

Abstract
Myeloid cells assume a wide range of phenotypes, some of which are protective against injury and infection while others promote cardiovascular disease. This heterogeneity is partially caused by switching of cell sources from local tissue resident macrophage proliferation to recruitment of circulating cells, and partially due to macrophages’ phenotypic plasticity. While long-lived tissue resident macrophages support development, tissue homeostasis and cardiac conduction, monocyte-derived cells may promote destruction of the arterial wall and the myocardium, leading to organ ischemia and heart failure. Influencing myeloid cell flux and phenotype shifts emerges as a therapeutic opportunity in many disease areas, including atherosclerosis, acute myocardial infarction, heart failure and stroke. However, it is currently unclear which cell subsets and drug targets are the most efficient and safest options. Here I review the neutrophil and macrophage supply chain and the cells’ emerging heterogeneity in the setting of atherosclerosis and ischemic heart disease.
Keywords: macrophage, monocyte, neutrophil, atherosclerosis, myocardial infarction
Introduction
Innate immune cells comprise neutrophils, monocytes, macrophages, mast cells, eosinophils and natural killer cells. Their key functions in the steady state are critical to host survival, as they defend against viral and bacterial infection by phagocytosis and rapid killing of bacteria and infected cells upon encounter. Many of these cells, with the exception of specific subsets that are protective, aggravate cardiovascular disease by damaging the integrity of the vascular wall, the heart and the brain. In this review, I focus on myeloid cells, the most frequent cells found in healthy and diseased cardiovascular organs, and their roles in atherosclerosis and organ ischemia, the most prevalent cardiovascular pathologies.
Myeloid cell functions in the steady state
Neutrophils are abundant in the blood stream (20–30% of white blood cells in healthy mice1 and ~40–60% in humans). They are rarely found in the healthy arterial wall and the myocardium. The same is true for monocytes which are, compared to neutrophils, somewhat longer lived and less frequent (2% of blood leukocytes in healthy mice1 and humans). For both myeloid cell types, subsets have been described that diverge in their functions. Human neutrophils can be subdivided according to their expression of CD177, and it is being discussed that the cells may assume different polarization states2,3 according to their environment and disease, a concept that is already well accepted for monocytes and macrophages.
Mouse monocytes are typically described as Ly6Chigh (classical, or inflammatory) and Ly6Clow, the latter converting from Ly6Chigh cells in the circulation. This process depends on the transcription factor Nur774. Human monocytes are frequently separated into three subsets that differ in their expression of CD16 and CD14. Whole genome comparison of mouse and human cells indicates broad conservation of subsets between both species and that Ly6Chigh mouse monocytes are most similar to the human CD16− subset5. Single cell RNA-sequencing studies suggest that perhaps there are even more monocyte subsets6; however, whether newly emerging subsets are functionally distinct remains to be determined. As is the case with neutrophils, monocytes are very sparse in healthy cardiovascular tissues. Both types of cells keep the blood free of pathogens, and patrol the vascular endothelium. Patrolling Ly6Clow monocytes scavenge endothelial cells that become apoptotic7. In addition to the cells in circulation, there are reservoirs of myeloid cells in the spleen8, the marginal pool and the bone marrow. These reservoirs can quickly supply additional cells if acute inflammation arises.
Tissue resident macrophages are present in all major organs, including the heart9,10, brain11 and arteries12. These cells arise mostly independent of circulating monocytes in the steady state, although monocyte recruitment increases slightly during normal aging of the heart13. Recent clinical data obtained in transplanted patients confirm that the human heart, similar to the mouse, does not rely on monocyte recruitment much14. Before birth, arterial macrophages derive from the mouse fetal liver12. Shortly after birth, for a brief period of time, the mouse aorta recruits monocytes that differentiate to macrophages and than again gain independence from recruitment12. Mouse cardiac monocytes derive from the fetal liver, take up residence in the organ a week before birth10, participate in the development of the heart’s vasculature15 and continue to proliferate locally10,16. The exception is the CCR2+ macrophage subset, a small macrophage pool that derives from blood monocytes10,14.
Typical steady state functions of resident macrophages include removal of apoptotic stromal cells and surveillance of the host tissue by constant sampling of their environment. Macrophages are well equipped for these tasks as they express an armamentarium of receptors that sense danger17. Some of the cells are positioned closely to the vascular lumen and are thus likely to encounter any pathogens that invade the stroma from the blood pool. Comparative gene expression studies of different tissue resident macrophages indicate that the cells are quite different depending on the organ they inhabit18–20. Transplant experiments indicate that tissue resident macrophages can rapidly adapt to a change in their environment18. These adaptions extent to macrophages’ reactions to plastic surfaces in the dish19, creating a clear limitation for in vitro experimentation with these cells. Tissue resident macrophages likely serve a dual role as cells that inhibit tissue inflammation if there is no danger, but rapidly alert the immune system if they encounter pathogens, signs of ischemic or any other stress. The cells are likely also involved in organizing the heart’s and arteries’ matrix scaffold by interacting with fibroblasts13 that produce collagen and by providing proteases21,22 that degrade extracellular matrix.
In a number of organs, macrophages were described to also pursue functions that are generally considered outside the mainstream leukocyte occupation. These organ specific functions may be a testament to the cells’ ability to adapt their phenotype to the microenvironment they live in. For instance, brain macrophages, i.e. microglia, regulate synaptic function by pruning during development23. In the hematopoietic system, CD169+ macrophages are part of the hematopoietic niche, and anchor blood progenitor cells via the adhesion molecule VCAM-124,25. Such a function may echo the proposed roles of macrophages in regeneration, where they are thought to instruct stromal progenitors during healing26.
