

Abstract
The fork-head box transcription factor FoxM1 is essential for hepatocellular carcinoma (HCC) development and its overexpression coincides with poor prognosis. Here, we show that the mechanisms by which FoxM1 drives HCC progression involve overcoming the inhibitory effects of the liver differentiation gene FoxA2. First, the expression patterns of FoxM1 and FoxA2 in human HCC are opposite. We show that FoxM1 represses expression of FoxA2 in G1 phase. Repression of FoxA2 in G1 phase is important, as it is capable of inhibiting expression of the pluripotency genes that are expressed mainly in S/G2 phases. Using a transgenic mouse model for oncogenic Ras-driven HCC, we provide genetic evidence for a repression of FoxA2 by FoxM1. Conversely, FoxA2 inhibits expression of FoxM1, and inhibits FoxM1-induced tumorigenicity. Also, FoxA2 inhibits Ras-induced HCC progression that involves FoxM1.
Keywords: DNMT3b, FoxA2, FoxM1, Hepatocellular carcinoma, Retinoblastoma protein
INTRODUCTION
Hepatocellular carcinoma (HCC) is the second most fatal malignancy in men worldwide (1,2). Development of HCC has been linked to viral hepatitis, alcohol abuse, as well as non-alcoholic steatohepatitis (3–5). Irrespective of its etiology, one-year survival with intervention is very low. That is partly due to high rate of recurrence of the cancer resulting from intrahepatic and extrahepatic metastasis (6,7). Recent studies have linked aggressive progression of HCC to over-expression of the fork-head box transcription factor FoxM1. For example, over-expression of FoxM1 has been shown to strongly correlate with poor prognosis and high-grade progression of HCC (8–10). Studies with mouse models provided strong causal link between FoxM1 and aggressive progression of HCC. It was shown that FoxM1 is essential for development of HCC in a chemical carcinogenesis model (11). Deletion of FoxM1 in the adult liver blocked Diethylnitrosamine (DEN)-induced HCC development. Moreover, in the same model of chemical carcinogenesis, deregulated FoxM1 drives highly aggressive, metastatic progression of HCC (12). In the absence of p19Arf, FoxM1 stimulates all steps of metastatic progression (12). Consequently, inhibition of FoxM1 impedes metastatic progression of HCC (12).
Similar observations were made with a transgenic mouse model expressing activated HRas in the liver, driven by the albumin promoter (10). In that model, HRas-induced HCC coincides with an increased expression of FoxM1. Conditional deletion of FoxM1 after HCC development causes inhibition of cancer progression. Hepatic progenitor cells for HCC have been characterized in the chemical carcinogenesis model. They express cell surface markers CD44 and EpCAM (13). Those cells were detected also in HRas-transgenic mouse model for HCC. They account for about 30 to 40% of the HCC cells in tumor sections (10). Interestingly, deletion of FoxM1 causes a preferential loss of those cells in the tumor nodules, indicating that FoxM1 is critical for the CD44 +ve and EpCAM +ve HCC cells (10). CD44 and EpCAM are expressed also by the human hepatic cancer stem cells. Moreover, the hepatic cancer stem cells in human HCC lines are dependent upon FoxM1, as deletion of FoxM1 causes a preferential loss of the cancer stem cells (10). In that regard, it is noteworthy that FoxM1 is a critical downstream factor of a variety of cancer signaling pathways, including Wnt/b-catenin signaling, that promote cancer stem cells (14). Moreover, FoxM1 stimulates expression of the pluripotency genes c-Myc, Oct4, Sox2 and Nanog (15), (16), (17).
Our recent studies on mammary luminal differentiation identified a transcriptional repression function of FoxM1 that is involved in regulation of the differentiation genes GATA3 (18). FoxM1 is expressed at high levels in the mammary stem and luminal progenitor cells. Deletion of FoxM1 decreases the population of the stem/progenitor cells and increases the differentiated luminal cells (10). Expression of FoxM1 has opposite effects in that it inhibits luminal differentiation (18). FoxM1 inhibits luminal differentiation by repressing the luminal differentiation gene GATA3. We showed that FoxM1 represses GATA3 expression by recruiting the retinoblastoma protein (Rb) and DNMT3b onto the GATA3 promoter (18). Moreover, in breast cancer, there is an inverse correlation between the expression of FoxM1 and GATA3 (18).
We sought to investigate whether the differentiation gene-repression function of FoxM1 is active in other systems where FoxM1 is over-expressed. FoxM1 is over-expressed in high-grade hepatocellular carcinoma (HCC) (10). The FoxA genes are important hepatic differentiation genes. For example, FoxA1 and FoxA2 were shown to be essential for liver development and hepatocyte differentiation (19). Interestingly, FoxA2 was shown to inhibit metastasis of the HCC cells (20). FoxM1, on the other hand, drives metastasis of HCC (21). Therefore, we investigated whether FoxM1 represses expression of FoxA2 in HCC. Here we show that, as in the case of GATA3 in breast cancer cells, FoxM1 inhibits FoxA2 in HCC cells by recruiting Rb and DNMT3b. Moreover, we show that FoxA2 inhibits FoxM1 expression, providing evidence for opposing roles of FoxM1 and FoxA2 in HCC.
MATERIALS AND METHODS
Cell Culture and Transfections
Human hepatocellular carcinoma Huh7, HepG2 and SNU449 cells were obtained from American Type Culture Collection. All experiments were performed within 8–10 passages. No further authentication of cells was performed. Cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (HyClone Laboratories Inc.) or 10% Tet System Approved FBS (Clontech, Mountain View, CA, Cat No. 631105) (for inducible cell lines) with 100 units of penicillin/ streptomycin were used to culture the cells at 37 °C with 5% CO2. Cells were transfected with plasmid DNA or siRNA using Lipofectamine™ 2000 (Invitrogen) in serum-free tissue culture medium following the manufacturer’s protocol. Six hours after transfection, cells were fed with complete Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum.
Immunohistochemistry and tissue microarray
Immunohistochemical staining was performed following standard procedure. Antigen retrieval was done using sodium citrate buffer and sections were then treated with antibodies overnight. Additional blocking step was performed using Avidin/biotin Vectastain kit following manufacturer’s protocol. Visualization was done using DAB and counterstained using Hematoxylin (Polyscientific, Bay Shore, NY). For antibodies of mouse origin, mouse on mouse (MOM) kit was used. All reagents are from Vector Labs (Burlingame, CA). Information about the antibodies is included in supplemental table 1.
Animal studies
All animal experiments were pre-approved by the UIC institutional animal care and use committee. Previously described Alb-H-ras12V mice were crossed with FoxM1fl/fl MxCre C57/BL6 mice to obtain FoxM1fl/fl MxCre Alb-H-ras12V and FoxM1+/+ MxCre Alb-H-ras12V mice. For deletion studies, eight months old male mice (FoxM1+/+ MxCre Alb-H-ras12V and FoxM1fl/fl MxCre Alb-H-ras12V) were subjected to five or ten intraperitoneal (IP) injections (every other day) with 250 µg of synthetic polyinosinic-polycytidylic acid (polyIpolyC) (Sigma-Aldrich, St. Louis, MO) to induce expression of the Mx-Cre transgene. The mice were sacrificed three weeks following the last injection, and the liver tissues and HCC nodules were harvested. For FoxA2 expression in mouse liver, tail vein injections (30-gauge needle) with 0.2 ml of adenovirus expressing FoxA2 or LacZ (1.7X1010 pfu/ml) in 10 months old Alb-H-Ras12V male mice were carried out under anesthesia.
