

Abstract
Aberrant regulation of EGFR is common in non-small cell lung carcinomas (NSCLC), and tumor resistance to targeted therapies has been attributed to emergence of other co-occurring oncogenic events, parallel bypass receptor tyrosine kinase pathways including IGF1R, and TNF-α-driven adaptive response via NF-κB. TNFAIP8, TNF-α-inducible protein 8, is an NF-κB-activated pro-survival and oncogenic molecule. TNFAIP8 expression protects NF-κB null cells from TNF-α-induced cell death by inhibiting caspase-8 activity. Here we demonstrate that knockdown of TNFAIP8 inhibited EGF and IGF-1-stimulated migration in NSCLC cells. TNFAIP8 knockdown cells showed decreased level of EGFR and increased expression of sorting nexin 1 (SNX1), a key regulator of the EGFR trafficking through the endosomal compartments, and treatment with SNX1 siRNA partially restored EGFR expression in these cells. TNFAIP8 knockdown cells also exhibited downregulation of IGF-1-induced pIGF1R and pAKT, and increased expression of IGF-1-binding protein 3 (IGFBP3), a negative regulator of the IGF-1/IGF1R signaling. Consistently, treatment of TNFAIP8 knockdown cells with IGFBP3 siRNA restored pIGF1R and pAKT levels. TNFAIP8 knockdown cells had enhanced sensitivities to inhibitors of EGFR, PI3K and AKT. Furthermore, immunohistochemical expression of TNFAIP8 was associated with poor prognosis in NSCLC. These findings demonstrate TNFAIP8-mediated regulation of EGFR and IGF1R via SNX1 and IGFBP3, respectively. We posit that TNFAIP8 is a viable, multi-pronged target downstream of the TNF-α/NF-κB axis, and silencing TNFAIP8 may overcome adaptive response in NSCLC.
Implication:
TNFAIP8 and its effectors SNX1 and IGFBP3 may be exploited to improve the efficacy of molecular targeted therapies in NSCLC and other cancers.
Introduction
Despite the fact that aberrant regulation of EGFR is quite frequently seen in non-small cell lung carcinoma (NSCLC), only a small percentage of NSCLC patients have responded to EGFR mutation-selective tyrosine kinase inhibitors (EGFR-TKIs) (1). Resistance to anti-EGFR therapies in advanced-stage NSCLC has been attributed to the secondary mutations or amplification of EGFR, emergence of co-occurring oncogenic events such as mutations in TP53, PIK3CA, BRAF, MET, CDK4 and CDK6, activation of parallel bypass signaling pathways via other receptor tyrosine kinases (RTKs) including AXL and IGF1R, and TNF-α-driven adaptive response (2–5). A large percentage of NSCLC (~ 30%) have gain-of-function mutations in KRAS associated with the primary resistance to EGFR-TKIs (6,7). Furthermore, most NSCLC patients do not respond to immune checkpoint inhibitor monotherapy (8,9). Multiple combination modalities, including agents targeting EGFR, ALK, immune checkpoints and/or immunosuppressive tumor microenvironment and chemotherapy are being tested; however, the long-term risks and benefits of these strategies in the treatment of NSCLC are currently unknown (10, 11). A better understanding of the mechanisms regulating EGFR expression and activity will advance the biology of naïve tumors, and inform rational strategies for the personalized, multimodality management of aggressive NSCLC.
TNF-α-inducible protein 8 (TNFAIP8) (aliases SCC-S2, GG2–1, NDED, TNFAIP8 variant 2) is an NF-κB-inducible, pro-survival, oncogenic and metastatic member of the TIPE family of proteins (12–19). The TIPE members have a highly conserved TIPE homology (TH) domain for binding to phosphoinositides and function as lipid transporters (20). TNFAIP8 expression is critical for inhibition of caspase-8 activity and evasion of drug-induced apoptosis by H1299 lung tumor cells expressing mutant p53 (K120R) (21). TNFAIP8 broadly represses wild type p53 in A549 lung cancer cells, and silencing of TNFAIP8 leads to enhanced p53 binding and induction of target gene expression, p53-dependent cell cycle arrest, and apoptosis in doxorubicin-treated lung cancer cells (22). Expression of transcriptional co-activator and a Hippo pathway effector YAP1 has been associated with resistance to TKI and BRAF inhibitors, upregulation of PD-L1, and poor survival in NSCLC (23, 24). TNFAIP8 has been shown to interact with LATS1, one of the Hippo core components, and promote nuclear localization of YAP and expression of downstream targets cyclin D1 and CDK6 in lung cancer cells (25).
The functional significance of TNFAIP8 in regulation of growth factor receptor tyrosine kinase signal transduction mechanisms remains unclear. Here we have investigated the effects of stable knockdown of TNFAIP8 on EGFR and IGF1R signaling primarily in KRAS mutant A549 NSCLC cells known to be relatively resistant to EGFR-TKIs (26). Our results demonstrate that depletion of TNFAIP8 results in loss of EGFR expression via upregulation of sorting nexin 1 (SNX1), previously shown to target EGFR to late endosomes/lysosomes (27–29). Similar observations were made in NRAS mutant H1299 NSCLC cells, KRAS mutant PANC-1 pancreatic cancer cells and MDA-MB-231 and LM2–4175 breast cancer cells, and C4–2B prostate cancer cells. We also demonstrate that knockdown of TNFAIP8 is associated with downregulation of IGF-1-inducible pIGF1R and pAKT levels via upregulation of IGF-1-binding protein 3 (IGFBP3), a known negative regulator of IGF-1/IGF1R signaling (30–32). Consistently, TNFAIP8 knockdown cells showed enhanced sensitivities to EGFR-TKI and the small molecule inhibitors of PI3K and AKT. In addition, our findings support the notion that TNFAIP8 expression is a poor prognosticator of NSCLC, warranting further study of TNFAIP8 in various molecular subtypes of NSCLC.
Materials and methods
Antibodies, growth factors and reagents used
Rabbit polyclonal anti-EGFR (D38B1), rabbit polyclonal anti-pEGFR Y1068 (D7A5), rabbit polyclonal anti-AKT (pan) (11E7), rabbit polyclonal anti-pAKT S473 (D9E), rabbit polyclonal anti-IGF-1 Receptorβ (IGF-1R), rabbit polyclonal anti-pIGF-1R Y1135 (DA7A8), rabbit polyclonal anti-ERK1/2 (p44/42 MAPK) (13F5), mouse monoclonal anti-pERK1/2 (p-p44/42 T202/Y204) (E10), rabbit monoclonal anti-Survivin (71G4B7), and rabbit polyclonal anti- Cleaved PARP (Asp214) antibodies were obtained from Cell Signaling (Danvers, MA). Mouse monoclonal anti-SNX1 and mouse monoclonal anti-EEA1 antibodies were obtained from BD Biosciences (Franklin Lakes, NJ). Rabbit polyclonal anti-IGFBP3 (H-98) and mouse monoclonal anti-β-ACTIN (AC-15) were obtained from Santa Cruz Biotechnology (Dallas, TX). Rabbit polyclonal anti-peptide TNFAIP8 antibody was custom made from Covance Antibody Products (Dedham, MA) as described earlier (15, 33). Conjugated secondary goat polyclonal anti-rabbit immunoglobulins-horseradish peroxidase (HRP) and goat polyclonal anti-mouse immunoglobulins-HRP were obtained from Dako (Carpinteria, CA). Alexa Fluor 488 goat anti-rabbit and Alexa Fluor 546 goat anti-mouse IgG1 (y1) were obtained from Life Technologies. Other reagents used were Selumetinib (AZD6244) (Selleck chemicals, Houston, TX), Erlotinib (Cell Signaling, Danvers, MA), LY294002 (Cell signaling, Danvers, MA), and MK-2206 2HCl (MK-2206) (Selleck chemicals, Houston, TX), and the XTT cell viability assay kit (Biotium, Fremont, CA).
Cell lines and culture conditions
Non-small cell lung cancer cell lines A549 (KRAS G12S) (Georgetown Lombardi Comprehensive Cancer Center Tissue Culture Shared Resource, TCSR) and H1299 (NRAS Q61K) (ATCC) were cultured in RPMI 1640 medium supplemented with heat-inactivated 10% fetal bovine serum (FBS) and 100 U/mL penicillin and 100 μg/mL streptomycin (P/S) (Life Technologies, Grand Island, NY). LM2–4175 cells, lung metastatic derivative of MD-MB-231 breast cancer cells (KRAS, G13D; BRAF, G464V; TP53, R280K), were a kind gift from Dr. Joan Massagué. Breast (MDA-MB-231 and LM2–4175), melanoma (MDA-MB-435), prostate (PC-3, TP53, K139fs31-del frame shift and C4–2B, TP53 wt) and pancreatic cancer cells (PANC-1, KRAS, G12D; TP53, R273H) (TCSR), and HEK293T cells (ATCC) were grown in Dulbecco’s modified essential medium (DMEM) with Glutamax and high glucose (Life Technologies) supplemented with heat-inactivated 10% FBS and P/S. Various cell lines were authenticated at TCSR using PowerPlex® 16 System (Promega). Mycoplasma testing at TCSR was done using the MycoAlert kit (Lonza). All cell lines were passaged and used in the laboratory within six months of receipt from TCSR or ATCC. All cultures were maintained in a humidified incubator at 37oC with 5% CO2.
TNFAIP8 shRNA lentivirus and stable transductions
We first performed TNFAIP8 shRNA transient transfections using five different TNFAIP8 shRNAs detailed in Supplementary materials, and identified TNFAIP8 shRNA #3 as the most potent sequence. Cancer cell lines stably expressing TNFAIP8 shRNA #3 (shT8) (5’- CCGGCATCAAGCTGGCCATTCTTTACTCGAGTAAAGAATGGCCAGCTTGATGTTTTTG-3’), scrambled shRNA (scr) (5’-CCTAAGGTTAAGTCGCCCTCGCTCGAGCGAGGGCGACTTAACCTTAGG-3’), or empty vector (pLKO.1) were established using the pLKO.1 lentiviral transduction procedure (Addgene) as detailed in Supplementary materials.