Recently, an organ-specific function was reported for cardiac macrophages27. The cells are more abundant in the conduction system when compared to the average in the left ventricle. Imaging of large heart tissue volumes in mice and humans revealed a heterogeneous density of macrophages, which can be found in higher numbers in the sinus and atrioventricular node27. Their density also increased in the vicinity of collagen and valves. Given the previously described involvement of microglia in neuronal information transfer, macrophages’ participation in cardiac conduction may not come as a surprise; however, our initial studies were triggered by a serendipitous finding. Using a mouse in which tissue resident macrophages can be ablated due to CD11b driven expression of the human diphtheria receptor, we planned to assay mice after macrophage depletion for abnormal heart function with cardiac magnetic resonance imaging. Typically, this diagnostic readout relies on the electrocardiogram to time data acquisition to the appropriate phase of the cardiac cycle. This imaging experiment turned out not to be possible because mice developed irregular heart rhythms after macrophage depletion, more specifically atrioventricular block, i.e. a prolonged time between atrial depolarization (the p wave in the ECG) and ventricular depolarization (the QRS complex). High grade heart block developed, indicated by completely independent atrial and ventricular activity27. Based on these observations, we initiated microscopic imaging studies of the normal mouse and human conduction system, which relied on optical clearing of hearts and fluorescence microcopy of tissue slabs containing the sinus node or the atrioventricular node. This imaging, in conjunction with gene expression studies, documented connexin-43+ connections of macrophages and conducting cells. Viewed together with patch clamp data obtained in macrophages cocultured with cardiomyocytes27, these data imply gap junction exchange of electrical charges between both cell types throughout the cardiac cycle. Deterioration of conduction after depletion of macrophages or deletion of connexin-43 from macrophages indicates that macrophages support steady state conduction27. At this point, it is less clear whether a changed macrophage number or altered phenotype of macrophages contribute to conduction abnormalities. First small studies associate macrophage numbers with atrial fibrillation in humans28,29, and macrophage depletion experiments30 suggest that the cells may be causally involved. Clearly, more work is needed in this emerging area of immuno-electrophysiology, given that there are many unknowns but pressing clinical needs, especially for atrial and ventricular fibrillation.
Myeloid cells in atherosclerosis and the ischemic heart
Activated by regional atherogenic lipid deposits, changes in sheer stress, circulating danger signals, higher sympathetic innervation and oxidative stress, arterial endothelial cells recruit circulating neutrophils and monocytes into atherosclerotic plaque. The involved cell adhesion molecules and chemokines are now well understood and described in detail in excellent reviews31–33. In progressing lesions, nearly all myeloid cells are recently recruited34, although some macrophage-like foam cells are thought to differentiate from smooth muscle cells that had previously inhabited the arteries’ media35. Neutrophils likewise enter the arterial wall, especially in the setting of plaque erosion36. In more mature mouse aortic lesions, recruited macrophages also proliferate in the plaque34, further increasing the local myeloid population, which undergoes profound phenotypic changes. The time course and dynamics of plaques in humans, leave alone plaque macrophages, is still poorly understood, partially because we lack non-invasive tools to follow individual vascular territories over time with sufficient spatial resolution and molecular sensitivity. Nevertheless, it is likely that some atherosclerotic lesions regress or perhaps even resolve completely. Understanding the mechanisms that determine whether a plaque regresses or progresses are of obvious therapeutic importance and therefore an important and vibrant research topic37. One intriguing hypothesis states that readjusting macrophage phenotypes, away from inflammatory actions that destroy the arterial wall integrity towards resolution cell phenotypes that lead to removal of pro-inflammatory lipid from the tissue, may promote shrinking or stabilization of atherosclerotic plaques. Observation of macrophage phenotypic changes in mouse models of atherosclerosis resolution supports that this may indeed be the case38. It will be instructive to answer the question whether those observed macrophage resolution phenotypes are merely a product of the changing microenvironment, or if an altered macrophage phenotype drives the resolution of inflammation in the arterial wall. The stimuli that propel macrophages towards disease-exacerbating actions includes cytokines, growth hormones, toll like receptor ligands, neutrophils, lymphocytes, oxidized LDL, ultimately promoting amplified inflammation, proteases destructing matrix, local cell death, swelling of the plaque, deterioration of the arterial wall integrity and thrombotic occlusion of the arterial lumen.
As a result, downstream brain and myocardium becomes ischemic, triggering death of neurons and cardiomyocytes. It also triggers a sharp activation of resident and recruited innate immune cells. Among other danger signals, this activation is triggered by DNA released from dying stromal cells which is processed by recruited macrophages39. The phenotypes of cells in the acute infarct change over time40. Initially highly inflammatory, macrophages transition to supporting repair and rebuilding the tissue destroyed by ischemia. If this phenotype transition does not occur, tissue recovery may be hindered, leading to left ventricular dilation41 or even rupture. Thus, studying factors that trigger phenotype transitions, which include intrinsic forces such a as transcription factors42 and post-phagocytotic adaptions43, is currently an area of intense research. Extrinsic stimuli, including interaction with other immune cells (T regulatory lymphocytes)44,45 are likely involved, as macrophages are highly sensitive to their microenvironment. Modulating this transition may be a therapeutic avenue for patients with large, non-reperfused infarcts which are not common in the era of rapid reperfusion therapy but these patients do exist and are at very high risk for developing heart failure. Developing treatment strategies for this patient group is a pressing clinical need. One avenue, supported by the clinical association of blood leukocyte numbers with mortality and remodeling46,47, may be curbing inflammatory cell recruitment, perhaps by blocking the action of the chemokine receptor CCR248.