RT-PCR, Western Blot, and Chromatin Immunoprecipitation
RNA was Trizol extracted (Invitrogen, Carlsbad, CA) and cDNA was synthesized using Bio-Rad reverse transcriptase (Bio-Rad, Hercules, CA). cDNA was amplified using SYBR Green (Bio-Rad, Hercules, CA) and analyzed via iCycler software. Western blots and chromatin-IPs were performed following previously described procedures (18). For chromatin-IPs, signals obtained with IgG and specific-antibodies were first normalized with signals obtained with those antibodies on a non-specific site in the GATA3 promoter (18). The normalized values were used to plot the fold enrichment with FoxM1-ab over IgG. For Rb-ChIP and DNMT3b-ChIP, we compared enrichments with same antibody, after normalization, in the presence and absence of FoxM1-siRNA, and the fold enrichments in control-siRNA over FoxM1-siRNA were plotted. All primer sequences and antibodies are included in supplemental table 1.
Isolation of genomic DNA
Genomic DNA (gDNA) was obtained from Huh7 cells and mouse tissue using Wizard™ Genomic DNA Purification kit as instructed by the manufacturers manual.
Bisulfite treatment and Quantitative methylation-specific PCR assay (qMSP)
Genomic DNA samples were treated with EZ DNA methylation™ kit (Zymo Research, Orange, CA) according to the manufacturer’s recommendation. The extent of methylation of desired gene was then measured by qPCR amplification with pairs of specific primers as mentioned in supplemental table-1 which were designed using MethPrimer MSP/BSP prediction and primer designing tool. Quantitative MSP was performed with using SYBR Green (Bio-Rad, Hercules, CA) and analyzed via iCycler software. Each reaction contained 20 ng of bisulfite-treated DNA as a template, 6.25 µl SYBR Green PCR (Bio-Rad, Hercules, CA) and 50 nM each forward and reverse primers in a total volume of 12.5 µl. The quantification cycle (Cq) was determined for each reaction with methylation-specific primers (MSP) the ratio of unmethylated to total amplifiable bisulfite-treated DNA was calculated.
Statistical analysis
Statistical significance was calculated by the Student’s t test (22) and Pearson correlation coefficient. Statistically significant changes were indicated with asterisks (*** p <0.001; **p<0.01;*p<0.05).
RESULTS
Opposite expression pattern of FoxM1 and FoxA2 in hepatocellular carcinoma
Expression of FoxM1 is low in non-tumor human liver sections, whereas FoxA2 is expressed at high levels in those sections (supplemental Fig. S1A). To investigate expression of FoxM1 and FoxA2 in HCC, we analyzed expression of the FoxM1 and FoxA2 proteins using tissue microarrays. We carried out immunohistochemical staining of tissue microarrays derived from consecutive sections of HCC specimens using specific antibodies against FoxM1 and FoxA2. There was an obvious difference in the expression pattern of FoxM1 and FoxA2. In the grade I samples, FoxA2 is vividly detectable, whereas the nuclear expression of FoxM1 is low (Fig. 1A-B). In the grade III specimens, on the other hand, expression of FoxA2 is low, but expression of FoxM1 is abundant. Pearson correlation analyses of FoxM1 and FoxA2 expressions in the consecutive TMAs indicated a strong negative correlation (Fig. 1C).
Figure 1: Expression of FoxM1 and FoxA2 in human HCC tumor tissue microarrays:
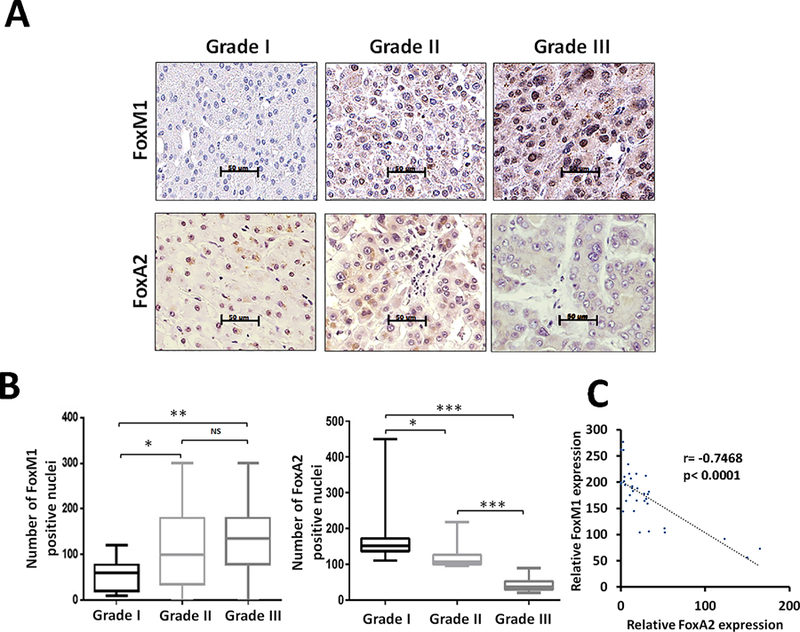
Human HCC tissue microarrays containing grade1 (n= 25), grade 2 (n= 19) and grade 3 (n= 27) were subjected to immunohistochemical staining using FoxM1 and FoxA2 antibodies. (A) The representative tissue cores from grades 1, 2 and 3 are shown. (B) Quantification of the imunohistochemical stains in the tissue cores corresponding to each of the grades was performed. (C) Pearson correlation between FoxM1 and FoxA2 expression is shown. Statistical calculations were performed using GraphPad. Statistical calculations were done using GraphPad Prism online tool for t-test and p values stated as *p≤0.05and **p≤0.001 and ***p≤0.0001. Image scale bar is 50µm.
Deletion of FoxM1 in a Ras-transgenic model for HCC causes accumulation of FoxA2
Recently, we studied the roles of FoxM1 in HCC progression using a transgenic mouse model that expresses oncogenic HRas in the liver (10). In that study, the floxed alleles of FoxM1 were deleted after HCC development using the MxCre deletion system, which is induced by injecting mice with double-stranded RNA (polyIpolyC). This system efficiently deletes floxed alleles in liver as well as in blood cells (23), and that somewhat mimics what would be expected from a drug that inhibits FoxM1. In those experiments, FoxM1-deletion in the HCC nodules were detected mainly in the HCC cells (10). Moreover, we showed that deletion of FoxM1 after HCC development inhibits HCC progression (10). Sections from those tumor nodules were analyzed for FoxA2 expression by immunohistochemistry. The HRas-derived tumor nodules without FoxM1-deletion exhibited very little expression of FoxA2 (Fig. 2A-B). But, in the FoxM1-deleted samples there was a significant increase in the FoxA2 (Fig. 2A-B). The observation was confirmed by western blot assays using extracts from tumor nodules with and without FoxM1-deletion (Figs. 2C). The increase in the expression of FoxA2 in the FoxM1-deleted samples provides in vivo genetic evidence that FoxM1 plays a role in the inhibition of FoxA2 in HCC. In normal mouse liver FoxM1 is expressed at a low level, whereas the expression of FoxA2 is abundant (supplemental Fig. S1B).
