the yields and stereochemical outcome was observed.
Scheme 124.
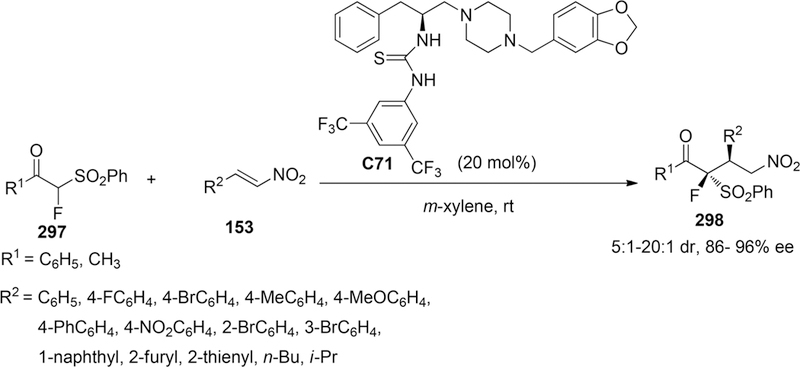
Asymmetric Michael Additions of α-Fluorinated-α-sulfonyl Ketones
α-Fluoroalkyl-phosphonates are widely represented in many biologically active compounds, in particular phosphorus analogues of amino acids.324–327 They also are good candidates for fluorinated nucleophiles and can react with Michael acceptors. In 2015, the Kim group applied α-fluoro ketophosphonates 299 as nucleophiles for the asymmetric Ni-catalyzed Michael addition reaction with nitrostyrenes (Scheme 125).328 The optimization of reaction conditions showed that chiral 1,2-cyclohexanediamine coordinated dicationic nickel complexes C72 was the best catalyst, and such catalytic combination promoted the reaction smoothly to give the expected Michael adducts in excellent yields, high stereochemical outcomes (10:1−50:1 dr, 93−99% ee). Increasing heating of the reaction to 50 °C resulted in the best yield (90%) without any reduction of diastereo- and enantioselectivity. The gram-scale synthesis of this method was also tried, and the same level of yield, dr and ee values was obtained.
Scheme 125.

Asymmetric Michael Additions of α-Fluoro β-Ketophosphonates
In 2015, the Lu group showed that 2-fluoro-1,3-diketones 301 could be successfully used as nucleophiles for the organocatalytic Michael addition reaction to nitroolefins (Scheme 126).329 After the scanning of organocatalysts, quinine-derived sulfonamide C72 bearing a 3,5-trifluoromethylphenyl substituent was demonstrated to be the best one for this asymmetric Michael reaction. Several nitro styrenes reacted very well with 2-fluoro-1,3-diketones to afford the corresponding Michael adducts 302 bearing C–F quaternary stereogenic centers in good to excellent yields (89 → 99%) and high diastereo- and enantioselectivities (2:1−8:1 dr, 94 → 99% ee). An isopropyl substituted nitroolefin substrate was examined in this system, but only moderate yield was observed with almost no stereoselectivity.
Scheme 126.

Michael Addition Reactions of 2-Fluoro-1,3-diketones with Nitroolefins
The asymmetric Michael addition of nitroolefins with fluorinated carbonyl compounds as nucleophiles provided an easy strategy for the synthesis of γ-nitro-α-fluorinated ketone/ester compounds. Although several related examples have been developed, α-fluoro-α-nitroalkanes as nucleophiles for the asymmetric Michael reaction to nitroolefins is rarely reported. In 2014, Lu and co-authors reported asymmetric organocatalytic Michael additions between α-fluoro-α-nitroalkanes 303 and nitroolefins (Scheme 127).330 The amino acid-incorporating multifunctional quinine-derived compound C74 was employed as an organocatalyst, which catalyzed the reaction to give the corresponding 1,3-dinitro compounds 304 bearing α-fluoro quaternary centers in good yields (71–95%) and good diastereo- and enantioselectivities (5:1−8:1 dr, 82–96% ee). The most interesting point was that the different diatereomers in most cases could be isolated by the regular flash silica gel chromatographic column. The hydrogenation reduction of the obtained 1,3-dinitro compound was conduct to afford 1,3-diamine compounds.
Scheme 127.

Asymmetric Michael Additions of α-Fluorinated Carbonyl Compounds with Nitroolefins
Recently, the Wennemers group successfully used fluorinated monothiomalonates 305 as nucleophiles for the organocatalytic Michael addition reaction with β-nitrostyrenes to provide a new way for the synthesis of chiral α-fluoro-γ-nitro thioesters 306 with fluorinated tetrasubstituted stereogenic centers (Scheme 128).331 The epi-cinchonine-urea C75 was identified as the best catalyst for this reaction after several types of catalyst screening. Under the optimized reaction conditions, fluorinated mono-thiomalonate reacted smoothly with several nitroolefins, resulting in high stereoselectivities (13:1 → 20:1 dr, 95−99% ee).
Scheme 128.
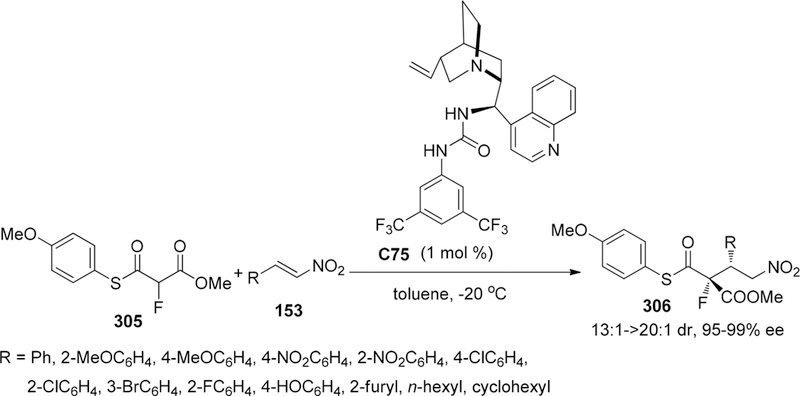
Asymmetric Organocatalytic Michael Addition Reactions of Fluorinated Monothiomalonates with β-Nitrostyrenes
3.2.4.2. Michael Addition Reactions with Other Reagents as Acceptors.
In 2009, Wong, Tan, and co-authors developed a new type of Michael acceptors, N-alkyl maleimides 307, for the organocatalytic asymmetric Michael reaction of α-fluoro-β-keto esters 263 (Scheme 129).332 They used (S,S)-bicyclic, guanidine C62 as the chiral organocatalysis for this reaction, which showed excellent stereocontrolled ability and only one diastereomer was obtained in almost all the cases. The substitution on the nitrogen atom of maleimides almost has no effect on the reaction efficiency, and ethyl, methyl, cyclohexyl, benzyl, n-hexyl, and t-butyl were all well tolerated in the current system. On the other hand, varieties of aryl keto-esters worked very well, resulting in excellent yields (80–99%) and high enantioselectivities (83–97% ee).
Scheme 129.
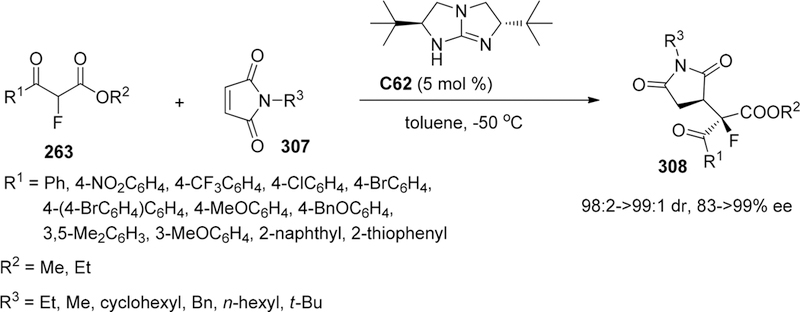
Organocatalytic Asymmetric Michael Reactions of α-Fluoro-β-keto Esters with N-Alkyl Maleimides
In 2010, the Lu group developed asymmetric Michael addition reactions with di-tert-butyl azodicarboxylate as acceptors to react with α-fluorinated β-keto esters 263, affording α-fluoro-α-amino-β-keto esters 310 with fluorine containing quaternary stereogenic centers (Scheme 130).333 Several α-fluorinated β-keto esters were tolerated in the reaction with chiral guanidines derived from cinchona alkaloids C76 as organocatalysts. The aliphatic keto ester substrates showed lower reactivity and stereoselectivity, and poor enantioselectivities were found (about 50% ee).
Scheme 130.

Asymmetric Michael Addition Reactions of α-Fluorinated β-Keto Esters with di-tert-Butyl Azodicarboxylates
Joseph, Delarue-Cochin, and co-workers reported an asymmetric Michael reaction with (S)-1-phenylethylamine as a chiral auxiliary (Scheme 131).334 They used 2-fluoroenaminoesters 311 derived from β-keto esters and (S)-1-phenylethylamine as chiral nucleophiles for the Michael addition reaction to α-substituted methyl acrylates under heating or refluxing conditions. The intermediary Michael adducts were obtained, which were directly hydrolysized in the presence of 10% aqueous AcOH at room temperature to give γ-substituted γ-fluoroglutamate 312 bearing a fluorinated quaternary carbon center. Three examples of α-substituted methyl acrylates were examined. The substituents showed much effect on the reaction. In the case of BocNH substituent, no expected adducts were obtained even when the reaction time were prolonged to 15 days. Interestingly, the two diastereomers could be easily separated by the regular silica gel chromatographic column through the thioketalization of the two diastereomers by 1,2-ethanedithiol.
Scheme 131.

Asymmetric Michael Reactions Using (S)-1-Phenylethylamine as the Chiral Auxiliary
In 2015, the Zhou group used fluorinated enol silyl ethers as nucleophiles for the asymmetric Michael addition to the isatylidene malononitrile 314 (Scheme 132).335 The chiral secondary amine phosphoramide C77 derived from 1,2-diaminocyclohexane was used as the organocatalyst for this Michael reaction, which could catalyze this reaction efficiently to give the corresponding adducts 315 in nearly quantitative yields and excellent diastereo- and enantioselectivities (4:1 → 20:1 dr, 84−94% ee). Both bis- and monofluorinated enol silyl ethers derived from α-fluorinated indanone or benzofuranone could be well tolerated in this reaction. Most interestingly, the obtained adducts could be easily reduced into the corresponding alcohols without any loss of enantioselectivity.
Scheme 132.
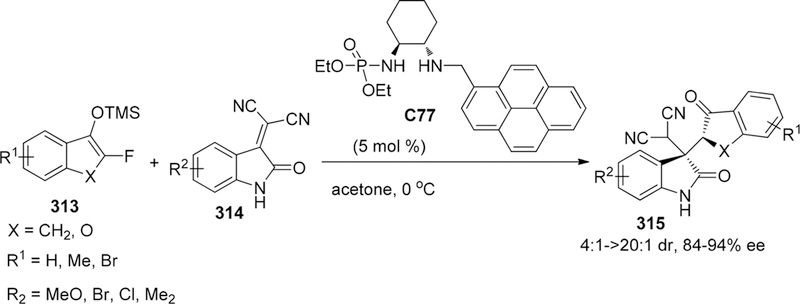
Asymmetric Michael Additions of Fluorinated Enol Silyl Ethers with Isatylidene Malononitriles
In 2014, the Lu group developed 2,3-butadienoates as Michael acceptors for the asymmetric addition reaction with 3-fluorooxindoles 233 for the synthesis of 3-fluoro-3-allyloxindoles containing a C–F quaternary center (Scheme 133).336 The l-threonine derived phosphine-amide C78 was demonstrated as the best choice of catalyst, which was able to catalyze the conversion of several 2,3-butadienoates 316 into the corresponding products with excellent yields (88−95%) and high enantioselectivities (83−94% ee). The ester group of 2,3-butadienoates almost have no effect on the reaction efficiency, and the groups including methyl, t-butyl, benzyl, and bulky anthryl were all well tolerated in the reaction, affording the same level of yields and enantioselectivities. The control experiments disclosed that the hydrogen bonding interactions between N-Boc of 3-fluoro-oxindoles and N-H group of the chiral catalyst led to the observed absolute configuration of the product.
Scheme 133.
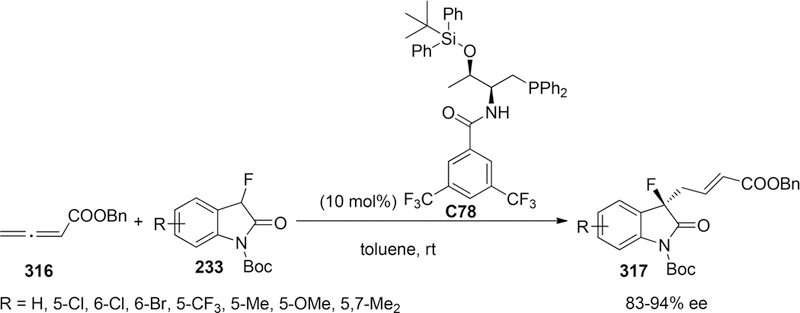
Asymmetric Michael Addition Reactions of 2,3-Butadienoates with 3-Fluorooxindoles
α,β-Unsaturated ketones have also been developed as Michael acceptors for the asymmetric addition reaction with malonates derivatives 319 by Zhao and co-authors for the construction of C–F quaternary stereogenic center (Scheme 134).337 They established a new catalytic Michael reaction by using dipeptide-derived multifunctional phosphonium salts C79 as chiral catalysts. Varieties of α,β-unsaturated ketones were tried in this catalytic system to react with malonates, resulting in excellent yields and high enantioselectivities. In their work, one example of fluorinated nucleophile, dimethyl 2-fluoromalonate, was used as substrate to react with (E)-chalcone, resulting in the product 320 with a C–F quaternary stereogenic center in 89% yield and 98% ee.
Scheme 134.

Asymmetric Michael Addition Reactions of α,β-Unsaturated Ketones with Malonates
In 2013, the Lu group developed the first example on the asymmetric organocatalytic Michael reaction with very active vinyl sulfones 321 as Michael acceptors (Scheme 135).338 The quinine-derived tertiary amine-thiourea C80 was used as the most efficient organocatalyst, which catalyzed the reaction of 3-fluorinated oxindoles 233 to give the expected products 322 with the best yield and enantioselectivity. Nine different substituted 3-fluorinated oxindoles were examined in the reaction, and the results showed that the substitution on the aromatic ring had almost no effect on the reaction, and >95% chemical yields were obtained for all the cases. The reaction was conducted under room temperature and completed within 1 h, which provides an easy way to sulfone substituted oxindoles containing a fluoro-quaternary stereogenic center.
Scheme 135.

Asymmetric Organocatalytic Michael Reactions Vinyl Sulfones with 3-Fluorinated Oxindoles
Very recently, Han, Soloshonok, and co-authors also developed an asymmetric Cu-catalyzed detrifluoroacetylation/Michael reaction by using cyclic fluorinated gem-diols 251 as nucleophiles (Scheme 136).339 They pointed out in this work that several types of Michael acceptors, such as α, β-unsaturated carbonyl derivatives, nitroolefins, and highly reactive N-(enoyl)-oxazolidinones,340–342 have been tried to react with the cyclic fluorinated gem-diols, which were not successful. Only the highly reactive 1-(1-(phenylsulfonyl)vinylsulfonyl)benzene 321 could react with cyclic fluorinated gem-diols well, resulting in the corresponding products 323 in excellent enantioselectivity and chemical yields. The combination of Cu(OTf)2 and (1S,2S)-1,2-diphenylethane-1,2-diamine L26 was applied as the chiral catalyst, which promoted the reactions of several types of 2-fluoro-1,3-diketones/hydrates to give the corresponding five-, six, and seven-membered rings and heterocyclic 3-fluoro-2,3-dihydrochromen-4-one derivatives bearing quaternary C–F stereogenic carbon. The authors demonstrated that chromatographic purification of the addition products should be conducted with particular care as a moderate SDE magnitude of 4% Δee343 was detected under the conditions of routine column chromatography.
Scheme 136.

Cu-Catalyzed Detrifluoroacetylative Michael Addition Reactions
3.2.5. Cross-Coupling Reactions.
Macrolides like erythromycin are widely prescribed as antibiotics. The development of new macrolide derivatives with activities against resistant pathogens has been the focus of study of several research groups. As a result, fluorine-substituted analogues of these macrolides have been synthesized. In 2004, the Beebe group reported the palladium-catalyzed Sonogashira coupling reaction of 2-fluoro-9-oxime ketolides 324 with several heteroaryl bromide reagents (Scheme 137).344 The use of the free alcohol at position 20 resulted in a convergent synthesis of analogues 325 in good yields. The fluorine atom was introduced in an earlier stage by reaction of the NaH-generated enolate with NFSI. Regarding the antibacterial activity, the 2-F analogues showed no improvement when compared to the 2-H compounds.
Scheme 137.

Palladium-Catalyzed Sonogashira Coupling Reactions of 2-Fluoro-9-oxime Ketolides
Later, the Beebe group employed a similar strategy to prepare the diazalides analogues 327 (Scheme 138).345 The 2-fluoro-6-O-propargyl diazalides 326 were used as the partner in the Sonogashira coupling with two different heteroaryl bromide reagents. The 20-O-protecting group was further removed by hydrolyses in hot methanol to afford the 6-O-heteroarylpropargyl diazalides 327. The introduction of the 2-fluoro group had no significant effect on the antibacterial activity when compared to the 2-H compounds.
Scheme 138.
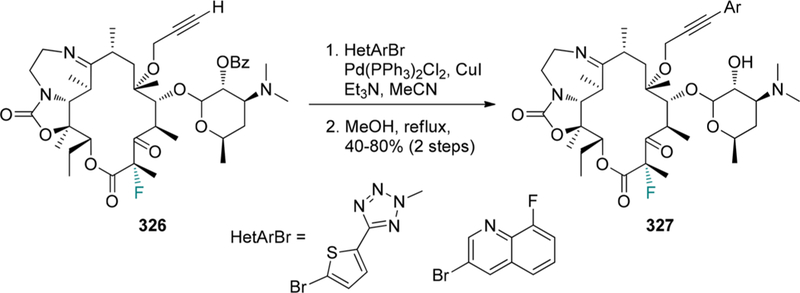
Sonogashira Coupling of 2-Fluoro-6-O-Propargyl Diazalides with Two Different Heteroaryl Bromide Reagents
In 2012, the Sugimoto group reported the synthesis of 6-O-(heteroaryl-isoxazolyl)propynyl 2-fluoro ketolides 329 in moderate yields via the palladium-catalyzed Sonogashira cross-coupling under copper-free conditions (Scheme 139).346 The coupling reaction was performed using the free alcohol 328, which was prepared by fluorination using NFSI, followed by 20-O-benzoyl group hydrolysis with methanol. The stereochemistry of fluorinated intermediate 328 was determined by X-ray analysis. The synthesized compounds exhibited promising activity against respiratory pathogens, although a comparison to the parent 2-H analogues was not presented.
Scheme 139.