Myeloid cell flux in cardiovascular disease
If inflammatory disease befalls the host, and cardiovascular disease is increasingly recognized to have a strong inflammatory component, the innate immune system activation profoundly alters myeloid cell phenotypes and dynamics. In homeostasis, there is little cell recruitment to the arterial wall and to the myocardium (Table 1). Both locations are inhabited by resident macrophages that promote immune quiescence in the absence of danger and non-self signals. These cells continue to exist in diseased tissues; however, they may change their phenotypes and are joined by cells recruited from the blood stream. The more acute the disease, the more pronounced are the alterations in cell number, life span and flux. For instance, while a steady state cardiac macrophage has a life span of 6–8 weeks16, monocyte derived macrophages in the infarct only live 20 hours before they die locally49. Plaque macrophages have somewhat longer life spans34, perhaps because the inflammation in the arterial wall is less acute when compared to ischemia which suddenly kills millions of heart and brain cells. The first week after onset of ischemia is a particularly turbulent time during which the number of myeloid cells rises an order of magnitude. Even the remote myocardium, which is not directly affected by reduced oxygen supply because it is perfused by a different coronary vessel, recruits myeloid cells from circulation in the first weeks after MI50. This recruitment continues to increase during left ventricular remodeling, while proliferation of cardiac macrophages also accelerates51. While shorter cell life spans certainly accelerate myeloid cell flux through tissues, exit of cells may also contribute to this phenomenon. Cell exit is notoriously difficult to quantify. In contrast to recruitment, where we can study presence of tagged cells in target organs after adoptive cell transfer, parabiosis or switching of fluorescent protein expression in genetic models, it is harder to follow cells that leave a plaque or inflamed organ because we have to work with much fewer cells to start with. It is wrong to assume that a reduction of cells in the tissue of interest means that cells have exited, because they may have died locally, which is a common fate of myeloid cells, or cell recruitment may have declined. We attempted to quantify myeloid cell exit in mouse myocardial infarction, because here cell numbers are still reasonably high (~106 per infarct) and the mouse heart can be transplanted with relative ease. Importantly, an isogeneic transplanted heart is well perfused through the coronaries and the ischemia time with skilled surgeons is below 15 minutes, reducing experimental confounders. Using two different labeling strategies, (i) radioactively labeled nanoparticles with high avidity to macrophages and (ii) FACS staining for divergent CD45 leukocyte surface markers in donors and recipients, we calculated that only 13% or 5% of myeloid cells exited the infarct, respectively49. The radio-isotope labeling method is particularly quantitative. After indium-111 labeled nanoparticle injection of the organ donor which underwent prior coronary ligation, we measured the gamma counts in the transplanted heart and in the recipient mouse. The radioactivity of the infarcted heart transplant was measured before implantation and 24 hrs later. The decline in heart gamma counts reported the exit of nanoparticles, and by extension macrophages, from the infarcted heart. The limitations of this experiment include that the transplantation cuts lymphatic drainage, and that the assumption that nanoparticles mirror cell migration with high fidelity is likely not precise. Nevertheless, both experiments were in good agreement on the magnitude of cell exit, which was relatively minor. We concluded that the majority of myeloid cells die locally in the infarct. If this is the case, why do we see so few apoptotic macrophages in histology (e.g. 2% of infarct macrophages on day 3)49? This is likely due to the fact that TUNEL staining and similar approaches only deliver a snapshot image that captures a very short time span. Death of a cell, or the phase during which typical death markers are available for staining, may be rather short. In vitro time lapse imaging reported that a macrophage displays annexin V for only 11 minutes before the cell death is complete49, at which time point the cell is removed by the next generation of infarct macrophages. A myeloid cell flux model that estimates numbers and rates for subacute mouse MI is provided in Fig. 1.
Table 1.
Origins of macrophages in steady state and diseased tissue.
| Organ | Macrophage source |
|---|---|
| Steady state myocardium | Mostly local proliferation, very little recruitment10,14,16 |
| Acute myocardial infarct | Exclusively recruitment from blood monocytes made in marrow and spleen8,16,49,63,75 |
| Chronically failing myocardium | Recruitment and local proliferation13 |
| Steady state arterial wall | Local proliferation, cells derive from a pool of monocytes that were recruited shortly after birth12 |
| Developing atherosclerotic plaque | Recruitment from blood, cells are produced in bone marrow and spleen34 |
| Mature atherosclerotic plaque | Local proliferation of cells that were recently recruited from blood34 |
| Steady state brain | Microglia depend exclusively on local proliferation; no recruitment11 |
| Acute stroke | Recruitment of neutrophils and monocytes; local proliferation and activation of microglia also contributes67 |
Figure 1: Model of monocyte flux on day 3–5 after intermediate sized infarct.
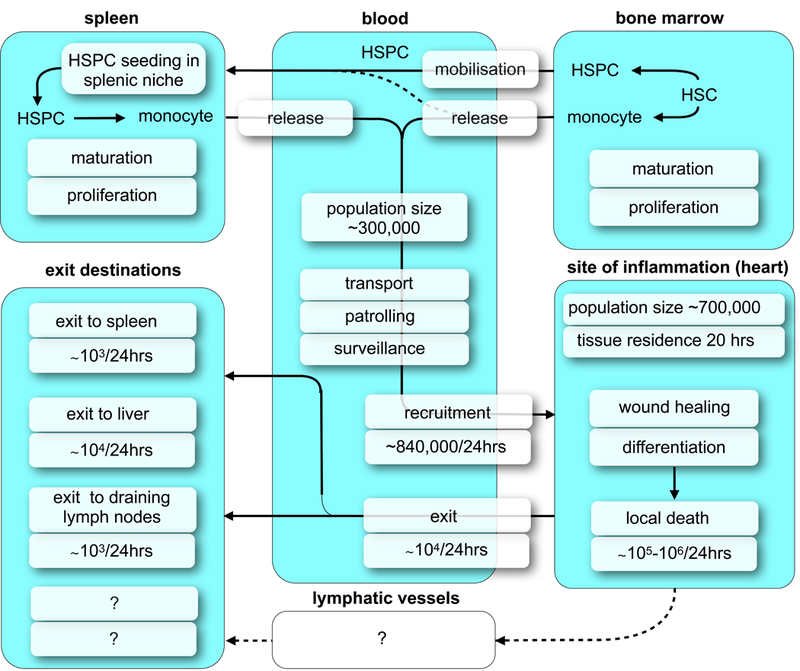
Adapted from49.