Growth factor treatments and immunoblotting
Stock solutions of EGF and IGF-1 were prepared at 500 μg/mL in phosphate buffered saline (PBS), and FGF-1 and VEGF were reconstituted in PBS containing 0.1% BSA at 25 μg/mL and 100 μg/mL, respectively. All stock solutions were aliquoted and stored at −80oC. Growth factor treatment conditions were followed as described before (30, 34, 35). In brief, logarithmically growing cells were seeded in 6-well tissue culture dishes (3×105 cells per well) in complete medium containing 10% FBS. At approximately 90% confluency (24 hr to 48 hr post-seeding), medium was replaced with SFM with P/S and incubations continued for 12 hr. Next day, SFM was replaced with fresh SFM containing desired final concentration of growth factor for indicated times. Cells were scraped in lysis buffer and the lysates were immediately processed for immunoblotting following the AbCAM Western Blotting guide (http://www.abcam.com/index.html?pageconfig=resource&rid=11375) as detailed in the Supplementary materials.
Detection of IGFBP3 in culture medium
To determine the level of IGFBP3 secreted into culture medium, logarithmically growing scr A549 and shT8 A549 cells were seeded in duplicate 6-well tissue culture dishes (0.3 × 106 cells/well) in complete medium containing 10% FBS. At approximately 90% confluency, medium was replaced with serum free RPMI 1640 with penicillin/streptomycin (SFM) (2 mL/well) for 12 hr. Culture media from scr A549 cells (scr-cm) and shT8 A549 cells (shT8-cm) were then collected in 15 mL tubes and stored on ice. For immunoblotting, normalized volume of culture medium (equivalent to approximately equal number of viable/adherent cells) was loaded per lane, followed by immunoblotting with anti-IGFBP3 antibody.
Culture medium treatments
Logarithmically growing cells were seeded in 6-well tissue culture dishes in complete medium containing 10% FBS. At approximately 90% confluency, medium was replaced with serum free RPMI 1640 with P/S (SFM) for 12 hr. Next day, medium was removed and replaced with 2 mL culture medium collected from scr A549 cells (scr-cm) or shT8 A549 cells (shT8-cm), or cells were left untreated (ut) for 30 min, followed by cell lysis and sequential immunoblotting with anti-pAKT and anti-β-ACTIN antibodies.
Immunofluorescence
Logarithmically growing cells were seeded at 1 × 105 cells on a sterile glass coverslip placed in a 12-well plate in complete medium. After 48 hr, cells were switched to SFM for 12 hr, and treated for various times with 50 ng/mL EGF or left untreated (-EGF). The immunofluorescence method is detailed in the Supplementary methods. In brief, cells were fixed in 4% paraformaldehyde and permeabilized in 0.1% Triton X-100 for 20 min, followed by blocking in 3% normal goat serum for 20 min. For immunostaining, combinations of two primary antibodies were mixed in 3% goat serum in PBS for a final dilution of 1:200 per antibody: rabbit anti-EGFR antibody plus mouse SNX1 antibody, and rabbit anti-EGFR antibody plus mouse anti-EEA1 antibody. The cells were incubated with a mixture of the two primary antibodies for 1 hr at RT, washed with PBS three times for 5 min each, followed by treatment with a mixture of two secondary antibodies Alexa Fluor 488 goat anti-rabbit A11034 (1/200 dilution), Alexa Fluor 546 goat anti-mouse IgG1 (y1) A21123 (1/200 dilution), and 1 μg/mL 4’,6-diamidino-2-phenylindole (DAPI) in 3% goat serum in PBS. After 1 hr, coverslips were washed in PBS and mounted on a glass slide in Fluorogel. The colocalization of EGFR and SNX1 (green and red) or EGFR and EEA1 (green and red) was detected by confocal immunofluorescence microscopy. Photomicrographs were visually scored for EGFR+ve puncta (green), EEA1+ve puncta (red), and EGFR+ve and EEA1+ve puncta (merge, yellow).
Wound healing assay
A549 cells stably transduced with TNFAIP8 shRNA (shT8 A549) or scrambled shRNA (scr A549) were seeded in 6-well plates at 0.3×106 cells/well and grown to 90% confluency in complete medium. Medium was replaced with serum-free medium (SFM) for 12 hr. A 200 μL pipette tip was used to scrape the cells through the center of the well longitudinally, and a line was drawn with a felt tip pen on the outside of the well perpendicular to the scratch as described (36). Immediately after marking the scratch, cells were rinsed in SFM, and medium was replaced with fresh SFM containing desired concentration of the growth factor. Phase contrast microscopy images of the wound at 10x magnification were captured immediately after wound generation, and at 6, 12, 18, and 24 hr post-scratch. Two images were captured per scratch per time point, above and below the felt marked line. At various times post-scratch, the wound area was quantified using ImageJ software (National Institutes of Health, Bethesda, MD) and data, mean ± standard deviation (SD), were plotted relative to the initial wound area (0 hr).
SNX1 siRNA and IGFBP3 siRNA transfections
A549 cells stably transduced with TNFAIP8 shRNA (shT8 A549) were seeded in 12-well tissue culture plates with 0.1 × 106 cells per well in complete medium. At 70% confluency, medium was replaced with 400 μL Optimem medium. Mixture of siRNA (0.25 μL of 100 μM stock) and LipofectAmine 2000 (4 μL) was resuspended in 100 μL Optimem medium and incubated at room temperature (RT) for 20 min. The mixture was added to the cells to a final concentration of 50 nM siRNA (siSNX1 #1, On-Targetplus SMARTpool; siSNX1 #2, 5’-AAGAACAAGACCAAGAGCCACUU-3’; siIGFBP3, On-Targetplus SMARTpool; or siRNA On-Targetplus SMARTpool control). After 6 hr, 500 μL of complete medium was added per well, and after additional 48 hr, culture medium was changed to SFM with or without 50 nM siRNA for 12 hr, followed by cell lysis and immunoblotting. For growth factor treatment, siRNA containing medium was replaced with fresh SFM and growth factor.
RT-PCR and qRT-PCR assays
Total RNA was extracted using RNeasy RNA miniprep kit according to manufacturer’s instructions (Qiagen), and 1 μg of RNA was reverse-transcribed to cDNA using SuperScriptIII First-Strand Synthesis SuperMix for qRT-PCR kit according to manufacturer’s protocol (Life Technologies) as detailed in Supplementary materials and reported earlier (37).
Cell viability assay
Sensitivities of shT8 A549 and scr A549 cells to Erlotinib, LY294002, and MK-2206 were tested using the XTT Cell Viability Assay Kit. Cells were seeded in DMEM complete medium supplemented with 10% FBS and P/S at a density of 1000 cells/100 μL/well (6 wells per data point) in a 96-well plate for approximately 16 hr. Next day medium was replaced with 100 μL complete medium containing indicated drug concentration for 72 hr. For vehicle control cells, medium containing 0.1% DMSO was used. Activated XTT reagent (2,3-Bis-(2-Methoxy-4-Nitro-5-Sulfophenyl)-2H-Tetrazolium-5-Carboxanilide) was added to each well (25μL/well) and incubations continued for additional 4hr. Absorbance was measured at 450 nm on a Victor III spectrophotometer plate reader (Perkin Elmer, Waltham, MA), and normalized to untreated cells. The experiment was repeated at least three times.
Tissue samples and immunohistochemistry
A total of 144 formalin-fixed, paraffin-embedded NSCLC specimens (122 primary tumor resections and 22 Tissue Microarrays samples) including 47 squamous cell carcinomas (SCC), 59 adenocarcinomas (AC), and 38 bronchioloalveolar carcinomas (BAC) were immunostained by a manual method using a custom made and validated anti-TNFAIP8 antibody as described earlier (33). The Tissue Microarrays (TMAs) were purchased from US Biomax, Inc. (Rockville, MD). The staining pattern was semi-quantitatively assessed based on staining intensity and distribution and the results were correlated with morphologic and prognostic variables.
Statistical analyses
Statistical significance was analyzed using the Student’s T-test. All means and confidence intervals were calculated using Microsoft Excel (Microsoft Corporation, Seattle, WA). Two-sided P-values were calculated and P < 0.05 was considered statistically significant. For statistical analysis of TNFAIP8 expression in clinical specimens, we used Pearson chi-square to test associations and Cox Proportional Hazard model for survival analysis.
Results
Inhibition of EGF and IGF-1-stimulated cell migration in TNFAIP8 knockdown lung cancer cells
TNFAIP8 knockdown (shT8) A549 lung cancer cells and scrambled (scr) control A549 cells were established using stable lentiviral transduction method as detailed in Materials and methods. Approximately 70% inhibitions of the TNFAIP8 protein and mRNA levels were seen in shT8 A549 cells relative to scr control (Fig. 1, A and B). In a time course experiment, significant decrease in cell migration (i.e. wound closure) in response to EGF was observed in shT8 A549 cells versus scr control (Fig. 1C, left panel). Under control SFM conditions, no significant difference was noted in wound closure in shT8 cells versus scr (all P-values > 0.05) (Fig. 1C, right panel). In an independent set of experiments, shT8 A549 cells showed a significantly delay in wound closure at 24 hr post-scratch as compared to scr control in the presence of four different growth factors tested, EGF, IGF-1, FGF-1, and VEGF (Fig. 1D and Fig. S1). These data demonstrate that knockdown of TNFAIP8 expression significantly decreases growth factor-induced cell migration in A549 lung cancer cells.
Figure 1. Stable knockdown of TNFAIP8 inhibits EGF and IGF-1-induced cell migration in A549 lung cancer cells.
