

Abstract
Low oxygen or hypoxia is a feature of all solid tumors, and has been associated with aggressive disease. Here, we describe a novel mechanism for the hypoxia-dependent degradation of the Ras-GTPase activating protein neurofibromin, by hypoxia-associated factor (HAF). We have previously characterized HAF as an oxygen-independent ubiquitin ligase for HIF-1α. Here, we show that HAF promotes neurofibromin ubiquitination and degradation independently of oxygen and pVHL, resulting in Ras-ERK pathway activation. Hypoxia enhanced HAF:neurofibromin binding independently of HAF-SUMOylation, whereas HAF knockdown increased neurofibromin levels primarily in hypoxia, supporting the role of HAF as a hypoxia-specific neurofibromin regulator. HAF overexpression increased p-ERK levels and promoted resistance of clear cell kidney cancer (CCRCC) cells to sorafenib and sunitinib in both normoxia and hypoxia. However, a greater-fold increase in sorafenib/sunitinib resistance was observed during hypoxia, particularly in pVHL-deficient cells. Intriguingly, HAF-mediated resistance was HIF-2α dependent in normoxia, but HIF-2α independent in hypoxia indicating two potential mechanisms of HAF-mediated resistance: a HIF-2α dependent pathway dominant in normoxia, and the direct activation of the Ras-ERK pathway through neurofibromin degradation dominant in hypoxia. CCRCC patients with high HAF transcript or protein levels showed significantly decreased overall survival compared to those with low HAF. Thus, we establish a novel, non-mutational pathway of neurofibromin inactivation through hypoxia-induced HAF-mediated degradation, leading to Ras-ERK activation and poor prognosis in CCRCC.
Implications:
We describe a novel mechanism of neurofibromin degradation induced by hypoxia which leads to activation of the pro-oncogenic Ras-ERK pathway and resistance to therapy.
Keywords: HAF, Neurofibromin, ERK, sorafenib, sunitinib, hypoxia, kidney cancer
Introduction
Neurofibromin is the protein product of the gene NF1, whose loss-of-function mutation(s) results in one of the most common inherited tumor predisposition syndromes - Neurofibromatosis Type 1 (NF1). The cardinal feature of NF1 is the development of multiple peripheral nerve sheath tumors, neurofibromas, and an increased predisposition for developing various other tumor types, which forms the leading cause of death from this disease (1,2). The NF1 gene is one of the largest genes in the human genome, with 60 exons spanning over 350kb of genomic DNA (3). Neurofibromin itself has an estimated molecular mass of 327 kDa and consists of 2818 amino acids. The tumor suppressor function of neurofibromin is largely attributed to a 360 amino acid region referred to as the GAP-related domain (GRD), with considerable similarity to ras-guanosine-triphosphate (GTP)ase activation proteins (GAPs) (1,4,5). Indeed, the best understood function of neurofibromin is in promoting the hydrolysis of active Ras-GTP to inactive Ras-GDP as the NF1 GRD is able to complement yeast IRA (inhibitory regulators of the Ras-cAMP pathway) mutants (4,6). The Ras-GAP function of neurofibromin is enhanced by protein kinase C (PKC) phosphorylation within the cysteine-serine rich domain (CSRD) within exons 11-17, which also harbors clusters of missense mutations among NF1 patients (7). Paradoxically, PKC activity has also been shown to promote neurofibromin degradation, which is particularly relevant to gliomas, which commonly show aberrant activation of PKC (8). In addition to its Ras-GAP function, the tumor suppressor function of neurofibromin has also been attributed to its binding to caveolin-1, which modulates of a variety of pathways regulating cell proliferation and differentiation such as Ras, Akt and focal adhesion kinase (9).
Reduction or loss of neurofibromin in NF1 disorder primarily leads to activation of the Ras pathway, resulting in the activation of a cascade of downstream signaling pathways, including the Ras-Raf-MEK-ERK (Ras-ERK), and PI3K-Akt-mTOR (PI3K-mTOR) pathways, which are commonly activated in cancer (4). In addition to genetically inherited NF1 disorder, somatic mutations in NF1 have been identified in a variety of neoplasms, particularly in glioblastoma and melanoma, where they play critical roles in tumor initiation and progression, and in the acquisition of drug resistance (10,11). Like many tumor suppressor proteins, neurofibromin can also be inactivated through non-genetic means, such as through the ubiquitin-proteasomal pathway (8,12,13).
Low oxygen or hypoxia is a feature of all solid tumors due to a combination of the diffusion limitation of oxygen, and the inability of the abnormal tumor vasculature to supply sufficient oxygen to rapidly proliferating tumor cells (14). Crucial mediators of the hypoxic response are the hypoxia-inducible factors (HIF) transcription factors, which activate transcription of hundreds of genes critical for the adaptation to hypoxia, and for tumor progression, such as those promoting aerobic glycolysis, angiogenesis, and metastasis (15–17). The HIFs are primarily regulated by the von Hippel-Lindau tumor suppressor protein (pVHL), which targets HIF-1/2α subunits for proteasomal degradation under aerobic conditions. In hypoxia, or in the presence of pVHL mutations, HIF-1/2α is stabilized and form active transcriptional complexes with HIF-1β. pVHL loss-of-function mutations are a frequent initiating event in clear cell renal cell carcinoma (CCRCC), which drives constitutive HIF activation associated CCRCC progression (18). In contrast to pVHL, which does not distinguish between HIF-1α or HIF-2α isoforms, the hypoxia-associated factor, HAF (also known as SART1) mediates the oxygen independent degradation of HIF-1α, while promoting HIF-2α transactivation, thus mediating HIF-α isoform-specific regulation (19–21).
In addition to the inhibition of pVHL-mediated proteasomal degradation, hypoxia also activates the ERK kinases, which promote HIF-1 transactivation activity (22). Hypoxia-induced ERK activation has been widely reported and has been attributed to a variety of context-specific mechanisms including activation of MEK or increased calcium signaling in hypoxia (22,23). However, a general mechanism by which the Ras-ERK pathway is regulated by hypoxia has not been identified.
Here, we describe a novel, conserved mechanism of hypoxia-induced neurofibromin degradation mediated by HAF, thus establishing a novel, non-mutational pathway of neurofibromin inactivation leading to Ras-ERK pathway activation in hypoxia.
Materials and Methods
Cell lines and reagents.
HEK293, ACHN, A498, RCC4, and 786-0 cells were from ATCC (Manasses, VA), and maintained in RPMI media (A498), Eagle’s MEM (HEK 293) or Dulbecco’s MEM (Life Technologies, ThermoFisher Scientific, Waltham MA), supplemented with 10% FBS. The identities of all cell lines were verified using STR fingerprinting and verified mycoplasma-free using the MycoAlert kit (Lonza, Alpharettem GA). Cells were maintained at 37°C in a humidified atmosphere with 5% CO2. Hypoxic incubations (1% O2/5% CO2) were performed using the INVIVO2 Hypoxia Workstation (Baker Ruskinn, Sanford, ME) or the Whitley H35 Hypoxystation (HypOxygen, Frederick, MO). Sorafenib tosylate was purchased from Astatech Inc. (Bristol, PA), MG132 was from Sigma-Aldrich (St Louis, MO) and SCH772984 was from Selleckchem (Houston, TX). HAF siRNA constructs used were ON-Target plus SMARTpool (Dharmacon L-017283, siHAF#1), Hs_SART1_1 FlexiTube siRNA (siHAF#2, Qiagen); and Control siRNA #2 (Ambion AM4613) at 10nM final concentrations.
Plasmid construction and transfections.
Stable cells lines overexpressing HAF and HAF DM (HAF K94R K141R) were generated by retroviral infection using pMX-IRES-GFP (20,24). 786-0 cells containing homozygous loss of HIF-2α (EPAS1): 786-0 EPAS1−/− #4, EPAS1 −/− #11, and control (Nic Con) clones were generated using plasmids targeting different regions within exon 2 (sc400647-NIC, sc400647-NIC-2, or control double nickase plasmid sc-437281 respectively, from Santa Cruz Biotechnology (Dallas, TX). HIF-2α loss was verified via sequencing of EPAS1 DNA, western blotting and luciferase assays (Supplemental Fig. S1). Transient transfections were performed using Lipofectamine 2000 (Life Technologies) for plasmid DNA (HA-ubiquitin) in ACHN cells and siRNA in HEK293 cells. Briefly, cells were transfected 24 hours after seeding, then lysed 48 hours or 72 hours after transfection for plasmid or siRNA transfections respectively.