Palladium-Catalyzed Sonogashira Cross-Coupling under Copper-Free Conditions
Very recently, the Liang group reported the palladium-catalyzed Heck (Scheme 140A) and Sonogashira coupling reactions (Scheme 140B) of allyl-(330) and propargyl-(332) 9-substituted oxime ketolides, respectively, in low yield.347 The final desired products 331 and 333 were obtained by subsequent hydrolysis of the 20-acetate (Scheme 140). The combined incorporation of aminopyridyl or carbamoylpyridyl and 2-fluorine atom resulted in high antibacterial activity.
Scheme 140.
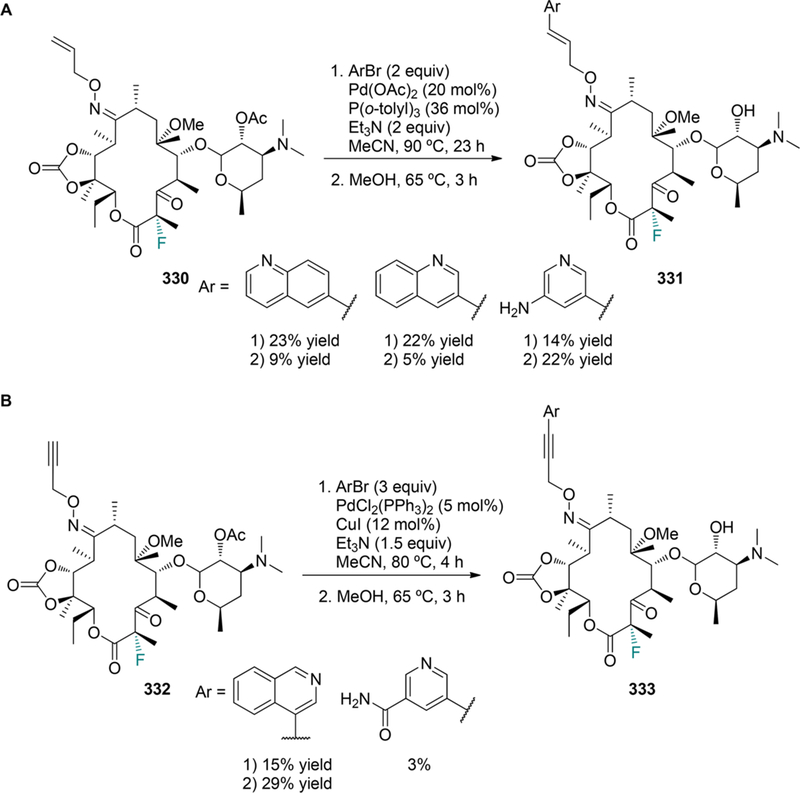
Palladium-Catalyzed Heck (A) and Sonogashira (B) Coupling Reactions
2′-Fluorine containing nucleosides have been synthesized and studied for their antiviral activity. The palladium-catalyzed cyanation, Suzuki, and Stille coupling reactions of 334 with the corresponding nucleophilic partners were reported for the synthesis of 7-heterocyclic substituted 7-deaza-adenine nucleo-sides 336−338, respectively (Scheme 141A).348 The coupling reactions were performed under harsh conditions, such as microwave irradiation and high temperatures, to yield the products in low to moderate yields. Furthermore, the phosphoramidate prodrug 341 was prepared from the 3-O-THP analogue 339 by using Suzuki coupling reaction conditions (Scheme 141B).
Scheme 141.

Palladium-Catalyzed Cyanation: Suzuki (A) and Stille (B) Coupling Reactions
Recently, Zhang, Tu, and co-authors reported the synthesis of several 2′-fluoro-7-deaza purine nucleoside derivatives 343−347 via cross-coupling reactions of iodide 342 with a wide range of nucleophilic parterns using Stille, Suzuki, Sonogashira, or Heck conditions (Scheme 142)349.
Scheme 142.

Cross-Coupling Reactions Using Stille, Suzuki, Sonogashira, or Heck Conditions
The Ando group studied the modification of α-bromo-α-fluoro-β-lactams. The Kumada coupling reaction of rac-348 with a wide range of aryl and heteroaryl Grignard reagents was achieved using a Ni/chiral bisoxazoline catalytic system. The corresponding anti-α-aryl-α-fluoro-β-lactams rac-349 were obtained in high diastereoselectivity and up to 98% yield using (R)-PhBOX L27 as ligand (Scheme 143).350,351
Scheme 143.

Kumada Coupling Reactions
Later, The Ando group reported the nickel-catalyzed Suzuki coupling reaction of the same type of substrates with a wide range of aryl-(9-BBN) reagents to yield the anti-isomer in up to 87% yield.352 The most efficient ligand was bipyridine derivative L28. It is worth noting that the chiral coupling product 351 (Scheme 144) was obtained without erosion of enantiopurity when the reaction was conducted with the enantioenriched α-bromo-α-fluoro-β-lactam 350.
Scheme 144.

Nickel-Catalyzed Suzuki Coupling Reactions
In both cross-coupling approaches, the respective alkyl reagents partners did not work. However, although not in a cross-coupling approach, the formation of the desired α-alkyl-α-fluoro-β-lactams was possible by alkylation and aldol reaction of these derivatives (Scheme 145).351,353
Scheme 145.
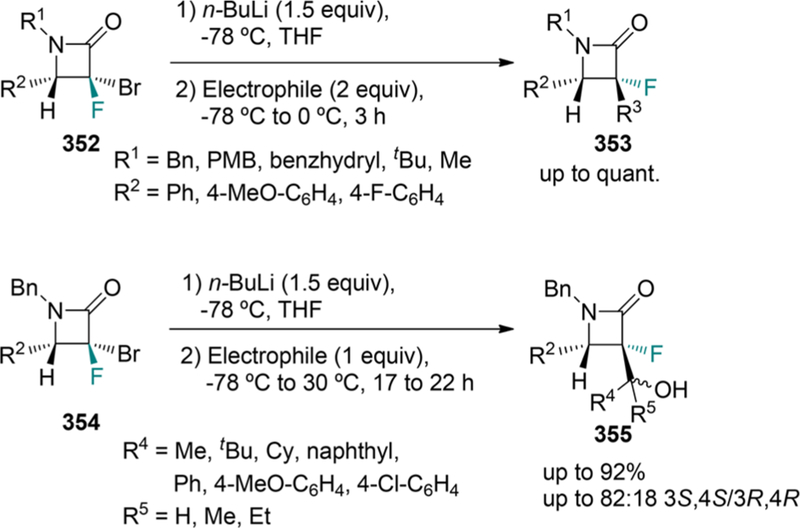
Cross-Coupling Approach for the Formation of α-Alkyl-α-fluoro-β-lactams
Dominguez prepared and studied the activity of several tetrasubstituted cyclopropane hydroxamic acid derivatives as class Ila histone deacetylase inhibitors.354 Fluorine substituted derivatives exhibited better biological activity. The final desired products containing different heterocyclic moieties were synthesized through a palladium-catalyzed Suzuki coupling using the fluorinated substrate as the nucleophilic partner and several heteroaryl halides (Scheme 146). The fluorine atom was introduced by reaction of the ketone with LDA followed by NFSI; subsequently, the enantiopure hydroxamic acid products 357 were obtained from chiral HPLC purification.
Scheme 146.
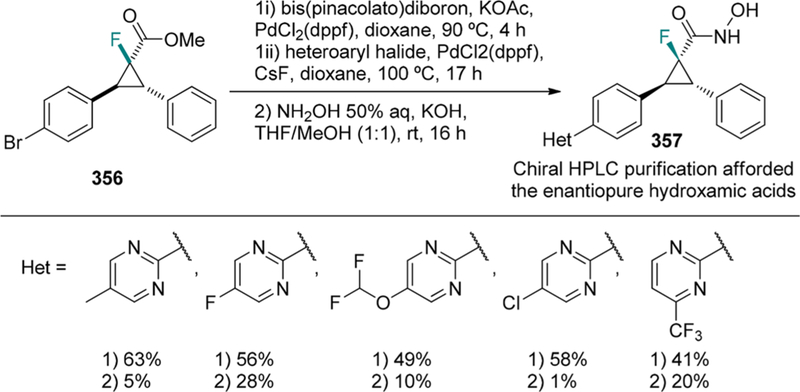
Palladium-Catalyzed Suzuki Coupling Reactions
The Magnus group reported the Suzuki coupling reaction of enantiopure iodo-sulfonamide 358 with 4-carboxylphenylboronic acid using palladium black as the catalyst (Scheme 147).355 The reaction afforded biphenyl product 359 as a single enantiomer in very good yield (88%). Further elaboration of the product yielded the API LY503430, an AMPA potentiator.
Scheme 147.
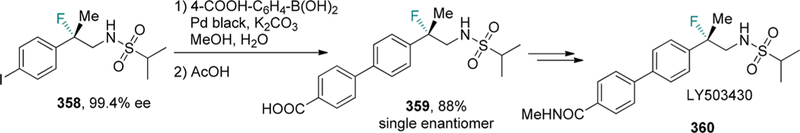
Suzuki Coupling Reactions of Iodo-sulfonamide with 4-Carboxylphenylboronic Acid Using Palladium Black as the Catalyst
3.3. Biocatalytic Approaches
Despite recent progress in the development of biological catalysts for the enantioselective introduction of fluorine,356 examples of biocatalytic approaches for the elaboration of quaternary F-containing substrates are mainly based on lipase-catalyzed hydrolysis or transesterification reactions for the kinetic resolution of target molecules. In 1959, the Rigler group reported the dehydrogenation of compound 362 into 21-deoxytriamcinolone 363 by using Nocardia coralline (Scheme 148).357 The desired product 398 was obtained in 53% yield after 11 h of fermentation.
Scheme 148.
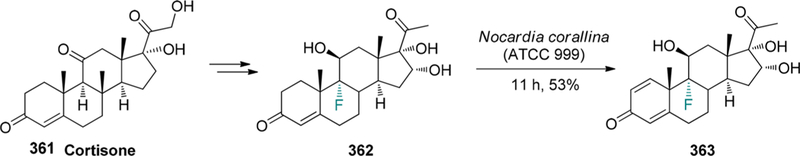
Lipase-Catalyzed Transesterification Reactions
In 1987, Kitazume and Yamamoto reported the hydrolysis of ester derivatives 364 into the corresponding carboxylic acids 365 by using the lipase-MY from Candida cylindracea.358 The target compounds were studied for their activity as inhibitors of angiotensin converting enzyme (Scheme 149).
Scheme 149.

Lipase-Catalyzed Hydrolysis Reactions
In 1985, Kamata, Nakamura, Susuki, and co-authors reported the oxidation of 366 into intermediate 367 in 17% yield by using the resting mycelium of Corynespora cassiicola (Scheme 150).359 The steroid derivative 368 exhibited strong binding affinity for the cytoplasmic mineralocorticoid receptor of rat kidney and good aldosterone antagonist activity in an in vivo assay.
Scheme 150.
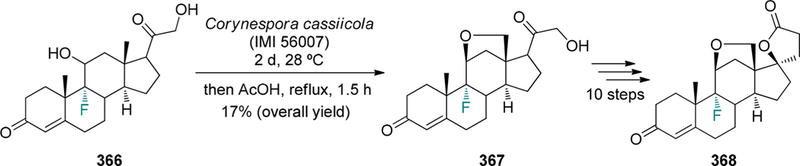
Biocatalytic Oxidation Reactions
In 1997, the Haufe group described the enzymatic kinetic resolution of 1-acetoxy-2-aryl-2-fluoroalkanes by hydrolysis of rac-369 (Scheme 151A) or by acet:ylation of the corresponding alcohol rac-370 (Scheme 151B).360 Lipase from Pseudomonas cepacia (Amano PS) was found to perform the best for both cases. In general, 61 mg of enzyme per mmol of substrate was used in the hydrolysis reaction. Furthermore, a lower selectivity was observed using the lipase from Candida cylindracea (CCL), which afforded the opposite enantiomer in the hydrolysis process. In general, the selectivity of the process increased with the increase in chain length of the substituent of the quaternary center. Introduction of i-Bu substituent on the para-position of the phenyl ring (resembling the ibuprofen structure) resulted in low selectivity; however, a lower quantity of enzyme was used.
Scheme 151.
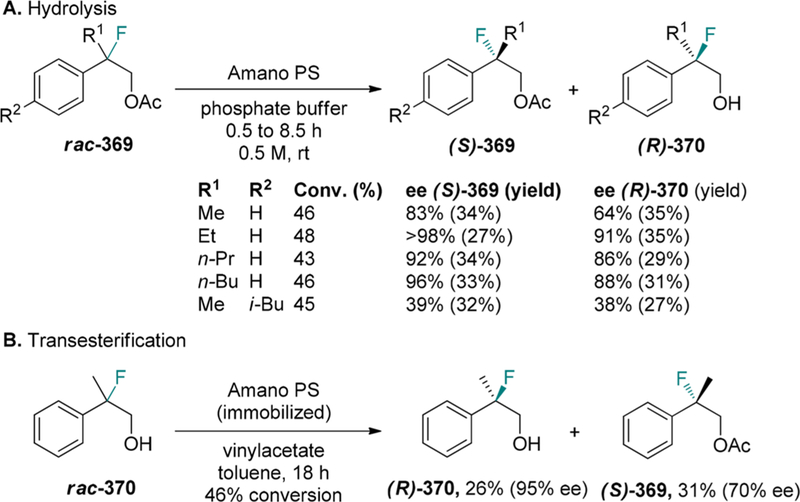
Enzymatic Kinetic Resolution of 1-Acetoxy-2-aryl-2-fluoroalkanes by Hydrolysis (A) and Transesterification (B)
In 1998, Hirai, Takeuchi and co-authors described the synthesis of chiral 2-fluorinated 372 by desymmetrization of glycol systems 371 and 373 (Scheme 152).361 The enantioselective hydrolysis of 2-fluorinated diacetates 371 catalyzed by the lipase from porcine pancreas (PPL) afforded the 372 in up to 96% ee (Scheme 152A). On the other hand, the acetylation of 2-fluorinated 1,3-propanediols 373 with vinyl acetate catalyzed by lipase PS afforded the corresponding monoalcohols 372 in up to 95% ee (Scheme 152b).
Scheme 152.
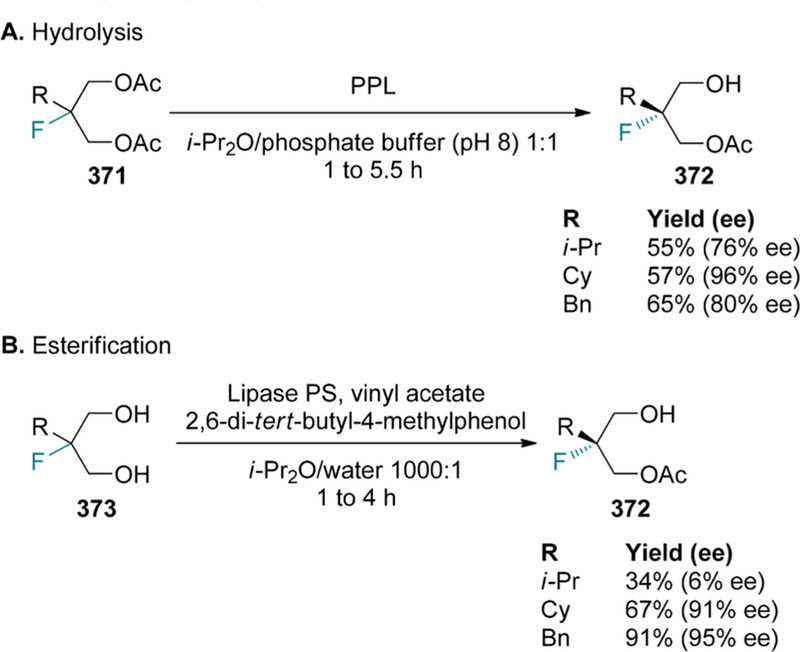
Desymmetrization of Glycol Systems by Biocatalytic Hydrolysis (A) and Esterification (B)
In 1998, Guanti, Narisano, and co-authors studied the desymmetrization of fluorinated polyfunctionalized C3 synthons by lipase mediated asymmetric monohydrolysis of 2-aryl-2-fluoromalonic acid diesters 373 (Scheme 153A) or monoacetylation of 2-aryl-2-fluoro-1,3-propanediols 376 (Scheme 153B).362,363 The most efficient lipase was PPL (lipase from porcine pancreas) supported on Celite (S-PPL), which afforded the chiral product in up to 96% ee.
Scheme 153.

Desymmetrization of Fluorinated Polyfunctionalized Synthons by Lipase Mediated Asymmetric Hydrolysis (A) and Esterification (B)
The Haufe group reported the enzymatic kinetic resolution of different racemic 2-fluoro-2-phenylcyclopropyl derivatives by lipase-catalyzed transesterification or hydrolysis (Scheme 154). The best results were achieved using Amano PS in tert-butyl methyl ether (MTBE) as solvent.364,365
Scheme 154.