Figure 2: Increase in FoxA2 protein level in mouse HCC samples upon FoxM1 deletion:
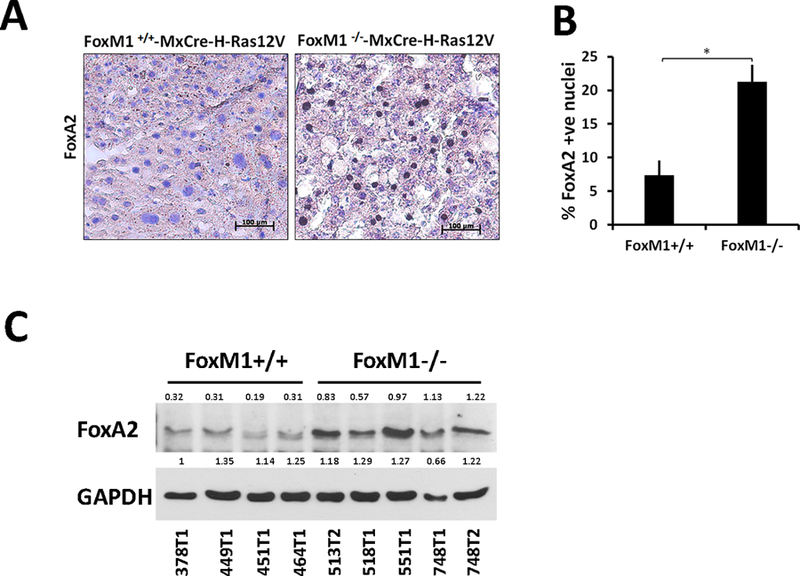
(A) Immunohistochemical staining of mouse HCC sections using FoxA2 antibody with and without deletion of FoxM1. HCC in those mice was driven by expression of oncogenic H-Ras. The mouse strains also harbored floxed alleles of FoxM1 and MxCre. FoxM1 deletion was induced following HCC development by injecting (5 times) the mice with polyIpolyC, as described before (10) (B) Graphs showing quantification of FoxA2 +ve HCC cells from 3 pairs of mice, and 5 random fields were chosen for analyses. The averaged SN ratio for FoxA2-mRNA expression is >3, which generates 80% statistical power with the number of mice used. (C) Comparison (0.1 mg of tumor extracts) of the FoxA2 protein-levels between FoxM1+/+ (4 different mice) and FoxM1 fl/fl (4 different mice, 2 different tumor nodules from one mouse) tumor tissue-extracts from mice injected with poly(I)poly(C) to delete the FoxM1 alleles, using western blots. The number above each band in FoxA2 western blots represents Image J quantification ratio calculated against GAPDH loading control. Whereas GAPDH blot shows intensity quantification relative to the first band set at 1. Statistical calculations were done using GraphPad Prism online tool for t-test and p values stated as *p≤0.05and **p≤0.001 and ***p≤0.0001. Image scale bar is 100µm.
FoxM1 directly inhibits expression of FoxA2 in HCC cells
Next, we determined the effects of FoxM1b over-expression and FoxM1-knockdown on the levels of FoxA2 using the widely used HCC cell lines HepG2, Huh7 and SNU449. The HepG2 cells express FoxM1 at low-levels (Fig. S2A), therefore, we used that line for over-expression experiments. The SNU449 cells express FoxM1 at high-levels, and we used that line for knock-down studies. Expression of FoxM1 in the Huh7 cells is in a range in between that observed in HepG2 and SNU449. Therefore, Huh7 was used for both knockdown and over-expression experiments. Expression of T7-tagged FoxM1b in Huh7 and HepG2 cells inhibited the levels of FoxA2 at both mRNA (Fig. 3A-B) and protein levels (Fig. S2C). GAPDH-mRNA values were used to calculate relative fold expression of FoxA2-mRNA. Knockdown of FoxM1 in Huh7 and SNU449 cells caused increase in the levels of FoxA2 in both mRNA (Fig. 3B-C) and protein levels (Fig. S2D-E). Also, we developed Huh7 stable cell lines in which FoxM1-shRNA can be expressed in an inducible manner by adding doxycycline in the culture medium. Expression of FoxM1-shRNA in three independent clones increased expression of FoxA2 proteins (Supplemental Fig. S2F).
Figure 3: FoxM1 inhibits expression of FoxA2 by recruiting Rb and DNMT3b onto the FoxA2 promoter:
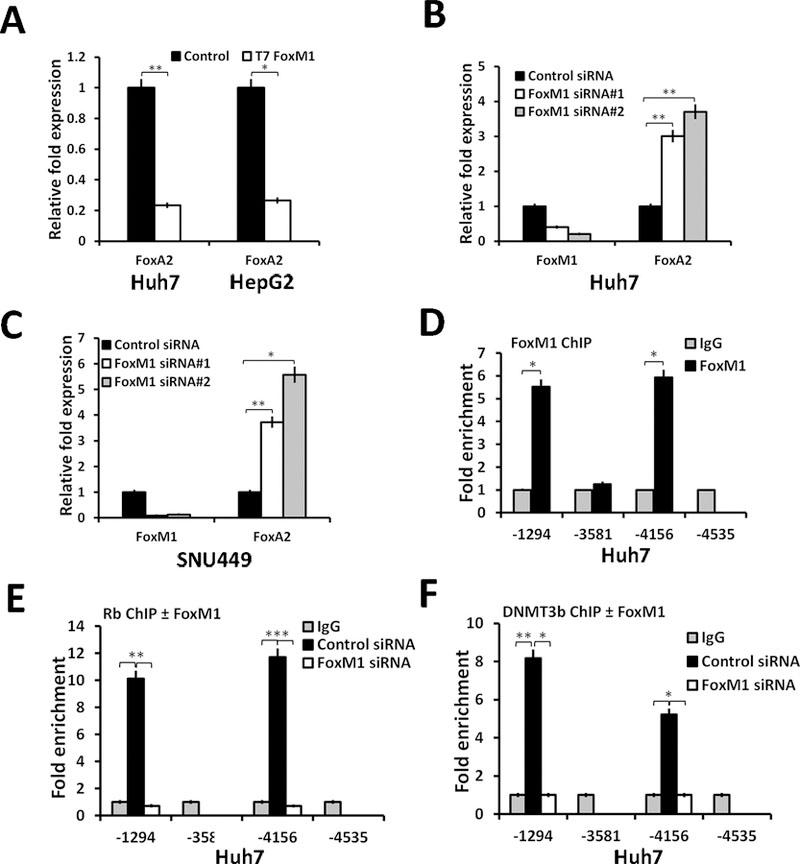
(A) Huh7 cells or HepG2 cells were transfected with empty vector (control) or vector expressing T7-FoxM1b. Forty-eight hours after transfection, cells were harvested for RNA assays. Total RNA (1µg) was analyzed by quantitative real time PCR (qRT-PCR) for expression of FoxA2 and GAPDH. Huh7 cells (B) or SNU449 cells (C) were transfected with control-siRNA or FoxM1-siRNAs. Seventy-two hours after siRNA transfection, cells were harvested for RNA (1 µg) assays. (D) Huh7 cells were subjected to crosslinking for chromatin-IP (18). The chromatin preparations were immunoprecipitated with FoxM1-ab or IgG. Enrichments of the FoxA2 promoter fragments were assayed by quantitative RT-PCR, and the relative enrichments with FoxM1-ab over that with IgG after normalization against a non-specific site are shown. (E-F) Huh7 cells transfected with control-siRNA or FoxM1 siRNA for 72h, and then processed for ChIP using Rb antibody (E) or DNMT3b-antibody. (F) Relative enrichments of the FoxA2 promoter fragments from control-siRNA transfected cells over those from the FoxM1-siRNA transfected cells are shown. Statistical calculations were done using GraphPad Prism online tool for t-test and p values stated as *p≤0.05and **p≤0.001 and ***p≤0.0001.