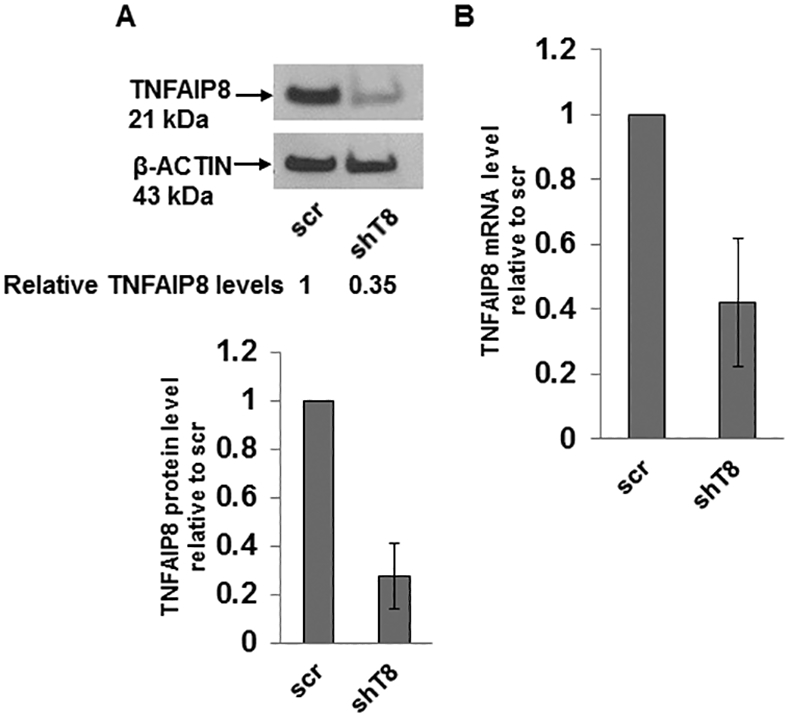
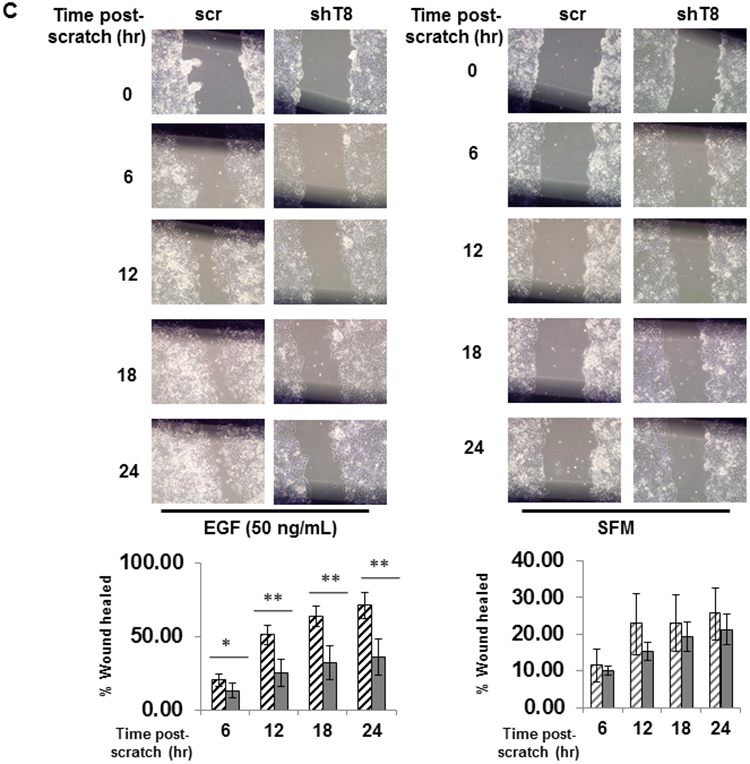
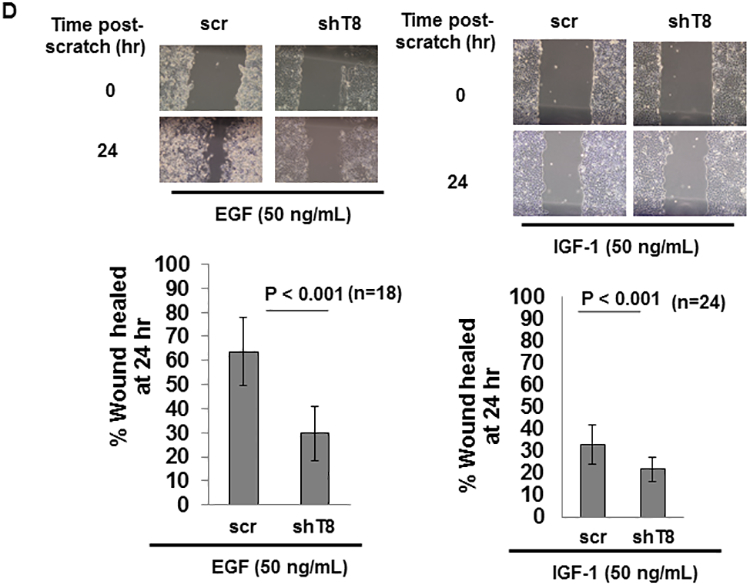
A. Upper panel, Immunoblot showing TNFAIP8 expression in scrambled shRNA (scr) and TNFAIP8 shRNA (shT8) transduced cells. Lower panel, Relative TNFAIP8 protein levels were quantified after normalization against the β-ACTIN signal in the corresponding lanes. Data shown are mean ± SD (n =3). B. qRT-PCR analysis of TNFAIP8 mRNA in shT8 cells versus scr. Data shown are mean ± SD (n =12). C. Wound healing assay showing decreased migration of shT8 cells in response to EGF. Top panels, Phase contrast microscopy images of wound closures at various time points post-scratch in the presence of EGF (left) or serum-free medium (SFM) (right). Bottom panels, Quantification data are shown as % of wound healed relative to 0 hr wound in scr cells (hatched bars) and shT8 cells (solid bars). Values shown are mean ± SD and each data point represents two fields per scratch per well (n =3). *, P-value < 0.05, **, P-value <0.001. D. Top panels, Phase contrast microscopy images of wound closures at 24 hr post-scratch in the presence of EGF (left) or IGF-1 (right). Bottom panels, Quantification data (mean ± SD) shown as % of wounds healed relative to 0 hr wounds in scr and shT8 cells.
Downregulation of EGFR expression and upregulation of vesicular trafficking protein SNX1 in TNFAIP8 knockdown cancer cells
We next investigated the effects of TNFAIP8 knockdown on expression and activity of EGFR in A549 cells. In the absence of EGF, moderate inhibition of EGFR expression (~ 40%) was observed in shT8 cells versus scr (Fig. 2A). No change in EGFR mRNA level was seen in shT8 A549 cells versus scr (Fig. S2). Upon treatment with various doses of EGF for 30 min, EGFR protein levels were further inhibited in shT8 cells versus scr. Representative data are shown in Fig. 2A. Consistent with decreased levels of total EGFR, pEGFR expression was also decreased in shT8 cells versus scr at all EGF doses tested (Fig. 2B). Basal expression of EGFR protein was also reduced by ~ 70% in TNFAIP8 knockdown H1299 cells by immunoblotting as compared to scrambled counterpart. Decreased basal levels of EGFR protein expression were also noted in the antibody microarrays analysis of three additional models of TNFAIP8 knockdown versus scrambled controls (MDA-MB-231, ~1.3-fold; PC-3, ~1.7-fold; MDA-MB-435, ~1.3-fold). Thus knockdown of TNFAIP8 expression was found to promote downregulation of EGFR expression in multiple cancer cell lines tested.
Figure 2. Decreased expression of EGFR and pEGFR and increased expression of SNX1 in TNFAIP8 knockdown A549 cells.