Kinetic Resolution of Racemic 2-Fluoro-2-phenylcyclopropyl Derivatives by Lipase-Catalyzed Transesterification or Hydrolysis
4. CONCLUSIONS
Over the past decade, fluoro-organic methodology has been growing at an extraordinary pace. Now it is commonly recognized that fluorine chemistry is in a league of its own, as fluorine and fluorine-containing groups cannot be considered as merely halogen substituents in terms of both chemical and biological properties. Thus, in most cases, synthesis of tailor-made fluoro-organics requires the discovery of rather unique, novel approaches or significant modifications of some general methods. This aspect of fluorine chemistry is particularly underscored by the invention of chiral electrophilic fluorination reagents, presented as discussed in the first part of this Review. Retrosynthetically attractive, the concept of electrophilic fluorination has received a great deal of attention, resulting in a reasonably good mechanistic validation for the design of new reagents and reaction conditions. Nevertheless, while methodologically very impressive, the electrophilic fluorination approach is still virtually exclusively limited to the fluorination in the α-position to a carbonyl group of structurally rigid aromatic/cyclic substrates. Moreover, with the exception of a handful of examples, the values of stereochemical outcome are still below the synthetically useful level. Most of the progress in this area has been made in catalyst design using more or less standard set of substrates. While this situation is rather logical and allows comparing the catalytic systems, the expansion of the substrate generality is highly desirable and expected in the near future. Some exciting progress in this direction has already been achieved with the invention of the tandem processes for functionalization of C=C bonds, offering innovative synthetic solution for previously inaccessible structurally complex and biologically relevant molecules. Asymmetric elaboration of C–F containing substrates, constituting the second major part of this Review, is a more matured area of fluorine chemistry, methodologically overlapping with general organic asymmetric synthesis. Thus, approaches based on application of chiral auxiliaries are traditionally reliable, offering synthetically useful solutions for preparation of target molecules featuring quaternary C–F stereogenic centers. Moreover, the possibility of purification of diastereomeric intermediates adds a certain advantage in the preparation of enantiomerically pure samples ready for biological applications. In the asymmetric catalysis area, some particular progress has been achieved in developing Mannich-type additions, allowing synthesis of highly biologically relevant fluorinated amino-compounds. Similar to electrophilic fluorination, this area is also still not very versatile in terms of substrates generality, mostly featuring various transformations around a rather reactive carbonyl group. In the area of biocatalytic transformations, one can notice a particular progress made in enzymatic desymmetrization of substrates containing quaternary C–F pro-chiral carbon. Considering a usually low cost-structure associated with of biocatalytic methods, this approach seems to be of great practical potential. In general, the research critically discussed in this Review represents one of the most intensely competitive areas in the current organic chemistry, oriented to discovery of new fluorinated structural motives and their application in the design of new generations of bioactive compounds.
ACKNOWLEDGMENTS
J.L.H. thanks the National Natural Science Foundation of China (21761132021). F.D.T. thanks the National Institute of Health (R35 GM118190), V.A.S. thanks IKERBASQUE, the Basque Foundation for Science, N.S. thanks JSPS KAKENHI (JP16H01142) in the Middle Molecular Strategy, JSPS KAKENHI (JP16H01017) in Precisely Designed Catalysts with Customized Scaffolding, the Advanced Catalytic Transformation (ACT-C) from the JST, and the Asahi Glass Foundation, and M.S. thanks RIKEN, JST, and JSPS for support of this work. J.A.S.C. thanks Fundacao para a Ciencia e a Tecnologia (SFRH/BPD/100433/2014) for a postdoctoral fellowship. M.S. thanks a graduate student, Cassandra J. Henderson, for help with the literature survey.
Biographies
Yi Zhu received his B.Sc. in organic chemistry from Jiangsu Normal University in 2013. Then he obtained his M.Sc. in organic chemistry from Nankai University in 2016. Currently, he is a Ph.D. student working under the supervision of Professor Yi Pan and Jianlin Han at Nanjing University. His research interests have centered on asymmetric synthesis of fluorine compounds.
Jianlin Han received his Ph.D. in organic chemistry in 2007 from Nanjing University under the supervision of Professor Yi Pan. He then carried out postdoctoral studies for one year at Texas Tech University on the research of asymmetric organic phosphorus chemistry. In 2008, he moved to the University of Okalahoma to continue postdoctoral research for nearly one year. In 2009, he took the position of Associate Professor at the Nanjing University. His major current research interests are fluorine chemistry, asymmetric synthesis, radical reactions, and organometallic chemistry.
Jiandong Wang received his B.Sc. (2012) andM.Sc. (2015) in chemistry from Beijing University of Chemical Technology. Currently, he is a Ph.D. student working under the supervision ofProfessor Norio Shibata at Nagoya Institute of Technology. His research interests have centered on C–F bond activation.
Norio Shibata is a Professor at the Nagoya Institute of Technology since 2008. He received a Ph.D. (1993) in pharmaceutical sciences from Osaka University under the direction of Professor Yasuyuki Kita. He worked at Dyson Perrins Laboratory (Professor Sir Jack. E. Baldwin), Oxford University (JSPS fellow, 1994–1996), Sagami Chemical Research Institute (Dr. Shiro Terashima, 1996), after which he was a lecturer at Toyama Medical & Pharmaceutical University (1997–2003), and an associate professor at the Nagoya Institute of Technology (2003–2008). He also acted as a visiting professor (2008, 2012) at the University of Rouen and Zhejiang Normal University (2017–2020), an academic visitor at the University of Oxford (2017) and University of Valencia (2017), a senior technical consultant at the National Engineering Technology Center of Fluoro Materials, Juhua Group Corporation (2017–2019). He is one of the Committee Members of the ACS Fluorine Division (2015–2017). He received the “Takeda Pharmaceutical Company Award in Synthetic Organic Chemistry, Japan 2000”, the “Fujifilm Award in Synthetic Organic Chemistry, Japan 2003”, the “Incentive Award in Synthetic Organic Chemistry, Japan ”, the “RSC Fluorine Prize (inaugural prize in 2005)”, the “20th Lecture Award for Young Chemists in Chemical Society of Japan”, the “Fluorine Chemistry Research Incentive Award in Research Foundation ITSUU Laboratory (inaugural prize in 2009)”, “The Pharmaceutical Society of Japan Award for Divisional Scientific Promotions (2010)”, and “Prizes for Science and Technology, The Commendation for Science and Technology by the Minister of Education, Culture, Sports, Science and Technology (2014)”, “CSJ Award for Creative Work in Chemical Society o f Japan (2015)”, and “Chinese Chemical Society, W.-Y. Huang Fluorine Prize (2015)”. He serves as the editor of Cogent Chemistry (2015–present), associate editor of Frontiers in Chemistry (2016–present), and on the editorial boards of Journal of Fluorine Chemistry (2013–present), ChemistryOpen (2012– present), ScienceOpen (2013–present), and Fluorine Notes (2014–present). His research interests are synthetic and medicinal fluorine chemistry.
Mikiko Sodeoka studied at Chiba University (Pharmaceutical Sciences) under the supervision of Prof. Toru Hino and Masako Nakagawa and received her B.Sc. (1981) andM.Sc. (1983). Then she worked with Prof. Masakatsu Shibasaki at Sagami Chemical Research Center (1983– 1986) and at Hokkaido University (1986–1990) and received her Ph.D. degree in 1889. She moved to Harvard University as a postdoctoral fellow and working with Prof. E. J. Corey and then Prof. Gregory L. Verdine (1990–1992). After working at the University of Tokyo, she started her independent career at Sagami Chemical Research Center in 1996. She became an associate professor of the University of Tokyo in 1999 and moved to Tohoku University as a full professor in 2000. Since 2006, she has been a chief scientist at RIKEN. She has also been appointed as a group director of Catalysis and Integrated Research Group at RIKEN Center for Sustainable Resource Science (2013– present). She is also acting as a visiting professor of Saitama University (2006–present), and Tokyo Medical and Dental University (2008–present). Her current research covers development of new reactions based on transition metal enolate chemistry and fluorine chemistry, design and synthesis of probe molecules based on natural products, and development of new methodologies for chemical biology research.
Vadim A. Soloshonok graduated from Kiev State University of Ukraine in 1983 and received his Ph.D. in 1987 from the Ukrainian Academy of Sciences. He continued his education in the area of asymmetric synthesis in collaboration with Prof. Y. Belokon (Moscow, USSR 1987–1990), Prof. P. Bravo (Milan, Italy, 1993), Prof. T. Hayashi (Sapporo, Japan, 1994–1995), and Prof. V. Hruby (Tucson, AZ, USA, 1998–2000). In 1987, he joined the Institute of Bioorganic Chemistry, Kiev, Ukraine, where he worked until 1995. From 1995 through 1999, he was senior researcher at the National Industrial Research Institute, Nagoya, Japan, and from 2001 to 2010 Professor of chemistry at the University of Oklahoma, USA. Currently he is the IKERBASQUE Research Professor at the University of the Basque Country, Donostia–San Sebastian, Spain. He is currently serving as a member of the international advisory editorial board of the Journal of Fluorine Chemistry (2003–present) and as Synthesis Field Editor of Amino Acids (2009–present). He serves as Past-Chair of the ACS Fluorine Division (2010), author of over 300 research papers with over 13000 citations and the h-index of 70. His major current research interests are fluorine chemistry, asymmetric synthesis, and the self-disproportionation of enantiomers.
Jaime A. S. Coelho obtained his B.Sc. and M.Sc. degrees in chemistry from Instituto Superior Tecnico, Portugal, and his Ph.D. (2014) from the Faculty of Pharmacy, University of Lisbon, Portugal, with Prof. Carlos A. M. Afonso. During his doctoral studies, he joined Prof. Nuno Maulide group as a visiting student (2013) at Max-Planck-Institut fur Kohlenforschung, Germany. He then completed postdoctoral studies with Prof. F. Dean Toste at the University of California, Berkeley, and Prof. Matthew S. Sigman at the University of Utah, United States. His research interests include asymmetric catalysis, development of new synthetic methodologies, and valorization of natural resources.
F. Dean Toste obtained his B.Sc and M.Sc. from the University of Toronto and his Ph.D. from Stanford University with Prof. Barry M. Trost. After a postdoctoral appointment with Prof. Robert H. Grubbs at the California Institute of Technology, he took a position as an Assistant Professor at the University of California, Berkeley, in 2002, where he was promoted to Associate Professor in 2007 and Professor of Chemistry in 2009. His honors include the Cope Scholar, E. J. Corey, and Creativity in Organic Synthesis Awards from the ACS, the OMCOS Award and Thieme Prize from IUPAC, the RSC Merck Award, the SSOCJ Mukaiyama Award, and the GDCh Horst-Pracejus Prize. In 2015, he was elected Fellow of the Academy of Science of the Royal Society of Canada.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Cameron AGW Abundances of the Elements in the Solar System. Space Sci. Rev 1973, 15, 121–146. [Google Scholar]
- (2).Morgan JW; Anders E Chemical Composition of Earth, Venus, and Mercury. Proc.. Natl. Acad. Sci. U. S. A 1980, 77, 6973–6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).O’Hagan D; Harper DB Fluorine-Containing Natural Products. J. Fluorine Chem 1999, 100, 127–133. [Google Scholar]
- (4).Andrews SM; Cooke JA; Johnson MS Distribution of Trace Element Pollutants in a Contaminated Ecosystem Established on Metalliferous Fluorspar Tailings. 3: Fluoride. Environ. Pollut 1989, 60, 165–179. [DOI] [PubMed] [Google Scholar]
- (5).Agarwal V; Miles ZD; Winter JM; Eustaquio AS; El Gamal AA; Moore BS Enzymatic Halogenation and Dehalogenation Reactions: Pervasive and Mechanistically Diverse. Chem. Rev 2017,117, 5619–5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Zhan C; Dixon DA Hydration of the Fluoride Anion: Structures and Absolute Hydration Free Energy from First-Principles Electronic Structure Calculations. J. Phys. Chem. A 2004, 108, 2020–2029. [Google Scholar]
- (7).Kim GH; Jang H; Yoon YJ; Jeong J; Park SY; Walker B; Jeon IY; Jo Y; Yoon H; Kim M; et al. Fluorine Functionalized Graphene Nano Platelets for Highly Stable Inverted Perovskite Solar Cells. Nano Lett 2017, 17, 6385–6390. [DOI] [PubMed] [Google Scholar]
- (8).Yun JH; Park S; Heo JH; Lee HS; Yoon S; Kang J; Im SH; Kim H; Lee W; Kim SB; et al. Enhancement of Charge Transport Properties of Small Molecule Semiconductors by Controlling Fluorine Substitution and Effects on Photovoltaic Properties of Organic Solar Cells and Perovskite Solar Cells. Chem. Sci 2016, 7, 6649–6661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Stuart AC; Tumbleston JR; Zhou H; Li W; Liu S; Ade H; You W Fluorine Substituents Reduce Charge Recombination and Drive Structure and Morphology Development in Polymer Solar Cells. J. Am. Chem. Soc 2013, 135, 1806–1815. [DOI] [PubMed] [Google Scholar]
- (10).Berger R; Resnati G; Metrangolo P; Weber E; Hulliger J Organic Fluorine Compounds: A Great Opportunity for Enhanced Materials Properties. Chem. Soc. Rev 2011, 40, 3496–3508. [DOI] [PubMed] [Google Scholar]
- (11).Treglia G; Dabbagh Kakhki VR; Giovanella L; Sadeghi R Diagnostic Performance of Fluorine-18-Fluorodeoxyglucose Positron Emission Tomography in Patients with Merkel Cell Carcinoma: A Systematic Review and Meta-Analysis. Am. J. Clin. Dermatol 2013, 14, 437–447. [DOI] [PubMed] [Google Scholar]
- (12).Brooks AF; Topczewski JJ; Ichiishi N; Sanford MS; Scott PJH Late-Stage [18F]fluorination: New Solutions to Old Problems. Chem. Sci 2014, 5, 4545–4553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Chen H; Viel S; Ziarelli F; Peng L 19F NMR: A Valuable Tool for Studying Biological Events. Chem. Soc. Rev 2013, 42, 7971–7982. [DOI] [PubMed] [Google Scholar]
- (14).Qiu X-L; Qing F-L Recent Advances in the Synthesis of Fluorinated Amino Acids. Eur.J. Org. Chem 2011, 2011, 3261–3278. [Google Scholar]
- (15).Acena JL; Sorochinsky AE; Soloshonok VA Recent Advances in the Asymmetric Synthesis of α-(Trifluoromethyl)-Containing α-Amino Acids. Synthesis 2012, 44, 1591–1602. [Google Scholar]
- (16).Kukhar VP; Sorochinsky AE; Soloshonok VA Practical Synthesis of Fluorine-Containing α- and β-Amino Acids: Recipes from Kiev, Ukraine. Future Med. Chem 2009, 1, 793–819. [DOI] [PubMed] [Google Scholar]
- (17).Mikami K; Fustero S; Sanchez-Rosello M; Acena JL; Soloshonok VA; Sorochinsky AE Synthesis of Fluorinated β-Amino Acids. Synthesis 2011, 2011, 3045–3079. [Google Scholar]