To determine whether a direct mechanism is in play, we sought to determine whether FoxM1 directly targets the FoxA2 promoter. The human FoxA2 gene contains at least four putative FoxM1-binding elements (Supplemental Fig. S3A). Chromatin-IP experiments using FoxM1-ab detected enrichment of DNA fragments encompassing the sites at −1294 and −4156 in the FoxA2 gene (Fig. 3D). The other sites in the FoxA2 upstream regions did not show any significant enrichment over that with the IgG (Fig. 3D). Previously, we showed that FoxM1 binds to both DNMT3b and Rb, forming a repressor complex in breast cancer cells (18). Immunoprecipitation of Huh7 cell extracts with a monoclonal antibody against FoxM1 (supplemental Fig. S3B) co-immunoprecipitated DNMT3b and Rb. Therefore, we carried out chromatin-IP experiments with Rb and DNMT3b antibodies using the Huh7 cells expressing control-siRNA or FoxM1-siRNA. Both Rb and DNMT3b bound to the same promoter-fragments that were enriched in chromatin-IP with FoxM1-ab (Fig. 3E-F). Moreover, knockdown of FoxM1 caused significant reduction in the bindings of Rb (Fig. 3E) and DNMT3b (Fig. 3F) onto the specific sites in the FoxA2 promoter. Chromatin-IP experiments with SNU449 cells further confirmed the binding of FoxM1, Rb and DNMT3b onto the FoxA2 promoter (supplemental Fig. S3C-E).
Recruitment of DNMT3b onto the FoxA2 promoter suggests that the repression by FoxM1 would involve methylation of CpG islands. To investigate that, Huh7 cells were transfected with FoxM1b-expresion vector or FoxM1-siRNA. Genomic DNAs from the transfected cells were treated with bisulfite followed by PCR using primers for the CpG islands in the FoxA2 promoter. Expression of FoxM1b caused an increase in CpG methylation near the FoxM1-binding sites in the FoxA2 gene (Fig. 4A and supplemental Fig. S3F). Moreover, knockdown of FoxM1 caused a decrease in CpG methylation at those sites in the FoxA2 promoter (Fig. 4B and Fig. S3G).
Figure 4: FoxM1 induces methylation of the FoxA2 promoters requiring Rb:
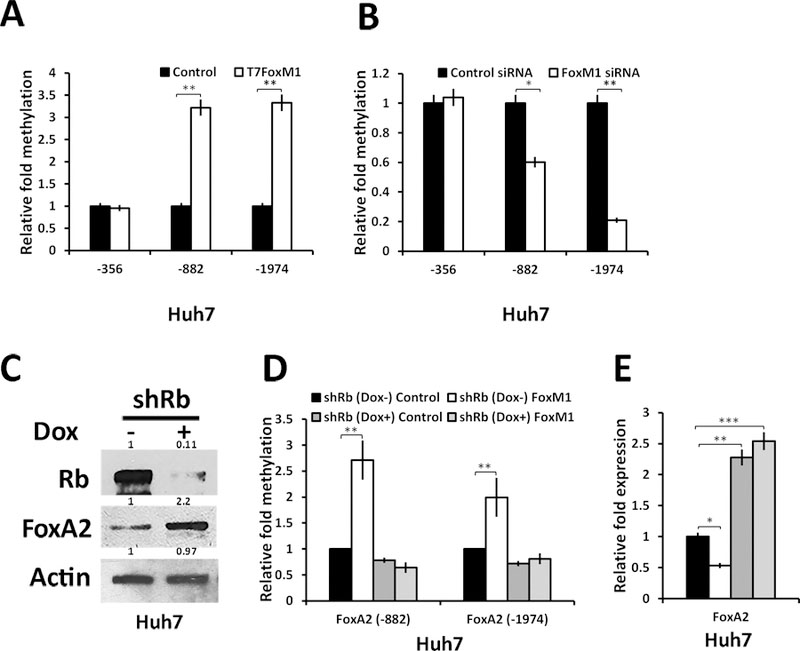
(A) Huh7 cells were transfected with empty vector (control) or T7-FoxM1b expressing vector. Genomic DNA was isolated and subjected to bisulphite treatment for CT conversion. Methylation of the FoxA2 upstream regions in control and FoxM1 transfected was assayed using qRT-PCR. (B) Bisulphite converted genomic DNAs isolated from Huh7 cells transfected with control- or FoxM1-siRNA were analyzed for methylation of the FoxA2 promoter by qRT-PCR. (C) Western blot (0.1 mg of cell-extracts) showing depletion of Rb in doxycycline-induced Huh7 cells and effect of Rb knock down on FoxA2 expression (The number above each band in western blots represents Image J quantification relative to control set at 1). (D) Huh7 cells stably expressing Dox inducible Rb-shRNA were transfected with control or T7-FoxM1 expression vector in the presence or absence of Dox as depicted. The genomic DNAs were isolated for CT conversion using bisulphite method and the difference in the promoter methylation of FoxA2 was assayed by qRT-PCR. (E) Quantification of FoxA2 expression using qRT-PCR in Huh7 cells expressing Dox inducible Rb-shRNA and transfected with either control vector or T7-FoxM1 in absence and presence of Dox. Statistical calculations were done using GraphPad Prism online tool for t-test and p values stated as *p≤0.05and **p≤0.001 and ***p≤0.0001.
Next, we employed an Rb-shRNA construct that allows inducible depletion of Rb in the presence of doxycycline (Fig. 4C). As shown in Fig. 4D, expression of FoxM1 increased methylation of the FoxA2 promoter in the presence of Rb (no doxycycline), but upon depletion of Rb (doxycycline) there was no increase in the CpG methylation at the indicated sites. Moreover, depletion of Rb caused increases in the expression of FoxA2 (Fig. 4E). Also, knockdown of DNMT3b increased expression of FoxA2 and reduced methylation of the FoxA2 promoter (Supplemental Fig. S4A-C). These observations demonstrate that the FoxM1/DNMT3b complex methylates and represses the FoxA2 promoters requiring Rb. The extent of FoxA2 repression by FoxM1b varied between 40 and 75%, which is likely due to variations in the levels of active Rb in the transfected cells. Changes in CpG methylation were detected also in the mouse HCC samples following deletion of mouse FoxM1 (Fig. S4D-E) that binds to Rb and DNMT3b (Fig. S4F).