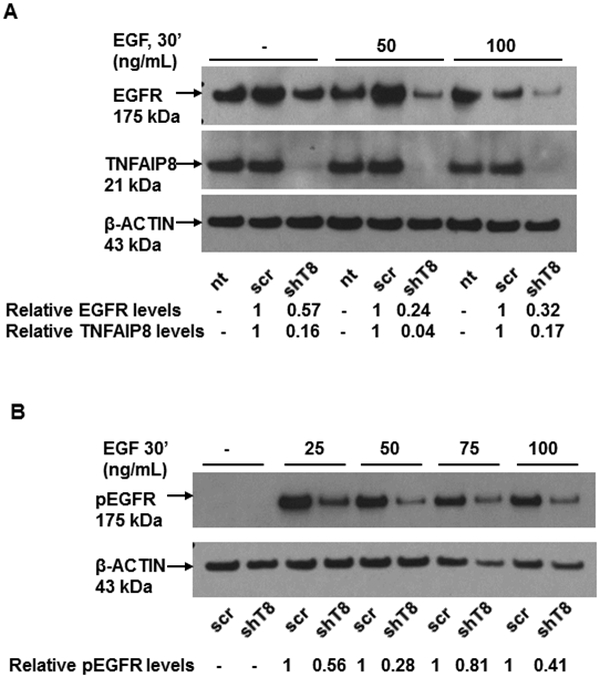
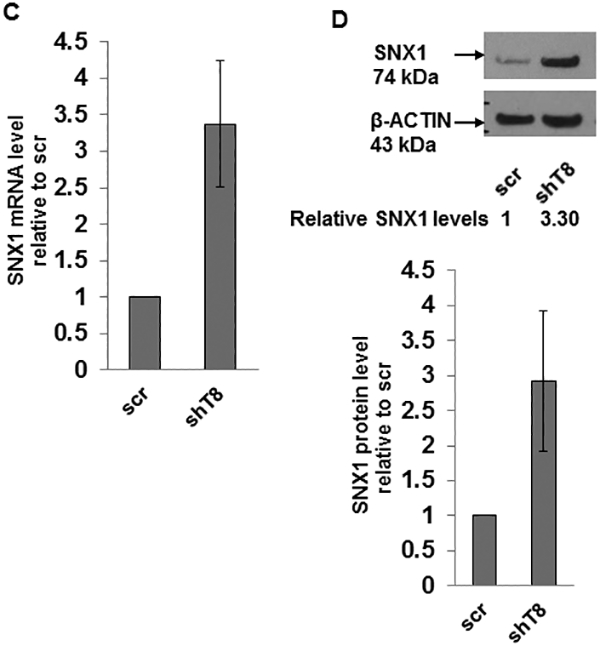
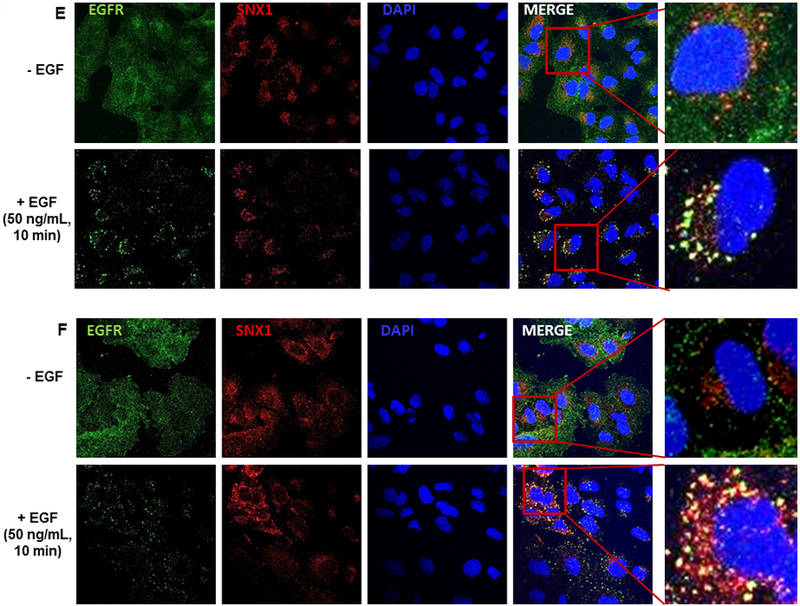
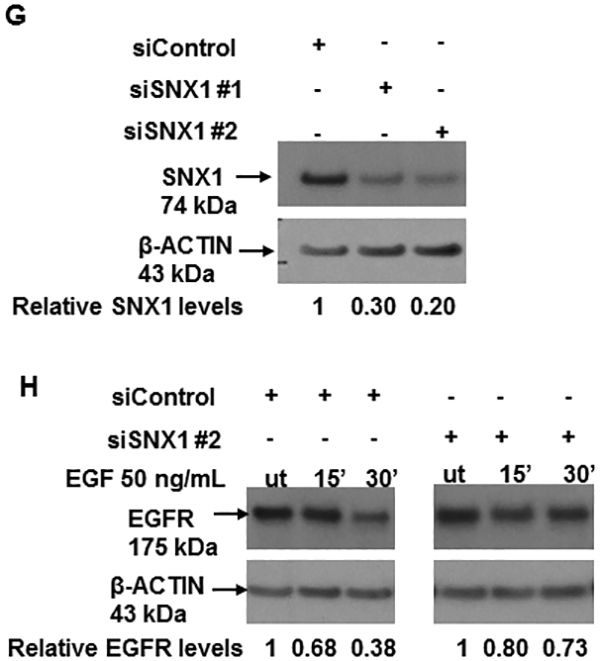
A. Downregulation of total EGFR expression in shT8 cells versus scr. The blot was sequentially probed with anti-EGFR, anti-TNFAIP8 and anti-β-ACTIN antibodies. Relative EGFR and TNFAIP8 protein levels were quantified after normalization against the β-ACTIN signal in the corresponding lanes. nt, non-transfected. B. Decreased expression of pEGFR expression in shT8 cells versus scr. The blot was sequentially probed with anti-pEGFR and anti-β-ACTIN antibodies. Relative pEGFR levels were quantified after normalization against the β-ACTIN signal in the corresponding lanes. C. qRT-PCR analysis showing increased expression of SNX1 mRNA in shT8 cells versus scr. SNX1 scores were normalized to ribosomal protein large, P0 (RPLP0) control. Quantification data shown are mean ± SD (n =24). D. Top panel, Increased expression of SNX1 in shT8 cells versus scr. Relative SNX1 levels were quantified after normalization against β-ACTIN signal in the corresponding lanes. Bottom panel, Quantification data shown are mean ± SD (n =8). E. Immunofluorescence staining showing co-localization of EGFR and SNX1 upon EGF treatment of scr A549 cells. F. Immunofluorescence staining showing co-localization of EGFR and SNX1 upon EGF treatment of shT8 A549 cells. G. SiRNA knockdown of SNX1 expression in shT8 A549 cells. shT8 A549 cells were transfected with 50 nM of indicated SNX1 siRNA (#1 or #2) or siControl for 12 hr, followed by cell lysis and sequential immunoblotting with anti-SNX1 and anti-β-ACTIN antibodies. Quantification data shown were normalized against β-ACTIN signal in the corresponding lanes. H. Partial restoration of EGFR expression in SNX1 siRNA knockdown shT8 A549 cells. For EGF treatment, SNX1 siRNA #2 containing medium was replaced with fresh serum-free medium containing EGF for indicated times, followed by cell lysis and sequential immunoblotting with anti-EGFR and anti-β-ACTIN antibodies. Quantification data shown were normalized against β-ACTIN signal in the corresponding lanes. ut, untreated.
Earlier reports have shown that overexpression of SNX1, a key vesicular trafficking protein associated with the early endosome-to-lysosome sorting of cell surface receptor, leads to direct binding of SNX1 with EGFR, accelerated degradation of EGFR through the endocytic-lysosomal pathway and cell death (27–29). Our results showed that mRNA and protein levels of SNX1 were increased by ~3-fold in shT8 A549 cells versus scr (Fig. 2, C and D). The RNA arrays profiling showed higher SNX1 mRNA levels (~1.4-fold) in TNFAIP8 knockdown PC-3 cells versus scrambled counterparts. Increased expression of SNX1 protein was also observed in three other shT8 models, H1299 NSCLC cells, PANC-1 pancreatic cancer cells and C4–2B prostate cancer cells (Fig. S3, A–C). The antibody microarrays expression profiling also showed higher SNX1 protein levels in two TNFAIP8 knockdown breast tumor cells versus scrambled counterparts (MDA-MB-231, ~1.5-fold; LM2–4175, ~1.5-fold). We have also detected enhanced expression of SNX1 in shT8 A549 cells versus scr, and demonstrated the co-localization of SNX1 and EGFR in response to EGF by the immunofluorescence method (Fig. 2, E and F). To verify that TNFAIP8 knockdown cells showed decreased levels of EGFR via upregulation of SNX1 expression, SNX1 was knocked down in shT8 A549 cells using SNX1 siRNA, siSNX1 #1 or siSNX1 #2 (Fig. 2G). As shown in Fig. 2H, in the siControl -treated shT8 A549 cells, EGFR expression was ~60% at 30 min of EGF treatment versus untreated cells (ut), whereas shT8 cells treated with siSNX1 #2 showed only ~ 30% inhibition of EGFR expression at 30 min post-EGF treatment, suggesting that siSNX1 knockdown partially restored EGFR expression in shT8 A549 cells.
The early endosomal association of SNX1 has been shown to be important in its targeting of internalized EGFR for lysosomal degradation (38, 39). To address whether the localization of EGFR to early endosomes following ligand stimulation was differentially regulated in TNFAIP8 knockdown cells versus scr control, immunofluorescence staining was performed in ligand-stimulated shT8 A549 cells and scr A549 cells using anti-EGFR antibody, and anti-EEA1 antibody to detect early endosome antigen 1 (EEA1), a marker of early endosomes. Following EGF treatment, EGFR and EEA1 showed strong co-localization at 10 min, and the co-localization of EGFR with EEA1 was reduced at 30 min in both shT8 A549 and scr A549 groups. However, the reduction in EGFR localized to early endosomes at 30 min was found to be significantly higher in shT8 A549 cells versus scr (Fig. 3A, and Fig. S4, A and B). These data suggest a relatively rapid exiting of EGFR from early endosomes in shT8 A549 cells versus scr. TNFAIP8 knockdown A549 cells also showed inhibition of EGF-induced pERK expression versus scr (Fig. 3B). Treatment of shT8 A549 cells with Selumetinib (AZD6244), a small molecule inhibitor of MEK resulted in increased expression of cleaved PARP relative to scr (Fig. 3C). In addition, EGFR-TKI erlotinib was found to be more cytotoxic in shT8 A549 cells as compared to scrambled control cells (Fig. 3D).
Figure 3. TNFAIP8 knockdown cells show rapid exit of EGFR from early endosomes, suppression of pERK expression, and enhanced sensitivity to EGFR-TKI, erlotinib.