- (18).Turcheniuk KV; Kukhar VP; Roschenthaler G-V; Acena JL; Soloshonok VA; Sorochinsky AE Recent Advances in the Synthesis of Fluorinated Aminophosphonates and Aminophosphonic Acids. RSCAdv 2013, 3, 6693–6716. [Google Scholar]
- (19).Acena JL; Sorochinsky AE; Moriwaki H; Sato T; Soloshonok VA Synthesis of Fluorine-Containing α-Amino Acids in Enantiomerically Pure Form via Homologation of Ni(II) Complexes of Glycine and Alanine Schiff Bases. J. Fluorine Chem 2013, 155, 21–38. [Google Scholar]
- (20).Marsh ENG; Suzuki Y Using 19F NMR to Probe Biological Interactions of Proteins and Peptides. ACS Chem. Biol 2014, 9, 1242–1250. [DOI] [PubMed] [Google Scholar]
- (21).Marsh ENG Fluorinated Proteins: From Design and Synthesis to Structure and Stability. Acc. Chem. Res 2014, 47, 2878–2886. [DOI] [PubMed] [Google Scholar]
- (22).Tirotta I; Dichiarante V; Pigliacelli C; Cavallo G; Terraneo G; Bombelli FB; Metrangolo P; Resnati G 19F Magnetic Resonance Imaging (MRI): From Design of Materials to Clinical Applications. Chem. Rev 2015, 115, 1106–1129. [DOI] [PubMed] [Google Scholar]
- (23).Zhang W Fluorous Linker-Facilitated Chemical Synthesis. Chem. Rev 2009, 109, 749–795. [DOI] [PubMed] [Google Scholar]
- (24).Zhang W Green Chemistry Aspects of Fluorous Techniques–Opportunities and Challenges for Small-Scale Organic Synthesis. Green Chem 2009, 11, 911–920. [Google Scholar]
- (25).Zhang W; Curran DP Synthetic Applications of Fluorous Solid-Phase Extraction (F-SPE). Tetrahedron 2006, 62, 11837–11865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Studer A; Hadida S; Ferritto R; Kim S-Y; Jeger P; Wipf P; Curran DP Fluorous Synthesis: A Fluorous-Phase Strategy for Improving Separation Efficiency in Organic Synthesis. Science 1997, 275, 823–826. [DOI] [PubMed] [Google Scholar]
- (27).Fujiwara T; O’Hagan D Successful Fluorine-Containing Herbicide Agrochemicals. J. Fluorine Chem 2014, 167, 16–29. [Google Scholar]
- (28).Jeschke P The Unique Role of Fluorine in the Design of Active Ingredients for Modern Crop Protection. ChemBioChem 2004, 5, 570–589. [DOI] [PubMed] [Google Scholar]
- (29).Theodoridis G Chapter 4 Fluorine-Containing Agrochemicals: An Overview of Recent Developments. Adv. Fluorine Sci 2006, 2, 121–175. [Google Scholar]
- (30).Jeschke P The Unique Role of Halogen Substituents in the Design of Modern Agrochemicals. Pest Manage. Sci 2010, 66, 10–27. [DOI] [PubMed] [Google Scholar]
- (31).Wang J; Sanchez-Rosello M; Acena JL; del Pozo C; Sorochinsky AE; Fustero S; Soloshonok VA; Liu H Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev 2014,114, 2432–2506. [DOI] [PubMed] [Google Scholar]
- (32).Zhou Y; Wang J; Gu Z; Wang S; Zhu W; Acena JL; Soloshonok VA; Izawa K; Liu H Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II–III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev 2016, 116, 422–518. [DOI] [PubMed] [Google Scholar]
- (33).Zhu W; Wang J; Wang S; Gu Z; Acena JL; Izawa K; Liu H; Soloshonok VA Recent Advances in the Trifluoromethylation Methodology and New CF3-Containing Drugs. J. Fluorine Chem 2014, 167,37–54. [Google Scholar]
- (34).McGrath NA; Brichacek M; Njardarson JT A Graphical Journey of Innovative Organic Architectures that Have Improved Our Lives. J. Chem. Educ 2010, 87, 1348–1349. [Google Scholar]
- (35).Izawa K; Acena JL; Wang J; Soloshonok VA; Liu H Small-Molecule Therapeutics for Ebola Virus (EBOV) Disease Treatment. Eur.J. Org. Chem 2016, 2016, 8–16. [Google Scholar]
- (36).Han J; Sorochinsky AE; Ono T; Soloshonok VA Biomimetic Transamination - a Metal-Free Alternative to the Reductive Amination. Application for Generalized Preparation of Fluorine-Containing Amines and Amino Acids. Curr. Org. Synth 2011, 8, 281–294. [Google Scholar]
- (37).Ahrens T; Kohlmann J; Ahrens M; Braun T Functionalization of Fluorinated Molecules by Transition-Metal-Mediated C–F Bond Activation to Access Fluorinated Building Blocks. Chem. Rev 2015,115, 931–972. [DOI] [PubMed] [Google Scholar]
- (38).Boltalina OV; Popov AA; Kuvychko IV; Shustova NB; Strauss SH Perfluoroalkylfullerenes. Chem. Rev 2015, 115, 1051–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Campbell MG; Ritter T Modern Carbon–Fluorine Bond Forming Reactions for Aryl Fluoride Synthesis. Chem. Rev 2015, 115, 612–633. [DOI] [PubMed] [Google Scholar]
- (40).Champagne PA; Desroches J; Hamel J-D; Vandamme M; Paquin J-F Monofluorination of Organic Compounds: 10 Years of Innovation. Chem. Rev 2015, 115, 9073–9174. [DOI] [PubMed] [Google Scholar]
- (41).Cresswell AJ; Davies SG; Roberts PM; Thomson JE Beyond the Balz–Schiemann Reaction: The Utility of Tetrafluoroborates and Boron Trifluoride as Nucleophilic Fluoride Sources. Chem. Rev 2015, 115, 566–611. [DOI] [PubMed] [Google Scholar]
- (42).Fustero S; Simon-Fuentes A; Barrio P; Haufe G Chem. Rev 2015, 115, 871–930. [DOI] [PubMed] [Google Scholar]
- (43).Gillis EP; Eastman KJ; Hill MD; Donnelly DJ; Meanwell NA J. Med. Chem 2015, 58, 8315–8359. [DOI] [PubMed] [Google Scholar]
- (44).Nenaj denko VG; Muzalevskiy VM; Shastin AV Polyfluorinated Ethanes as Versatile Fluorinated C2-Building Blocks for Organic Synthesis. Chem. Rev 2015, 115, 973–1050. [DOI] [PubMed] [Google Scholar]
- (45).Ni C; Hu M; Hu J Good Partnership between Sulfur and Fluorine: Sulfur-Based Fluorination and Fluoroalkylation Reagents for Organic Synthesis. Chem. Rev 2015, 115, 765–825. [DOI] [PubMed] [Google Scholar]
- (46).Shao X; Xu C; Lu L; Shen Q Shelf-Stable Electrophilic Reagents for Trifluoromethylthiolation. Acc. Chem. Res 2015, 48, 1227–1236. [DOI] [PubMed] [Google Scholar]
- (47).Xu X-H; Matsuzaki K; Shibata N Synthetic Methods for Compounds Having CF3–S Units on Carbon by Trifluoromethylation, Trifluoromethylthiolation, Triflylation, and Related Reactions. Chem. Rev 2015, 115, 731–764. [DOI] [PubMed] [Google Scholar]
- (48).Chatterjee T; Iqbal N; You Y; Cho EJ Controlled Fluoroalkylation Reactions by Visible-Light Photoredox Catalysis. Acc. Chem. Res 2016, 49, 2284–2294. [DOI] [PubMed] [Google Scholar]
- (49).Ni C; Hu J The Unique Fluorine Effects in Organic Reactions: Recent Facts and Insights into Fluoroalkylations. Chem. Soc. Rev 2016, 45, 5441–5454. [DOI] [PubMed] [Google Scholar]
- (50).Sorochinsky AE; Soloshonok VA Asymmetric Synthesis of Fluorine-Containing Amines, Amino Alcohols, α-and β-Amino Acids Mediated by Chiral Sulfinyl Group. J. Fluorine Chem 2010, 131, 127–139. [Google Scholar]
- (51).Yerien DE; Bonesi S; Postigo A Fluorination Methods in Drug Discovery. Org. Biomol. Chem 2016, 14, 8398–8427. [DOI] [PubMed] [Google Scholar]
- (52).Shibata N Development of Shelf-Stable Reagents for Fluoro-Functionalization Reactions. Bull. Chem. Soc.Jpn 2016, 89,1307–1320. [Google Scholar]
- (53).Yerien DE; Barata-Vallejo S; Postigo A Difluoromethylation Reactions of Organic Compounds. Chem. - Eur. J 2017, 23, 14676–14701. [DOI] [PubMed] [Google Scholar]
- (54).Mei H; Xie C; Han J; Soloshonok VA N-tert-Butylsulfinyl-3,3,3-trifluoroacetaldimine: Versatile Reagent for Asymmetric Synthesis of Trifluoromethyl-Containing Amines and Amino Acids of Pharmaceutical Importance. Eur.J. Org. Chem 2016, 2016, 5917–5932. [Google Scholar]
- (55).Egami H; Sodeoka M Trifluoromethylation of Alkenes with Concomitant Introduction of Additional Functional Groups. Angew. Chem., Int. Ed 2014, 53, 8294–8303. [DOI] [PubMed] [Google Scholar]
- (56).Sha W; Zhu Y; Mei H; Han J; Soloshonok VA; Pan Y Catalytic Enantioselective Cyano-Trifluoromethylation of Styrenes. ChemistrySelect 2017, 2, 1129–1132. [Google Scholar]
- (57).Kirk KL Fluorination in Medicinal Chemistry: Methods, Strategies, and Recent Developments. Org. Process Res. Dev 2008, 12, 305–321. [Google Scholar]
- (58).Shibata N; Mizuta S; Kawai H Recent Advances in Enantioselective Trifluoromethylation Reactions. Tetrahedron: Asymmetry 2008, 19, 2633–2644. [Google Scholar]
- (59).Shibata N; Matsnev A; Cahard D Shelf-Stable Electrophilic Trifluoromethylating Reagents: A Brief Historical Perspective. Beilstein J. Org. Chem 2010, 6, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Tomashenko OA; Grushin VV Aromatic Trifluoromethylation with Metal Complexes. Chem. Rev 2011, 111, 4475–4521. [DOI] [PubMed] [Google Scholar]
- (61).Studer AA Renaissance in Radical Trifluoromethylation. Angew. Chem., Int. Ed 2012, 51, 8950–8958. [DOI] [PubMed] [Google Scholar]
- (62).Barata-Vallejo S; Lantano B; Postigo A Recent Advances in Trifluoromethylation Reactions with Electrophilic Trifluoromethylating Reagents. Chem. - Eur.J 2014, 20, 16806–16829. [DOI] [PubMed] [Google Scholar]
- (63).Chu L; Qing F-L Oxidative Trifluoromethylation and Trifluoromethylthiolation Reactions Using (Trifluoromethyl)-trimethylsilane as a Nucleophilic CF3 Source. Acc. Chem. Res 2014, 47, 1513–1522. [DOI] [PubMed] [Google Scholar]
- (64).Alonso C; Martinez de Marigorta E; Rubiales G; Palacios F Carbon Trifluoromethylation Reactions of Hydrocarbon Derivatives and Heteroarenes. Chem. Rev 2015, 115, 1847–1935. [DOI] [PubMed] [Google Scholar]
- (65).Huang Y-Y; Yang X; Chen Z; Verpoort F; Shibata N Catalytic Asymmetric Synthesis of Enantioenriched Heterocycles Bearing a C–CF3 Stereogenic Center. Chem. - Eur. J 2015, 21, 8664–8684. [DOI] [PubMed] [Google Scholar]
- (66).Liu X; Xu C; Wang M; Liu Q Trifluoromethyltrimethylsilane: Nucleophilic Trifluoromethylation and Beyond. Chem. Rev 2015, 115, 683–730. [DOI] [PubMed] [Google Scholar]
- (67).Sato A; Han J; Ono T; Wzorek A; Acena JL; Soloshonok VA Introducing a New Radical Trifluoromethylation Reagent. Chem. Commun 2015, 51, 5967–5970. [DOI] [PubMed] [Google Scholar]
- (68).France S; Weatherwax A; Lectka T Recent Developments in Catalytic, Asymmetric α-Halogenation: A New Frontier in Asymmetric Catalysis. Eur. J. Org. Chem 2005, 2005, 475–479. [Google Scholar]
- (69).Shibata N; Ishimaru T; Nakamura S; Toru T New Approaches to Enantioselective Fluorination: Cinchona Alkaloids Combinations and Chiral Ligands/Metal Complexes. J. Fluorine Chem 128, 469–483. [Google Scholar]
- (70).Brunet VA; O’Hagan D Catalytic Asymmetric Fluorination Comes of Age. Angew. Chem., Int. Ed 2008, 47, 1179–1182. [DOI] [PubMed] [Google Scholar]
- (71).Ma J-A; Cahard D Update 1 of: Asymmetric Fluorination, Trifluoromethylation, and Perfluoroalkylation Reactions. Chem. Rev 108, PR1–PR43. [DOI] [PubMed] [Google Scholar]
- (72).Lectard S; Hamashima Y; Sodeoka M Recent Advances in Catalytic Enantioselective Fluorination Reactions. Adv. Synth. Catal 2010, 352, 2708–2732. [Google Scholar]
- (73).Zhao Y; Pan Y; Sim S-BD; Tan C-H Enantioselective Organocatalytic Fluorination Using Organofluoro Nucleophiles. Org. Biomol. Chem 2012, 10, 479–485. [DOI] [PubMed] [Google Scholar]
- (74).Yang X; Wu T; Phipps RJ; Toste FD Advances in Catalytic Enantioselective Fluorination, Mono-, Di-, and Trifluoromethylation, and Trifluoromethylthiolation Reactions. Chem. Rev 2015, 115, 826–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Neel AJ; Milo A; Sigman MS; Toste FD Enantiodivergent Fluorination of Allylic Alcohols: Data Set Design Reveals Structural Interplay between Achiral Directing Group and Chiral Anion. J. Am. Chem. Soc 2016, 138, 3863–3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Fried J; Sabo EF Synthesis of 17α-Hydroxycorticosterone and Its 9α-Halo Derivatives from 11-Epi-17α-hydroxycorticosterone. J. Am. Chem. Soc 1953, 75, 2273–2274. [Google Scholar]
- (77).Fried J; Sabo EF 9α-Fluoro Derivatives of Cortisone and Hydrocortisone. J.Am. Chem. Soc 1954, 76, 1455–1456. [Google Scholar]
- (78).WHO Model List ofEssential Medicines (19th List); World Health Organization: Geneva, April 2015. [Google Scholar]
- (79).Calverley PMA; Anderson JA; Celli B; Ferguson GT; Jenkins C; Jones PW; Yates JC; Vestbo J Salmeterol and Fluticasone Propionate and Survival in Chronic Obstructive Pulmonary Disease. N. Engl. J. Med 2007, 356, 775–789. [DOI] [PubMed] [Google Scholar]
- (80).Mulki L; Foster CS Difluprednate for Inflammatory Eye Disorders. Drugs Today 2011, 47, 327–333. [DOI] [PubMed] [Google Scholar]
- (81).Jamal KN; Callanan DG The Role of Difluprednate Ophthalmic Emulsion in Clinical Practice. Clin. Ophthalmol 2009, 3, 381–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Yamaguchi M; Yasueda S; Isowaki A; Yamamoto M; Kimura M; Inada K; Ohtori A Formulation of an Ophthalmic Lipid Emulsion Containing an anti-Inflammatory Steroidal Drug, Difluprednate. Int. J. Pharm 2005, 301, 121–128. [DOI] [PubMed] [Google Scholar]
- (83).Gardi R; Vitali R; Falconi G; Ercoli A Antiinflammatory Activities of 17, 21-Methyl Ortho Esters, 17-Mono-and 17, 21-Diesters of 6a,9α-Difluorocorticosteroids. J. Med. Chem 1972, 15, 556–558. [DOI] [PubMed] [Google Scholar]
- (84).Ercoli A; Gardi R; Brianza C 17-Butyrate, 21-Ester Derivatives of 6a,9α-Difluoroprednisolone, Compositions and Use. U.S. Patent US3780177A, 1973.
- (85).Reinert RR Clinical Efficacy of Ketolides in the Treatment of Respiratory Tract infections. J. Antimicrob. Chemoth 2004, 53, 918–927. [DOI] [PubMed] [Google Scholar]
- (86).Woosley LN; Castanheira M; Jones R N. CEM-101 Activity against Gram-Positive Organisms. Antimicrob. Agents Chemother 2010, 54, 2182–2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Llano-Sotelo B; Dunkle J; Klepacki D; Zhang W; Fernandes P; Cate JHD; Mankin AS Binding and Action of CEM-101, ANew Fluoroketolide Antibiotic that Inhibits Protein Synthesis. Antimicrob. Agents Chemother 2010, 54, 4961–4970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Banks RE; Mohialdin-Khaffaf SN; Lal GS; Sharif I; Syvret RG 1-Alkyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane Salts: A Novel Family of Electrophilic Fluorinating agents. J. Chem. Soc., Chem. Commun 1992, 1992, 595–596. [Google Scholar]
- (89).Chatalova-Sazepin C; Hemelaere R; Paquin JF; Sammis GM Recent Advances in Radical Fluorination. Synthesis 2015, 47, 2554–2569. [Google Scholar]
- (90).Manral L Selectfluor (F-TEDA-BF4) C7H14B2ClF9N2. Synlett 2006, 2006, 0807–0808. 4 7 14 2 9 2 [Google Scholar]
- (91).John JP; Colby DA Synthesis of α-Halo-α,α-difluoromethyl Ketones by a Trifluoroacetate Release/Halogenation Protocol. J. Org. Chem 2011, 76, 9163–9168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Wang S; Wang Y; Wang J; Sato T; Izawa K; Soloshonok VA; Liu H The Second-Generation of Highly Potent Hepatitis C Virus (HCV) NS¾A Protease Inhibitors: Evolutionary Design Based on Tailor-Made Amino Acids, Synthesis and Major Features ofBio-activity. Curr. Pharm. Des 2017, 23, 4493–4554. [DOI] [PubMed] [Google Scholar]
- (93).Sato T; Izawa K; Acena JL; Liu H; Soloshonok VA Tailor-Made α-Amino Acids in the Pharmaceutical Industry: Synthetic Approaches to (1R,2S)-1-Amino-2-vinylcyclopropane-1-carboxylic Acid (Vinyl-ACCA). Eur. J. Org. Chem 2016, 2016, 2757–2774. [Google Scholar]
- (94).Sofia MJ; Bao D; Chang W; Du J; Nagarathnam D; Rachakonda S; Reddy PG; Ross BS; Wang P; Zhang H-R; et al. Discovery of a β-D-2′-Deoxy-2′-α-fluoro-2′-β-C-methyluridine Nucleotide Prodrug (PSI-7977) for the Treatment of Hepatitis C Virus. J. Med. Chem 2010, 53, 7202–7218. [DOI] [PubMed] [Google Scholar]
- (95).Keating GM; Vaidya A Sofosbuvir: First Global Approval. Drugs 2014, 74, 273–282. [DOI] [PubMed] [Google Scholar]
- (96).Clark JL; Hollecker L; Mason JC; Stuyver LJ; Tharnish PM; Lostia S; McBrayer TR; Schinazi RF; Watanabe KA; Otto MJ; et al. Design, Synthesis, and Antiviral Activity of 2′-Deoxy-2′-fluoro-2′-C-methylcytidine, a Potent Inhibitor of Hepatitis C Virus Replication. J. Med. Chem 2005, 48, 5504–5508. [DOI] [PubMed] [Google Scholar]