Another method for measuring cell exit from tissues is direct observation by intravital microscopy. While technically challenging due to motion and light penetration issues, recent concepts are solving these problems52,53. Because cell exit may not be a frequent event, it remains challenging to image long enough to observe such an event in the fairly small field of view of microscopic imaging systems, in particular when considering the surgically exposed heart or atherosclerotic plaque. On this note, a clever experiment recently examined the exit of dendritic cells from the mouse aorta by investigating the eluate of the isolated vessel with flow cytometry54. While this in vitro wash out setup has limitations such as severance of lymphatic connections, the method is able to enumerate relatively rare events. By FACS, the overall macrophage number in an excised aorta, which depends on disease stage, is below 10,000, several orders of magnitude lower than the number of macrophages in the acute MI. Since FACS is a technology that can reliably quantify rare cells, it may therefore be particularly helpful. The question of the cells’ destination after tissue exit, however, will have to be addressed in vivo. This, in addition to quantifying the contribution of cell departure to an overall reduction of cells in plaque, is an unresolved question in atherosclerosis. In steady state, aged neutrophils return to the bone marrow where they influence the hematopoietic niche55. Such returning cells may transfer information from a site of inflammation to hematopoietic and lymphatic organs. Examining these types of myeloid cell related feed back loops in cardiovascular disease is undoubtedly enticing but a challenging task.
Altered production of immune cells in cardiovascular disease
In the steady state, circulating myeloid cells are produced in the bone marrow. Hematopoietic progenitor cells with myeloid bias diverge from the lymphoid lineage and give rise to neutrophils and monocytes. These fate decisions are governed by specific transcription and growth factors, which are reviewed elsewhere56. There is also a growing appreciation of heterogeneity in hematopoietic niches (see excellent reviews on the topic57–59). These niches, which are formed by a stromal cell repertoire, may have a bias to host quiescent versus proliferating HSPC60, and myeloid versus lymphoid cell lineages. Cell production and departure are tightly regulated, including by circadian rhythms61, and many of the key factors have been well defined. For instance, the chemokine MCP-1 regulates monocyte62 and HSPC departure63 form the marrow, while SDF-1 signaling retains neutrophils64 and HSPC65 in the niche. SDF-1, also known as CXCL12, acts as a major quiescence-inducing signal in the marrow57. A number of cardiovascular risk factors (reviewed in more detail here66) increase hematopoiesis. This includes classical risk factors such as hyperlipidemia and psychosocial stress. In addition, cardiovascular events such as stroke67 and myocardial infarction68 profoundly influence blood stem cell behavior. In mice with middle cerebral artery occlusion67 or coronary ligation68, increased sympathetic nervous signaling in the marrow decreases the production of SDF-1, which leads to enhanced HSPC proliferation and leukocyte departure from the marrow. The most upstream point of activation, i.e. the least differentiated cells with increased proliferative rates, are CCR2+ SLAM HSC63. Heart failure, either with reduced51 or preserved ejection13 fraction, likewise increases sympathetic nervous signaling in the marrow. Interestingly, myocardial infarction also triggers blood cell production in the spleen. Increased traffic of CCR2+ HSPC to the spleen, where they are retained by VCAM-1 expressing CD169+ macrophages, initiates the outsourced myeloid production which continues in mice with heart failure25. In mice, the contribution of splenic monocytes to the infarct population is substantial, and the spleen changes its volume and cell count dynamically. Initially, a reservoir is mobilized which leads to smaller spleens in the first days after coronary ligation8. When splenic myelopoiesis peaks, the organ’s size is above the steady state49. In ApoE−/− mice with atherosclerosis, spleen size is even more increased. These changes in organ volume are likely not just due to differences in monocyte numbers and myelopoiesis; rather, other immune cell fluctuations contribute. While it is well known that the human spleen enlarges in endocarditis and disorders of the hematopoietic system, it is currently unknown if the spleen changes its size in humans with atherosclerosis or myocardial infarction. However, clinical PET studies documented that there are significant increases in splenic glucose uptake after MI69.