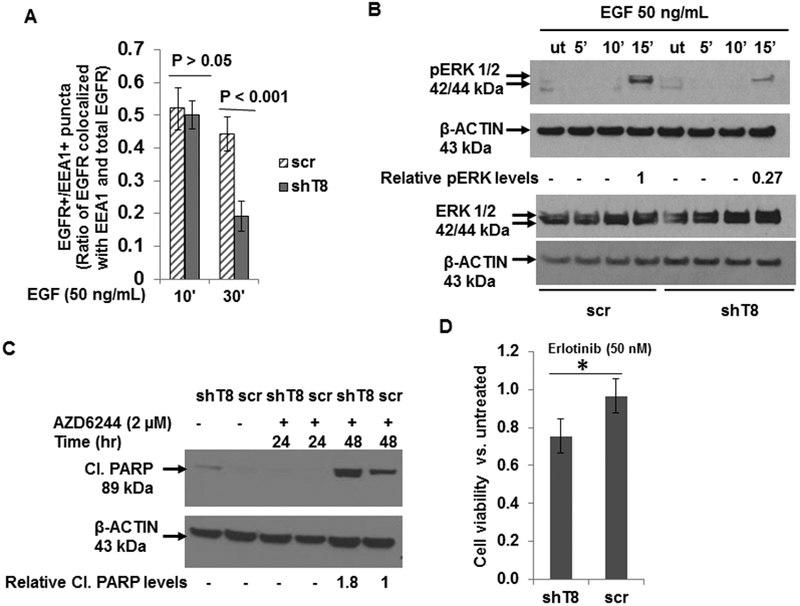
A. Quantification data of immunofluorescence staining using anti-EGFR and anti-EEA antibodies showing decreased localization of EGFR in the early endosomal compartment in response to EGF in shT8 A549 cells versus scr. Three to five fields (~20 – 60 cells per treatment group) were scored for EGFR+/EEA+ puncta after immunofluorescent staining. B. Decreased expression of pERK in shT8 A549 cells versus scr cells. The same cell lysates were probed with pERK and ERK antibodies, followed by reprobing with anti-β-ACTIN antibody. C. Enhanced expression of cleaved PARP in shT8 A549 cells treated with a MEK inhibitor AZD6244. Quantification data shown were normalized against anti-β-ACTIN signal in the corresponding lanes. D. XTT cell viability assay showing enhanced cytotoxicity of erlotinib in shT8 A549 cells versus scr control. Cells were treated with the drug for 72 hr. The cell viability was measured after the addition of activated XTT reagent for 4hr. Values shown are mean ± SD and represent 4 independent experiments, each experiment had 5–6 replicates per data point. *P = 0.017.
Downregulation of pIGF1R and upregulation of IGFBP3 in TNFAIP8 knockdown lung cancer cells
In dose-response and time- course experiments, IGF-1 treatment was found to have no effect on expression of total IGF1R in shT8 A549 cells and scr. However, following treatment with IGF-1 (10 ng/mL, 50 ng/mL, 75 ng/mL or 100 ng/mL, for 30 min), shT8 A549 cells showed suppressed levels of pIGF1R relative to scr control. Representative data are shown in Fig. 4 (A and B) and Fig. S5 (A and B). In addition, IGF-1-stimulated pAKT expression was also found to be suppressed in shT8 A549 cells versus scr (Fig. 4, C and D).
Figure 4. TNFAIP8 knockdown cells show suppression of pIGF1R and pAKT expression.
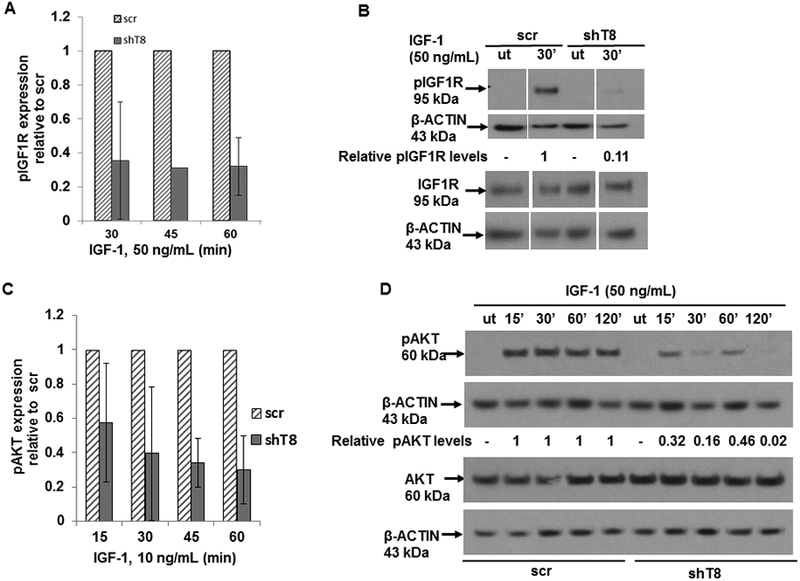
A. Quantification data showing suppression of pIGF1R expression in shT8 A549 cells versus scr cells at various times of treatment with IGF-1 (50 ng/mL). Values shown are mean ± SD from one (45 min) or two independent experiments (30 min and 60 min). B. Immunoblot showing decreased expression of pIGF1R in shT8 A549 cells versus scr cells in response to IGF-1. The same cell lysates were probed with pIGF1R and IGF1R antibodies, followed by reprobing with anti-β-ACTIN antibody. Gaps within the same immunoblot indicate non-relevant lanes that were digitally removed. C. Quantification data showing suppression of pAKT expression in shT8 A549 cells versus scr cells at 45 min and 60 min of treatment with low dose IGF-1. Values shown are mean ± SD from two independent experiments. D. Downregulation of pAKT expression in shT8 A549 cells versus scr cells in response to high dose IGF-1. The same cell lysates were probed with anti-pAKT and anti-AKT antibodies, followed by reprobing with anti-β-ACTIN antibody. ut, untreated.
IGF1R signaling is mitigated by IGF-binding proteins (IGFBPs), a family of secreted proteins, through their high affinity binding to IGF-1 and IGF-2 (31, 32). Our results showed that IGFBP3 protein level was increased by ~ 5-fold in shT8 A549 cells versus scr (Fig. 5A). The antibody array profiling showed that IGFBP3 expression was also increased by ~ 2-fold in shT8 PC-3 cells relative to scr control. IGFBP3 mRNA level was also found to be higher in shT8 A549 cells relative to scr (Fig. S6, left panel). Similar observations were made in shT8 C4–2B prostate cancer cells by the RNA array profiling (~ 4 to 5-fold) and validated by qRT-PCR (Fig. S6, right panel). In three independent experiments, the levels of IGFBP3 present in the culture medium from shT8 A549 cells (shT8-cm) were found to be relatively higher than in the culture medium from scr A549 cells (scr-cm). Representative data are shown in Fig S7A. In addition, treatment of scr A549 and shT8 A549 cells with shT8-cm led to decrease in basal expression of pAKT relative to scr-cm (Fig. S7, B-D). To verify that increased expression of IGFBP3 directly led to downregulation of IGF-1-induced pIGF1R and pAKT expression in shT8 A549 cells, IGFBP3 expression was transiently silenced in shT8 A549 cells using IGFBP3 siRNA, siIGFBP3 (Fig. 5B). In the presence of siIGFBP3, pIGF1R and pAKT levels in shT8 A549 cells were higher both at 30 min and 60 min of IGF-1 exposure relative to siControl (Fig. 5, C-E). Furthermore, shT8 A549 cells treated with a PI-3K inhibitor LY294002 showed decreased expression of an inhibitor of apoptosis protein Survivin (Fig. 5F) and enhanced cell death (Fig. 5G). TNFAIP8 knockdown cells were also more sensitive to a pan-AKT inhibitor MK-2206 relative to scrambled control (Fig. 5H).
Figure 5. Upregulation of IGFBP3 levels in TNFAIP8 knockdown A549 cells and enhanced sensitivities to inhibitors of PI3K and AKT.