- (97).Clark JL; Mason JC; Hobbs AJ; Hollecker L; Schinazi RF Synthesis of 2-Deoxy-2-Fluoro-2-C-Methyl-D-Ribofuranoses. J. Carbohydr. Chem 2006, 25, 461–470. [Google Scholar]
- (98).Ross BS; Ganapati Reddy PG; Zhang H-R; Rachakonda S; Sofia MJ Synthesis of Diastereomerically Pure Nucleotide Phosphoramidates. J. Org. Chem 2011, 76, 8311–8319. [DOI] [PubMed] [Google Scholar]
- (99).Wang P; Chun B-K; Rachakonda S; Du J; Khan N; Shi J; Stec W; Cleary D; Ross BS; Sofia MJ An Efficient and Diastereoselective Synthesis of PSI-6130: A Clinically Efficacious Inhibitor of HCV NS5B Polymerase. J. Org. Chem 2009, 74, 6819–6824. [DOI] [PubMed] [Google Scholar]
- (100).Middleton WJ New Fluorinating Reagents. Dialkylamino-sulfur Fluorides. J. Org. Chem 1975, 40, 574–578. [Google Scholar]
- (101).Markovskij LN; Pashinnik VE; Kirsanov AV Aplication of Dialkylaminosulfur Trifluorides in the Synthesis of Fluoroorganic Compounds. Synthesis 1973, 1973, 787–789. [Google Scholar]
- (102).Ma H; Jiang WR; Robledo N; Leveque V; Ali S; Lara-Jaime T; Masjedizadeh M; Smith DB; Cammack N; Klumpp K; et al. Characterization of the Metabolic Activation of Hepatitis C Virus Nucleoside Inhibitor β-d-2′-Deoxy-2′-fluoro-2′-C-methylcytidine (PSI-6130) and Identification of a Novel Active 5′-Triphosphate Species. J. Biol. Chem 2007, 282, 29812–29820. [DOI] [PubMed] [Google Scholar]
- (103).Differding E; Lang RW New Fluorinating Reagents-I. The First Enantioselective Fluorination Reaction. Tetrahedron Lett 1988,29, 6087–6090. [Google Scholar]
- (104).Xue X-S; Wang Y; Li M; Cheng J-P Comprehensive Energetic Scale for Quantitatively Estimating the Fluorinating Potential of N–F Reagents in Electrophilic Fluorinations. J. Org. Chem 2016, 81, 4280–4289. [DOI] [PubMed] [Google Scholar]
- (105).Yang J-D; Wang Y; Xue X-S; Cheng J-P A Systematic Evaluation of the N–F Bond Strength of Electrophilic N–F Reagents: Hints for Atomic Fluorine Donating Ability. J. Org. Chem 2017, 82, 4129–4135. [DOI] [PubMed] [Google Scholar]
- (106).Shibata N; Suzuki E; Takeuchi Y A Fundamentally New Approach to Enantioselective Fluorination Based on Cinchona Alkaloid Derivatives/Selectfluor Combination. J. Am. Chem. Soc 2000, 122, 10728–10729. [Google Scholar]
- (107).Cahard D; Audouard C; Plaquevent J-C; Roques N Design, Synthesis, and Evaluation of A Novel Class of Enantioselective Electrophilic Fluorinating Agents: N-Fluoro Ammonium Salts of Cinchona Alkaloids (F-CA-BF4). Org. Lett 2000, 2, 3699–3701. [DOI] [PubMed] [Google Scholar]
- (108).Hintermann L; Togni A Catalytic Enantioselective Fluorination of β-Ketoesters. Angew. Chem., Int. Ed 2000, 39, 4359–4362. [DOI] [PubMed] [Google Scholar]
- (109).Baba D; Fuchigami T Electrolytic Partial Fluorination of Organic Compounds. Part 61: The First Example of Direct α-Fluorination of Protected α-Amino Acids. Tetrahedron Lett 2002, 43, 4805–4808. [Google Scholar]
- (110).Lu D-F; Liu G-S; Zhu C-L; Yuan B; Xu H Iron (II)-Catalyzed Intramolecular Olefin Aminofluorination. Org. Lett 2014,16, 2912–2915. [DOI] [PubMed] [Google Scholar]
- (111).Vo NT; Pace RDM; O’Har F; Gaunt MJ An Enantioselective Organocatalytic Oxidative Dearomatization Strategy. J. Am. Chem. Soc 2008, 130, 404–405. [DOI] [PubMed] [Google Scholar]
- (112).Suzuki S; Kamo T; Fukushi K; Hiramatsu T; Tokunaga E; Dohi T; Kita Y; Shibata N Iodoarene-Catalyzed Fluorination and Aminofluorination by an Ar-I/HF’ pyridine/mCPBA System. Chem. Sci 2014, 5, 2754–2760. [Google Scholar]
- (113).Mohar B; Sterk D; Ferron L; Cahard D Enantioselective and Diastereoselective Synthesis of Fluorinated Dipeptides by Late Electrophilic Fluorination. Tetrahedron Lett 2005, 46, 5029–5031. [Google Scholar]
- (114).Kodama T; Matsuda A; Shuto S Synthesis of 1′-Fluorouracil Nucleosides as Potential Antimetabolites. Tetrahedron 2006, 62, 10011–10017. [Google Scholar]
- (115).Genet J-P; Durand J-O; Roland S; Savignac M; Jung F Synthesis of 3-Fluoro Azetidinone by Electrophilic Fluorination. Tetrahedron Lett 1997, 38, 69–72. [Google Scholar]
- (116).Ihara M; Kawabuchi T; Tokunaga Y; Fukumoto K Enantiocontrolled Synthesis of Chiral Propane-1,3-diol Derivatives Possessing Fluorinated Quaternary Stereogenic Centers. Tetrahedron: Asymmetry 1994, 5, 1041–1050. [Google Scholar]
- (117).Davis FA; Zhou P; Murphy CK Asymmetric Fluorination of Enolates with N-Fluoro 2,10-(3,3-dichlorocamphorsultam). Tetrahedron Lett 1993, 34, 3971–3974. [Google Scholar]
- (118).Davis FA; Zhou P; Murphy CK; Sundarababu G; Qi H; Han W; Przeslawski RM; Chen B-C; Carroll PJ Asymmetric Fluorination of Enolates with Nonracemic N-Fluoro-2,10-Camphorsultams. J. Org. Chem 1998, 63, 2273–2280. [Google Scholar]
- (119).Takeuchi Y; Suzuki T; Satoh A; Shiragami T; Shibata N N-Fluoro-3-cyclohexyl-3-methyl-2,3-dihydrobenzo[1,2-d]isothiazole 1,1-Dioxide: An Efficient Agent for Electrophilic Asymmetric Fluorination of Enolates. J. Org. Chem 1999, 64, 5708–5711. [DOI] [PubMed] [Google Scholar]
- (120).Liu Z; Shibata N; Takeuchi Y Novel Methods for the Facile Construction of 3,3-Disubstituted and 3,3-Spiro-2H,4H-benzo[e]1,2-thiazine-1,1-diones: Synthesis of (11S,12R,14R)-2-Fluoro-14-methyl-11-(methylethyl)spiro[4H-benzo[e]-1,2-thiazine-3,2′-cyclohexane]-1,1-dione, an Agent for the Electrophilic Asymmetric Fluorination of Aryl Ketone Enolates. J. Org. Chem 2000, 65, 7583–7587. [DOI] [PubMed] [Google Scholar]
- (121).Shibata N; Liu Z; Takeuchi Y Novel Enantioselective Fluorinating Agents, (R)-and (S)-N-fluoro-3-tert-butyl-7-nitro-3,4-dihydro-2H-benzo[e][1,2]thiazine 1,1-dioxides. Chem. Pharm. Bull 2000, 48, 1954–1958. [DOI] [PubMed] [Google Scholar]
- (122).Thierry B; Audouard C; Plaquevent J-C; Cahard D Polystyrene-Bound Cinchona Alkaloids: Application to Enantioselective Electrophilic Fluorination of Silyl Enol Ethers. Synlett 2004, 2004, 0856–0860. [Google Scholar]
- (123).Greedy B; Paris J-M; Vidal T; Gouverneur V Regio- and Enantioselective Synthesis of Allylic Fluorides by Electrophilic Fluorodesilylation of Allyl Silanes. Angew. Chem., Int. Ed 2003, 42, 3291–3294. [DOI] [PubMed] [Google Scholar]
- (124).Shibata N; Suzuki E; Asahi T; Shiro M Enantioselective Fluorination Mediated by Cinchona Alkaloid Derivatives/Selectfluor Combinations: Reaction Scope and Structural Information for N-Fluorocinchona Alkaloids. J. Am. Chem. Soc 2001, 123, 7001–7009. [DOI] [PubMed] [Google Scholar]
- (125).Yamamoto T; Suzuki Y; Ito E; Tokunaga E; Shibata N Asymmetric Synthesis of Both Mirror Images of 3 -Fluorothalidomide by Enantiodivergent Fluorination Using a Single, Cinchona Alkaloid. Org. Lett 2011, 13, 470–473. [DOI] [PubMed] [Google Scholar]
- (126).Mohar B; Baudoux J; Plaquevent J-C; Cahard D Electrophilic Fluorination Mediated by Cinchona Alkaloids: Highly Enantioselective Synthesis of α-Fluoro-α-phenylglycine Derivatives. Angew. Chem., Int. Ed 2001, 40, 4214–4216. [DOI] [PubMed] [Google Scholar]
- (127).Zhu C-L; Maeno M; Zhang F-G; Shigehiro T; Kagawa T; Kawada K; Shibata N; Ma J-A; Cahard D Chiral N-Fluorodibenze-nesulfonimide Analogues for Enantioselective Electrophilic Fluorination and Oxidative Fluorination. Eur. J. Org. Chem 2013, 2013, 6501–6505. [Google Scholar]
- (128).Wolstenhulme JR; Rosenqvist J; Lozano O; Ilupeju J; Wurz N; Engle KM; Pidgeon GW; Moore PR; Sandford G; Gouverneur V Asymmetric Electrophilic Fluorocyclization with Carbon Nucleophiles. Angew. Chem., Int. Ed 2013, 52, 9796–9800. [DOI] [PubMed] [Google Scholar]
- (129).Shibata N; Ishimaru T; Suzuki E; Kirk KL Enantioselective Fluorination Mediated by N-Fluoroammonium Salts of Cinchona Alkaloids: First Enantioselective Synthesis ofBMS-204352 (MaxiPost). J. Org. Chem 2003, 68, 2494–2497. [DOI] [PubMed] [Google Scholar]
- (130).Zoute L; Audouard C; Plaquevent J-C; Cahard D Enantioselective Synthesis of BMS-204352 (MaxiPost) Using N-Fluoroammonium Salts of Cinchona Alkaloids (F–CA–BF4). Org. Biomol. Chem 2003, 1, 1833–1834. [DOI] [PubMed] [Google Scholar]
- (131).Shibata N; Ishimaru T; Nakamura M; Toru T 20-Deoxy-20-fluorocamptothecin: Design and Synthesis of Camptothecin Isostere. Synlett 2004, 2004, 2509–2512. [Google Scholar]
- (132).Fukuzumi T; Shibata N; Sugiura M; Nakamura S; Toru T Enantioselective Fluorination Mediated by Cinchona Alkaloids/Selectfluor Combinations: A Catalytic Approach. J. Fluorine Chem 2006, 127, 548–551. [Google Scholar]
- (133).Ishimaru T; Shibata N; Horikawa T; Yasuda N; Nakamura S; Toru T; Shiro M Cinchona Alkaloid Catalyzed Enantioselective Fluorination of Allyl Silanes, Silyl Enol Ethers, and Oxindoles. Angew. Chem., Int. Ed 2008, 47, 4157–4161. [DOI] [PubMed] [Google Scholar]
- (134).Yasui H; Yamamoto T; Ishimaru T; Fukuzumi T; Tokunaga E; Akikazu K; Shiro M; Shibata. N,N-Fluoro-(3,5-di-tert-butyl-4-methoxy)benzenesulfonimide (NFBSI): A Sterically Demanding Electrophilic Fluorinating Reagent for Enantioselective Fluorination. J. Fluorine Chem 2011, 132, 222–225. [Google Scholar]
- (135).Zhang Y; Yang X-J; Xie T; Chen G-L; Zhu W-H; Zhang X-Q; Yang X-Y; Wu X-Y; He X-P; He H-M Comparative Studies on the Enantioselective Fluorination of Oxindoles with Structurally Modified N-Fluorobenzenesulfonimides. Tetrahedron 2013, 69, 4933–4937. [Google Scholar]
- (136).Zhu W; Hu X; Wang F; Yang X; Wu X A Convenient Preparation of Fluorinating Reagent F-TEDA Bearing Bisphenylsulfonylimide Counterion and Its Fluorination to Oxindoles. Chin. J. Chem 2015, 33, 220–224. [Google Scholar]
- (137).Lozano O; Blessley G; Martinez del Campo T; Thompson AL; Giuffredi GT; Bettati M; Walker M; Borman R; Gouverneur V Organocatalyzed Enantioselective Fluorocyclizations. Angew. Chem., Int. Ed 2011, 50, 8105–8109. [DOI] [PubMed] [Google Scholar]
- (138).Gilli P; Pretto L; Bertolasi V; Gilli G Predicting Hydrogen-Bond Strengths from Acid–Base Molecular Properties. The pKa Slide Rule: Toward the Solution of a Long-Lasting Problem. Acc. Chem. Res 2009, 42, 33–44. [DOI] [PubMed] [Google Scholar]
- (139).Zhang H; Wang B; Cui L; Bao X; Qu J; Song Y Organocatalytic Asymmetric Fluorination of 4-Substituted Isoxazoli-nones. Eur. J. Org. Chem 2015, 2015, 2143–2147. [Google Scholar]
- (140).Bao X; Wei S; Zou L; Song Y; Qu J; Wang B Asymmetric Fluorination of 4-Substituted Pyrazolones Catalyzed by Quinine. Tetrahedron: Asymmetry 2016, 27, 436–441. [Google Scholar]
- (141).Marigo M; Fielenbach D; Braunton A; Kjβrsgaard A; J0rgensen KA Enantioselective Formation of Stereogenic Carbon–Fluorine Centers by a Simple Catalytic Method. Angew. Chem., Int. Ed 2005, 44, 3703–3706. [DOI] [PubMed] [Google Scholar]
- (142).Steiner DD; Mase N; Barbas CF Direct Asymmetric α-Fluorination of Aldehydes. Angew. Chem., Int. Ed 2005, 44,3706–3710. [DOI] [PubMed] [Google Scholar]
- (143).Shibatomi K; Yamamoto H Stereoselective Synthesis of α, α-Chlorofluoro Carbonyl Compounds Leading to the Construction of Fluorinated Chiral Quaternary Carbon Centers. Angew. Chem., Int. Ed 2008, 47, 5796–5798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (144).Shibatomi K; Okimi T; Abe Y; Narayama A; Nakamura N; Iwasa S Organocatalytic Asymmetric Fluorination of α-Chloroaldehydes Involving Kinetic Resolution. Beilstein J. Org. Chem 2014, 10, 323–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (145).Hayes MD; Rodriguez-Alvarado M; Brenner-Moyer SE An Organocascade Approach to α,α-Chlorofluoroalcohols. Tetrahedron Lett 2015, 56, 4718–4720. [Google Scholar]
- (146).Fjelbye K; Marigo M; Clausen RP; Juhl K Diastereodivergent Access to Syn and Anti 3,4-Substituted β-Fluoropyrrolidines: Enhancing or Reversing Substrate Preference. Org. Lett 2016,18, 1170–1173. [DOI] [PubMed] [Google Scholar]
- (147).Fjelbye K; Marigo M; Clausen RP; Juhl K Enantioselective Fluorination of Spirocyclic β-Prolinals Using Enamine Catalysis. Synlett 2017, 28, 425–428. [Google Scholar]
- (148).Bures J; Armstrong A; Blackmond DG Kinetic Correlation between Aldehyde/Enamine Stereoisomers in Reactions between Aldehydes with α-Stereocenters and Chiral Pyrrolidine-Based Catalysts. Chem. Sci 2012, 3, 1273–1277. [Google Scholar]
- (149).Emma MG; Lombardo M; Trombini C; Quintavalla A The Organocatalytic α-Fluorination of Chiral γ-Nitroaldehydes: the Challenge of Facing the Construction of a Quaternary Fluorinated Stereocenter. Eur. J. Org. Chem 2016, 2016, 3223–3232. [Google Scholar]
- (150).Brandes S .; Niess B; Bella M; Prieto A; Overgaard J; J0rgensen KA Non-Biaryl Atropisomers in Organocatalysis. Chem. - Eur.J 2006, 12, 6039–6052. [DOI] [PubMed] [Google Scholar]
- (151).Witten MR; Jacobsen EN A Simple Primary Amine Catalyst for Enantioselective α-Hydroxylations and α-Fluorinations of Branched Aldehydes. Org. Lett 2015, 17, 2772–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (152).Sanchez D; Bastida D; Burés J; Isart C; Pineda O; Vilarrasa J Relative Tendency of Carbonyl Compounds To Form Enamines. Org. Lett 2012, 14, 536–539. [DOI] [PubMed] [Google Scholar]
- (153).Shibatomi K; Kitahara K; Okimi T; Abe Y; Iwasa S Enantioselective Fluorination of α-Branched Aldehydes and Subsequent Conversion to α-Hydroxyacetals via Stereospecific C–F Bond Cleavage. Chem. Sci 2016, 7, 1388–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (154).Mazaleyrat J-P; Gaucher A; Wakselman M; Tchertanov L; Guilhem J A New Chiral α-Aminoacid with Only Axial Dissymmetry: Synthesis and X-Ray Analysis of a 1,1′-Binaphthyl-Substituted α-Aminoisobutyric Acid (Bin) and of Its Biphenyl Analogue (Bip). Tetrahedron Lett 1996, 37, 2971–2974. [Google Scholar]
- (155).You Y; Zhang L; Luo S Reagent-Controlled Enantioselectivity Switch for the Asymmetric Fluorination of β-Ketocarbonyls by Chiral Primary Amine Catalysis. Chem. Sci 2017, 8, 621–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (156).Zhang L; Fu N; Luo S Pushing the Limits of Aminocatalysis: Enantioselective Transformations of α-Branched β-Ketocarbonyls and Vinyl Ketones by Chiral Primary Amines. Acc. Chem. Res 2015, 48, 986–997. [DOI] [PubMed] [Google Scholar]
- (157).Shang JY; Li L; Lu Y; Yang KF; Xu LW Enantionselective Fluorination Reaction of β-ketoester-Catalyzed Chiral Primary Amine-Based Multifunctional Catalyst Systems. Synth. Commun 2014, 44, 101–114. [Google Scholar]
- (158).Brak K; Jacobsen EN Asymmetric Ion-Pairing Catalysis. Angew. Chem., Int. Ed 2013, 52, 534–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).Jew S.-s.; Park H.-g. Cinchona-Based Phase-Transfer Catalysts for Asymmetric Synthesis. Chem. Commun 2009, 7090–7103. [DOI] [PubMed]
- (160).Kim DY; Park EJ Catalytic Enantioselective Fluorination of β-Keto Esters by Phase-Transfer Catalysis Using Chiral Quaternary Ammonium Salts. Org. Lett 2002, 4, 545–547. [DOI] [PubMed] [Google Scholar]
- (161).Park EJ; Kim HR; Joung CU; Kim Y Catalytic Enantioselective Fluorination of α-Cyano Esters by Phase-Transfer Catalysis Using Chiral Quaternary Ammonium Salts. Bull. Korean Chem. Soc 2004, 25, 1451–1452. [Google Scholar]
- (162).Luo J; Wu W; Xu L-W; Meng Y; Lu Y Enantioselective Direct Fluorination and Chlorination of cyclic β-Ketoesters Mediated by Phase-Transfer Catalysts. Tetrahedron Lett 2013, 54, 2623–2626. [Google Scholar]
- (163).Poulsen TB; Bernardi L; Bell M; J0rgensen KA Organocatalytic Enantioselective Nucleophilic Vinylic Substitution. Angew. Chem., Int. Ed 2006, 45, 6551–6554. [DOI] [PubMed] [Google Scholar]
- (164).Dijkstra GDH; Kellogg RM ; Wynberg H; Svendsen JS; Marko I; Sharpless KB Conformational Study of Cinchona Alkaloids. A Combined NMR, Molecular Mechanics and x-Ray Approach. J. Am. Chem. Soc 1989, 111, 8069–8076. [Google Scholar]
- (165).Dijkstra GDH; Kellogg RM; Wynberg H Conformational Study of Cinchona Alkaloids. A Combined NMR and Molecular Orbital Approach. J. Org. Chem 1990, 55, 6121–6131. [Google Scholar]