Western blots and Immunoprecipitations.
Cells were lysed in cold cell lysis buffer with protease and phosphatase inhibitors, and western blots were performed as described (20). Primary antibodies against phosphorylated or total proteins (EGFR, AKT, VEGFR, MEK, ERK, HIF-1α, HIF-2α) were from Cell Signaling Technology (Danvers, MA). Neurofibromin and FLAG antibodies were from Bethyl Laboratories (Montgomery, TX) and Sigma-Aldrich respectively, whereas the polyclonal HAF antibody has been described (19). Immunoprecipitations were performed using the neurofibromin and FLAG antibodies as described (19). Gel images were obtained using the Fluorchem M imaging system, and quantitation was performed using AlphaView software (Protein Simple, San Jose, CA).
Cell viability assays.
Cells were seeded in 96-well plates at 3,000 cells/well for ACHN, RCC4, and 786-0 cells in growth media supplemented with 10% FBS. 24 hours after seeding, cells were treated with sorafenib using equal final concentrations of DMSO for 72 hours in normoxia or hypoxia. Cell viability was determined using XTT Cell Viability Assay (Biotium, Fremont, CA). IC50 values were calculated using GraphPad Prism 7.0 software (La Jolla, CA). 3D tumor spheroids were generated using Cultrex 10X Spheroid Formation ECM (Trevigen, Gaithersburg, MD). Briefly, 6000 cells were seeded in 100µl media/per well in 96-well U-bottom plates, and spheroids allowed to form for 72 hours, after which an additional 100µl media containing 2X concentration of vehicle, sorafenib, or sorafenib + SCH772984 were added for a further 72 hours. Cell viability was quantitated using Resazurin Cell viability assay (Biotium).
Active Ras pull-down assays.
This was performed using the Active RAS Pull-down and Detection Kit (ThermoFisher Scientific). Briefly, 1×106 vector or HAF overexpressing 786-0 cells were seeded in T75 flasks. 24 hours after seeding, cells were incubated for 2 hours in normoxia or hypoxia, lysed and GTP bound Ras (Ras-GTP) was pulled down with GST-Raf1-RBD according to the manufacturer’s protocol. Ras-GTP was detected by western blot analysis and quantified.
CCRCC tumor microarray (TMA) preparation and TCGA data analysis.
TMAs were prepared from patients with metastatic CCRCC enrolled in phase II trials at the U.T.M.D. Anderson Cancer Center and were treated with sorafenib or sorafenib plus interferon (25). Three cores were obtained per patient and stained using HAF monoclonal antibody (20). Images were scanned using an Aperio AT2 slide scanner, and HAF nuclear staining was quantitated using Aperio Image analysis software (Leica Biosystems Inc, Buffalo Grove, IL). TCGA data was accessed using OncoLnc (26). Both datasets were stratified into two equal groups of high or low HAF transcript or protein. Kaplan Meier survival curves, p-values and hazard ratios were calculated using Prism 7.0.
Results
Neurofibromin is degraded during hypoxia in a variety of cell lines
To investigate the impact of hypoxia on Ras-ERK signaling, we exposed a panel of cell lines to increasing durations of hypoxia (1% oxygen). Hypoxic exposure resulted in marked decreases in neurofibromin and increases in ERK phosphorylation (p-ERK) in ACHN (pVHL wild-type renal cancer cells), MIAPaCa-2 (pancreatic cancer cells with mutant KRas), HEK293 (human embryonic kidney cells), U87 (glioblastoma cells expressing wild-type NF1) and RCC4 (pVHL mutant renal cancer cells). LN229 cells that harbor a total NF1 gene deletion showed an attenuated induction of P-ERK in hypoxia, which may be due to hypoxia-dependent activation of the SDF-1-CXCR7 pathway, previously reported in this cell line (Fig. 1A) (8,27). However, levels of the p120 Ras GTPase activating protein (RasGAP), another Ras-GTPase activating protein was unaffected by hypoxia (Fig. 1A) (28). Since neurofibromin is dynamically regulated by the ubiquitin-proteasome pathway, we investigated the role of the proteasome in the hypoxia-induced decrease in neurofibromin (12). Pre-treatment with the proteasome inhibitor, MG132 attenuated the hypoxia-induced decrease in neurofibromin in pVHL wild-type HEK293 and ACHN cells, and in pVHL mutant RCC4 cells, suggesting that the hypoxia-induced decrease in neurofibromin was proteasomal-dependent and pVHL-independent (Fig. 1B). The effects of MG132 on p-ERK levels were difficult to ascertain as MG132 also systemically perturbs the intracellular phosphoproteome, potentially impacting ERK signaling at multiple levels (29). Thus, we identify a novel mechanism of neurofibromin regulation through hypoxia-induced proteasomal degradation. This appears to be a general mechanism observed in multiple cell types irrespective of KRas and pVHL status.
Figure 1: Hypoxia promotes proteasomal-dependent neurofibromin degradation and increased ERK phosphorylation.
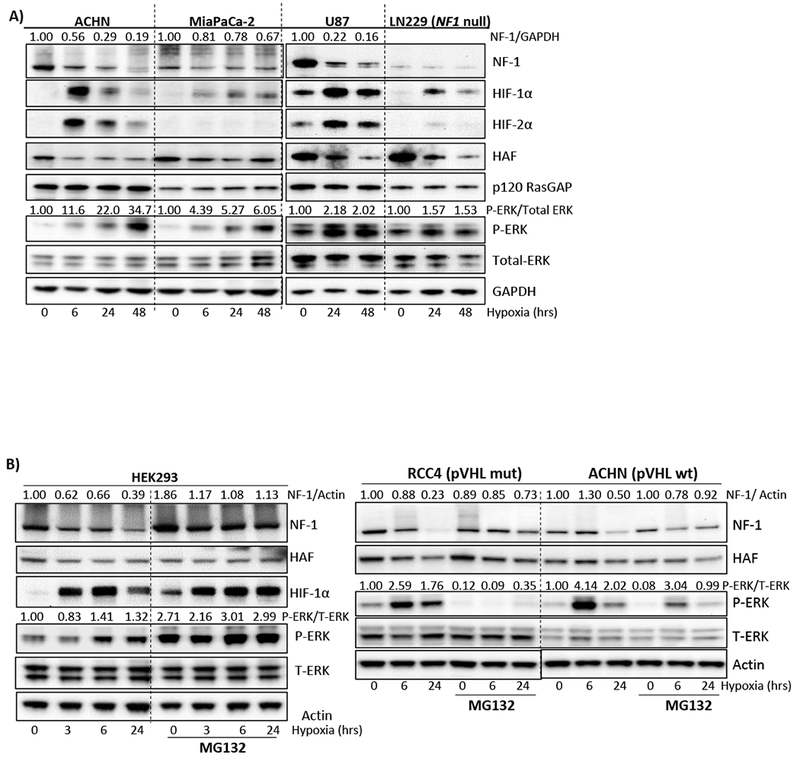
A) Impact of increasing durations of hypoxia (1% O2) on levels of neurofibromin (NF-1), HIFs and components of the Ras-ERK pathway in a panel of cell lines. B) Impact of treatment with the proteasomal inhibitor, MG132 on the hypoxia-induced decrease in neurofibromin in indicated cells. Normalized densitometry values of neurofibromin normalized to GAPDH or actin are shown above blots.