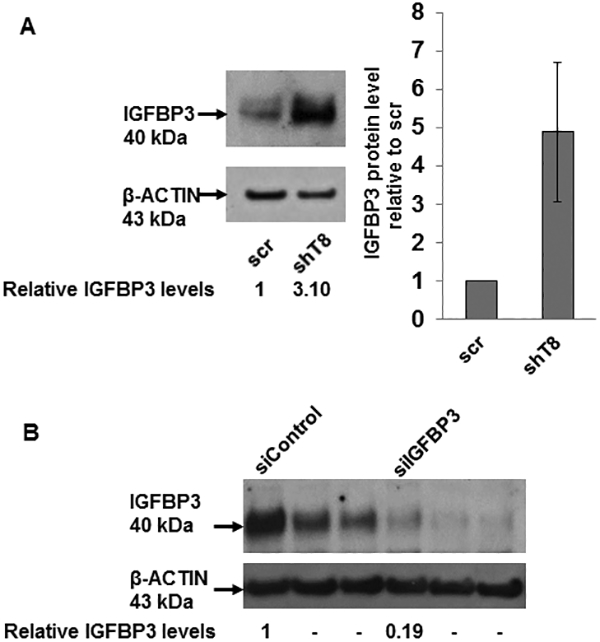
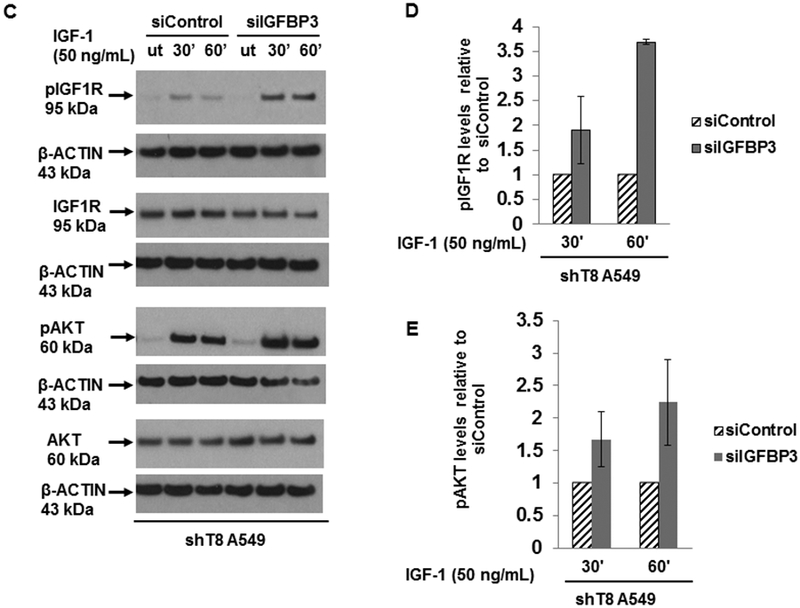
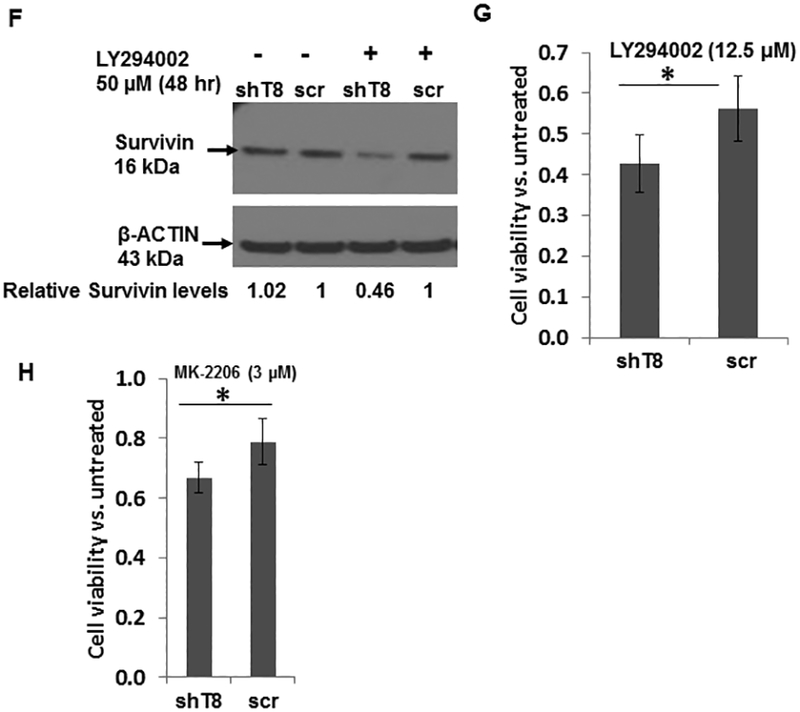
A. Left Panel, Increased expression of IGFBP3 protein in shT8 A549 cells versus scr cells. Right panel, Quantification data showing relative levels of IGFBP3. Values shown are mean ± SD (n =4). B. SiRNA silencing of IGFBP3 expression in shT8 A549 cells. shT8 A549 cells were transfected with 50 nM control siRNA (siControl) or IGFBP3 siRNA (siIGFBP3), followed by cell lysis and sequential immunoblotting with anti-IGFBP3 and anti-β-ACTIN antibodies. Quantification data shown were normalized against β-ACTIN signal in the corresponding lanes. C. Silencing of IGFBP3 in shT8 A549 cells resulted in increased IGF-1-induced expression of pIGF1R and pAKT. A549 cells stably expressing TNFAIP8 shRNA (shT8) were transfected with indicated 50 nM siRNA. After 6 hr, 500 μL of complete medium was added per well. After 48 hr, culture medium was changed to SFM for 12 hr. The siRNA containing medium was then replaced with fresh SFM and IGF-1 for indicated times, followed by cell lysis and immunoblotting with anti-pIGF1R and anti-IGF1R antibodies, and reprobing with anti-β-ACTIN antibody. The same cell lysates were also probed with anti-pAKT and anti-AKT antibodies, followed by reprobing with anti-β-ACTIN antibody. ut, untreated. D and E. Histograms showing quantification of pIGF1R (D) and pAKT expression (E) in siIGFBP3 treated shT8 A549 cells relative to siControl cells. The values shown are mean ± SD from two independent experiments. F. Decreased expression of Survivin in response to PI-3K inhibitor LY294002 in shT8 A549 cells. shT8 A549 and scr A549 cells were treated with the drug as indicated, followed by cell lysis and sequential immunoblotting with anti-Survivin and anti- β-ACTIN antibodies. Quantification data shown were first normalized against β-ACTIN signal in the corresponding lanes. G. XTT cell viability assay showing enhanced cytotoxicity of LY294002 in shT8 A549 cells versus scr. Cells were treated with the drug for 72 hr. The cell viability was measured after the addition of activated XTT reagent for 4hr. Values shown are mean ± SD and represent 7 independent experiments, each experiment had 6 replicates per data point. *p < 0.05. H. XTT cell viability assay showing enhanced cytotoxicity of MK-2206 in shT8 A549 cells versus scr control. Cells were treated with the drug for 72 hr. The cell viability was measured after the addition of activated XTT reagent for 4hr. Values shown are mean ± SD and represent 6 independent experiments, each experiment had 6 replicates per data point. *P = 0.01.
Immunohistochemical expression of TNFAIP8 portends poor prognosis in NSCLC
In a total of 144 NSCLC specimens tested, TNFAIP8 immunoreactivity was found to be predominantly cytoplasmic (Fig. 6). TNFAIP8 protein overexpression was noted in 84/144 (58%) tumors and correlated with tumor type [45/59 (76)% lung adenocarcinomas (AC) versus 29/47 (62%) lung squamous cell carcinomas versus 10/38 (26%) bronchioloalveolar carcinomas, p<0.0001], overall with advanced tumor stage [24/31 (77%) advanced versus 60/113 (53%) early, p=0.015], and lymph node positive status [31/41 (76%) LN+ versus 53/103 (52%) LN-, p=0.008]. Within the AC subgroup, there was a trend toward correlation with advanced tumor stage [13/14 (93%) advanced versus 32/45 (71%) early, p=0.095] and lymph node positive status [18/20 (90%) LN+ vs 27/39 (69%) LN-, p=0.076]. On multivariate analysis, LN+ status independently predicted shortened survival. We conclude that TNFAIP8 immunohistochemical expression portends poor prognosis in NSCLC, warranting further study of TNFAIP8 in relation to known molecular subtypes of NSCLC.
Figure 6. Immunohistochemical expression of TNFAIP8 in representative NSCLC specimens.
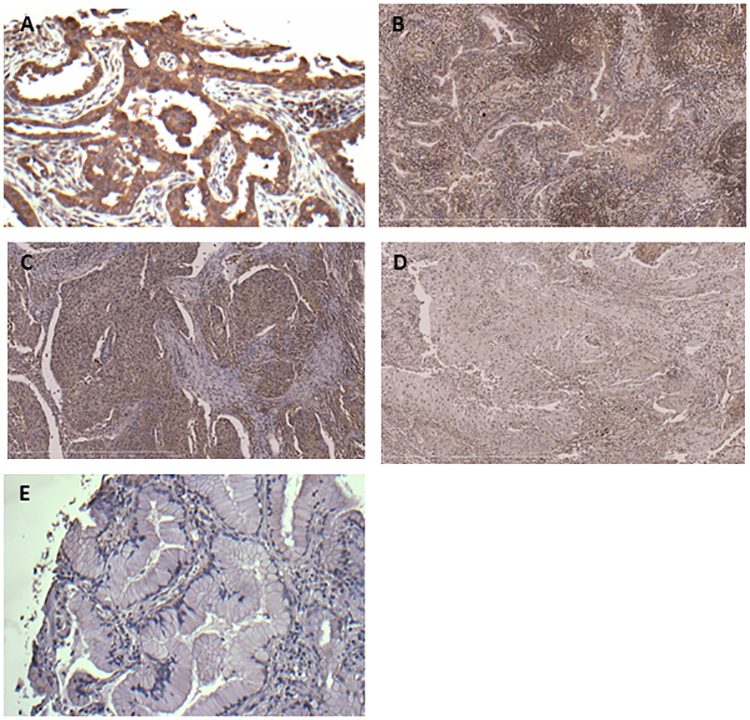
A. Lung adenocarcinoma showing intense diffuse cytoplasmic TNFAIP8 immunoreactivity (advanced stage, LN+). B. Lung adenocarcinoma showing moderate diffuse cytoplasmic TNFAIP8 immunoreactivity (early stage, LN-). C. Lung squamous cell carcinoma with intense diffuse cytoplasmic TNFAIP8 immunoreactivity (advanced stage, LN+). D. Lung squamous cell carcinoma with weak diffuse cytoplasmic TNFAIP8 immunoreactivity (early stage, LN-). E. Bronchioloalveolar carcinoma with no cytoplasmic TNFAIP8 immunoreactivity (early stage, LN-).
Discussion
This study demonstrates for the first time the mechanistic significance of TNFAIP8 in regulation of EGFR, one of the topmost growth factor RTKs implicated in NSCLC and many other malignancies. Our data show that TNFAIP8 knockdown cells exhibited significant inhibition of cell motility in response to EGF as compared to scrambled control cells. The early endosomal association of SNX1 via its 3-phosphoinositide-binding phox homology (PX) domain is required for SNX1-mediated targeting of internalized EGFR for lysosomal degradation (38, 39). How SNX1 expression may be regulated in tumor cells has not been addressed thus far. In this study, we found that the constitutive levels of SNX1 mRNA and protein were high in TNFAIP8 knockdown tumor cells. The siRNA knockdown of SNX1 in TNFAIP8 knockdown cells was found to restore, in part, EGFR expression. The co-localization of EGFR and SNX1 was seen within 10 min of EGF treatment in both the TNFAIP8 knockdown and scrambled control A549 cells; however, consistent with a relatively high level of SNX1, the internalized EGFR seemed to exit early endosomes rather rapidly in TNFAIP8 knockdown A549 cells versus control cells. These data suggest that transcriptional upregulation of SNX1 is a consequence of depletion of TNFAIP8, leading to enhanced SNX1-directed endosomal/lysosomal targeting of EGFR.