- (166).Tanzer E-M; Schweizer WB; Ebert M-O; Gilmour R Designing Fluorinated Cinchona Alkaloids for Enantioselective Catalysis: Controlling Internal Rotation by a Fluorine-Ammonium Ion gauche Effect (ϕNCCF). Chem. - Eur. J 2012, 18, 2006–2013. [DOI] [PubMed] [Google Scholar]
- (167).Wang X; Lan Q; Shirakawa S; Maruoka K Chiral Bifunctional Phase Transfer Catalysts for Asymmetric Fluorination of β-Keto Esters. Chem. Commun 2010, 46, 321–323. [DOI] [PubMed] [Google Scholar]
- (168).Kaneko S; Kumatabara Y; Shirakawa S A New Generation of Chiral Phase-Transfer Catalysts. Org. Biomol. Chem 2016, 14, 5367–5376. [DOI] [PubMed] [Google Scholar]
- (169).Zhu C-L; Fu XY; Wei AJ; Cahard D; Ma J-A P-Spiro Phosphonium Salts Catalyzed Asymmetric Fluorination of 3-Substituted Benzofuran-2(3H)-ones. J. Fluorine Chem 2013, 150, 60–66. [Google Scholar]
- (170).Zhu C-L; Zhang F-G; Meng W; Nie J; Cahard D; Ma J-A Enantioselective Base-Free Electrophilic Amination of Benzofuran-2 (3H)-ones: Catalysis by Binol-Derived P-Spiro Quaternary Phosphonium Salts. Angew. Chem., Int. Ed 2011, 50, 5869–5872. [DOI] [PubMed] [Google Scholar]
- (171).Phipps RJ; Hamilton GL; Toste FD The Progression of Chiral Anions from Concepts to Applications in Asymmetric Catalysis. Nat. Chem 2012, 4, 603–614. [DOI] [PubMed] [Google Scholar]; (b) Mahlau M; List, B. Asymmetric Counteranion-Directed Catalysis: Concept, Definition, and Applications. Angew. Chem., Int. Ed 2013, 52, 518–533. [DOI] [PubMed] [Google Scholar]
- (172).Hamilton GL; Kanai T; Toste FD Chiral Anion-Mediated Asymmetric Ring Opening ofmeso-Aziridinium and Episulfonium Ions. J. Am. Chem. Soc 2008, 130, 14984–14986. [DOI] [PubMed] [Google Scholar]
- (173).Yang X; Phipps RJ ; Toste FD Asymmetric Fluorination of α-Branched Cyclohexanones Enabled by a Combination of Chiral Anion Phase-Transfer Catalysis and Enamine Catalysis using Protected Amino Acids. J. Am. Chem. Soc 2014, 136, 5225–5228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (174).Xu J; Hu Y; Huang D; Wang K-H; Xu C; Niu T Thiourea-Catalyzed Enantioselective Fluorination of β-Keto Esters. Adv. Synth. Catal 2012, 354, 515–526. [Google Scholar]
- (175).Novacek J; Waser M Syntheses and Applications of (Thio) Urea-Containing Chiral Quaternary Ammonium Salt Catalysts. Eur. J. Org. Chem 2014, 2014, 802–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (176).Li F; Sun L; Teng Y; Yu P; Zhao CGJ; Ma J-A Highly Diastereo- and Enantioselective Organocatalytic One-Pot Sequential 1,4-Addition/Dearomative Fluorination Transformation. Chem. - Eur. J 2012, 18, 14255–14260. [DOI] [PubMed] [Google Scholar]
- (177).Meng W-T; Zheng Y; Nie J; Xiong H-Y; Ma J-A Organocatalytic Asymmetric One-Pot Sequential Conjugate Addition/Dearomative Fluorination: Synthesis of Chiral Fluorinated Isoxazol-5(4H)-ones. J. Org. Chem 2013, 78, 559–567. [DOI] [PubMed] [Google Scholar]
- (178).Wang H-F; Cui H-F; Chai Z; Li P; Zheng C-W; Yang Y-Q; Zhao G Asymmetric Synthesis of Fluorinated Flavanone Derivatives by an Organocatalytic Tandem Intramolecular Oxa-Michael Addition/Electrophilic Fluorination Reaction by Using Bifunctional Cinchona Alkaloids. Chem. - Eur. J 2009, 15, 13299–13303. [DOI] [PubMed] [Google Scholar]
- (179).Bao X; Wang B; Cui L; Zhu G; He Y; Qu J; Song Y An Organocatalytic Asymmetric Friedel–Crafts Addition/Fluorination Sequence: Construction of Oxindole–Pyrazolone Conjugates Bearing Vicinal Tetrasubstituted Stereocenters. Org. Lett 2015,17, 5168–5171. [DOI] [PubMed] [Google Scholar]
- (180).Zhao YM; Cheung MS; Lin Z; Sun J Enantioselective Synthesis of β,γ-Unsaturated α-Fluoroesters Catalyzed by N-Heterocyclic Carbenes. Angew. Chem., Int. Ed 2012, 51, 10359–10363. [DOI] [PubMed] [Google Scholar]
- (181).Zou L; Bao X; Zhang H; Song Y; Qu J; Wang B Novel Tartrate-Based Guanidines for Enantioselective Fluorination of 1,3-Dicarbonyl and α-Cyano Carbonyl Compounds. Aust.J. Chem 2014,67, 1115–1118. [Google Scholar]
- (182).Mori K; Miyake A; Akiyama T Enantioselective Fluorination of β-Ketoesters Catalyzed by Chiral Sodium Phosphate: Remarkable Enhancement of Reactivity by Simultaneous Utilization of Metal Enolate and Metal Phosphate. Chem. Lett 2014, 43, 137–139. [Google Scholar]
- (183).Lee SY; Neufeind S; Fu GC Enantioselective Nucleophile-Catalyzed Synthesis of Tertiary Alkyl Fluorides via the α-Fluorination of Ketenes: Synthetic and Mechanistic Studies. J. Am. Chem. Soc 2014, 136, 8899–8902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (184).Wolstenhulme JR; Gouverneur V Asymmetric Fluorocyclizations of Alkenes. Acc. Chem. Res 2014, 47, 3560–3570. [DOI] [PubMed] [Google Scholar]
- (185).Zhang C; Li Z; Zhu L; Yu L; Wang Z; Li C Silver-Catalyzed Radical Phosphonofluorination of Unactivated Alkenes. J. Am. Chem. Soc 2013, 135, 14082–14085. [DOI] [PubMed] [Google Scholar]
- (186).Nie J; Zhu H-W; Cui H-F; Hua M-Q; Ma J-A Catalytic Stereoselective Synthesis of Highly Substituted Indanones via Tandem Nazarov Cyclization and Electrophilic Fluorination Trapping. Org. Lett 2007, 9, 3053–3056. [DOI] [PubMed] [Google Scholar]
- (187).Wang L; Meng W; Zhu C-L; Zheng Y; Nie J; Ma J-A The Long-Arm Effect: Influence of Axially Chiral Phosphoramidite Ligands on the Diastereo- and Enantioselectivity of the Tandem 1,4-Addition/Fluorination. Angew. Chem., Int. Ed 2011, 50, 9442–9446. [DOI] [PubMed] [Google Scholar]
- (188).Rauniyar V; Lackner AD; Hamilton GL; Toste FD Asymmetric Electrophilic Fluorination Using an Anionic Chiral Phase-Transfer catalyst. Science 2011, 334, 1681–1684. [DOI] [PubMed] [Google Scholar]
- (189).Phipps RJ; Hiramatsu K; Toste FD Asymmetric Fluorination of Enamides: Access to α-Fluoroimines Using an Anionic Chiral Phase-Transfer catalyst. J.Am. Chem. Soc 2012,134, 8376–8379. [DOI] [PubMed] [Google Scholar]
- (190).Honjo T; Phipps RJ; Rauniyar V; Toste FD A Doubly Axially Chiral Phosphoric Acid Catalyst for the Asymmetric Tandem Oxyfluorination of Enamides. Angew. Chem., Int. Ed 2012, 51, 9684–9688. [DOI] [PubMed] [Google Scholar]
- (191).Shunatona HP; Fruh N; Wang Y-M; Rauniyar V; Toste FD Enantioselective Fluoroamination: 1,4-Addition to Conjugated Dienes Using Anionic Phase-Transfer Catalysis. Angew. Chem., Int. Ed 2013, 52, 7724–7727. [DOI] [PubMed] [Google Scholar]
- (192).Wu J; Wang Y-M; Drljevic A; Rauniyar V; Phipps RJ; Toste FD A Combination of Directing Groups and Chiral Anion Phase-Transfer Catalysis for Enantioselective Fluorination of Alkenes. Proc. Nati. Acad. Sci. U. S. A 2013, 110, 13729–13733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (193).Zi W; Wang Y-M; Toste FD An In Situ Directing Group Strategy for Chiral Anion Phase-Transfer Fluorination of Allylic Alcohols. J. Am. Chem. Soc 2014, 136, 12864–12867 23h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (194).Phipps RJ ; Toste FD Chiral Anion Phase-Transfer Catalysis Applied to the Direct Enantioselective Fluorinative Dearomatization of Phenols. J.Am. Chem. Soc 2013, 135, 1268–1271. [DOI] [PubMed] [Google Scholar]
- (195).Liang X-W; Liu C; Zhang W; You S-L Asymmetric Fluorinative Dearomatization of Tryptamine Derivatives. Chem. Commun 2017, 53, 5531–5534. [DOI] [PubMed] [Google Scholar]
- (196).Hamashima Y; Yagi K; Takano H; Tamas L; Sodeoka M An Efficient Enantioselective Fluorination of Various β-Ketoesters Catalyzed by Chiral Palladium Complexes. J. Am. Chem. Soc 2002, 124, 14530–14531. [DOI] [PubMed] [Google Scholar]
- (197).Bertogg A; Hintermann L; Huber DP; Perseghini M; Sanna M; Togni A Substrate Range of the Titanium TADDOLate Catalyzed Asymmetric Fluorination ofActivated Carbonyl Compounds. Helv. Chim. Acta 2012, 95, 353–403. [Google Scholar]
- (198).Hintermann L; Perseghini M; Togni A Development of the Titanium–TADDOLate-Catalyzed Asymmetric Fluorination of Ketoesters. Beilstein J. Org. Chem 2011, 7, 1421–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (199).Toullec PY; Bonaccorsi C; Mezzetti A; Togni A Expanding the Scope of Asymmetric Electrophilic Atom-Transfer Reactions: Titanium- and Ruthenium-Catalyzed Hydroxylation of β-Ketoesters. Proc. Nati. Acad. Sci. U. S. A 2004, 101, 5810–5814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (200).Piana S; Devillers I; Togni A; Rothlisberger U The Mechanism of Catalytic Enantioselective Fluorination: Computational and Experimental Studies. Angew. Chem., Int. Ed 2002, 41, 979–982. [DOI] [PubMed] [Google Scholar]
- (201).Perseghini M; Massaccesi M; Liu Y; Togni A Structural and Stereochemical Aspects of the Enantioselective Halogenation of 1,3-Dicarbonyl Compounds Catalyzed by Ti(TADDOLato) Complexes. Tetrahedron 2006, 62, 7180–7190. [Google Scholar]
- (202).Frantz R; Hintermann L; Perseghini M; Broggini D; Togni A Titanium-Catalyzed Stereoselective Geminal Heterodihalogenation of β-Ketoesters. Org. Lett 2003, 5, 1709–1712. [DOI] [PubMed] [Google Scholar]
- (203).Cho MJ; Kang YK; Lee NR; Kim DY Catalytic Asymmetric Fluorination of α-Chloro-β-Ketoesters in the Presence of Chiral Palladium Complexes. Bull. Korean Chem. Soc 2007, 28, 2191–2192. [Google Scholar]
- (204).Hamashima Y; Suzuki T; Takano H; Shimura Y; Tsuchiya Y; Moriya K; Goto T; Sodeoka M Highly Enantioselective Fluorination Reactions of β-Ketoesters and β-Ketophosphonates Catalyzed by Chiral Palladium Complexes. Tetrahedron 2006, 62, 7168–7179. [Google Scholar]
- (205).Hamashima Y; Takano H; Hotta D; Sodeoka M Immobilization and Reuse of Pd Complexes in Ionic Liquid: Efficient Catalytic Asymmetric Fluorination and Michael Reactions with Ketoesters. Org. Lett 2003, 5, 3225–3228. [DOI] [PubMed] [Google Scholar]
- (206).Hamashima Y; Hotta D; Sodeoka M Direct Generation of Nucleophilic Palladium Enolate from 1,3-Dicarbonyl Compounds: Catalytic Enantioselective Michael Reaction with Enones. J. Am. Chem. Soc 2002, 124, 11240–11241. [DOI] [PubMed] [Google Scholar]
- (207).Hamashima Y; Hotta D; Umebayashi N; Tsuchiya Y; Suzuki T; Sodeoka M Catalytic Enantioselective Michael Reaction of 1,3-Dicarbonyl Compounds via Formation of Chiral Palladium Enolate. Adv. Synth. Catal 2005, 347, 1576–1586. [Google Scholar]
- (208).Hayamizu K; Terayama N; Hashizume D; Dodo K; Sodeoka M Unique Features of Chiral Palladium Enolates Derived from β-Ketoamide: Structure and Catalytic Asymmetric Michael and Fluorination Reactions. Tetrahedron 2015, 71, 6594–6601. [Google Scholar]
- (209).Suzuki T; Goto T; Hamashima Y; Sodeoka M Enantioselective Fluorination of Tert-Butoxycarbonyl Lactones and Lactams Catalyzed by Chiral Pd(II)-Bisphosphine Complexes. J. Org. Chem 2007, 72, 246–250. [DOI] [PubMed] [Google Scholar]
- (210).Sodeoka M; Hamashima Y Synthesis of Optically Active Heterocyclic Compounds Uusing Pd-catalyzed Asymmetric Reactions as a Key Step. Pure Appl. Chem 2008, 80, 763–776. [Google Scholar]
- (211).Kwon SJ; Kim DY Enantioselective Fluorination of Ketoamides in the Presence of Chiral Palladium Complexes. J. Fluorine Chem 2015, 180, 201–207. [Google Scholar]
- (212).Curtis NR; Davies SH; Gray M; Leach SG; McKie RA ; Vernon LE; Walkington AJ Asymmetric Fluorination Approach to the Scalable Synthesis of a SYK Inhibitor. Org. Process Res. Dev 2015,19, 865–871. [Google Scholar]
- (213).Hamashima Y; Suzuki T; Shimura Y; Shimizu T; Umebayashi N; Tamura T; Sasamoto N; Sodeoka M An Efficient Catalytic Enantioselective Fluorination of β-Ketophosphonates Using Chiral Palladium Complexes. Tetrahedron Lett 2005, 46, 1447–1450. [Google Scholar]
- (214).Kim SM; Kim HR; Kim DY Catalytic Enantioselective Fluorination and Amination of β-Keto Phosphonates Catalyzed by Chiral Palladium Complexes. Org. Lett 2005, 7, 2309–2311. [DOI] [PubMed] [Google Scholar]
- (215).Lee NR; Kim SM; Kim DY Enantioselective Fluorination of β-Keto Phosphonates and β-Ketoesters Catalyzed by Chiral Palladium Complexes. Bull. Korean Chem. Soc 2009, 30, 829–836. [Google Scholar]
- (216).Woo SB; Suh CW; Koh KO; Kim DY Enantioselective Fluorination of α-Chloro- β-Keto Phosphonates in the Presence of Chiral Palladium Complexes. Tetrahedron Lett 2013, 54, 3359–3362. [Google Scholar]
- (217).Kim SM; Kang YK; Lee KS; Mang JY; Kim DY Asymmetric Electrophilic Fluorination of β-Ketophosphonates in Ionic Liquid Media Catalyzed by Chiral Palladium Complexes. Bull. Korean Chem. Soc 2006, 27, 423–425. [Google Scholar]
- (218).Moriya K; Hamashima Y; Sodeoka M Pd(II)-Catalyzed Asymmetric Fluorination of α-Aryl-α-Cyanophosphonates with the Aid of 2,6-Lutidine. Synlett 2007, 2007, 1139–1142. [Google Scholar]
- (219).Kang YK; Cho MJ; Kim SM; Kim DY Asymmetric Electrophilic Fluorination of α-CyanoalkylphosphonatesCatalyzed by Chiral Palladium Complexes. Synlett 2007, 2007, 1135–1138. [Google Scholar]
- (220).Kim SM; Kang YK; Cho MJ; Mang JY; Kim DY Catalytic Enantioselective Fluorination Reactions of α-Cyano Acetates and α-Cyanophosphonates Using Chiral Palladium Complexes. Bull. Korean Chem. Soc 2007, 28, 2435–2441. [Google Scholar]
- (221).Kim HR; Kim DY Catalytic Enantioselective Fluorination of α-Cyano Acetates Catalyzed by Chiral Palladium Complexes. Tetrahedron Lett 2005, 46, 3115–3117. [DOI] [PubMed] [Google Scholar]
- (222).Kwon BK; Mang JY; Kim DY Catalytic Enantioselective Fluorination of α-Cyanosulfones in the Presence of Chiral Palladium Complexes. Bull. Korean Chem. Soc 2012, 33, 2481–2482. [Google Scholar]
- (223).Jacquet O; Coment ND; Blanco C; Belmonte MM; Benet-Buchholz J; van Leeuwen PWNM SPANphos Ligands in Palladium-Catalyzed Asymmetric Fluorination. Eur. J. Org. Chem 2012, 2012, 4844–4852. [Google Scholar]
- (224).Hamashima Y; Suzuki T; Takano H; Shimura Y; Sodeoka M Catalytic Enantioselective Fluorination of Oxindoles. J. Am. Chem. Soc 2005, 127, 10164–10165. [DOI] [PubMed] [Google Scholar]
- (225).Wang F; Li J; Hu Q; Yang X; Wu X-Y; He H Enantioselective Fluorination of 2-Oxindoles by Structure-Micro-Tuned N-Fluorobenzenesulfonamides. Eur. J. Org. Chem 2014, 2014, 3607–3613. [Google Scholar]
- (226).Zhang R; Wang D; Xu Q; Jiang J; Shi M Axially Chiral C2-Symmetric N-Heterocyclic Carbene (NHC) Palladium Complex-Catalyzed Asymmetric Fluorination and Amination of Oxindoles. Chin. J. Chem 2012, 30, 1295–1304. [Google Scholar]
- (227).Suzuki S; Kitamura Y; Lectard S; Hamashima Y; Sodeoka M Catalytic Asymmetric Mono-Fluorination ofα-Keto Esters: Synthesis of Optically Active β-Fluoro-α-Hydroxy and β-Fluoro-α-Amino Acid Derivatives. Angew. Chem, Int. Ed 2012, 51, 4581–4585. [DOI] [PubMed] [Google Scholar]
- (228).Katcher MH; Doyle AG Palladium-Catalyzed Asymmetric Synthesis of Allylic Fluorides. J. Am. Chem. Soc 2010, 132, 17402–17404. [DOI] [PubMed] [Google Scholar]
- (229).Katcher MH; Sha A; Doyle AG Palladium-Catalyzed Regio- and Enantioselective Fluorination of Acyclic Allylic Halides. J. Am. Chem. Soc 2011, 133, 15902–15905. [DOI] [PubMed] [Google Scholar]
- (230).Talbot EPA; Fernandes T. de A.; McKenna JM; Toste FD Asymmetric Palladium-Catalyzed Direct Intermolecular Fluorination of Styrenes. J.Am. Chem. Soc 2014, 136, 4101–4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (231).Zhu R-Y; Tanaka K; Li G-C; He J; Fu H-Y; Li S-H; Yu J-Q Ligand-Enabled Stereoselective β-C(sp3)-H Fluorination: Synthesis of Unnatural Enantiopure anti-β-Fluoro-α-amino Acids. J. Am. Chem. Soc 2015, 137, 7067–7070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (232).Shibata N; Ishimaru T; Nagai T; Kohno J; Toru T First Enantio-Flexible Fluorination Reaction Using Metal-Bis(oxazoline) Complexes. Synlett 2004, 2004, 1703–1706. [Google Scholar]