HAF promotes neurofibromin ubiquitination and degradation
Since HAF functions as an oxygen-independent E3 ubiquitin ligase for HIF-1α, we investigated whether neurofibromin could be a target for HAF (19). We evaluated HAF and the HAF SUMOylation deficient mutant HAF K94R K141R (HAF double mutant, DM), which is deficient in HIF-2α binding and activation while retaining HIF-1α E3 ligase activity (20). RCC4, 786-0 and ACHN cells overexpressing either wild-type HAF or HAF DM showed decreased levels of neurofibromin in both normoxia and hypoxia, and frequently showed increased levels of a high molecular weight smear above the expected neurofibromin band, often indicative of increased ubiquitination (Fig. 2A). The neurofibromin decrease in HAF overexpressing cells was associated with increased p-ERK primarily under hypoxic conditions in RCC4 and 786-0 cells, but was also observed in normoxia (Fig. 4A-B), and was observed in both normoxia and hypoxia in pVHL wild type ACHN cells (Fig. 2A). Consistent with a proteasomal-dependent mechanism of degradation, neurofibromin loss was reversible by treatment with MG132 (Fig. 2B). Intriguingly, HAF knockdown using siRNA increased neurofibromin levels only in hypoxia, suggesting that endogenous HAF may function as a ubiquitin ligase for neurofibromin primarily under hypoxic conditions (Fig. 2C). We also observed a reduction in HAF levels in hypoxia and induction of HIF-1α with HAF knockdown (Fig. 2C), both of which we have previously reported (19). Thus, HAF overexpression promotes neurofibromin proteasomal degradation primarily under hypoxic conditions without requiring HAF SUMOylation.
Figure 2: HAF overexpression promotes neurofibromin ubiquitination and degradation, and increases ERK phosphorylation.
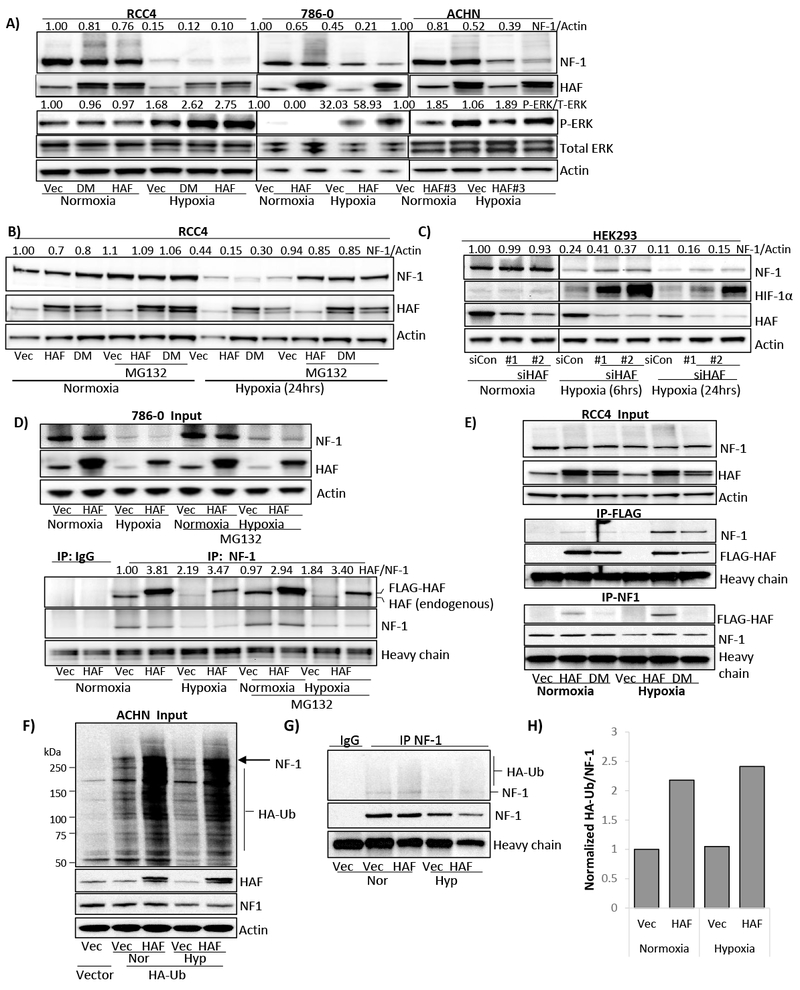
A-B) Effect of stable HAF overexpression on levels of neurofibromin and p-ERK in a panel of cell lines exposed to normoxia or hypoxia for 24 hours (A), or on neurofibromin levels in RCC4 cells in normoxia and hypoxia ± 5µM MG132 (B). C) Effects of HAF knockdown using two separate siRNAs on neurofibromin levels in normoxia and hypoxia. D) Input (top panel) and immunoprecipitations (bottom panel) using neurofibromin or isotype control antibodies in vector or HAF overexpressing 786-0 cells after exposure to 24 hours’ normoxia or hypoxia ± 5µM MG132. E) Inputs (top panel) and immunoprecipitation using FLAG (middle panel) or neurofibromin (bottom panel) antibodies in RCC4 cells overexpressing HAF or HAF DM exposed to normoxia or hypoxia for 6 hours (+ 5µM MG132). F) HA-ubiquitin incorporation in vector or HAF overexpressing ACHN cells exposed to normoxia or hypoxia for 24 hours (with 5µM MG132 added for the last 6 hours). Arrow indicates the expected molecular weight for neurofibromin. G) HA-ubiquitin incorporation into immunoprecipitated neurofibromin with densitometric quantitation shown in (H).
To characterize the relationship between HAF and neurofibromin, we performed immunoprecipitation experiments using 786-0 cells exposed to normoxia or hypoxia for 24 hours, with MG132 added for the last 6 hours. Although both endogenous and overexpressed FLAG-HAF bound endogenous neurofibromin, the binding of endogenous HAF to neurofibromin was increased approximately 2-fold in hypoxia regardless of whether cells were MG132 treated (Fig. 2D lower panel). By contrast, the binding of FLAG-HAF to neurofibromin was not similarly affected by hypoxia, likely due to saturation of neurofibromin with high levels of FLAG-HAF (Fig. 2D lower panel). However, immunoprecipitation of FLAG-HAF or FLAG-HAF DM in RCC4 cells pulled down neurofibromin with equivalent affinity, with binding dramatically increased in hypoxia (Fig. 2E middle panel). Similarly, neurofibromin immunoprecipitation also showed a marked increase in the pulldown of FLAG-HAF in hypoxia (Fig. 2E bottom panel). Intriguingly, we noted that pulldown of FLAG-HAF DM with neurofibromin was substantially weaker than FLAG-HAF (Fig. 2E bottom panel). By contrast, Cul3, an E3 ubiquitin ligase which degrades neurofibromin in response to growth factors, was not detected in any of our neurofibromin immunoprecipitations (not shown) (13). To determine the impact of HAF on neurofibromin ubiquitination, we transiently transfected HA-tagged ubiquitin into ACHN cells overexpressing HAF, then exposed cells to normoxia or hypoxia for 24 hours, with MG132 added for the last 6 hours. We observed an overall increase in HA-Ub incorporation with HAF overexpression, and found increased ubiquitination of immune-precipitated neurofibromin in both normoxia and hypoxia (Fig. 2F-H). Thus, HAF binds neurofibromin with increased affinity under hypoxic conditions, and promotes neurofibromin ubiquitination in both normoxia and hypoxia.
HAF regulates neurofibromin turnover
To investigate the impact of HAF on neurofibromin turnover, we used cycloheximide to inhibit protein translation. We found that in normoxia, the half-life of neurofibromin was decreased from 48 hours in the vector cells to 15 hours in the HAF overexpressing cells, whereas in hypoxia, this was decreased from 2.5 hours to 45 minutes (Fig. 3A, B, E). Intriguingly, cycloheximide treatment in hypoxia did not cause any further decrease in neurofibromin levels when compared to vehicle-treated cells, suggesting that neurofibromin translation is inhibited under hypoxic conditions (Fig. 3B, E). Additionally, the drastic decrease in neurofibromin half-life in hypoxia compared to normoxia, even under cycloheximide treatment suggests a hypoxia-specific post-translational degradation mechanism that is largely inactive in normoxia, which is supported by our MG132 data (Fig. 1B).
Figure 3: HAF regulates neurofibromin turnover.