FoxM1 inhibits FoxA2 in G1 and stimulates pluripotency genes in S/G2/M phases
Given that Rb is required for FoxM1 mediated repression of FoxA2, we predicted that repression occurs mainly in the G1 phase in which Rb is in the underphosphorylated form and is most active. Also, it is the underphosphorylated form of Rb that was shown to associate with FoxM1 (24). The doxycycline-inducible FoxM1-shRNA cells were treated with vehicle or doxycycline (300 ng/ml) for 96h, and then treated with Hoechst that allows fluorescent staining of DNA in live-cells. The cells were then fractionated using a cell-sorter to obtain cell-populations enriched for G1, S or G2/M cells. As expected, expression of the FoxM1-shRNA (Fig. 5A) caused an increase in the G1 population and reductions in the S and G2/M cells (Fig. 5B-C). Clearly, depletion of FoxM1 (open bars in Fig. 5D) caused increase in the expression of FoxA2 in G1 phase. Expressions of the pluripotency genes, on the other hand, were inhibited in the FoxM1-depleted cells, and the inhibition was observed mainly in the S and G2/M cells (Fig. 5E-F). There was no significant difference in FoxA2 expression in the G2/M phases (Fig. 5F) because FoxM1 is phosphorylated by Plk1, which blocks binding to Rb (25). We repeated this experiment with three independent clones of FoxM1-shRNA cells and observed similar results. The changes in gene expression were not indirect effects of G1-inhibition because similar changes were not observed for Oct-4 and Nanog in G1 cells. Also, an unrelated gene Fyn did not show any significant changes in the G1 and S phase cells. Doxycycline treatment alone, in the absence of FoxM1-shRNA, did not exhibit any significant effect on FoxM1 or FoxA2 expression in the parental Huh7 cells (supplemental Fig.S5).
Figure 5: FoxM1 inhibits the expression of FoxA2 in G1 phase of the cell cycle:
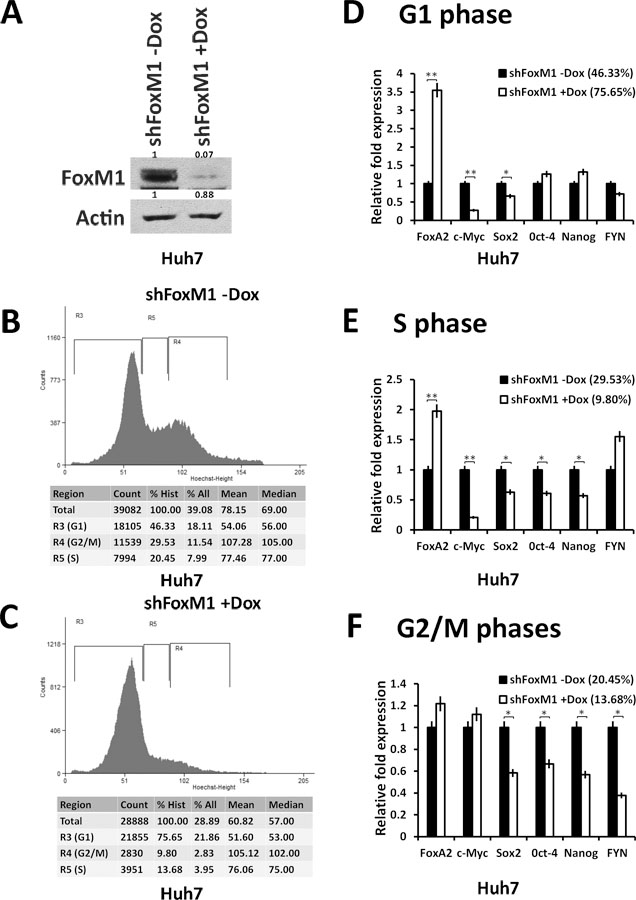
Doxycycline inducible Huh7-pINDUCER-shFoxM1 cells were either mock treated or treated with 300ng/ml of Doxycycline for 4 days to knock down of FoxM1. Cells were treated with Hoechest 33342 for one hour and then sorted to fractionate cells enriched for G1, S or G2/M phases of the cell cycle. (A) Whole cell extracts (100µg) were assayed for FoxM1 knock down by western blotting (The number above each band in western blots represents Image J quantification relative to control set at 1). (B) Representative cell cycle histogram of Hoechest 33342 treated Doxycycline inducible Huh7-pINDUCER-shFoxM1 cells mock treated and (C) treated with Doxycycline (300ng/ml). (D) Messenger-RNAs of the indicated genes from G1-phase cells, (E) S-phase cells and from (F) G2/M cells were assayed by quantitative real time PCR. Statistical calculations were done using GraphPad Prism online tool for t-test and p values stated as *p≤0.05and **p≤0.001 and ***p≤0.0001.
Ectopic expression of FoxA2 inhibits the pluripotency genes and blocks auto-activation of FoxM1
Expression of FoxM1 in HepG2 cells caused a significant increase in the number of spheres when cells were plated in sphere formation media (Fig. 6A-B). Expression of FoxA2, on the other hand, instead of increasing the number of spheres, caused decreases in the number of spheres in both HepG2 (Fig. 6A) and SNU449 cells (Fig. S6A). That is also consistent with the observation that expression of FoxA2 caused inhibition of the pluripotency genes along with FoxM1 and FoxM1 target genes (Fig. 6C and Fig. S6B, C). Moreover, expression of FoxA2 caused increases in the expression of the hepatocyte differentiation markers ALB, AAT and HNF4α (Fig. 6D). It is noteworthy that inhibition of FoxM1 also increased expression of those differentiation genes (supplemental Fig. S6D).