- (233).Shibata N; Kohno J; Takai K; Ishimaru T; Nakamura S; Toru T; Kanemasa S Highly Enantioselective Catalytic Fluorination and Chlorination Reactions of Carbonyl Compounds Capable of Two-Point Binding. Angew. Chem., Int. Ed 2005, 44, 4204–4207. [DOI] [PubMed] [Google Scholar]
- (234).Shibatomi K; Tsuzuki Y; Nakata S; Sumikawa Y; Iwasa S Synthesis of Axially Chiral Cyclic Amine-Substituted 2-(Oxazolinyl)-pyridine Ligands for Catalytic Asymmetric Fluorination of β-Keto Esters. Synlett 2007, 2007, 0551–0554. [Google Scholar]
- (235).Shibatomi K; Tsuzuki Y; Iwasa S Lewis Acid-catalyzed Asymmetric Fluorinations of β-Keto Esters: Dramatic Improvement in Enantioselectivities by Changing the Operation Sequence. Chem. Lett 2008, 37, 1098–1099. [Google Scholar]
- (236).Deng Q-H; Wadepohl H; Gade LH The Synthesis of a New Class of Chiral Pincer Ligands and Their Applications in Enantioselective Catalytic Fluorinations and the Nozaki-Hiyama-Kishi Reaction. Chem. - Eur.J 2011, 17, 14922–14928. [DOI] [PubMed] [Google Scholar]
- (237).Kang SH; Kim DY Catalytic Enantioselective Fluorination of α-Chloro-β-Keto Esters in the Presence of Chiral Nickel Complexes. Adv. Synth. Catal 2010, 352, 2783–2786. [Google Scholar]
- (238).Niu T; Han X; Huang D; Wang K-H; Su Y; Hu Y; Fu Y Enantioselective Fluorination of β-Ketoesters Catalysed by Complexes of New Mono-Oxazoline Ligands. J. Fluorine Chem 2015, 175, 6–11. [Google Scholar]
- (239).Jacquet O; Clement ND; Freixa Z; Ruiz A; Claver C; van Leeuwen PWNM SPANamine Derivatives in the Catalytic Asymmetric α-Fluorination of β-Keto Esters. Tetrahedron: Asymmetry 2011, 22, 1490–1498. [Google Scholar]
- (240).Suzuki T; Hamashima Y; Sodeoka M Asymmetric Fluorination of α-Aryl Acetic Acid Derivatives with the Catalytic System NiCl2–Binap/R3SiOTf/2,6-Lutidine. Angew. Chem., Int. Ed 2007, 46, 54325–5439. 3 [DOI] [PubMed] [Google Scholar]
- (241).Ishimaru T; Shibata N; Reddy DS; Horikawa T; Nakamura S; Toru T DBFOX-Ph/metal Complexes: Evaluation as Catalysts for Enantioselective Fluorination of 3-(2-Arylacetyl)-2-Thiazolidinones. Beilstein J. Org. Chem 2008, 4, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (242).Reddy DS; Shibata N; Horikawa T; Suzuki S; Nakamura S; Toru T; Shiro M A DBFOX-Ph-Based Combinatorial Catalyst for Enantioselective Fluorination of Aryl Acetyl and 3-Butenoyl Thiazolidinones. Chem. - Asian J 2009, 4, 1411–1415. [DOI] [PubMed] [Google Scholar]
- (243).Ma J-A; Cahard D Copper(II) Triflate-Bis(oxazoline)-Catalyzed Enantioselective Electrophilic Fluorination of β-Ketoesters. Tetrahedron: Asymmetry 2004, 15, 1007–1011. [Google Scholar]
- (244).Frings M; Bolm C Enantioselective Halogenation of β-Oxo Esters Catalyzed by a Chiral Sulfoximine-Copper Complex. Eur. J. Org. Chem 2009, 2009, 4085–4090. [Google Scholar]
- (245).Balaraman K; Vasanthan R; Kesavan V Enantioselective Fluorination of β-Ketoesters Using Tartrate Derived Bidentate Bioxazoline-Cu(II) Complexes. Tetrahedron: Asymmetry 2013, 24, 919–924. [Google Scholar]
- (246).Narayama A; Shibatomi K; Soga Y; Muto T; Iwasa S Copper(II)-Catalyzed Enantioselective Fluorination of β-Keto Esters Using Chiral Spiro Oxazoline Ligands. Synlett 2013, 24, 375–378. [Google Scholar]
- (247).Peng J; Du D-M Efficient Enantioselective Fluorination of Keto Esters/amides Catalysed by Diphenylamine-Linked bis-(thiazoline)–Cu(OTf)2 Complexes. RSC Adv 2014, 4, 2061–2067. [Google Scholar]
- (248).Peng J; ZHao B-L; Du D-M A combination of Metal and Organic Catalysis: Highly Diastereo- and Enantioselective Construction of Fluorinated 2-Aminocyclopenta[b]pyran Derivatives. Adv. Synth. Catal 2015, 357, 3639–3547. [Google Scholar]
- (249).Wang Y; Wang H; Jiang Y; Zhang C; Shao J; Xu D Fast, Solvent-Free and Highly Enantioselective Fluorination of β-Keto Esters Catalyzed by Chiral Copper Complexes in a Ball Mill. Green Chem 2017, 19, 1674–1677. [Google Scholar]
- (250).Zheng L-S; Wei Y-L; Jiang K-Z; Deng Y; Zheng Z-J; Xu L-W Enantioselective Fluorination of β-Ketoamides Catalyzed by Ar-BINMOL-Derived Salan-Copper Complex. Adv. Synth. Catal 2014, 356, 3769–3776. [Google Scholar]
- (251).Assalit A; Billard T; Chambert S; Langlois BR; Queneau Y; Coe D 2,2′-Bipyridine-3,3′-dicarboxylic Carbohydrate Esters and Amides. Synthesis and Preliminary Evaluation as ligands in Cu(II)-Catalyzed Enantioselective Electrophilic Fluorination. Tetrahedron: Asymmetry 2009, 20, 593–601. [Google Scholar]
- (252).Shibata N; Yasui H; Nakamura S; Toru T DNA-Mediated Enantioselective Carbon-Fluorine Bond Formation. Synlett 2007, 2007, 1153–1157. [Google Scholar]
- (253).Ibrahim H; Togni A Enantioselective Halogenation Reactions. Chem. Commun 2004, 2004, 1147–1155. [Google Scholar]
- (254).Bonaccorsi C; Althaus M; Becker C; Togni A; Mezzetti A Chiral Ru/PNNP Complexes in Catalytic and Stoichiometric Electrophilic O- and F-atom Transfer to 1,3-Dicarbonyl Compounds. Pure Appl. Chem 2006, 78, 391–396. [Google Scholar]
- (255).Althaus M; Becker C; Togni A; Mezzetti A Ruthenium-Catalyzed Electrophilic Fluorination of 1,3-Dicarbonyl Compounds. Organometallics 2007, 26, 5902–5911. [Google Scholar]
- (256).Althaus M; Togni A; Mezzetti A Asymmetric Oxidative α-Fluorination of 2-Alkylphenylacetaldehydes with AgHF2 and ruthenium/PNNP Catalysts. J. Fluorine Chem 2009, 130 (8), 702–707. [Google Scholar]
- (257).Ma J-A; Cahard D Screening of Chiral Catalysts for Enantioselective Electrophilic Fluorination of β-Ketoesters. J. Fluorine Chem 2004, 125, 1357–1361. [Google Scholar]
- (258).Bernardi L .; J0rgensen KA Enantioselective Chlorination and Fluorination of β-Keto Phosphonates Catalyzed by Chiral LEwis Acids. Chem. Commun 2005, 2005, 1324–1326. [DOI] [PubMed] [Google Scholar]
- (259).Reddy DS; Shibata N; Nagai J; Nakamura S; Toru T; Kanemasa S Desymmetrization-like Catalytic Enantioselective Fluori-nation of Malonates and Its Application to Pharmaceutically Attractive Molecules. Angew. Chem, Int. Ed 2008, 47, 164–168. [DOI] [PubMed] [Google Scholar]
- (260).Suzuki S; Furuno H; Yokoyama Y; Inanaga J Asymmetric Fluorination of β-Keto Esters Catalyzed by Chiral Rare Earth Perfluorinated Organophosphates. Tetrahedron: Asymmetry 2006, 17, 504–507. [Google Scholar]
- (261).Li J; Cai Y; Chen W; Liu X; Lin L; Feng X Highly Enantioselective Fluorination of Unprotected 3-Substituted Oxindoles: One-Step Synthesis ofBMS 204352 (MaxiPost). J. Org. Chem 2012, 77, 9155. [DOI] [PubMed] [Google Scholar]
- (262).Kawatsura M; Hayashi S; Komatsu Y; Hayase S; Itoh T Enantioselective α-Fluorination and Chlorination of β-Ketoesters by Cobalt Catalyst. Chem. Lett 2010, 39, 466–467. [Google Scholar]
- (263).Gu X; Zhang Y; Xu Z-J; Che C-M Iron(iii)–salan Complexes Catalysed Highly Enantioselective Fluorination and Hydroxylation of β-Keto Esters and N-Boc Oxindoles. Chem. Commun 2014, 50, 7870–7873. [DOI] [PubMed] [Google Scholar]
- (264).Arai S; Oku M; Ishida T; Shioiri T Asymmetric Alkylation Reaction of α-Fluorotetralone under Phase-Transfer Catalyzed Conditions. Tetrahedron Lett 1999, 40, 6785–6789. [Google Scholar]
- (265).Ding C; Maruoka K Enantioselective Alkylation of α-Fluoro-β-Keto Esters by Asymmetric Phase-Transfer Catalysis. Synlett 2009, 2009, 664–666. [Google Scholar]
- (266).Jiao Z; Beiger JJ; Jin Y; Ge S; Zhou JS; Hartwig JF Palladium-Catalyzed Enantioselective α-Arylation of α-Fluoroketones. J.Am. Chem. Soc 2016, 138, 15980–15986. [DOI] [PubMed] [Google Scholar]
- (267).Jin Y; Chen M; Ge S; Hartwig JF Palladium-Catalyzed, Enantioselective α-Arylation of α-Fluorooxindoles. Org. Lett 2017, 19, 1390–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (268).Belanger E; Cantin K; Messe O; Tremblay M; Paquin J-F Enantioselective Pd-Catalyzed Allylation Reaction of Fluorinated Silyl Enol Ethers. J.Am. Chem. Soc 2007, 129, 1034–1035. [DOI] [PubMed] [Google Scholar]
- (269).Yan L; Han Z; Zhu B; Yang C; Tan C-H; Jiang Z Asymmetric Allylic Alkylation of Morita–Baylis–Hillman Carbonates with α-Fluoro- β-Keto Esters. Beilstein J. Org. Chem 2013, 9,1853–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (270).Wang W; Shen H; Wan XL; Chen QY; Guo Y Enantioselective Pd-Catalyzed Allylation of Acyclic α-Fluorinated Ketones. J. Org. Chem 2014, 79, 6347–6353. [DOI] [PubMed] [Google Scholar]
- (271).Balaraman K; Wolf C Catalytic Enantioselective and Diastereoselective Allylic Alkylation with Fluoroenolates: Efficient Access to C3-Fluorinated and All-Carbon Quaternary Oxindoles. Angew. Chem., Int. Ed 2017, 56, 1390–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (272).Nakamura M; Hajra A; Endo K; Nakamura E Synthesis of Chiral α-Fluoroketones through Catalytic Enantioselective Decarboxylation. Angew. Chem., Int. Ed 2005, 44, 7248–7251. [DOI] [PubMed] [Google Scholar]
- (273).Bel anger E; Houze C; Guimond N; Cantin K; Paquin J-F Unexpected Effect of the Fluorine Atom on the Optimal Ligand-to-Palladium Ratio in the Enantioselective Pd-Catalyzed Allylation Reaction of Fluorinated Enol Carbonates. Chem. Commun 2008, 2008, 3251–3253. [DOI] [PubMed] [Google Scholar]
- (274).Liang Y; Fu GC Catalytic Asymmetric Synthesis of Tertiary Alkyl Fluorides: Negishi Cross-Couplings of Racemic α,α-Dihaloketones. J.Am. Chem. Soc 2014, 136, 5520–5524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (275).Xie C; Wu L; Mei H; Soloshonok VA; Han J; Pan Y Generalized Access to Fluorinated β-Keto Amino Compounds through Asymmetric Additions of α,α-Difluoroenolates to CF3-Sulfinylimine. Org. Biomol. Chem 2014, 12, 7836–7843. [DOI] [PubMed] [Google Scholar]
- (276).Xie C; Dai Y; Mei H; Han J; Soloshonok VA; Pan Y Asymmetric Synthesis of Quaternary α-Fluoro-β-keto-amines via Detrifluoroacetylative Mannich Reactions. Chem. Commun 2015, 51, 9152. [DOI] [PubMed] [Google Scholar]
- (277).Mei H; Xie C; Acena JL; Soloshonok VA; Roschenthaler G-V; Han J Recent Progress in the in situ Detrifluoroacetylative Generation of Fluoro Enolates and Their Reactions with Electrophiles. Eur.J. Org. Chem 2015, 2015, 6401–6412. [Google Scholar]
- (278).Zhang L; Zhang W; Sha W; Mei H; Han J; Soloshonok VA Detrifluoroacetylative generation and chemistry of fluorine containing tertiary enolates. J. Fluorine Chem 2017, 198, 2–9. [Google Scholar]
- (279).Xie C; Zhang L; Sha W; Soloshonok VA; Han J; Pan Y Detrifluoroacetylative in Situ Generation of Free 3-Fluoroindolin-2-one-Derived Tertiary Enolates: Design, Synthesis, and Assessment of Reactivity toward Asymmetric Mannich Reactions. Org. Lett 2016, 18, 3270–3273. [DOI] [PubMed] [Google Scholar]
- (280).Xie C; Zhang L; Mei H; Han J; Soloshonok VA; Pan Y Development and Evaluation of Different Methods for Preparation of Fluorine-Containing (R)- and (S)-N-tert-Butanesulfinyl-aldimines. ChemistrySelect 2016, 1, 4435–4439. [Google Scholar]
- (281).Mei H; Xie C; Wu L; Soloshonok VA; Han J; Pan Y Asymmetric Mannich reactions of imidazo[2,1-b]-thiazole-derived nucleophiles with (SS)-N-tertbutanesulfinyl-(3,3,3)-trifluoroacetaldimine. Org. Biomol. Chem 2013, 11, 8018–8021. [DOI] [PubMed] [Google Scholar]
- (282).Zhang W; Sha W; Zhu Y; Han JL; Soloshonok VA; Pan Y Asymmetric Synthesis of Quaternary β-Perfluorophenyl-β-amino-indolin-2-ones. Eur.J. Org. Chem 2017, 2017, 1540–1546. [Google Scholar]
- (283).Soloshonok VA; Hayashi T Gold(I)-Catalyzed Asymmetric Aldol Reaction of Fluorinated Benzaldehydes with α-Isocyanoacetamide. Tetrahedron: Asymmetry 1994, 5, 1091–1094. [Google Scholar]
- (284).Soloshonok VA; Hayashi T Gold(I)-Catalyzed Asymmetric Aldol Reaction of Methyl Isocyanoacetate with Fluorinated Benzaldehydes. Tetrahedron Lett 1994, 35, 2713–2716. [Google Scholar]
- (285).Soloshonok VA; Kacharov AD; Hayashi T Gold(I)-Catalyzed Asymmetric Aldol Reactions of Isocyanoacetic Acid Derivatives with Fluoroaryl Aldehydes. Tetrahedron 1996, 52, 245–254. [Google Scholar]
- (286).Xie C; Sha W; Zhu Y; Han JL; Soloshonok VA; Pan Y Asymmetric Synthesis of C–F Quaternary α-Fluoro- β-amino-indolin-2-ones via Mannich Addition Reactions; Facets of Rreactivity, Structural Generality and Stereochemical Outcome. RSC Adv 2017, 7, 5679–5683. [Google Scholar]
- (287).Shang H; Li Y; Li X; Ren X Diastereoselective Addition of Metal α-Fluoroenolates of Carboxylate Esters to N-tert-Butylsulfinyl Imines: Synthesis of α-Fluoro-β-amino Acids. J. Org. Chem 2015, 80, 8739–8747. [DOI] [PubMed] [Google Scholar]
- (288).Li X; Li Y; Shang H A Diastereoselective Mannich-Type Reaction of α-Fluorinated Carboxylate Esters: Synthesis of β-Amino Acids Containing α-Quaternary Fluorinated Carbon Centers. Org. Biomol. Chem 2016, 14, 6457–6462. [DOI] [PubMed] [Google Scholar]
- (289).Li Y; Li X; Shang H; Chen X; Ren X Diastereoselective Mannich Reactions Using Fluorinated Ketones: Synthesis of Stereogenic Carbon-Fluorine Units. J. Org. Chem 2016, 81, 9858–9866. [DOI] [PubMed] [Google Scholar]
- (290).Han X; Kwiatkowski J; Xue F; Huang KW; Lu Y Asymmetric Mannich Reaction of Fluorinated Ketoesters with a Tryptophan-Derived Bifunctional Thiourea Catalyst. Angew. Chem., Int. Ed 2009, 48, 7604–7607. [DOI] [PubMed] [Google Scholar]
- (291).Kang YK; Kim DY Catalytic Asymmetric Mannich-Type Reactions of Fluorinated Ketoesters with N-Boc Aldimines in the Presence of Chiral Palladium Complexes. Tetrahedron Lett 2011, 52, 2356–2358. [Google Scholar]
- (292).Cosimi E; Engl OD; Saadi J; Ebert MO; Wennemers H Stereoselective Organocatalyzed Synthesis of α-Fluorinated β-Amino Thioesters and Their Application in Peptide Synthesis. Angew. Chem., Int. Ed 2016, 55, 13127–13131. [DOI] [PubMed] [Google Scholar]
- (293).Pan Y; Zhao Y; Ma T; Yang Y; Liu H; Jiang Z; Tan CH Enantioselective Synthesis of α-Fluorinated β-Amino Acid Derivatives by an Asymmetric Mannich Reaction and Selective Deacylation/Decarboxylation Reactions. Chem. - Eur.J 2010, 16, 779–782. [DOI] [PubMed] [Google Scholar]
- (294).Zhao Y; Pan Y; Liu H; Yang Y; Jiang Z; Tan CH Fluorinated Aromatic Ketones as Nucleophiles in the Asymmetric Organocatalytic Formation of C–C and C–N Bonds: A Facile Route to the Construction of Fluorinated Quaternary Stereogenic Centers. Chem. - Eur. J 2011, 17, 3571–3574. [DOI] [PubMed] [Google Scholar]
- (295).Trost BM; Saget T; Lerchen A; Hung CI Catalytic Asymmetric Mannich Reactions with Fluorinated Aromatic Ketones: Efficient Access to Chiral β-Fluoroamines. Angew. Chem., Int. Ed 2016, 55, 781–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (296).Vaithiyanathan V; Kim MJ; Liu y.; Yan HL; Song CE Direct Access to Chiral β-Fluoroamines with Quaternary Stereogenic Center through Cooperative Cation-Binding Catalysis. Chem. - Eur. J 2017, 23, 1268–1272. [DOI] [PubMed] [Google Scholar]
- (297).Liu X; Zhang J; Zhao L; Ma S; Yang D; Yan W; Wang R Construction of Vicinal Tetrasubstituted Stereocenters with a C–F Bond through a Catalytic Enantioselective Detrifluoroacetylative Mannich Reaction. J. Org. Chem 2015, 80, 12651–12658. [DOI] [PubMed] [Google Scholar]
- (298).Han C; Kim EH; Colby DA Cleavage of Carbon-Carbon Bonds through the Mild Release of Trifluoroacetate: Generation of α, α-Difluoroenolates for Aldol Reactions. J. Am. Chem. Soc 2011, 133, 5802–5805. [DOI] [PubMed] [Google Scholar]
- (299).Zhang P; Wolf C Synthesis of Pentafluorinated β-Hydroxy Ketones. J. Org. Chem 2012, 77, 8840–8844. [DOI] [PubMed] [Google Scholar]
- (300).Zhang P; Wolf C Catalytic Enantioselective Difluoroalkylation of Aldehydes. Angew. Chem., Int. Ed 2013, 52, 7869–7873. [DOI] [PubMed] [Google Scholar]
- (301).Qian J; Yi W; Huang X; Jasinski JP; Zhang W 2,2-Difluoro-1,3-diketones as gem-Difluoroenolate Precusors for Asymmetric Aldol Addition with N-Benzylisatins. Adv. Synth. Catal 2016, 358, 2811–2816. [Google Scholar]