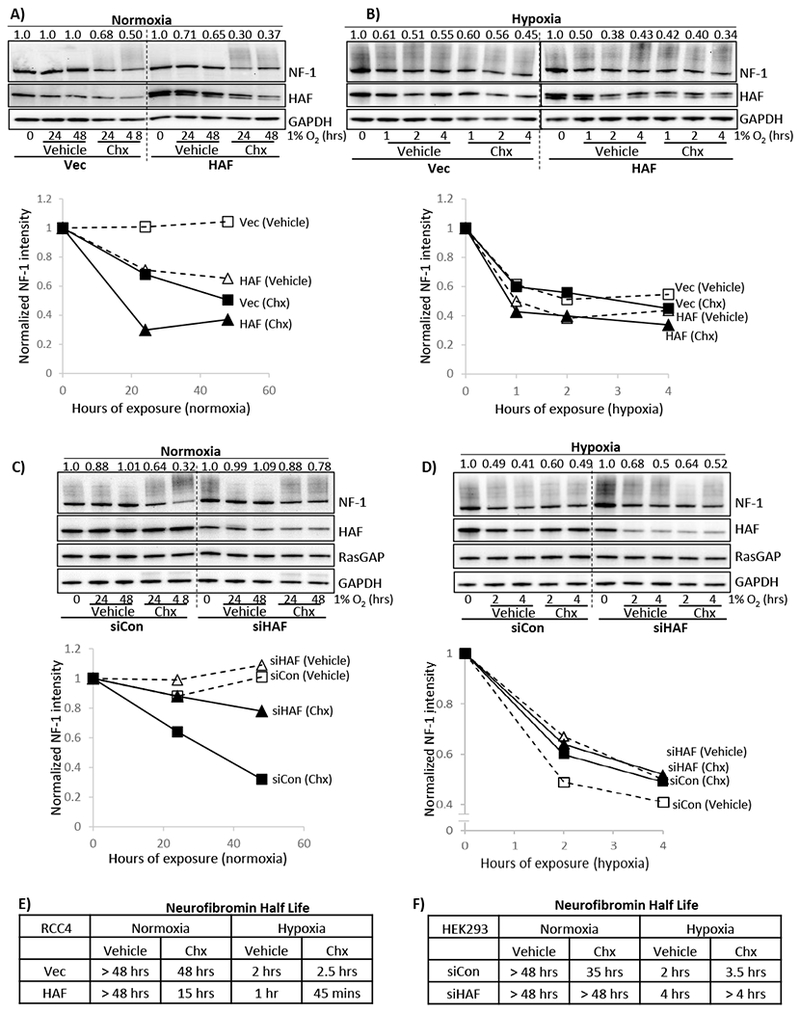
A,B) Effects of indicated durations of normoxia (A) or hypoxia (B) ± 50µg/ml cycloheximide on levels of neurofibromin in RCC4 cells stably overexpressing empty vector or HAF. C, D) Effects of indicated durations of normoxia (C) or hypoxia (D) ± 20µg/ml cycloheximide on levels of neurofibromin in HEK293 cells transiently transfected with non-targeting (siCon) or siHAF siRNA. For A-D, normalized densitometry values of neurofibromin normalized to GAPDH are shown above blots with plots shown below blots. E-F) Tables summarizing neurofibromin half-lives determined in in A,B and C,D are shown in E-F respectively.
By contrast, HAF knockdown increased neurofibromin half-life in normoxia from 35 hours in control cells to >48 hours with HAF siRNA transfection (Fig. 3C, F). However, differences in neurofibromin half-life were less clear with cycloheximide in hypoxia, which may have been caused by excessive cell death due to the combined stresses of siRNA transfection, hypoxia and cycloheximide treatment (Fig. 3D, F). Nevertheless, in the absence of cycloheximide, HAF knockdown increased neurofibromin half-life in hypoxia from 2 hours to 4 hours (Fig. 3D, F). Thus, we show that HAF overexpression increased, whereas HAF knockdown decreased, neurofibromin turnover in both normoxia and hypoxia.
HAF activates the Ras-ERK pathway in normoxia and hypoxia
To characterize the impact of HAF on the Ras-ERK pathway, we exposed HAF overexpressing RCC4 cells to increasing durations of hypoxia. Exposure to hypoxia for as little as 20 minutes induced neurofibromin degradation and p-ERK in both the vector and HAF overexpressing cells (Fig. 4A). As expected, neurofibromin levels were lower in HAF-overexpressing cells compared to the vector cells in both normoxia and hypoxia (Fig. 4A). Additionally, hypoxia-induced p-ERK was decreased in the vector cells after 4 hours of hypoxic exposure, whereas p-ERK in the HAF overexpressing cells was maintained (Fig. 4A) and was still apparent after 24 hours of hypoxia (Fig. 2A). To determine whether the effect of HAF on p-ERK was mediated through Ras activation, we performed active Ras pulldown assays, to quantitate the amount of active GTP-bound Ras (Ras-GTP). Indeed, HAF overexpression increased levels of active Ras in both normoxia and hypoxia (Fig. 4B). Hypoxia itself induced a 2.7-fold increase in levels of Ras-GTP compared to normoxia (Fig. 4B). Thus, HAF enhances p-ERK in both normoxia and hypoxia by directly promoting Ras activation.
Figure 4: HAF promotes increased Ras-ERK pathway activation and is associated with resistance to sorafenib.
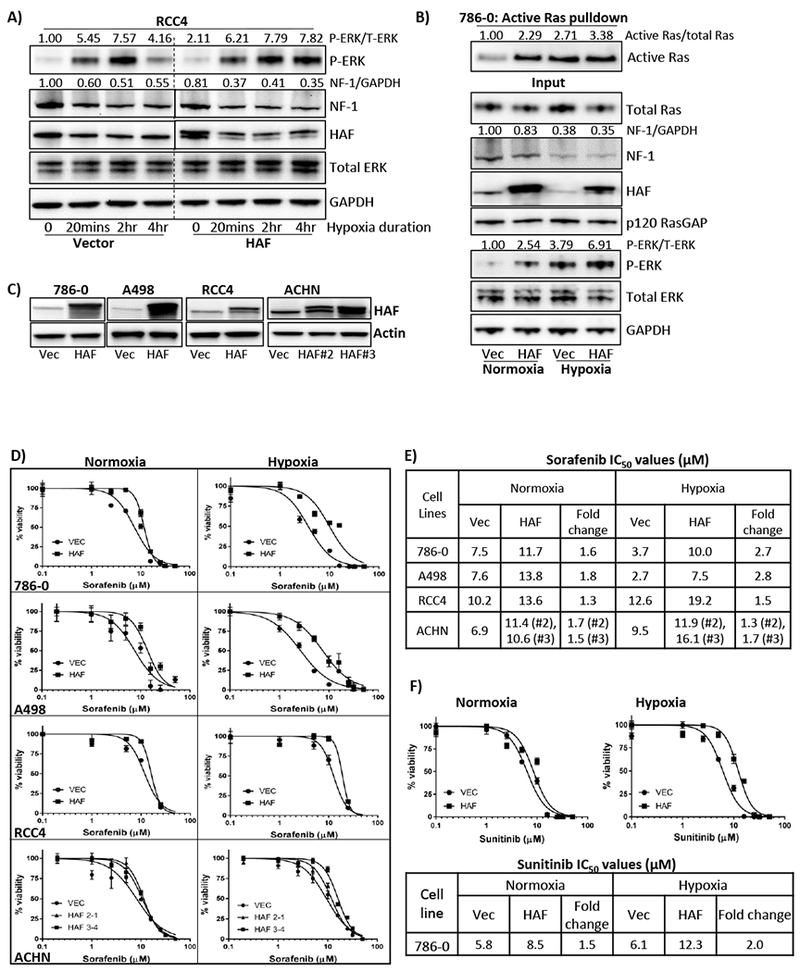
A) Impact of increasing duration of hypoxia on levels of neurofibromin and p-ERK in vector or HAF overexpressing RCC4 cells. Normalized densitometric values for p-ERK and neurofibromin relative to total ERK and GAPDH respectively are indicated. B) Results of active Ras pulldown assay in 786-0 cells stably expressing empty vector or HAF exposed to normoxia or hypoxia for 2 hours. Normalized densitometric values for active GTP-bound Ras relative to total Ras are indicated. C) Western blot showing HAF levels in a panel of stably generated vector or HAF overexpressing CCRCC cell lines. D-F) XTT viability assays of indicated cell lines after 72 hours’ continuous treatment with sorafenib (D), or sunitinib (F) with calculated IC50 values in (E-F). Data shown are representative of at least two independent experiments.