Figure 6: FoxA2 inhibits FoxM1 induced sphere formation by competing for FoxM1 promoter:
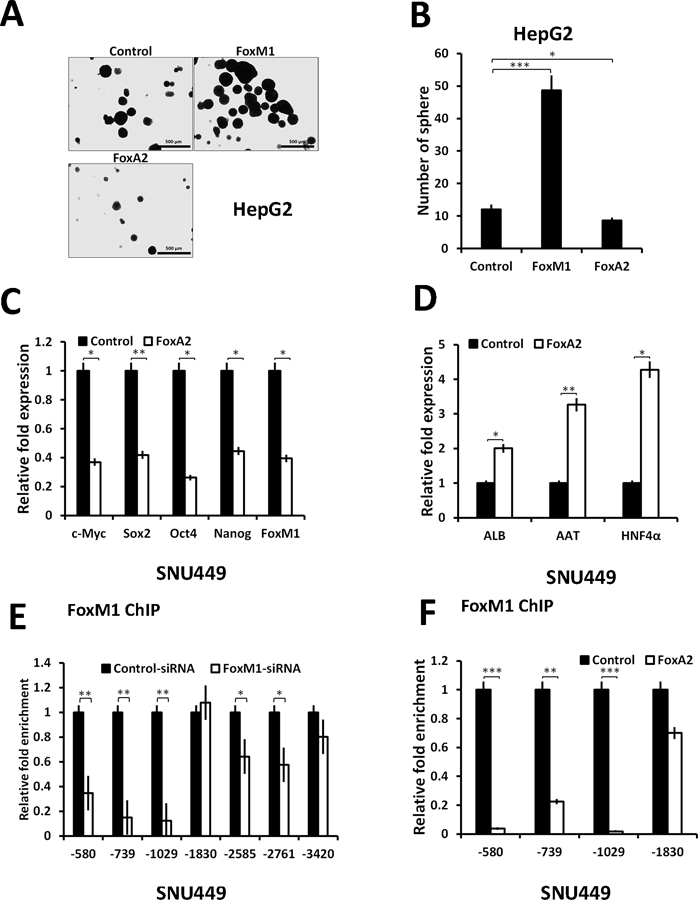
(A) HepG2 cells stably transfected with empty vector, FoxM1 or FoxA2 expression plasmid as indicated in the figure. Two hundred cells were then seeded in sphere formation medium in low attachment petri dishes. Cells were allowed to grow for a week. Cells were counted and representative picture was captured under microscope. (B) Shows the quantification of sphere formation assay. (C) SNU449 cells were transfected with Empty vector or FoxA2 expression vector. Cells were harvested 48h post-transfection and RNA was isolated to assay for stem cell marker genes and FoxM1 as indicated in figure. (D) Shows the effect of FoxA2 on the indicated liver differentiation genes. (E) SNU449 cell transfected with control siRNA and FoxM1 siRNA were harvested 72h post transfection. Cells were subjected for ChIP assay using FoxM1 antibody. Binding of FoxM1 on FoxM1 promoter were analyzed by qRT-PCR. (F) SNU449 cells were either transfected with control vector or with FoxA2 plasmid. Forty-eight hours post-transfection cells were harvested and processed for ChIP. FoxM1 binding was analyzed using qRT PCR. Statistical calculations were done using GraphPad Prism online tool for t-test and p values stated as *p≤0.05and **p≤0.001 and ***p≤0.0001. Image scale bar is 500µm.
The inhibition of FoxM1 by FoxA2 is interesting because that could be the mechanism by which FoxA2 inhibits the pluripotency genes. We did not detect an interaction between Rb and FoxA2. Therefore, we considered other possibilities. For example, FoxM1 was shown to auto-activate its own transcription (26). In chromatin-IP assays we detected interactions of FoxM1 with multiple sites in the FoxM1 promoter (Fig. 6E). Since FoxA2 bind to similar cognate DNA-elements, we considered the possibility that FoxA2 could compete with FoxM1 and inhibit its binding. Consistent with that notion we observed strong inhibitions of FoxM1-binding to its own promoter when FoxA2 was over-expressed (Fig. 6F). We analyzed the promoter-proximal binding sites because the enrichments on those sites were dependent upon FoxM1.
FoxA2 inhibits FoxM1b-induced clonogenicity and soft agar colony formation
FoxM1 is a pro-proliferation transcription factor that also inhibits apoptosis, and drives aggressive progression of cancers when over-expressed (21). If repression of FoxA2 is important, the prediction is that expression of FoxA2 would inhibit the FoxM1 pathways in HCC cells. We observed that expression of FoxM1 led to significant increases in clonogenicity of the Huh7 cells (Supplemental Fig. S7A-B). Expression of FoxA2 alone did not show any significant effect over control, but when expressed in combination with FoxM1 they strongly inhibited the FoxM1-induced increased clonogenicity of the Huh7 cells (Supplemental Fig. S7A-B). Similarly, co-expression of FoxA2 inhibited FoxM1-induced increase in soft agar colonies (Supplemental Fig. S7C-D). As expected from the observation that FoxA2 inhibit auto-activation of FoxM1, we consistently observed that expression of FoxA2 affected the levels of total FoxM1. FoxA2 did not have any significant effect on the co-expressed Flag tagged FoxM1b levels (Supplemental Fig. S7E, Flag panel). However, expression of Flag-FoxM1b increased the levels of the endogenous FoxM1 (Supplemental Fig. S7E, top panel), and co-expression of FoxA2 inhibited the increase of the endogenous FoxM1 (Supplemental Fig. S7E, top panel). The results are consistent with a model in which FoxM1 and FoxA2 have opposite regulatory effects on each other.
FoxA2 inhibits FoxM1 and Ras-induced HCC
We showed that FoxM1 is essential for progression of Ras-induced HCC (10). Based on the observation that FoxA2 inhibits FoxM1 expression, we predicted that FoxA2 would inhibit Ras-induced HCC progression. We tested that by tail vein injection of FoxA2-expressing adenovirus (Ad-FoxA2) in 10-months old Alb-Hras12V transgenic mice harboring HCC. Five male mice were injected with Ad-FoxA2 or Ad-LacZ twice. Second injection was done 12-days after the first and the livers were harvested 10-days after the second injection. Expression of FoxA2 caused significant reductions in the number of tumor nodules and tumor burden. Representative livers with HCC nodules are shown in Fig. 7A, and quantifications are shown in Fig. 7B-C. The inhibition of HCC progression was associated with inhibition of FoxM1 expression as well as Ki67+ve cells (Fig. 7D-F). These observations provide in vivo evidence that FoxA2 inhibits FoxM1 and Ras-induced HCC progression.
Figure. 7: Adenoviral Foxa2 expression inhibits HCC progression and FoxM1 expression.
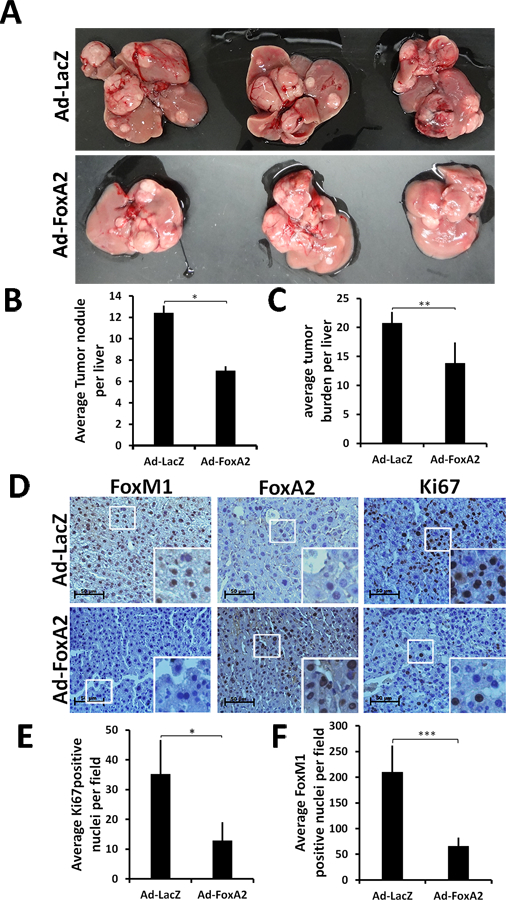
Alb-H-Ras12V male mice (n=5 per group) at 10 months of age were injected via the tail vein with purified Ad-FoxA2 or Ad-LacZ (0.2 ml at 1.7X1010 pfu per ml). Twelve days following first injection the mice were injected again with the same virus and sacrificed after 10 days. (A) Livers from three mice of each group are shown. (B) Average number of tumor nodules and (C) tumor burden are plotted. (D) IHC staining for FoxM1, FoxA2 and Ki67 are shown. (E) Average Ki67+ve nuclei/field and (F) average FoxM1+ve nuclei/field from 20 different fields of sections from 5 mice are plotted. The mouse experiment had 80% statistical power with 2-fold inhibition of the number of tumor nodules. Statistical calculations were done using GraphPad Prism online tool for t-test and p values stated as *p≤0.05, **p≤0.001 and ***p≤0.0001. Image scale bar is 50µm.