Amplifications of both the wild-type and mutant EGFR have been associated with the acquired resistance of NSCLC to EGFR-TKIs (40–42). Deletions or mutations within exons 18–21 of EGFR spanning amino acid residues 688–875 within the core EGFR tyrosine kinase domain (residues 663–958) have also been associated with resistance or sensitivity of NSCLC to EGFR-TKIs (43). Approximately 30% of NSCLC have mutant KRAS with the highest mutational frequency at codons 12 and 13 (7), and there are no druggable targets in KRAS mutant cancers. Moreover, KRAS mutations have been associated with the primary resistance of NSCLC to EGFR-TKIs (6). An important question is whether SNX1-directed lysosomal degradation of EGFR translates into enhanced sensitivity of distinct molecular subtypes of NSCLCs to EGFR-TKIs. A549 cells express wild-type EGFR and mutant KRAS (G12S) and TNFAIP8 knockdown in these cells was found to increase sensitivity to EGFR-TKI erlotinib. TNFAIP8 knockdown was also found to increase SNX1 expression in H1299 NSCLC cells (NRAS, Q61K), PANC-1 pancreatic cancer cells (KRAS, G12D) and two breast cancer cell lines with mutant KRAS (G13D) MDA-MB-231 and LM2–4175. SNX1 binds to a 15-amino acid domain of EGFR (residues 943 to 957 containing the predicted lysosomal targeting signal) distally within the tyrosine kinase domain (27). Thus, it is plausible that TNFAIP8 downregulation and concomitant increase in SNX1 levels may also increase EGFR-TKI sensitivities of NSCLC cell models expressing deletion mutant EGFR (ΔE746-A750) (e.g. HCC827 and PC-9). As expected, TNFAIP8 knockdown A549 cells also showed decreased expression of pERK upon EGF stimulation. In addition, enhanced level of cleaved PARP was seen in TNFAIP8 knockdown cells in the presence of a MEK inhibitor AZD6244. Taken together, we conclude that TNFAIP8 knockdown results in enhanced SNX1 expression and endosomal/lysosomal trafficking and downregulation of EGFR as well as enhanced sensitivity to EGFR-TKI.
We have also observed decreased motility of TNFAIP8 knockdown lung cancer cells in response to IGF-1. While IGF-1 treatment had no effect on total IGF1R expression, decreased expression of pIGF1R and pAKT was seen in IGF-1-treated TNFAIP8 knockdown cells as compared to scrambled control cells. Interestingly, TNFAIP8 knockdown led to increased steady state levels of IGFBP3 mRNA and protein. Furthermore, silencing of IGFBP3 resulted in restoration of IGF-1-stimulated pIGF1R and pAKT levels in TNFAIP8 knockdown cells. We have also ascertained upregulation of IGFBP3 level in the culture medium collected from TNFAIP8 knockdown cells, and observed downregulation of pIGF1R and pAKT in cells treated with this culture medium. These data demonstrate that TNFAIP8 regulates IGF-1-induced pIGF1R and pAKT levels via modulation of IGFBP3 expression in lung cancer cells. Consistently, TNFAIP8 knockdown A549 cells treated with PI3K inhibitor LY294002 showed reduced expression of Survivin and an enhanced loss of cell viability as compared to scrambled control. TNFAIP8 knockdown A549 cells were also found to be sensitive to pan-AKT inhibitor MK-2206. Earlier, silencing of transcription factor FOXA1 has been associated with an increased expression of IGFBP3 (44), and FOXA1 expression has been found to be significantly decreased in TNFAIP8 knockdown cancer cells (37). Therefore, one possibility is that upregulation of IGFBP3 in TNFAIP8 knockdown cells may be due to downregulation of FOXA1.
Cytoplasmic and/or nuclear expression of TNFAIP8 has been correlated with adverse prognosis in several malignancies (17, 18, 33, 45). Data presented here extend these findings in NSCLC specimens representing early or advanced stage of lung adenocarcinomas, lung squamous cell carcinomas, or bronchioloalveolar carcinomas. All specimens were primary tumors collected at initial diagnosis. The mutational status of these tumors was unknown since these archival cases were diagnosed before mutation testing was recommended and routinely performed. In an early study, TNFAIP8 expression seems to positively correlate with EGFR expression in pancreatic cancer specimens and progression of pancreatic cancer (46). Conversely, here TNFAIP8 knockdown PANC-1 pancreatic cancer cells showed upregulation of SNX1 expression, indicative of decreased EGFR expression. Clearly, further studies are necessary in order to establish a direct relationship between TNFAIP8 expression in NSCLC primary tumors and markers of EGFR and IGF1R activation.
Recent report suggests that rapid increase in TNF-α level is a universal adaptive response resulting in resistance to EGFR inhibition in lung cancers regardless of whether EGFR is wild type or mutant (5). NF-κB appears to be a major downstream effector of TNF-α in lung cancer, and activation of NF-κB leads to an increased transcription of TNF-α in a feed-forward manner (5). How may high TNF-α levels protect lung cancer cells from TKI? TNFAIP8 mRNA and protein levels are induced by TNF-α (13), and TNFAIP8 expression protects NF-κB null cells from TNF-α-induced cell death by inhibiting caspase-8 activity (14). Furthermore, TNFAIP8 is critical for evasion of drug-induced apoptosis in lung tumor cells expressing mutant or wild type P53 (21, 22). Thus TNFAIP8 is a pro-survival molecule along the TNF-α/NF-κB axis. Based on the present work, it is tempting to speculate that TNF-α-driven adaptive response in lung cancer cells may be overcome by silencing TNFAIP8.
In conclusion, activation of EGFR and IGF1R and prominent downstream effectors pERK and pAKT are hallmarks of NSCLC and many other cancers. Moreover, bypass activation of IGF1R has been reported in TKI-resistant NSCLC (4, 34, 35, 47). Our data demonstrate that TNFAIP8 knockdown promotes trafficking and degradation of EGFR via increased expression of SNX1, and downregulation of pIGF1R and pAKT via increased level of IGFBP3 (Fig. 7). These observations provide a strong rationale for future preclinical validation of TNFAIP8-mediated regulation of EGFR and IGF1R, and testing the usefulness of SNX1 and circulating IGFBP3 as potential biomarkers in the naïve and matched TKI-sensitive and resistant models of NSCLC.
Figure 7. Schematic of how TNFAIP8 regulates EGFR trafficking and activity and IGF1R activity in NSCLC cells.

.
Supplementary Material
Acknowledgements
We thank the Lung Cancer Biospecimen Resource Network (LCBRN) Coordination Center at The University of Virginia funded by the Congressionally Directed Medical Research Programs of the Department of Defense for providing some of the human lung cancer specimens used in this study. This study was funded, in part, by the grants from the National Institutes of Health (CA68322, CA74175, and CA186944) and Department of Defense (PC074171). Timothy F. Day was supported, in part, by the pre-doctoral fellowship award from the Department of Defense (W81XWH-10-1-0107), and a portion of the work presented here was carried out by TFD toward his Ph.D. dissertation. Shared resources of the Georgetown Lombardi Comprehensive Cancer Center used in this study were supported by the National Institutes of Health Grant P30CA51008.
Financial support: This study was funded, in part, by the grants from the National Institutes of Health (CA68322, CA74175, and CA186944) and Department of Defense (PC074171). Timothy Day was supported, in part, by the pre-doctoral fellowship award from the Department of Defense (W81XWH-10-1-0107).
Footnotes
Conflicts of Interest: The authors declare no potential conflicts of interest.