While it is quite clear that there is heterogeneity in hematopoietic tissues on a microscopic level (e.g. quiescent versus non-quiescent microenvironments, adjacent to arterioles or sinusoids, respectively), whether or not the marrow, which expands through many bones in our bodies, differs depending on location is not known. The current concept entails that the entire marrow reacts homogeneously to systemic stimuli. We challenged this concept by examining the hypothesis that the skull marrow reacts differently to brain inflammation, due to its proximity, than tibial marrow. Indeed, neutrophils labeled with a membrane dye in different locations showed a divergent propensity to travel to the inflamed CNS70. Neutrophils labeled in skull marrow cavities were more likely to enter ischemic brain tissue. The quiescence promoting hematopoietic factor SDF-1 decreased more steeply in skull marrow after stroke, and this led to a sharper decline of neutrophils in the skull when compared to the tibia. When looking for regional vascular connections between the skull marrow and the meninges, which cover the surface of the brain, we found that the inner skull cortex is traversed by numerous microscopic channels (Fig. 2) that are clad with endothelial cells. These channels contain leukocytes. In mice, the channel diameter measures about 22µm, and in the human skull 5-fold larger. Such channels also exist in the tibia of mice. Neutrophil traffic through channels increased after stroke or induction of aseptic meningitis. In steady state, blood flow was directed away from the brain, with leukocytes traveling towards the bone marrow cavity. However, in inflammatory settings, neutrophils crawled from the marrow cavity towards the meninges. The precise connection of these channels to the systemic circulation is currently incompletely resolved, i.e. we still have to determine whether they originate from arteries or veins. Their morphology is not suggestive of lymphatic vessels, which are also generally presumed to be absent in the bone marrow. Perhaps the blood flow differs from channel to channel or depends on the pressure inside the skull. Thus, while the channels’ presence is intriguing and suggestive of direct marrow — CNS communication, the relative importance of these vascular connections needs to be investigated next. Hypothetically, the channels may serve as a leukocyte migration route that, if CNS inflammation is present, bypasses the systemic blood pool. Likewise, soluble factors such as growth factors, interleukins or danger signals originating from injured brain tissue may reach skull marrow via these channels, which connect the meninges with hematopoietic tissue. Our observations of differences in leukocyte release depending on bone location raise the possibility that the hematopoietic marrow does not always act homogeneously. Spatial proximity to an inflammatory site may introduce a bias, or certain marrow areas may pursue specific, yet to be determined functions.
Figure 2: Channels connecting the skull marrow with the CNS surface.
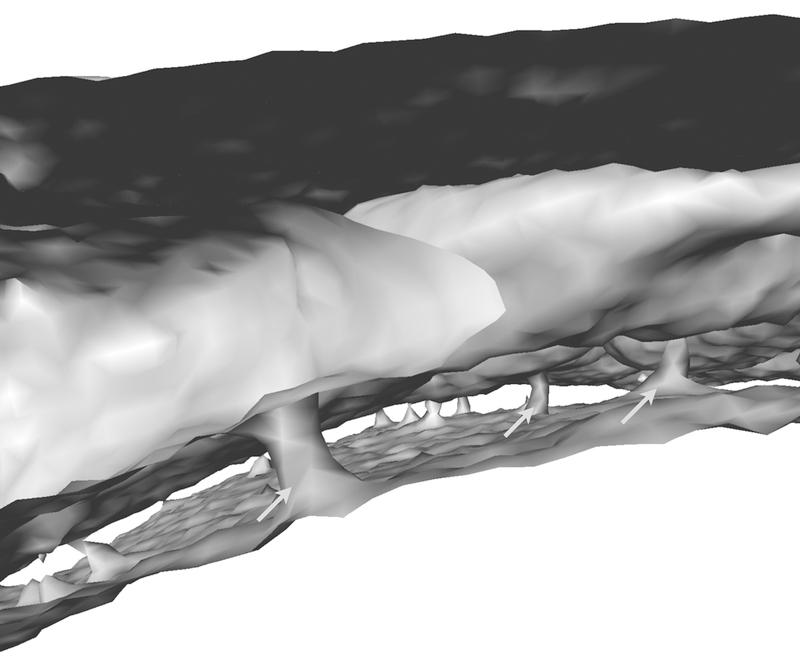
Micro X-ray computed tomography reconstruction of the mouse skull shows ~20µm diameter channels which are frequented by neutrophils70.
System interactions
The heart and its immune cells are in constant exchange with systemic information traveling in the autonomic nervous system and the systemic circulation, which delivers cells, danger signals and nutrients (Fig. 3). Thus, the heart reacts and is subject to any systemic challenge that meets the host. Cardiac immune cells, especially during disease, are recruited from the circulation after expanding in lymphatic and hematopoietic organs. Such recruited macrophages derive from monocytes made in the bone marrow and spleen. This supply of circulating cells to the heart can be viewed as information traveling with cells, and is influenced by factors that enhance hematopoiesis, among them cardiovascular risk factors such as hyperlipidemia, diabetes, and lifestyle factors66. Psychosocial stress71 and myocardial infarction itself68 enhances bone marrow activity via the sympathetic nervous system. Sympathetic innervation in the heart can also influence endothelial cells’ expression of adhesion molecules and thus influence cell recruitment72. Sympathetic signaling may directly act on leukocytes, which express receptors for neurotransmitters (www.Immgen.org20). How increased sympathetic tone influences the immune cells residing in the heart is currently unknown. Further, it is tempting to speculate that arterial and cardiac macrophages store information epigenetically, i.e. that prior contact with metabolic or inflammatory stress elicits ‘trained immunity’ effects and alters the cells’ phenotypes in steady state and recall situations. This type of ‘trained immunity’ affects monocytes and their progenitors, enhancing atherosclerosis73 (Fig. 4). Similar immune cell training may influence the myocardial leukocyte repertoire in the steady state and after injury. Thus, how the organ deals with biological stress, either chronic such as hypertension or acute such as myocardial infarction or cardiac surgery, may depend on co-morbidities that influence the immune system.
Figure 3: Systems interaction.