DISCUSSION
Results presented here are significant in several ways. First, our observations provide insights into the mechanisms by which FoxM1 drives accumulation of poorly differentiated cancer cells during high-grade progression of HCC. We show that FoxM1 suppresses expression of the hepatocyte-differentiation genes FoxA2 in G1 phase, and that suppression is important for expression of the pluripotency genes in S/G2 phases of the cell cycle. Also, we show that FoxA2 regulates FoxM1 expression by blocking its auto-activation mechanism. Our results suggest that it is the levels of FoxM1 versus FoxA2 that determine the differentiation state of the HCC cells.
Reduced expression of FoxA2 coincides with over-expression of FoxM1 (Fig. 1). The need for over-expression of FoxM1 might be related to low-abundance of active Rb in HCC cells. High levels of FoxM1 would be needed to seek out active under-phosphorylated Rb protein because it is the under-phosphorylated Rb that binds to FoxM1 (24). We suspect that the FoxM1 mediated inhibition of FoxA2 might be involved in de-differentiation of the HCC cells or maintenance of poorly differentiated HCC cells. Alternatively, in the event that the low- and high-grade HCCs develop from different progenitors, we speculate that the FoxM1 mediated repression of FoxA2 is important for the progenitors that give rise to high-grade HCC. It is noteworthy that, although we show regulation at the RNA-levels, TCGA datasets did not show opposite expression patterns of FoxM1 and FoxA2, which is likely related to RNA contributions from other cell types in the HCC nodules.
The involvement of Rb in the suppression of FoxA2 also suggests that the mechanism might be more active in G1 phase where under-phosphorylated Rb is abundant. Consistent with that, depletion of FoxM1 caused accumulation of FoxA2 mainly in G1 phase. Interestingly, studies on embryonic stem (ES) cell differentiation indicated that G1 phase is the phase in which the chromatin is available for the differentiation mechanisms, and that the ES cells retain pluripotency by suppressing differentiation mechanisms in G1 and increasing expression of the pluripotency genes in the S/G2 phases (27). In the light of those observations in ES cells, our observations that FoxA2 inhibits expression of the pluripotency genes and that FoxM1 inhibits FoxA2 are interesting because they explain how FoxM1 over-expression maintain poorly differentiated state of cells in high-grade HCC. The involvement of Rb in the suppression of the FoxA2 gene is surprising because it suggests that Rb participates in progression of high-grade HCC. We show that FoxM1 recruits Rb and DNMT3b onto the promoter of FoxA2, and increases methylation of the CpG islands in that promoter. Moreover, depletion of Rb blocks FoxM1-mediated increase in promoter-methylation and suppression of the FoxA2 gene. Together, the results suggest that, in the context of over-expressed FoxM1, there is a gain of function for Rb, and that function is related to the suppression of differentiation genes, which is likely involved in high-grade progression of HCC.
We provide evidence for a new regulatory loop in which over-expression of FoxA2 inhibits expression of FoxM1 and the FoxM1 target genes in HCC cells. TCGA database analyses revealed that FoxA2 is rarely mutated in HCC. Therefore, an understanding of how FoxM1 level increases in the presence of FoxA2 during HCC progression will require further analyses. FoxM1 enhancer/promoter are activated by multiple signaling pathways and cancer-relevant transcription factors, including Myc (28), HIF1 (29) and AP1 (30). Also, FoxM1 is regulated at the protein level. For example, CDK4 was shown to stabilize FoxM1 (31). Consistent with that, FoxM1 is expressed at high levels in HCC developed in the H-Ras12V transgenic mice, and that could be related to activation of the JNK1-AP1 as well as the Cyclin D/Cdk4 pathways by activated Ras (32) (33). Also, the mechanism by which FoxA2 inhibits FoxM1 is unclear. It remains possible that a repression-partner of FoxA2 is down-regulated during HCC progression. We speculate that a combination of those pathways overrides inhibition by FoxA2 to increase expression of FoxM1 during high-grade progression of HCC.
Supplementary Material
Implication:
The observations provide strong genetic evidence for an opposing role of FoxM1 and FoxA2 in HCC progression. Moreover, FoxM1 drives high grade HCC progression partly by inhibiting the hepatocyte differentiation gene FoxA2.
ACKNOWLEDGMENT
Authors thank Dr. S. Elledge (Harvard Medical School, Boston) for sharing the Rb-shRNA construct.
Financial Support: The work was supported by grants from National Institute of Health (CA 177655 and CA 175380) to PR. PR is supported also by a Merit Review Grant (BX000131) from the Veteran Administration.