HAF promotes increased resistance to sorafenib and sunitinib in vitro
Since ERK is the major downstream target of a variety of kinase inhibitors used for cancer therapy, we investigated whether HAF, by activating the Ras-ERK pathway, could modulate treatment response. We investigated the impact of HAF on response to sorafenib, a Raf kinase inhibitor that also inhibits a variety of upstream receptor tyrosine kinases (RTKs) such as vascular endothelial growth factor receptor (VEGFR) and and platelet-derived growth factor receptor (PDGFR) (30). As previously reported, there were no significant differences between the growth rates of empty vector or HAF overexpressing cells (Fig. S2A and (19)). HAF overexpression was associated with increased resistance to sorafenib in both normoxia and hypoxia, independently of pVHL status (Fig. 4C-E). Similar to the effects of HAF on p-ERK, HAF-mediated sorafenib resistance was more pronounced in hypoxia, especially in the pVHL deficient A498 and 786-0 cells, which express only HIF-2α and no HIF-1α (Fig 2A, 4D-E). Comparable results were obtained with sunitinib, a multi-kinase inhibitor, with increased resistance also observed in hypoxia (Fig. 4F) (31). Thus, HAF overexpression was associated with sorafenib and sunitinib resistance in normoxia and hypoxia, with increased resistance associated with hypoxia in pVHL deficient CCRCC cells.
HIF-2α is required for HAF-mediated p-ERK elevation and sorafenib resistance in normoxia, but is dispensable for HAF-mediated p-ERK elevation and sorafenib resistance in hypoxia
HAF promotes HIF-2 transactivation through a mechanism requiring hypoxia-dependent HAF SUMOylation, which is deficient in the HAF DM cells (20). HIF-2α has also been associated with sorafenib resistance in hepatocellular carcinoma (32). Our findings suggest that both HAF and HAF DM are comparable in their effects on inducing neurofibromin degradation (Fig. 2A). To determine the effects of HAF overexpression on HIF-2α and downstream kinase signaling in the context of sorafenib treatment, we exposed 786-0 cells to varying concentrations of sorafenib for 72 hours. Here, we observed marked induction of p-ERK in HAF or HAF DM overexpressing cells compared to vector cells in normoxia (Fig. 5A). We noted that some concentrations of sorafenib resulted in elevated p-ERK, consistent with the activation of negative feedback loops frequently associated with MAPK pathway inhibition (33). Additionally, exposure to 72 hours’ hypoxia resulted in a ~3.8-fold induction in p-ERK compared to normoxia, which was further increased by HAF or HAF DM overexpression (Fig. 5A). We observed induction of HIF-2α with sorafenib treatment as previously reported, but only under normoxic conditions (32). Additionally, HAF or HAF DM overexpression and/or higher sorafenib concentrations were associated with increases in HIF-2α, and in phosphorylated epidermal growth factor receptor (p-EGFR) and p-VEGFR, both primarily HIF-2α target genes, again, only during normoxia (32,34). Indeed, prolonged hypoxia decreased HIF-2α levels (also seen in Fig. 1A) and suppressed phosphorylation of EGFR, VEGFR, and MEK, with no apparent induction by HAF or HAF DM overexpression (Fig. 5A, Supplemental Fig. S2B). PDGFR was undetectable in 786-0 cells (Fig. S3). Thus, HAF overexpression and/or high concentrations of sorafenib increased HIF-2α, p-EGFR and p-VEGFR only under normoxic conditions. Since HAF overexpression increased p-ERK levels in both normoxia and hypoxia, the data suggest that HAF-induced p-ERK in hypoxia is largely HIF-2α/RTK independent.
Figure 5: HAF-mediated sorafenib resistance is associated with increased P-ERK and is dependent on HIF-2α only in normoxia.
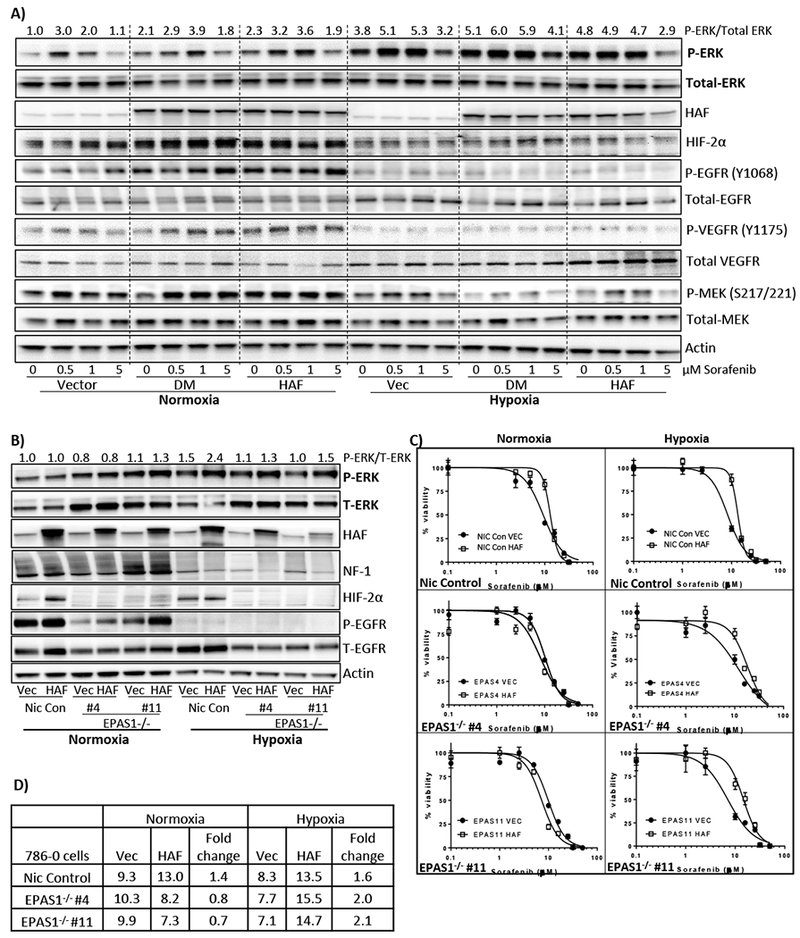
A) Effect of sorafenib treatment on indicated proteins in 786-0 cells overexpressing empty vector, HAF or HAF K94R K141R (double mutant, DM) for 72 hours’ in normoxia or hypoxia. Densitometric quantitation of p-ERK normalized to total ERK is shown above blots. B) Effect of HAF overexpression on indicated proteins in 786-0 nickase vector control (Nic Con) cells or cells bearing HIF-2α deletion (EPAS1−/− clones #4 and #11) in normoxia and hypoxia (24 hours). C) XTT viability assays of cells in (B) after 72 hours’ continuous treatment with sorafenib in normoxia or hypoxia with calculated IC50 values in (D).
To determine whether HIF-2α is required for HAF-mediated p-ERK activation and/or resistance to sorafenib, we generated 786-0 cells with HIF-2α (EPAS1) deletion using a Crispr/Cas9 double nickase strategy. Cells with deletions at two distinct sites within exon 2 of the EPAS1 gene (EPAS1#4 and EPAS1#11) were generated, verified (Fig. S1), after which cells were virally transduced with HAF or empty vector plasmid. Consistent with the previously reported role of HIF-2α in EGFR translation, we observed a marked decrease in the levels of total EGFR in the HIF-2α-deleted cells, primarily under hypoxic conditions (Fig. 5B) (35). We also saw increased levels of total ERK with HIF-2α deletion although the significance of this is unclear. Intriguingly, HAF-induced p-ERK was maintained in hypoxia in the absence of HIF-2α, whereas no induction of P-ERK was observed in normoxia irrespective of HIF-2α status (Fig. 5B). Similar to Fig. 2A, we observed the HAF-induced neurofibromin decrease primarily under hypoxic conditions, which occurred independently of HIF-2α (Fig. 5B). Furthermore, in the absence of HIF-2α, HAF overexpression promoted sorafenib resistance only in hypoxia, whereas HAF overexpression in HIF-2α proficient cells promoted sorafenib resistance in both normoxia and hypoxia (Fig. 5C-D). As in Fig. 5A, we again observed increased p-EGFR with HAF overexpression, but only in normoxia, and this occurred independently of HIF-2α (Fig. 5B). Thus, HAF-mediated sorafenib resistance is HIF-2α-dependent in normoxia, but HIF-2α-independent in hypoxia.