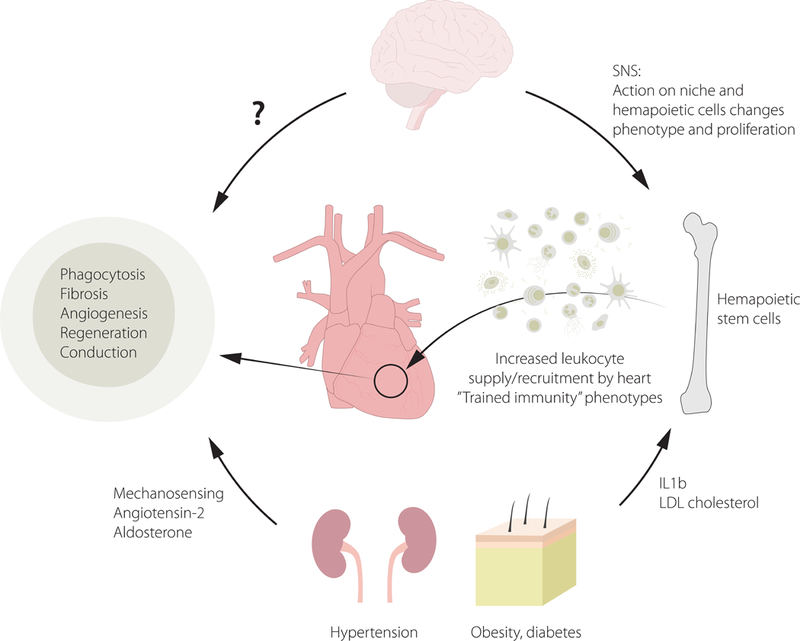
This cartoon shows inflammatory comorbidities and risk factors that act on the heart’s immune system and cause pathologies.
Figure 4: Trained immunity in cardiovascular disease.
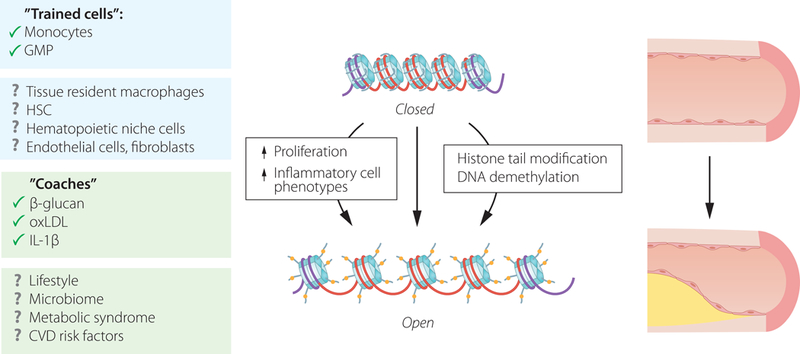
Trained immunity describes an imprinted memory in innate immune cells that leads to an enhanced inflammatory recall response. The “training” phenomenon is linked to epigenetic changes triggered by exposure to β-glucan. First described for monocytes82, the concept has been expanded to granulocyte-macrophage progenitors (GMP)73,83 and other stimuli, including IL-1β and oxidized low density lipoprotein (oxLDL)84.
Enabling technologies
Several recent technological developments provide considerable advances in the cellular resolution and breadth of data acquisition for studying immune cells in the heart. These technological advances allow us to analyze immune cells in the steady-state and diseased myocardium: i) Flow cytometry analysis and cell sorting of non-cardiomyocytes after tissue disruption with proteases was first introduced for the aorta by Galkina and Ley74 and later adapted for the mouse and human heart. Cardiac single cell solutions are stained like blood75, allowing for precise quantification and characterization of leukocytes, endothelial cells, and fibroblasts according to their surface markers76. ii) Bulk and single cell RNA sequencing6,27,39,77,78 of the above cell preparations obtained from mice and humans enable unbiased discovery of disease-relevant cells, their subsets, and their protein expression. iii) Intravital microscopy of the mouse heart53,79 and the arteries52 follows dynamic immune cell (inter)actions with other cells in vivo. iv) Tissue clearing and subsequent ex vivo microscopy provides three-dimensional tissue structures of large myocardial volumes with cellular resolution27. This is now possible on all relevant tissues including the brain, heart, vasculature and hematopoietic organs. v) In vivo molecular and cellular imaging of murine and human arteries and heart is carried out with immune cell targeted imaging agents such as nanoparticles, small molecules and antibodies (reviewed in reference80).
Conclusions
Our understanding of myeloid cell contributions to pathology in atherosclerosis and organ ischemia is rapidly evolving, and propelled by two primary forces. First, new fundamental data on myeloid cell fate, behavior, sources and phenotypes in the steady state are currently emerging, and second, methodological advances now provide precise cellular resolution, a large step forward from measuring inflammatory activity in the entire plaque or myocardium. In addition, emerging clinical data motivate this work. Increasingly, associations between clinical outcomes and myeloid cell actions become available. A first large clinical trial demonstrated convincingly that interfering with inflammation reduces cardiovascular events in patients80. Hopefully, these developments will result in new therapeutics that help to deal with the increasing number of patients that suffer from cardiovascular disease.
Acknowledgements
This work was funded in part by federal funds from the National Institutes of Health NS084863, HL139598, HL128264, HL117829, HL096576 and HL131495; the European Union’s Horizon 2020 research and innovation program under grant agreement No 667837; the Global Research Lab (GRL) program (NRF-2015K1A1A2028228) of the National Research Foundation by the Korean government and the MGH Research Scholar Program.
Footnotes
The author declares no competing financial interests.