Footnotes
Conflict of interest statement: The authors declare no potential conflicts of interest
REFERENCES
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 2011;61(2):69–90 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Siegel R, Ward E, Hao YP, Xu JQ, Murray T, et al. Cancer statistics, 2008. Ca-Cancer J Clin 2008;58(2):71–96 doi 10.3322/Ca.2007.0010. [DOI] [PubMed] [Google Scholar]
- 3.Blonski W, Kotlyar DS, Forde KA. Non-viral causes of hepatocellular carcinoma. World J Gastroenterol 2010;16(29):3603–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sanyal A, Poklepovic A, Moyneur E, Barghout V. Population-based risk factors and resource utilization for HCC: US perspective. Curr Med Res Opin 2010;26(9):2183–91 doi 10.1185/03007995.2010.506375. [DOI] [PubMed] [Google Scholar]
- 5.Tsai WL, Chung RT. Viral hepatocarcinogenesis. Oncogene 2010;29(16):2309–24 10.1038/onc.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Masuda T, Beppu T, Ishiko T, Horino K, Baba Y, Mizumoto T, et al. Intrahepatic dissemination of hepatocellular carcinoma after local ablation therapy. J Hepatobiliary Pancreat Surg 2008;15(6):589–95 doi 10.1007/s00534-007-1288-4. [DOI] [PubMed] [Google Scholar]
- 7.Utsunomiya T, Shimada M, Imura S, Morine Y, Ikemoto T, Mori M. Molecular signatures of noncancerous liver tissue can predict the risk for late recurrence of hepatocellular carcinoma. J Gastroenterol 2010;45(2):146–52 doi 10.1007/s00535-009-0164-1. [DOI] [PubMed] [Google Scholar]
- 8.Sun H, Teng M, Liu J, Jin D, Wu J, Yan D, et al. FOXM1 expression predicts the prognosis in hepatocellular carcinoma patients after orthotopic liver transplantation combined with the Milan criteria. Cancer Lett 2011;306(2):214–22 10.1016/j.canlet.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 9.Sun HC, Li M, Lu JL, Yan DW, Zhou CZ, Fan JW, et al. Overexpression of Forkhead box M1 protein associates with aggressive tumor features and poor prognosis of hepatocellular carcinoma. Oncol Rep 2011;25(6):1533–9 doi 10.3892/or.2011.1230. [DOI] [PubMed] [Google Scholar]
- 10.Kopanja D, Pandey A, Kiefer M, Wang Z, Chandan N, Carr JR, et al. Essential roles of FoxM1 in Ras-induced liver cancer progression and in cancer cells with stem cell features. J Hepatol 2015;63(2):429–36 doi 10.1016/j.jhep.2015.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kalinichenko VV, Major ML, Wang X, Petrovic V, Kuechle J, Yoder HM, et al. Foxm1b transcription factor is essential for development of hepatocellular carcinomas and is negatively regulated by the p19ARF tumor suppressor. Genes Dev 2004;18(7):830–50 doi 10.1101/gad.120070418/7/830 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park HJ, Gusarova G, Wang Z, Carr JR, Li J, Kim KH, et al. Deregulation of FoxM1b leads to tumour metastasis. EMBO Mol Med 2011;3(1):21–34 doi 10.1002/emmm.201000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He G, Dhar D, Nakagawa H, Font-Burgada J, Ogata H, Jiang Y, et al. Identification of liver cancer progenitors whose malignant progression depends on autocrine IL-6 signaling. Cell 2013;155(2):384–96 doi 10.1016/j.cell.2013.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gong A, Huang S. FoxM1 and Wnt/beta-catenin signaling in glioma stem cells. Cancer Res 2012;72(22):5658–62 doi 10.1158/0008-5472.CAN-12-0953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xie Z, Tan G, Ding M, Dong D, Chen T, Meng X, et al. Foxm1 transcription factor is required for maintenance of pluripotency of P19 embryonal carcinoma cells. Nucleic Acids Res 2010;38(22):8027–38 doi gkq715 [pii] 10.1093/nar/gkq715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bella L, Zona S, Nestal de Moraes G, Lam EW. FOXM1: A key oncofoetal transcription factor in health and disease. Semin Cancer Biol 2014;29:32–9 doi 10.1016/j.semcancer.2014.07.008. [DOI] [PubMed] [Google Scholar]
- 17.Wang Z, Park HJ, Carr JR, Chen YJ, Zheng Y, Li J, et al. FoxM1 in tumorigenicity of the neuroblastoma cells and renewal of the neural progenitors. Cancer Res 2011;71(12):4292–302 doi 0008-5472.CAN-10-4087 [pii] 10.1158/0008-5472.CAN-10-4087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carr JR, Kiefer MM, Park HJ, Li J, Wang Z, Fontanarosa J, et al. FoxM1 regulates mammary luminal cell fate. Cell Rep 2012;1(6):715–29 doi 10.1016/j.celrep.2012.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee CS, Friedman JR, Fulmer JT, Kaestner KH. The initiation of liver development is dependent on Foxa transcription factors. Nature 2005;435(7044):944–7 doi 10.1038/nature03649. [DOI] [PubMed] [Google Scholar]
- 20.Wang J, Zhu CP, Hu PF, Qian H, Ning BF, Zhang Q, et al. FOXA2 suppresses the metastasis of hepatocellular carcinoma partially through matrix metalloproteinase-9 inhibition. Carcinogenesis 2014;35(11):2576–83 doi 10.1093/carcin/bgu180. [DOI] [PubMed] [Google Scholar]
- 21.Raychaudhuri P, Park HJ. FoxM1: A Master Regulator of Tumor Metastasis. Cancer Res 2011;71(13):4329–33 10.1158/0008-5472.CAN-11-0640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aguilera KY, Brekken RA. Hypoxia Studies with Pimonidazole in vivo. Bio Protoc 2014;4(19). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuhn R, Schwenk F, Aguet M, Rajewsky K. Inducible Gene Targeting in Mice. Science 1995;269(5229):1427–9 doi DOI 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- 24.Major ML, Lepe R, Costa RH. Forkhead box M1B transcriptional activity requires binding of Cdk-cyclin complexes for phosphorylation-dependent recruitment of p300/CBP coactivators. Mol Cell Biol 2004;24(7):2649–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mukhopadhyay NK, Chand V, Pandey A, Kopanja D, Carr JR, Chen YJ, et al. Plk1 Regulates the Repressor Function of FoxM1b by inhibiting its Interaction with the Retinoblastoma Protein. Sci Rep-Uk 2017;7 10.1038/srep46017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Halasi M, Gartel AL. A novel mode of FoxM1 regulation Positive auto-regulatory loop. Cell Cycle 2009;8(12):1966–7 doi DOI 10.4161/cc.8.12.8708. [DOI] [PubMed] [Google Scholar]
- 27.Gonzales KA, Liang H, Lim YS, Chan YS, Yeo JC, Tan CP, et al. Deterministic Restriction on Pluripotent State Dissolution by Cell-Cycle Pathways. Cell 2015;162(3):564–79 doi 10.1016/j.cell.2015.07.001. [DOI] [PubMed] [Google Scholar]
- 28.Wierstra I, Alves J. FOXM1, a typical proliferation-associated transcription factor. Biol Chem 2007;388(12):1257–74 doi 10.1515/BC.2007.159. [DOI] [PubMed] [Google Scholar]
- 29.Raghavan A, Zhou GF, Zhou QY, Ibe JCF, Ramchandran R, Yang QW, et al. Hypoxia-Induced Pulmonary Arterial Smooth Muscle Cell Proliferation Is Controlled by Forkhead Box M1. Am J Respir Cell Mol Biol 2012;46(4):431–6 doi 10.1165/rcmb.2011-0128OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petrovic V, Costa RH, Lau LF, Raychaudhuri P, Tyner AL. FoxM1 regulates growth factor-induced expression of kinase-interacting stathmin (KIS) to promote cell cycle progression. J Biol Chem 2008;283(1):453–60 10.1074/jbc.M705792200. [DOI] [PubMed] [Google Scholar]
- 31.Anders L, Ke N, Hydbring P, Choi YJ, Widlund HR, Chick JM, et al. A Systematic Screen for CDK4/6 Substrates Links FOXM1 Phosphorylation to Senescence Suppression in Cancer Cells. Cancer Cell 2011;20(5):620–34 doi 10.1016/j.ccr.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park HJ, Carr JR, Wang Z, Nogueira V, Hay N, Tyner AL, et al. FoxM1, a critical regulator of oxidative stress during oncogenesis. Embo J 2009;28(19):2908–18 10.1038/emboj.2009.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aktas H, Cai H, Cooper GM. Ras links growth factor signaling to the cell cycle machinery via regulation of cyclin D1 and the Cdk inhibitor p27(KIP1). Mol Cell Biol 1997;17(7):3850–7 doi Doi 10.1128/Mcb.17.7.3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.