Inhibition of p-ERK abrogates HAF-mediated resistance to sorafenib
Since tumor cells reside in a three-dimensional structure exposed to oxygen gradients, we generated tumor spheroids that more closely model this tumor architecture. HAF overexpressing cells tended to form larger spheroids (Fig. 6A-B) then vector cells, consistent with the role of the Ras-ERK pathway in promoting anchorage independent growth (36). Similar to the 2D assays, 3D spheroids derived from HAF overexpressing cells were resistant to sorafenib treatment compared to vector cells (IC50s: Vec = 32µM; HAF > 50µM; Fig 6B). When spheroids were exposed to hypoxia (likely creating an anoxic environment within their cores), the viability of the vector spheroids was significantly reduced compared to the HAF spheroids, and sensitivity of both cell types to sorafenib was greatly enhanced (IC50s: Vec = 1.2µM; HAF = 5.0µM; Fig. 6A,C). Co-treatment of 25µM sorafenib with the ERK inhibitor SCH772984 (Fig. S4) was sufficient to reverse HAF-mediated resistance in the 786-0 spheroids in a concentration-dependent manner (Fig. 6D-E) (37). Thus, p-ERK is required for HAF mediated resistance to sorafenib.
Figure 6: HAF-mediated sorafenib resistance is enhanced in 3D tumor spheroids and abrogated by inhibition of ERK activation.
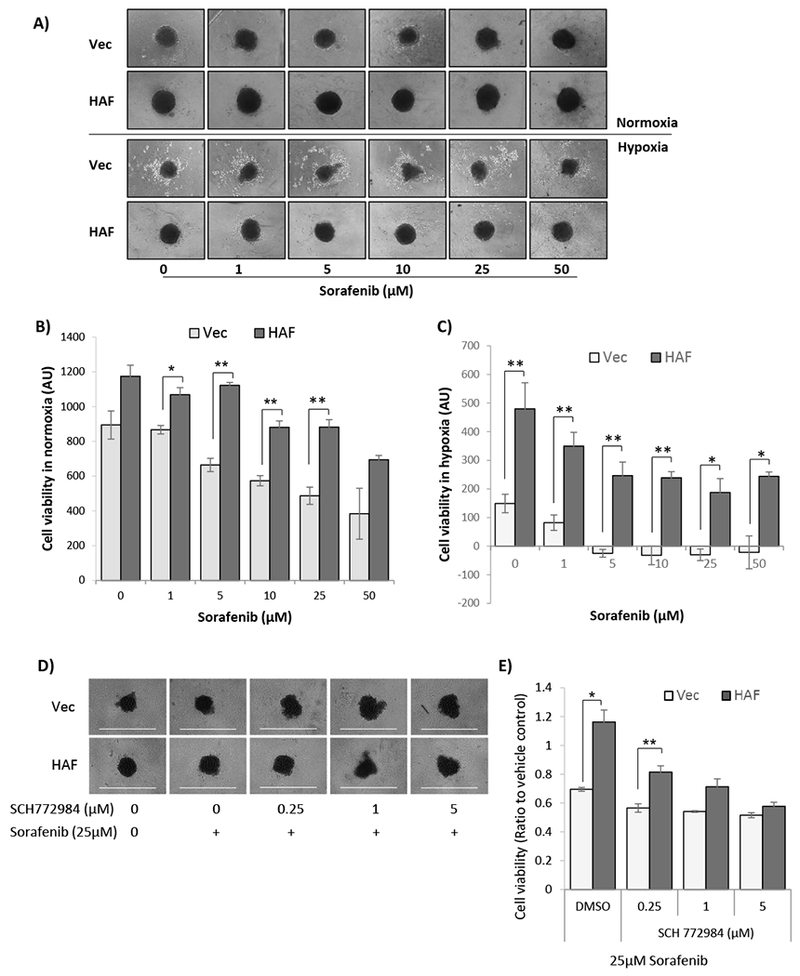
A) Photomicrographs showing 3D tumor spheroids generated from vector or HAF overexpressing 786-0 cells treated with sorafenib for 72 hours in normoxia or hypoxia. B, C) Cell viability quantitation of spheroids grown in normoxia (B), or hypoxia (C), determined using resazurin. Data shown are the averages of 4 replicate wells ± SEM. D) Photomicrographs showing vector or HAF overexpressing 786-0 spheroids after treatment with vehicle, or 25µM sorafenib ± indicated concentrations of the ERK inhibitors SCH772984 for 72 hours in normoxia, with cell viability quantitation shown in E). * p < 0.05; ** p< 0.01.
High HAF (SART1) transcript or protein levels correlate to decreased overall survival in CCRCC patients
To investigate the relationship between HAF and patient outcome, we stratified the 522 patients from The Cancer Genome Atlas Kidney Renal Clear Cell Carcinoma (TCGA-KIRC) database into two equal groups of low and high HAF. Intriguingly, patients with high SART1 transcript showed significantly decreased overall survival compared to those with low SART1 in TCGA-KIRC (p = 0.0007; Fig. 7A, S6). We obtained similar results with TMAs stained for HAF protein from patients with grade III/IV metastatic CCRCC treated with sorafenib: patients with higher HAF had significantly reduced overall survival compared to those with low HAF (p=0.048; Fig. 7B). Representative images and associated data are shown in (Fig. S5, S6). Thus, HAF significantly correlates with poorer overall survival in CCRCC patients.
Figure 7: HAF predicts for poor survival in CCRCC.
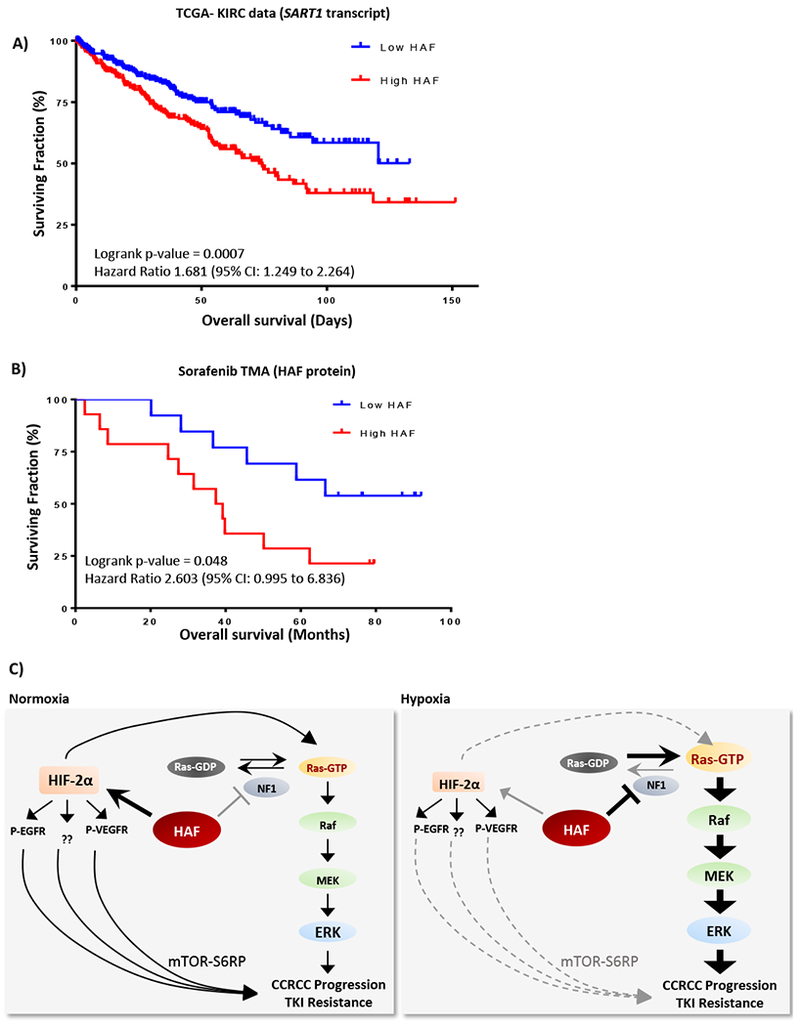
A) Kaplan-Meier plots showing overall survival of patients with (A), high or low HAF (SART1) transcript levels from the TCGA-KIRC dataset (n = 522); or (B), high or low HAF protein levels from a TMA of Grade III/IV metastatic CCRCC patients treated with sorafenib (n=27). C) Diagram depicting distinct mechanisms by which HAF drives CCRCC progression and/or TKI resistance in normoxia and hypoxia in pVHL deficient CCRCC cells.