References
- 1.O’Connell KE et al. Practical murine hematopathology: a comparative review and implications for research. Comp Med 65, 96–113 (2015). [PMC free article] [PubMed] [Google Scholar]
- 2.Cuartero MI et al. N2 neutrophils, novel players in brain inflammation after stroke: modulation by the PPARgamma agonist rosiglitazone. Stroke 44, 3498–3508 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Ma Y et al. Temporal neutrophil polarization following myocardial infarction. Cardiovasc Res 110, 51–61 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanna RN et al. The transcription factor NR4A1 (Nur77) controls bone marrow differentiation and the survival of Ly6C-monocytes. Nat Immunol 12, 778–785 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ingersoll MA et al. Comparison of gene expression profiles between human and mouse monocyte subsets. Blood 115, e10–9 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Villani AC et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 356, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carlin LM et al. Nr4a1-dependent Ly6C(low) monocytes monitor endothelial cells and orchestrate their disposal. Cell 153, 362–375 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Swirski FK et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science 325, 612–616 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pinto AR et al. An abundant tissue macrophage population in the adult murine heart with a distinct alternatively-activated macrophage profile. PLoS One 7, e36814 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Epelman S et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 40, 91–104 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ginhoux F et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841–845 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ensan S et al. Self-renewing resident arterial macrophages arise from embryonic CX3CR1(+) precursors and circulating monocytes immediately after birth. Nat Immunol 17, 159–168 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Hulsmans M et al. Cardiac macrophages promote diastolic dysfunction. J Exp Med 215, 423–440 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bajpai G et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat Med (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leid J et al. Primitive Embryonic Macrophages are Required for Coronary Development and Maturation. Circ Res 118, 1498–1511 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heidt T et al. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ Res 115, 284–295 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Medzhitov R Toll-like receptors and innate immunity. Nat Rev Immunol 1, 135–145 (2001). [DOI] [PubMed] [Google Scholar]
- 18.Lavin Y et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 159, 1312–1326 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gosselin D et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 159, 1327–1340 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gautier EL et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol 13, 1118–1128 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nahrendorf M et al. Dual channel optical tomographic imaging of leukocyte recruitment and protease activity in the healing myocardial infarct. Circ Res 100, 1218–1225 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Meschiari CA et al. Macrophage overexpression of matrix metalloproteinase-9 in aged mice improves diastolic physiology and cardiac wound healing after myocardial infarction. Am J Physiol Heart Circ Physiol 314, H224–H235 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paolicelli RC et al. Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Chow A et al. CD169(+) macrophages provide a niche promoting erythropoiesis under homeostasis and stress. Nat Med 19, 429–436 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dutta P et al. Macrophages retain hematopoietic stem cells in the spleen via VCAM-1. J Exp Med 212, 497–512 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Godwin JW, Pinto AR & Rosenthal NA Macrophages are required for adult salamander limb regeneration. Proc Natl Acad Sci U S A 110, 9415–9420 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hulsmans M et al. Macrophages Facilitate Electrical Conduction in the Heart. Cell 169, 510–522.e20 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen MC et al. Increased inflammatory cell infiltration in the atrial myocardium of patients with atrial fibrillation. Am J Cardiol 102, 861–865 (2008). [DOI] [PubMed] [Google Scholar]
- 29.Smorodinova N et al. Analysis of immune cell populations in atrial myocardium of patients with atrial fibrillation or sinus rhythm. PLoS One 12, e0172691 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun Z et al. Cross-talk between macrophages and atrial myocytes in atrial fibrillation. Basic Res Cardiol 111, 63 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Galkina E & Ley K Immune and inflammatory mechanisms of atherosclerosis (*). Annu Rev Immunol 27, 165–197 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weber C & Noels H Atherosclerosis: current pathogenesis and therapeutic options. Nat Med 17, 1410–1422 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Moore KJ & Tabas I Macrophages in the pathogenesis of atherosclerosis. Cell 145, 341–355 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robbins CS et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat Med 19, 1166–1172 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shankman LS et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med 21, 628–637 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Franck G et al. Roles of PAD4 and NETosis in Experimental Atherosclerosis and Arterial Injury: Implications for Superficial Erosion. Circ Res (2018). [DOI] [PMC free article] [PubMed]
- 37.Rahman K & Fisher EA Insights From Pre-Clinical and Clinical Studies on the Role of Innate Inflammation in Atherosclerosis Regression. Front Cardiovasc Med 5, 32 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rahman K et al. Inflammatory Ly6Chi monocytes and their conversion to M2 macrophages drive atherosclerosis regression. J Clin Invest 127, 2904–2915 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.King KR et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat Med 23, 1481–1487 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mouton AJ et al. Mapping macrophage polarization over the myocardial infarction time continuum. Basic Res Cardiol 113, 26 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Panizzi P et al. Impaired infarct healing in atherosclerotic mice with Ly-6C(hi) monocytosis. J Am Coll Cardiol 55, 1629–1638 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Courties G et al. In vivo silencing of the transcription factor IRF5 reprograms the macrophage phenotype and improves infarct healing. J Am Coll Cardiol 63, 1556–1566 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.DeBerge M et al. Efferocytosis and Outside-In Signaling by Cardiac Phagocytes. Links to Repair, Cellular Programming, and Intercellular Crosstalk in Heart. Front Immunol 8, 1428 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dobaczewski M, Xia Y, Bujak M, Gonzalez-Quesada C & Frangogiannis NG CCR5 signaling suppresses inflammation and reduces adverse remodeling of the infarcted heart, mediating recruitment of regulatory T cells. Am J Pathol 176, 2177–2187 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weirather J et al. Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ Res 115, 55–67 (2014). [DOI] [PubMed] [Google Scholar]