- (302).Saidalimu I; Fang X; He XP; Liang J; Yang X; Wu F Highly Enantioselective Construction of 3-Hydroxy Oxindoles through a Decarboxylative Aldol Addition ofTrifluoromethyl α-Fluorinated gem-Diols to N-Benzyl Isatins. Angew. Chem., Int. Ed 2013, 52, 5566–5570. [DOI] [PubMed] [Google Scholar]
- (303).Xie C; Wu L; Han J; Soloshonok VA; Pan Y Assembly of Fluorinated Quaternary Stereogenic Centers through Catalytic Enantioselective Detrifluoroacetylative Aldol Reactions. Angew. Chem., Int. Ed 2015, 54, 6019–6023. [DOI] [PubMed] [Google Scholar]
- (304).Soloshonok VA; Roussel C; Kitagawa O; Sorochinsky AE Self-Disproportionation of Enantiomers via Achiral Chromatography: a Warning and Extra Dimension in Optical Purifications. Chem. Soc. Rev 41,4180–4188. [DOI] [PubMed] [Google Scholar]
- (305).Soloshonok VA; Berbasov DO Self-Disproportionation of Enantiomers on Achiral Phase Chromatography. One More Example of Fluorine’s Magic Powers. Chim. Oggi/Chemistry Today 2006,24,44–47. [Google Scholar]
- (306).Ueki H; Yasumoto M; Soloshonok VA Rational application of Self-Disproportionation of Enantiomers via Sublimation–a Novel Methodological Dimension for Enantiomeric purifications. Tetrahedron: Asymmetry 2010, 21, 1396–1400. [Google Scholar]
- (307).Han J; Nelson DJ; Sorochinsky AE; Soloshonok VA Self-Disproportionation of Enantiomers via Sublimation; New and Truly Green Dimension in Optical Purification. Curr. Org. Synth 2011, 8, 310–317. [Google Scholar]
- (308).Sorochinsky AE; Katagiri T; Ono T; Wzorek A; Acena JL; Soloshonok VA Optical Purifications via Self-Disproportionation of Enantiomers by Achiral Chromatography; Case Study of a Series of α-CF3-Containing Secondary Alcohols. Chirality 2013, 25, 365–368. [DOI] [PubMed] [Google Scholar]
- (309).Sorochinsky AE; Acena JL; Soloshonok VA Self-Disproportionation of Enantiomers of Chiral, Non-Racemic Fluoroorganic Compounds: Role of Fluorine as Enabling Element. Synthesis 45, 141–152. [Google Scholar]
- (310).Yasumoto M; Ueki H; Ono T; Katagiri T; Soloshonok VA Self-Disproportionation of Enantiomers via Sublimation: Isopropyl 3,3,3-(Trifluoro)-Lactate. J. Fluorine Chem 2010, 131, 535–539. [Google Scholar]
- (311).Yasumoto M; Ueki H; Soloshonok VA Self-Disproportionation of Enantiomers of Trifluoro Lactic Acid Amides via Sublimation. J. Fluorine Chem 2010, 131, 266–269. [Google Scholar]
- (312).Zhang L; Xie C; Dai Y; Mei H; Han J; Soloshonok VA; Pan Y Catalytic Asymmetric Detrifluoroacetylative Dldol Reactions of Aliphatic Aldehydes for Construction of C–F Quaternary Stereogenic Centers. J. Fluorine Chem 2016, 184, 28–35. [Google Scholar]
- (313).Zhang L; Zhang W; Mei H; Han J; Soloshonok VA; Pan Y Catalytic Asymmetric Aldol Addition Reactions of 3-Fluoroindolinone Derived Enolates. Org. Biomol. Chem 2017, 15, 311–315. [DOI] [PubMed] [Google Scholar]
- (314).Soloshonok VA; Berbasov DO Self-Disproportionation of Enantiomers of (R)-Ethyl 3-(3,5-Dinitrobenzamido)-4,4,4-trifluorobutanoate on Achiral Silica Gel Stationary Phase. J. Fluorine Chem 2006, 127, 597–603. [Google Scholar]
- (315).Sha W; Zhang L; Wu X; Mei H; Han J; Soloshonok VA; Pan Y Detrifluoroacetylative Cascade Reactions of Bicyclic Fluoro-Enolates with ortho-Phthalaldehyde: Aspects of Reactivity, Diastereo- and Enantioselectivity. J. Fluorine Chem 2017, 196, 14–23. [Google Scholar]
- (316).Sha W; Zhang L; Zhang W; Mei H; Soloshonok VA; Han J; Pan Y Catalytic Cascade Aldol–Cyclization of Tertiary Ketone Enolates for Enantioselective Synthesis of Keto-Esters with a C–F Quaternary Stereogenic Center. Org. Biomol. Chem 2016, 14, 7295–7303. [DOI] [PubMed] [Google Scholar]
- (317).Liu YL; Liao FM; Niu YF; Zhao XL; Zhou J Highly Stereoselective Construction of Adjacent Tetrasubstituted Carbon Stereogenic Centres via an Organocatalytic Mukaiyama-Aldol Reaction of Monofluorinated Silyl Enol Ethers to Isatins. Org. Chem. Front 2014, 1, 742–747. [Google Scholar]
- (318).Saadi J; Wennemers H Enantioselective Aldol Reactions with Masked Fluoroacetates. Nat. Chem 2016, 8, 276–280. [DOI] [PubMed] [Google Scholar]
- (319).Han X; Luo J; Liu C; Lu Y Asymmetric Generation of Fluorine-Containing Quaternary Carbons Adjacent to Tertiary Stereocenters: Uses of Fluorinated Methines as Nucleophiles. Chem. Commun 2009, 2009, 2044–2046. [DOI] [PubMed] [Google Scholar]
- (320).Li H; Zhang S; Yu C; Song X; Wang W Organocatalytic Asymmetric Synthesis of Chiral Fluorinated Quaternary Ccarbon Containing β-Ketoesters. Chem. Commun 2009, 2009, 2136–2138. [DOI] [PubMed] [Google Scholar]
- (321).Oh Y; Kim SM; Kim DY Organocatalytic Synthesis of Quaternary Stereocenter Bearing a Fluorine Atom: Enantioselective Conjugate Addition of α-Fluoro-β-ketoesters to Nitroalkenes. Tetrahedron Lett 2009, 50, 4674–4676. [Google Scholar]
- (322).Lu Y; Zou G; Zhao G Primary-Secondary Diamines Catalyzed Michael Reaction to Generate Chiral Fluorinated Quaternary Carbon Centers. Tetrahedron 2015, 71, 4137–4144. [Google Scholar]
- (323).Cui HF; Li P; Wang XW; Zhu SZ; Zhao G Asymmetric Michael Addition of α-Fluoro-α-phenylsulfonyl Ketones to Nitroolefins Catalyzed by Phenylalanine-Based Bifunctional Thioureas. J. Fluorine Chem 2012, 133, 120–126. [Google Scholar]
- (324).Kukhar’ VP; Soloshonok VA; Solodenko VA Asymmetric Synthesis of Phosphorus Analogs of Amino Acids. Phosphorus, Sulfur Silicon Relat. Elem 1994, 92, 239–264. [Google Scholar]
- (325).Roschenthaler G-V; Kukhar VP; Kulik IB; Belik MY; Sorochinsky AE; Rusanov EB; Soloshonok VA Asymmetric Synthesis of Phosphonotrifluoroalanine and Its Derivatives using N-tert- Butanesulfinyl Imine Derived from Fluoral. Tetrahedron Lett 2012, 53, 539–542. [Google Scholar]
- (326).Kukhar’ VP; Svistunova NY; Solodenko VA; Soloshonok VA Asymmetric Synthesis of Fluorine and Phosphorus-Conaining Amino Acids. Russ. Chem. Rev 1993, 62, 261–278. [Google Scholar]
- (327).Turcheniuk KV; Poliashko KO; Kukhar VP; Rozhenko AB; Soloshonok VA; Sorochinsky AE Efficient asymmetric synthesis of trifluoromethylated β-aminophosphonates and their incorporation into dipeptides. Chem. Commun 2012, 48, 11519–11521. [DOI] [PubMed] [Google Scholar]
- (328).Sung HJ; Mang JY; Kim DY Catalytic Asymmetric Conjugate Addition of α-Fluoro β-Ketophosphonates to Nitroalkenes in the Presence of Nickel Complexes. J. Fluorine Chem 2015, 178, 40–46. [Google Scholar]
- (329).Kwiatkowski J; Lu Y Highly Enantioselective Michael Addition of 2-Fluoro-1,3-diketones to Nitroalkenes. Eur. J. Org. Chem 2015, 2015, 320–324. [Google Scholar]
- (330).Kwiatkowski J; Lu Y Asymmetric Michael Addition of α-Fluoro- α-nitroalkanes to Nitroolefins: Facile Preparation of Fluorinated Amines and Tetrahydropyrimidines. Chem. Commun 2014, 50, 9313–9316. [DOI] [PubMed] [Google Scholar]
- (331).Cosimi E; Saadi J; Wennemers H Stereoselective Synthesis of α-Fluoro-γ-nitro Thioesters under Organocatalytic Conditions. Org. Lett 2016, 18, 6014–6017. [DOI] [PubMed] [Google Scholar]
- (332).Jiang Z; Pan Y; Zhao Y; Ma T; Lee R; Yang Y; Huang KW; Wong MW; Tan CH Synthesis of a Chiral Quaternary Carbon Center Bearing a Fluorine Atom: Enantio- and Diastereoselective Guanidine-Catalyzed Addition of Fluorocarbon Nucleophiles. Angew. Chem., Int. Ed 2009, 48, 3627–3631. [DOI] [PubMed] [Google Scholar]
- (333).Han X; Zhong F; Lu Y Highly Enantioselective Amination Reactions of Fluorinated Keto Esters Catalyzed by Novel Chiral Guanidines Derived from Cinchona Alkaloids. Adv. Synth. Catal 2010, 352, 2778–2782. [Google Scholar]
- (334).Drege E; Guillaume A; Boukhedimi N; Marrot J; Troufflard C; Tran Huu-Dau M-E; Joseph D; Delarue-Cochin S A Facile and Stereocontrolled Synthesis of γ-Substituted γ-Fluoroglutamates by Conjugate Addition: Conflicting Effect between Fluorinated Enami-noester and Hinderered Michael Acceptor. J. Org. Chem 2010, 75, 7596–7604. [DOI] [PubMed] [Google Scholar]
- (335).Yu JS; Liao FM; Gao WM; Liao K; Zuo RL; Zhou J Michael Addition Catalyzed by Chiral Secondary Amine Phosphor-amide Using Fluorinated Silyl Enol Ethers: Formation of Quaternary Carbon Stereocenters. Angew. Chem., Int. Ed 2015, 54, 7381–7385. [DOI] [PubMed] [Google Scholar]
- (336).Wang T; Hoon DL; Lu X Enantioselective Synthesis of 3-Fluoro-3-allyloxindoles via Phosphine-Catalyzed Asymmetric γ-Addition of 3-Fluoro-oxindoles to 2,3-Butadienoates. Chem. Commun 2015, 51, 10186–10189. [DOI] [PubMed] [Google Scholar]
- (337).Cao D; Fang G; Zhang J; Wang H; Zheng C; Zhao G Enantioselective Michael Addition of Malonates to Chalcone Derivatives Catalyzed by Dipeptide-derived Multifunctional Phosphonium Salts. J. Org. Chem 2016, 81, 9973–9982. [DOI] [PubMed] [Google Scholar]
- (338).Dou X; lu Y Enantioselective Conjugate Addition of 3-Fluorooxindoles to Vinyl Sulfone: an Organocatalytic Access to Chiral Fluoro-3-substituted Oxindoles. Org. Biomol. Chem 2013, 11, 5217–5221. [DOI] [PubMed] [Google Scholar]
- (339).Zhu Y; Zhang W; Mei H; Han JL; Soloshonok VA; Pan Y Catalytic Enantioselective Michael Addition Reactions of Tertiary Enolates Generated by Detrifluoroacetylation. Chem. - Eur. J 2017, 23, 11221–11225. [DOI] [PubMed] [Google Scholar]
- (340).Soloshonok VA; Cai C; Hruby VJ Asymmetric Michael Addition Reactions of Chiral Ni(II) Complex of Glycine with N-(Enoyl)oxazolidinones: Improved Reactivity and Stereochemical Outcome. Tetrahedron: Asymmetry 1999, 10, 4265–4269. [Google Scholar]
- (341).Soloshonok VA; Cai C; Hruby VJ (S)- or (R)-N-(E-enoyl)-phenyl-1,3-oxazolidin-2-ones: Ideal Michael Acceptors to Afford a Virtually Complete Control of Simple and Face Diastereoselectivity in Addition Reactions with Glycine Derivatives. Org. Lett 2000, 2, 747–750. [DOI] [PubMed] [Google Scholar]
- (342).Cai C; Soloshonok VA; Hruby VJ Michael Addition Reactions Between Chiral Ni(II) Complex of Glycine and 3-(trans-enoyl)oxazolidin-2-ones. A Case of Electron Donor-Acceptor Attractive Interactions-Controlled Face Diastereoselectivity. J. Org. Chem 2001, 66, 1339–1350. [DOI] [PubMed] [Google Scholar]
- (343).Soloshonok VA; Klika KD Terminology Related to the Phenomenon ‘Self-Disproportionation of Enantiomers’ (SDE). Helv. Chim. Acta 2014, 97, 1583–1589. [Google Scholar]
- (344).Beebe X; Yang F; Bui MH; Mitten MJ; Ma ZK; Nilius AM; Djuric SW Synthesis and Antibacterial Activity of 6-O-Arylpropargyl-9-oxime-11,12-carbamate Ketolides. Bioorg. Med. Chem. Lett 2004, 14, 2417–2421. [DOI] [PubMed] [Google Scholar]
- (345).Yong H; Gu YG; Clark RF; Marron T; Ma Z; Soni N; Stone GG; Nilius AM; Marsh K; Djuric SW Design, Synthesis and Structure-Activity Relationships of 6-O-Arylpropargyl Diazalides with Potent Activity Against Multidrug-Resistant Streptococcus Pneumoniae. Bioorg. Med. Chem. Lett 2005, 15, 2653–2658. [DOI] [PubMed] [Google Scholar]
- (346).Sugimoto T; Shimazaki Y; Manaka A; Tanikawa T; Suzuki K; Nanaumi K; Kaneda Y; Yamasaki Y; Sugiyama H Synthesis and Antibacterial Activity of 6-O-(Heteroaryl-isoxazolyl)propynyl 2-Fluoro Ketolides. Bioorg. Med. Chem. Lett 2012, 22, 5739–5743. [DOI] [PubMed] [Google Scholar]
- (347).Tian JC; Han X; Lv W; Li YX; Wang H; Fan BZ; Cushman M; Liang JH Design, Synthesis and Structure-Bactericidal Activity Relationships of Novel 9-Oxime Ketolides and Reductive Epimers of a Acylides. Bioorg. Med. Chem. Lett 2017, 27, 1513–1524. [DOI] [PubMed] [Google Scholar]
- (348).Di Francesco ME; Avolio S; Pompei M; Pesci S; Monteagudo E; Pucci V; Giuliano C; Fiore F; Rowley M; Summa V Synthesis and Antiviral Properties of Novel 7-Heterocyclic Substituted 7-Deaza-adenine Nucleoside Inhibitors of Hepatitis C NS5B Polymerase. Bioorg. Med. Chem 2012, 20, 4801–4811. [DOI] [PubMed] [Google Scholar]
- (349).Lin C; Sun CH; Liu X; Zhou YQ; Hussain M; Wan JT; Li MK; Li X; Jin RL; Tu ZC; et al. Design, Synthesis, and in vitro Biological Evaluation of Novel 6-Methyl-7-substituted-7-deaza Purine Nucleoside Analogs as Anti-influenza A Agents. Antiviral Res 2016,129, 13–20. [DOI] [PubMed] [Google Scholar]
- (350).Tarui A; Kondo S; Sato K; Omote M; Minami H; Miwa Y; Ando A Ni-Catalyzed α-Arylation of Secondary α-Bromo- α-fluoro-β-lactam: Cross-Coupling of a Secondary Fluorine-Containing Electrophile. Tetrahedron 2013, 69, 1559–1565. [Google Scholar]
- (351).Tarui A; Nishimura H; Ikebata T; Tahira A; Sato K; Omote M; Minami H; Miwa Y; Ando A Ligand-Promoted Asymmetric Imino-Reformatsky Reaction of Ethyl Dibromofluoroacetate. Org. Lett 2014, 16, 2080–2083. [DOI] [PubMed] [Google Scholar]
- (352).Tarui A; Miyata E; Tanaka A; Sato K; Omote M; Ando A Stereoselective Suzuki Coupling Reaction of an α-Bromo- α fluoro-β-lactam. Synlett 2015, 26, 55–58. [Google Scholar]
- (353).Tarui A; Kawashima N; Kawakita T; Sato K; Omote M; Ando A Direct and Diastereoselective Alkylation and Aldol Reactions of α-Bromo-α-fluoro-β-lactams. J. Org. Chem 2013, 78, 7903–7911. [DOI] [PubMed] [Google Scholar]
- (354).Luckhurst CA; Breccia P; Stott AJ; Aziz O; Birch HL; Burli RW; Hughes SJ; Jarvis RE; Lamers M; Leonard PM; et al. Potent, Selective, and CNS-Penetrant Tetrasubstituted Cyclopropane Class IIa Histone Deacetylase (HDAC) Inhibitors. ACS Med. Chem. Lett 2016, 7, 34–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (355).Magnus NA; Aikins JA; Cronin JS; Diseroad WD; Hargis AD; LeTourneau ME; Parker BE; Reutzel-Edens SM; Schafer JP; Staszak MA; et al. Diastereomeric Salt Resolution Based Synthesis of LY503430, an AMPA (α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic Acid) Potentiator. Org. Process Res. Dev 2005, 9, 621–628. [Google Scholar]
- (356).Walker MC; Chang MCY Natural and Engineered Biosynthesis of Fluorinated Natural Products. Chem. Soc. Rev 2014, 43, 6527–6536. [DOI] [PubMed] [Google Scholar]
- (357).Bernstein S; Brown JJ; Feldman LI; Rigler NE 16-Hydroxylated Steroids. X.1a The Synthesis of 21-Deoxytriamcinolone. J. Am. Chem. Soc 1959, 81, 4956–4962. [Google Scholar]
- (358).Kitazume T; Yamamoto T A Synthetic Approach to F-Analogues of Angiotensin Converting Enzyme Inhibitors. J. Fluorine Chem 1987, 35, 467–476. [Google Scholar]
- (359).Kamata S; Haga N; Mitsugi T; Kondo E; Nagata W; Nakamura M; Miyata K; Odaguchi K; Shimizu T; Kawabata T; Suzuki T; Ishibashi M; Yamada F Aldosterone Antagonists. 1. Synthesis and Biological Activities of 11β,18-Epoxypregnane Derivatives. J. Med. Chem 1985, 28, 428–433. [DOI] [PubMed] [Google Scholar]
- (360).Goj O; Burchardt A; Haufe G A Versatile Approach to Optically Active Primary 2-Fluoro-2-phenylalkanols through Lipase-Catalyzed Transformations. Tetrahedron: Asymmetry 1997, 8, 399–408. [Google Scholar]
- (361).Yokoyama H; Hyodo R; Nakada A; Yamaguchi S; Hirai Y; Kometani T; Goto M; Shibata N; Takeuchi Y Asymmetric Synthesis of Chiral 2-Fluorinated 1,3-Propanediols and Its Application to the Preparation ofMonofluorinated Chiral Synthon. Tetrahedron Lett 1998, 39, 7741–7744. [Google Scholar]
- (362).Guanti G; Narisano E; Riva R Lipase Mediated Preparation of Differently Protected Homochiral 2-Aryl-2-fluoro-1,3-propanediols. Tetrahedron: Asymmetry 1998, 9, 1859–1862. [Google Scholar]
- (363).Narisano E; Riva R Chemoenzymatic Synthesis of 2-Substituted 2-Fluoro-1,3-dioxygenated Chiral Building Blocks. Tetrahedron: Asymmetry 1999, 10, 1223–1242. [Google Scholar]
- (364).Meyer OGJ; Frohlich R; Haufe G Asymmetric Cyclopropanation of Vinyl Fluorides: Access to Enantiopure Monofluorinated Cyclopropane Carboxylates. Synthesis 2000, 2000, 1479–1490. [Google Scholar]
- (365).Rosen TC; Haufe G Synthesis of Enantiopure Monofluorinated Phenylcyclopropanes by Lipase-Catalyzed Kinetic Resolution. Tetrahedron: Asymmetry 2002, 13, 1397–1405. [Google Scholar]