Discussion
The Ras oncogene family and their downstream signaling pathways play a central role in cellular proliferation, differentiation and survival, and are thus heavily implicated in cancer development and resistance to therapy (38). The tumor suppressor role of neurofibromin through inhibition of the Ras-ERK pathway is supported by biochemical evidence of hyperactive Ras in NF1 patients, as well as elevated GTP-bound Ras in somatic NF1 aberrations in solid tumors (1,39). Here, we describe a novel mechanism of neurofibromin inactivation through hypoxia- and HAF-mediated proteasomal degradation, which drives Ras-ERK pathway activation specifically during hypoxia. We also show that hypoxia is a potent stimulus for neurofibromin degradation across cancer cell types. Moreover, since most solid tumors contain hypoxic regions, this suggests that hypoxia-induced neurofibromin degradation and concomitant Ras-ERK activation may be prevalent in solid tumors.
CCRCC typically lack mutations such as KRAS, BRAF or NF1 that commonly drive Ras-ERK or PI3K-mTOR pathway activation in solid tumors, yet tyrosine kinase inhibitors (TKIs) targeting these pathways still provide treatment benefit, underscoring their importance in CCRCC progression (40). Thus, the inhibition of HAF-mediated neurofibromin degradation may be of therapeutic value in CCRCC and in other solid tumor types. The role of HAF-mediated neurofibromin degradation also implicates HAF as a potential biomarker for predicting resistance to the TKIs.
Sorafenib and sunitinib are both approved for the treatment of metastatic renal cell carcinoma (31,41). However, the high likelihood of disease progression remains a major challenge and therefore, the understanding of their resistance mechanisms is critical for the identification of more effective treatment strategies (30,42). Combination regimens of immune checkpoint inhibitors with TKIs have shown great promise, indicating that TKIs, either as single agents or in combination, will continue to be a mainstay for CCRCC therapy for the foreseeable future (43–45). The antitumor activity of sorafenib and sunitinib have been mainly attributed to their anti-angiogenic properties in endothelial cells and pericytes (46,47). They also have direct anti-proliferative and/or pro-apoptotic effects on tumor cells, resulting in tumor cell apoptosis prior to, and in addition to disruption of the tumor vasculature (48,49).
Our studies show that the protective effect of HAF was enhanced in hypoxia despite hypoxia-associated inhibition of p-VEGFR and p-EGFR, supporting the notion of an RTK-independent mechanism of resistance, consistent with our proposed HAF-mediated neurofibromin degradation pathway (Fig. 4D-E, Fig. 2D-E). HIF-2α deletion abrogated HAF-mediated sorafenib resistance only in normoxia, suggesting the existence of distinct pathways of HAF-mediated sorafenib resistance (Fig. 5C-D). In this regard, HAF-mediated sorafenib resistance was observed in normoxia despite lack of p-ERK elevation (Fig. 5B), suggesting a p-ERK independent resistance mechanism in normoxia (Fig. 7C). Indeed, we observed elevated levels of phosphorylated ribosomal S6 protein (P-S6RP) with HAF overexpression primarily in normoxic 786-0 cells (Fig. S7A). This was abrogated by treatment with rapamycin without affecting P-ERK (Fig. S7B). Since P-S6RP is HIF-2α-dependent in 786-0 cells, this suggests a potential HIF-2α-driven pathway of sorafenib resistance via the mTOR-S6RP axis (50). However, during hypoxia, HAF mediated Ras-ERK activation through neurofibromin degradation is likely to override any HIF-2α-mediated effect on sorafenib response. Studies are ongoing to map the interacting domains of HAF and neurofibromin, and to identify the mechanisms driving the hypoxia-dependent increase in binding affinity.
In conclusion, we highlight a new, non-mutational mechanism of neurofibromin inactivation during hypoxia, which leads to Ras-ERK pathway activation. This new mechanism may be significant during physiological hypoxia such as during embryonic development and wound healing, but may also play a driving role in the development and progression of hypoxia-associated pathophysiology such as cancer.
Supplementary Material
Acknowledgments
Supported by NIH/NCI grants CA181106 and CA217905 to MYK
Footnotes
Conflict of interest: MYK is founder and has equity in Kuda Therapeutics Inc. All other authors declare no conflict of interest.
References
- 1.Yap YS, McPherson JR, Ong CK, Rozen SG, Teh BT, Lee AS, et al. The NF1 gene revisited - from bench to bedside. Oncotarget 2014;5:5873–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jett K, Friedman JM. Clinical and genetic aspects of neurofibromatosis 1. Genet Med 2010;12:1–11 [DOI] [PubMed] [Google Scholar]
- 3.Cawthon RM, Weiss R, Xu GF, Viskochil D, Culver M, Stevens J, et al. A major segment of the neurofibromatosis type 1 gene: cDNA sequence, genomic structure, and point mutations. Cell 1990;62:193–201 [DOI] [PubMed] [Google Scholar]
- 4.Xu G, O’Connell P, Viskochil D, Cawthon R, Robertson M, Culver M, et al. The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell 1990;62:599–608 [DOI] [PubMed] [Google Scholar]
- 5.Ballester R, Marchuk D, Boguski M, Saulino A, Letcher R, Wigler M , et al. The NF1 locus encodes a protein functionally related to mammalian GAP and yeast IRA proteins. Cell 1990;63:851–9 [DOI] [PubMed] [Google Scholar]
- 6.Bollag G, Clapp DW, Shih S, Adler F, Zhang YY, Thompson P , et al. Loss of NF1 results in activation of the Ras signaling pathway and leads to aberrant growth in haematopoietic cells. Nat Genet 1996;12:144–8 [DOI] [PubMed] [Google Scholar]
- 7.Mattocks C, Baralle D, Tarpey P, ffrench-Constant C, Bobrow M, Whittaker J. Automated comparative sequence analysis identifies mutations in 89% of NF1 patients and confirms a mutation cluster in exons 11-17 distinct from the GAP related domain. J Med Genet 2004;41:e48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McGillicuddy LT, Fromm JA, Hollstein PE, Kubek S, Beroukhim R, De Raedt T, et al. Proteasomal and genetic inactivation of the NF1 tumor suppressor in gliomagenesis. Cancer Cell 2009;16:44–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boyanapalli M, Lahoud OB, Messiaen L, Kim B, Anderle de Sylor MS, Duckett SJ, et al. Neurofibromin binds to caveolin-1 and regulates ras, FAK, and Akt. Biochem Biophys Res Commun 2006;340:1200–8 [DOI] [PubMed] [Google Scholar]
- 10.Philpott C, Tovell H, Frayling IM, Cooper DN, Upadhyaya M. The NF1 somatic mutational landscape in sporadic human cancers. Hum Genomics 2017;11:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Whittaker SR, Theurillat JP, Van Allen E, Wagle N, Hsiao J, Cowley GS, et al. A genome-scale RNA interference screen implicates NF1 loss in resistance to RAF inhibition. Cancer Discov 2013;3:350–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cichowski K, Santiago S, Jardim M, Johnson BW, Jacks T. Dynamic regulation of the Ras pathway via proteolysis of the NF1 tumor suppressor. Genes Dev 2003;17:449–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hollstein PE, Cichowski K. Identifying the Ubiquitin Ligase complex that regulates the NF1 tumor suppressor and Ras. Cancer Discov 2013;3:880–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hockel M, Vaupel P. Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J Natl Cancer Inst 2001;93:266–76 [DOI] [PubMed] [Google Scholar]