- 46.Engstrom G, Melander O & Hedblad B Leukocyte count and incidence of hospitalizations due to heart failure. Circ Heart Fail 2, 217–222 (2009). [DOI] [PubMed] [Google Scholar]
- 47.Tsujioka H et al. Impact of heterogeneity of human peripheral blood monocyte subsets on myocardial salvage in patients with primary acute myocardial infarction. J Am Coll Cardiol 54, 130–138 (2009). [DOI] [PubMed] [Google Scholar]
- 48.Majmudar MD et al. Monocyte-directed RNAi targeting CCR2 improves infarct healing in atherosclerosis-prone mice. Circulation 127, 2038–2046 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leuschner F et al. Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J Exp Med 209, 123–137 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee WW et al. PET/MRI of inflammation in myocardial infarction. J Am Coll Cardiol 59, 153–163 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sager HB et al. Proliferation and Recruitment Contribute to Myocardial Macrophage Expansion in Chronic Heart Failure. Circ Res 119, 853–864 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McArdle S, Chodaczek G, Ray N & Ley K Intravital live cell triggered imaging system reveals monocyte patrolling and macrophage migration in atherosclerotic arteries. J Biomed Opt 20, 26005 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee S, Courties G, Nahrendorf M, Weissleder R & Vinegoni C Motion characterization scheme to minimize motion artifacts in intravital microscopy. J Biomed Opt 22, 36005 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roufaiel M et al. CCL19-CCR7-dependent reverse transendothelial migration of myeloid cells clears Chlamydia muridarum from the arterial intima. Nat Immunol 17, 1263–1272 (2016). [DOI] [PubMed] [Google Scholar]
- 55.Casanova-Acebes M et al. Rhythmic modulation of the hematopoietic niche through neutrophil clearance. Cell 153, 1025–1035 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pittet MJ, Nahrendorf M & Swirski FK The journey from stem cell to macrophage. Ann N Y Acad Sci 1319, 1–18 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Morrison SJ & Scadden DT The bone marrow niche for haematopoietic stem cells. Nature 505, 327–334 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Birbrair A & Frenette PS Niche heterogeneity in the bone marrow. Ann N Y Acad Sci 1370, 82–96 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mendelson A & Frenette PS Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nat Med 20, 833–846 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kunisaki Y et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 502, 637–643 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mendez-Ferrer S, Lucas D, Battista M & Frenette PS Haematopoietic stem cell release is regulated by circadian oscillations. Nature 452, 442–447 (2008). [DOI] [PubMed] [Google Scholar]
- 62.Serbina NV & Pamer EG Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol 7, 311–317 (2006). [DOI] [PubMed] [Google Scholar]
- 63.Dutta P et al. Myocardial Infarction Activates CCR2(+) Hematopoietic Stem and Progenitor Cells. Cell Stem Cell 16, 477–487 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Suratt BT et al. Role of the CXCR4/SDF-1 chemokine axis in circulating neutrophil homeostasis. Blood 104, 565–571 (2004). [DOI] [PubMed] [Google Scholar]
- 65.Aiuti A, Webb IJ, Bleul C, Springer T & Gutierrez-Ramos JC The chemokine SDF-1 is a chemoattractant for human CD34+ hematopoietic progenitor cells and provides a new mechanism to explain the mobilization of CD34+ progenitors to peripheral blood. J Exp Med 185, 111–120 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nahrendorf M Myeloid cell contributions to cardiovascular health and disease. Nat Med 24, 711–720 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Courties G et al. Ischemic stroke activates hematopoietic bone marrow stem cells. Circ Res 116, 407–417 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dutta P et al. Myocardial infarction accelerates atherosclerosis. Nature 487, 325–329 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Emami H et al. Splenic metabolic activity predicts risk of future cardiovascular events: demonstration of a cardiosplenic axis in humans. JACC Cardiovasc Imaging 8, 121–130 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Herisson F et al. Direct vascular channels connect skull bone marrow and the brain surface enabling myeloid cell migration. Nat Neurosci 21,1209–1217 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Heidt T et al. Chronic variable stress activates hematopoietic stem cells. Nat Med 20, 754–758 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sager HB et al. RNAi targeting multiple cell adhesion molecules reduces immune cell recruitment and vascular inflammation after myocardial infarction. Sci Transl Med 8, 342ra80 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Christ A et al. Western Diet Triggers NLRP3-Dependent Innate Immune Reprogramming. Cell 172, 162–175.e14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Galkina E et al. Lymphocyte recruitment into the aortic wall before and during development of atherosclerosis is partially L-selectin dependent. J Exp Med 203, 1273–1282 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nahrendorf M et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med 204, 3037–3047 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pinto AR et al. Revisiting Cardiac Cellular Composition. Circ Res 118, 400–409 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cochain C et al. Single-Cell RNA-Seq Reveals the Transcriptional Landscape and Heterogeneity of Aortic Macrophages in Murine Atherosclerosis. Circ Res 122, 1661–1674 (2018). [DOI] [PubMed] [Google Scholar]
- 78.Skelly DA et al. Single-Cell Transcriptional Profiling Reveals Cellular Diversity and Intercommunication in the Mouse Heart. Cell Rep 22, 600–610 (2018). [DOI] [PubMed] [Google Scholar]
- 79.Lee S et al. Real-time in vivo imaging of the beating mouse heart at microscopic resolution. Nat Commun 3, 1054 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vandoorne K & Nahrendorf M Multiparametric Imaging of Organ System Interfaces. Circ Cardiovasc Imaging 10, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ridker PM et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med 377, 1119–1131 (2017). [DOI] [PubMed] [Google Scholar]
- 82.Saeed S et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 345, 1251086 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mitroulis I et al. Modulation of Myelopoiesis Progenitors Is an Integral Component of Trained Immunity. Cell 172, 147–161.e12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bekkering S et al. Innate immune cell activation and epigenetic remodeling in symptomatic and asymptomatic atherosclerosis in humans in vivo. Atherosclerosis 254, 228–236 (2016). [DOI] [PubMed] [Google Scholar]