References
- 1. Roy S, Herbst DM, Boshoff C. The biology and management of non-small cell lung cancer, Nature 2018; 553:446–54. [DOI] [PubMed] [Google Scholar]
- 2. Blakely CM, Watkins TBK, Wu W, Gini B, Chabon JJ, McCoach CE, et al. Evolution and clinical impact of co-occurring genetic alterations in advanced-stage EGFR-mutant lung cancers. Nat Genet 2017; 49:1693–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rotow J, Bivona TG. Understanding and targeting resistance mechanisms in NSCLC. Nat Rev Can 2017;17:637–58. [DOI] [PubMed] [Google Scholar]
- 4. Cortot AB, Repellin CE, Shimamura T, Capelletti M, Zejnullahu K, Ercan D, et al. Resistance to irreversible EGF receptor tyrosine kinase inhibitors through a multistep mechanism involving the IGF1R pathway. Cancer Res 2012;73:834–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gong K, Guo G, Gerber DE, Gao B, Peyton M, Huang C, et al. TNF-driven adaptive response mediates resistance to EGFR inhibition in lung cancer. J Clin Invest 2018; 10.1172/JCI96148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Skoulidis F, Byers LA, Diao L, Papadimitrakopoulou VA, Tong P, Izzo J et al. Co-occurring genomic alterations define major subsets of KRAS - mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov 2015; 5: 860–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Simanshu DK, Nissley DV, McCormick F. RAS proteins and their regulators in human disease. Cell 2017; 170:17–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rizvi H, Sanchez-Vega F, La K, Chatila W, Jonsson P, Halpenny D, et al. Molecular determinants of response to anti–Programmed Cell Death (PD)-1 and anti–Programmed Death-Ligand 1 (PD-L1) blockade in patients with non–small-cell lung cancer profiled with targeted next-generation sequencing. J Clin Oncol 2018;36:633–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lin JJ, Gainor JF. ATLANTIC: a sea change in immunotherapy for oncogene-driven lung cancer? Lancet Oncology 2018;19:438–39. [DOI] [PubMed] [Google Scholar]
- 10. Tong CWS, Wu WKK, Loong HHF, Cho WCS, To KKW. Drug combination approach to overcome resistance to EGFR tyrosine kinase inhibitors in lung cancer. Cancer Letters 207:405:100–10. [DOI] [PubMed] [Google Scholar]
- 11. Marmarelis ME, Aggarwal C. Combination immunotherapy in non-small cell lung cancer. Current Oncology Reports 2018; 10.1007/s11912-018-0697-7. [DOI] [PubMed] [Google Scholar]
- 12. Horrevoets AJ, Fontijn RD, van Zonneveld AJ, de Vries CJ, ten Cate JW, Pannekoek H. Vascular endothelial genes that are responsive to tumor necrosis factor-alpha in vitro are expressed in atherosclerotic lesions, including inhibitor of apoptosis protein-1, stannin, and two novel genes. Blood 1999;93:3418–31. [PubMed] [Google Scholar]
- 13. Kumar D, Whiteside TL, Kasid U. Identification of a novel tumor necrosis factor-alpha-inducible gene, SCC-S2, containing the consensus sequence of a death effector domain of fas-associated death domain-like interleukin- 1beta-converting enzyme-inhibitory protein. J Biol Chem 2000;275:2973–78. [DOI] [PubMed] [Google Scholar]
- 14. You Z, Ouyang H, Lopatin D, Polver PJ, Wang CY. Nuclear factor-kappa B-inducible death effector domain-containing protein suppresses tumor necrosis factor-mediated apoptosis by inhibiting caspase-8 activity. J Biol Chem 2001;276:26398–404. [DOI] [PubMed] [Google Scholar]
- 15. Kumar D, Gokhale P, Broustas C, Chakravarty D, Ahmad I, Kasid U. Expression of SCC-S2, an antiapoptotic molecule, correlates with enhanced proliferation and tumorigenicity of MDA-MB 435 cells. Oncogene 2004;23:612–16. [DOI] [PubMed] [Google Scholar]
- 16. Zhang C, Chakravarty D, Sakabe I, Mewani RR, Boudreau HE, Kumar D, et al. Role of SCC-S2 in experimental metastasis and modulation of VEGFR-2,MMP-1, and MMP-9 expression. Mol Ther 2006;13:947–55. [DOI] [PubMed] [Google Scholar]
- 17. Lou Y, Liu S. The TIPE (TNFAIP8) family in inflammation, immunity, and cancer. Mol Immunol 2011;49:4–7. [DOI] [PubMed] [Google Scholar]
- 18. Goldsmith JR, Chen YH. Regulation of inflammation and tumorigenesis by the TIPE family of phospholipid transfer proteins. Cell Mol Immunology 2017; 14:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Padmavathi G, Banik K, Monisha J, Bordoloi D, Bano S, Arfuso F, et al. Novel tumor necrosis factor-α induced protein eight (TNFAIP8/TIPE) family: Functions and downstream targets involved in cancer progression. Cancer Letters 2018; doi: 10.1016/j.canlet.2018.06.017. [DOI] [PubMed] [Google Scholar]
- 20. Fayngerts SA, Wu J, Oxley CL, Liu AV, Cathopoulis T, Wang Z, et al. TIPE3 is the transfer protein of lipid second messengers that promote cancer. Cancer Cell 2014; 26:465–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Monteith JA, Mellert H, Sammons MA, Kuswanto LA, Sykes SM, Resnick-Silverman L, et al. A rare DNA contact mutation in cancer confers p53 gain-of-function and tumor cell survival via TNFAIP8 induction. Mol Oncology 2016; 10:1207–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lowe JM, Nguyen TA, Grimm SA, Gabor KA, Peddada SD, Li L, et al. The novel p53 target TNFAIP8 variant 2 is increased in cancer and offsets p53-dependent tumor suppression. Cell Death and Differentiation 2017;24:181–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lin L, Sabnis AJ, Chan E, Olivas V, Cade L, Pazarentzos E, et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat Genet 2015; 47:250–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee BS, Park DI, Lee DH, Lee JE, Yeo MK, Park YH, et al. Hippo effector YAP directly regulates the expression of PD-L1 transcripts in EGFR-TKI-resistant lung adenocarcinoma. Biochem Biophy Res Comm 2017;491:493–99. [DOI] [PubMed] [Google Scholar]
- 25. Han Y, Tang ZP, Zhao Y, Li Q, Wang E. TNFAIP8 regulates Hippo pathway through interacting with LATS1 to promote cell proliferation and invasion in lung cancer. Mol Carcinogenesis 2018; 57:159–66. [DOI] [PubMed] [Google Scholar]
- 26. Dong S, Zhang XC, Cheng H, Zhu JQ, Chen ZH, Zhang YF, et al. Everolimus synergizes with gefitinib in non-small-cell lung cancer cell lines resistant to epidermal growth factor receptor tyrosine kinase inhibitors. Cancer Chemother Pharmacol 2012; 70:707–16. [DOI] [PubMed] [Google Scholar]
- 27. Kurten RC, Cadena DL, Gill GN. Enhanced degradation of EGF receptors by a sorting nexin, SNX1, Science 1996; 272:1008–10. [DOI] [PubMed] [Google Scholar]
- 28. Worby CA, Dixon JE. Sorting out the cellular functions of sorting nexins. Nat Rev Mol Cell Biol 2002;3:919–31. [DOI] [PubMed] [Google Scholar]
- 29. Tomas A, Futter CE, Eden ER. EGF receptor trafficking: consequences for signaling and cancer. Trends Cell Biol 2014;24:26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guix M, Faber AC, Wang SE, Olivares MG, Song Y, Qu S, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGF-binding proteins. J Clin Invest 2008;118:2609–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Baxter RC. IGF binding proteins in cancer: mechanistic and clinical insights. Nat Rev Cancer 2014;14:329–41. [DOI] [PubMed] [Google Scholar]
- 32. Wang YA,Sun Y, Palmer J, Solomides C, Huang LC, Shyr Y, et al. IGFBP3 modulates lung tumorigenesis and cell growth through IGF1 signaling. Mol Cancer Res 2017;15:896–904. [DOI] [PubMed] [Google Scholar]
- 33. Zhang C, Kallakury BV, Ross JS, Mewani RR, Sheehan CE, Sakabe T, et al. The significance of TNFAIP8 in prostate cancer response to radiation and docetaxel and disease recurrence. Int J Cancer 2013;33:31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Morgillo F, Woo JK, Kim ES, Hong WK, Lee HY. Heterodimerization of insulin-like growth factor receptor/epidermal growth factor receptor and induction of survivin expression counteract the antitumor action of erlotinib. Cancer Res 2006; 66:10100–111. [DOI] [PubMed] [Google Scholar]
- 35. Morgillo F, Kim WY, Kim ES, Ciardiello F, Hong WK, Lee HY. Implication of the insulin-like growth factor-IR pathway in the resistance of non-small cell lung cancer cells to treatment with gefitinib. Clin Cancer Res 2007;13:2795–2803. [DOI] [PubMed] [Google Scholar]
- 36. Kim JI, Lakshmikanthan V, Frilot N, Daaka Y. Prostaglandin E2 promotes lung cancer cell migration via EP4-betaArrestin1-c-Src signalsome. Mol Cancer Res 2010;8:569–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Day TF, Mewani RR, Starr J, Li X, Chakravarty D, Ressom H, et al. Transcriptome and proteome analyses of TNFAIP8 knockdown cancer cells reveal new insights into molecular determinants of cell survival and tumor progression. Methods in Mol Biol Cancer Gene Networks 2017;1513:83–100. [DOI] [PubMed] [Google Scholar]
- 38. Cozier GE, Carlton J, McGregor AH, Gleeson PA, Teasdale RD, Mellor H, et al. The Phox homology (PX) domain-dependent, 3-Phosphoinositide-mediated association of sorting nexin-1 with an early sorting endosomal compartment is required for its ability to regulate epidermal growth factor receptor degradation. J Biol Chem 2002; 277:48730–36. [DOI] [PubMed] [Google Scholar]
- 39. Zhong Q, Lazar CS, Tronchère H, Sato T, Meerloo T, Yeo M, et al. Endosomal localization and function of sorting nexin 1. Proc Natl Acad Sci 2002;99:6767–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nukaga S, Yasuda H, Tsuchihara K, Hamamoto J, Masuzawa K, Kawada I et al. Amplification of EGFR wild-type alleles in non-small cell lung cancer cells confers acquired resistance to mutation-selective EGFR tyrosine kinase inhibitors. Cancer Res 2017; 77:2078–89. [DOI] [PubMed] [Google Scholar]
- 41. Ercan D, Zejnullahu K, Yonesaka K, Xiao Y, Capelletti M, Rogers A et al. Amplification of EGFR T790M causes resistance to an irreversible EGFR inhibitor. Oncogene 2010; 29: 2346–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Piotrowska Z, Niederst MJ, Karlovich CA, Wakelee HA, Neal JW, Mino-Kenudson M, et al. , Heterogeneity underlies the emergence of EGFRT790 wild-type clones following treatment of T790M-positive cancers with a third-generation EGFR inhibitor. Cancer Discov 2015;5:713–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer 2007;7:169–81. [DOI] [PubMed] [Google Scholar]
- 44. Imamura Y, Sakamoto S, Endo T, Utsumi T, Fuse M, Suyama T, et al. FOXA1 promotes tumor progression in prostate cancer via the insulin-like growth factor binding protein 3 pathway. PLoS One 2012;7:e42456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Xiao M, Xu QY, Lou C, Qin Y, Ning XM, Liu TB, et al. Overexpression of TNFAIP8 is associated with tumor aggressiveness and poor prognosis in patients with invasive ductal breast carcinoma. Human Pathology 2017;62:40–49. [DOI] [PubMed] [Google Scholar]
- 46. Liu K, Qin CK, Wang ZY, Liu SX, Cui XP, Zhang DY. Expression of tumor necrosis factor-alpha-induced protein 8 in pancreas tissues and its correlation with epithelial growth factor receptor levels.. Asian Pac J Cancer Prev 2012;13:847–50. [DOI] [PubMed] [Google Scholar]
- 47. Niederst MJ, Engelman JA. Bypass mechanisms of resistance to receptor tyrosine kinase inhibition in lung cancer. Sci Signaling 2013;6:re6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.