- 15.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A 1995;92:5510–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer 2011;11:393–410 [DOI] [PubMed] [Google Scholar]
- 17.Koh MY, Powis G. Passing the baton: the HIF switch. Trends Biochem Sci 2012;37:364–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaelin WG, Jr. The von Hippel-Lindau tumor suppressor gene and kidney cancer. Clin Cancer Res 2004;10:6290S–5S [DOI] [PubMed] [Google Scholar]
- 19.Koh MY, Darnay BG, Powis G. Hypoxia-Associated Factor, a Novel E3-Ubiquitin Ligase, Binds and Ubiquitinates Hypoxia-Inducible Factor 1α, Leading to Its Oxygen-Independent Degradation. Molecular and Cellular Biology 2008;28:7081–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koh MY, Nguyen V, Lemos R, Darnay BG, Kiriakova G, Abdelmelek M, et al. Hypoxia-Induced SUMOylation of E3 Ligase HAF Determines Specific Activation of HIF2 in Clear-Cell Renal Cell Carcinoma. Cancer Research 2015;75:316–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koh MY, Lemos R, Jr., Liu X, Powis G. The Hypoxia-Associated Factor Switches Cells from HIF-1{alpha}- to HIF-2{alpha}-Dependent Signaling Promoting Stem Cell Characteristics, Aggressive Tumor Growth and Invasion. Cancer Res 2011;71:4015–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Minet E, Arnould T, Michel G, Roland I, Mottet D, Raes M, et al. ERK activation upon hypoxia: involvement in HIF-1 activation. FEBS Lett 2000;468:53–8 [DOI] [PubMed] [Google Scholar]
- 23.Mottet D, Michel G, Renard P, Ninane N, Raes M, Michiels C. Role of ERK and calcium in the hypoxia-induced activation of HIF-1. J Cell Physiol 2003;194:30–44 [DOI] [PubMed] [Google Scholar]
- 24.Lamothe B, Besse A, Campos AD, Webster WK, Wu H, Darnay BG. Site-specific Lys-63-linked tumor necrosis factor receptor-associated Factor 6 auto-ubiquitination is a critical determinant of IkappaB kinase activation. J Biol Chem 2007;282:4102–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jonasch E, Corn P, Pagliaro LC, Warneke CL, Johnson MM, Tamboli P, et al. Upfront, randomized, phase 2 trial of sorafenib versus sorafenib and low-dose interferon alfa in patients with advanced renal cell carcinoma: clinical and biomarker analysis. Cancer 2010;116:57–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anaya J OncoLnc: linking TCGA survival data to mRNAs, miRNAs, and lncRNAs. PeerJ Computer Science 2016;2:e67 [Google Scholar]
- 27.Esencay M, Sarfraz Y, Zagzag D. CXCR7 is induced by hypoxia and mediates glioma cell migration towards SDF-1alpha. BMC Cancer 2013;13:347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ratner N, Miller SJ. A RASopathy gene commonly mutated in cancer: the neurofibromatosis type 1 tumour suppressor. Nat Rev Cancer 2015;15:290–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cirit M, Grant KG, Haugh JM. Systemic perturbation of the ERK signaling pathway by the proteasome inhibitor, MG132. PLoS One 2012;7:e50975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Staehler M, et al. Sorafenib for Treatment of Renal Cell Carcinoma: Final Efficacy and Safety Results of the Phase III Treatment Approaches in Renal Cancer Global Evaluation Trial. Journal of Clinical Oncology 2009;27:3312–8 [DOI] [PubMed] [Google Scholar]
- 31.Mendel DB, Laird AD, Xin X, Louie SG, Christensen JG, Li G, et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res 2003;9:327–37 [PubMed] [Google Scholar]
- 32.Zhao D, Zhai B, He C, Tan G, Jiang X, Pan S, et al. Upregulation of HIF-2alpha induced by sorafenib contributes to the resistance by activating the TGF-alpha/EGFR pathway in hepatocellular carcinoma cells. Cell Signal 2014;26:1030–9 [DOI] [PubMed] [Google Scholar]
- 33.Lake D, Correa SA, Muller J. Negative feedback regulation of the ERK1/2 MAPK pathway. Cell Mol Life Sci 2016;73:4397–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wiesener MS, Turley H, Allen WE, Willam C, Eckardt KU, Talks KL, et al. Induction of endothelial PAS domain protein-1 by hypoxia: characterization and comparison with hypoxia-inducible factor-1alpha. Blood 1998;92:2260–8 [PubMed] [Google Scholar]
- 35.Uniacke J, Holterman CE, Lachance G, Franovic A, Jacob MD, Fabian MR, et al. An oxygen-regulated switch in the protein synthesis machinery. Nature 2012;486:126–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends in Biochemical Sciences 2011;36:320–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morris EJ, Jha S, Restaino CR, Dayananth P, Zhu H, Cooper A, et al. Discovery of a Novel ERK Inhibitor with Activity in Models of Acquired Resistance to BRAF and MEK Inhibitors. Cancer Discovery 2013;3:742. [DOI] [PubMed] [Google Scholar]
- 38.Fernandez-Medarde A, Santos E. Ras in cancer and developmental diseases. Genes Cancer 2011;2:344–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weiss B, Bollag G, Shannon K. Hyperactive Ras as a therapeutic target in neurofibromatosis type 1. Am J Med Genet 1999;89:14–22 [PubMed] [Google Scholar]
- 40.Gattenlöhner S, Etschmann B, Riedmiller H, Müller-Hermelink HK. Lack of KRAS and BRAF Mutation in Renal Cell Carcinoma. European Urology 2009;55:1490–1 [DOI] [PubMed] [Google Scholar]
- 41.Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res 2004;64:7099–109 [DOI] [PubMed] [Google Scholar]
- 42.Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, et al. Sorafenib in Advanced Clear-Cell Renal-Cell Carcinoma. New England Journal of Medicine 2007;356:125–34 [DOI] [PubMed] [Google Scholar]
- 43.Motzer RJ, Tannir NM, McDermott DF, Arén Frontera O, Melichar B, Choueiri TK, et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. New England Journal of Medicine 2018;378:1277–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med 2015;373:1803–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Atkins MB, Plimack ER, Puzanov I, Fishman MN, McDermott DF, Cho DC, et al. Axitinib in combination with pembrolizumab in patients with advanced renal cell cancer: a non-randomised, open-label, dose-finding, and dose-expansion phase 1b trial. Lancet Oncol 2018;19:405–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adnane L, Trail PA, Taylor I, Wilhelm SM. Sorafenib (BAY 43‐9006, Nexavar®), a Dual‐Action Inhibitor That Targets RAF/MEK/ERK Pathway in Tumor Cells and Tyrosine Kinases VEGFR/PDGFR in Tumor Vasculature. Methods in Enzymology 2006;407:597–612 [DOI] [PubMed] [Google Scholar]
- 47.Huang D, Ding Y, Li Y, Luo WM, Zhang ZF, Snider J, et al. Sunitinib acts primarily on tumor endothelium rather than tumor cells to inhibit the growth of renal cell carcinoma. Cancer Res 2010;70:1053–62 [DOI] [PubMed] [Google Scholar]
- 48.Xin H, Zhang C, Herrmann A, Du Y, Figlin R, Yu H. Sunitinib inhibition of Stat3 induces renal cell carcinoma tumor cell apoptosis and reduces immunosuppressive cells. Cancer Res 2009;69:2506–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu L, Cao Y, Chen C, Zhang X, McNabola A, Wilkie D, et al. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res 2006;66:11851–8 [DOI] [PubMed] [Google Scholar]
- 50.Elorza A, Soro-Arnaiz I, Melendez-Rodriguez F, Rodriguez-Vaello V, Marsboom G, de Carcer G, et al. HIF2alpha acts as an mTORC1 activator through the amino acid carrier SLC7A5. Mol Cell 2012;48:681–91 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.