

Abstract
Aims/Introduction
Non‐alcoholic steatohepatitis (NASH), which occurs in association with insulin resistance and hepatic fat accumulation, is characterized by chronic liver injury and fibrosis. NASH onset and progression is closely related to hepatic inflammation, which is partly regulated by the vagus nerve through the α7 nicotinic acetylcholine receptor (α7nAchR). Hepatic α7nAchR action is impeded in obesity and insulin resistance. In the present study, using α7nAchR knockout (α7KO) mice, we elucidated the effect of α7nAchR deficiency on NASH‐related inflammation and fibrosis.
Materials and Methods
α7KO mice were fed an atherogenic high‐fat diet (AD) for 32 weeks or methionine/choline‐deficient diet (MCD) for 6 weeks, both of which induce NASH. Mice were then examined for the degree of NASH‐related inflammation and fibrosis by hepatic gene expression analysis and Sirius red histological staining.
Results
Hepatic triglyceride accumulation and elevated plasma transaminase levels were observed in both AD and MCD mice, but the plasma transaminase level increase was higher in α7KO mice than in control mice. α7KO mice fed an AD showed significant upregulation of the Col1a1 gene encoding alpha‐1 type I collagen, which is involved in liver fibrosis, and the Ccl2 gene encoding C‐C motif chemokine ligand 2, a pro‐inflammatory chemokine; α7KO mice fed an MCD had significant upregulation of the Col1a1 gene and the Tnf gene, an inflammatory cytokine. Histological analysis showed that AD and MCD exacerbated liver fibrosis in α7KO mice.
Conclusions
The results of this study suggest that α7nAchR deficiency exacerbates hepatic inflammation and fibrosis in a diet‐induced mouse model of NASH.
Keywords: Fibrosis, Inflammation, Non‐alcoholic steatohepatitis
Introduction
Non‐alcoholic fatty liver disease (NAFLD), which occurs in association with insulin resistance and is characterized by fat accumulation in the liver, is a disease spectrum that begins with simple fatty liver and progresses to non‐alcoholic steatohepatitis (NASH) accompanied by cell injury/fibrosis and eventually cirrhosis1. In the spectrum of NAFLD, the part of the liver with simple steatosis progresses to NASH/cirrhosis, with inflammation playing an important role in the process. Indeed, hepatic inflammation is upregulated in NASH, with increases in the number of hepatic macrophages and the expression of pro‐inflammatory cytokines, such as tumor necrosis factor‐α (TNFα), interleukin‐6 and C‐C motif chemokine ligand 2 (CCL2)2, 3. Macrophages undergo polarized activation to pro‐inflammatory M1 or anti‐inflammatory M2 states, which involve the expression of CD11c or CD206 and CD163, respectively4. In NASH, the hepatic macrophage polarization shifts to the M1 state5, 6. Furthermore, NASH is exacerbated by inflammatory inducers, such as pro‐inflammatory cytokines, bacterial cell components derived from intestinal microbiota and saturated fatty acids1, 7. In contrast, anti‐inflammatory factors, such as adiponectin and unsaturated fatty acids, suppress the progression to NASH, suggesting their potential as candidate drugs for the prevention and treatment of NASH8, 9.
The vagus nerve regulates inflammation through the action of acetylcholine10. In a mouse study, electrical stimulation of the vagus nerve decreased the blood levels of TNFα and interleukin‐611. We have also shown the upregulation of hepatic inflammatory responses, such as increased interleukin‐6 gene expression in the liver after vagotomy12. The vagus nerve regulates inflammatory responses in macrophages and Kupffer cells through α7 nicotinic acetylcholine receptor (α7nAchR)12, 13. Indeed, α7nAchR‐knockout (α7KO) mice have high levels of pro‐inflammatory cytokines in the blood during endotoxemia, and in these mice, electrical stimulation of the vagus nerve fails to decrease plasma TNFα levels13. The vagal regulation of inflammation through α7nAchR is closely associated with the regulation of glucose metabolism. α7KO mice develop insulin resistance when fed a high‐fat diet (HFD) that induces obesity14. We have also shown that the vagal α7nAchR action plays an important role in the brain‐mediated regulation of hepatic glucose production12. The brain detects elevated plasma insulin and amino acid levels, resulting in the decrease in hepatic glucose production through the vagal regulation of inflammation through α7nAchR12, 15, 16.
Along with the development of insulin resistance, which is an inducer of NASH, vagal fluctuation disappears, which is induced in response to changes in nutrient signals, such as plasma insulin and amino acid levels, and the action of the vagus nerve continues to weaken in the liver12. Given that vagotomy induces a hepatic inflammatory response, impaired vagal action through α7nAchR as a result of insulin resistance might be involved in the aggravation of NASH through the exacerbation of hepatic inflammation. However, the role of vagal α7nAchR action in the onset and exacerbation of NASH remains to be fully elucidated.
In the present study, α7KO mice were fed an atherogenic high‐fat diet (AD) or methionine/choline‐deficient diet (MCD), both of which induce NASH, and the effect of vagal α7nAchR impairment on the exacerbation of NASH‐related inflammation and fibrosis was investigated. AD induces hepatic inflammation and fibrosis in addition to mild obesity and insulin resistance2, 17, whereas MCD induces a NASH‐like pathology accompanied by hepatic inflammation and fibrosis, but not insulin resistance, by impairing the secretion of very low‐density lipoprotein (VLDL) for hepatic release of triglycerides, and thus triggering their accumulation in the liver2, 17, 18. These diet‐induced NASH animal models have shown that α7nAchR deficiency exacerbates NASH‐related inflammation and fibrosis.
Methods
Animals
All animal experiments were approved by the Animal Ethics Committee of Kanazawa University (approval number AP‐132743), carried out according to the Animal Ethics Committee guidelines for the care and use of laboratory animals at Kanazawa University, and carried out following the national guidelines and the relevant national laws on the protection of animals. C57BL/6J Slc mice were purchased from Japan SLC (Shizuoka, Japan) and α7KO mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA), and maintained in a temperature‐controlled environment with a 12‐h light/dark cycle and free access to food and water under specific pathogen‐free conditions in the Institute for Experimental Animals of Kanazawa University. Male α7KO mice were obtained by mating heterogeneous α7KO mice, and wild‐type littermates of α7KO mice were used as controls. Sample sizes are stated in the figure legends.
Diets and sample collection
Mice were fed an AD (D06061403; Research Diet, New Brunswick, NJ, USA) or MCD (A02082002B; Research Diet) for 32 or 6 weeks from 7 weeks‐of‐age, respectively. The AD provides 61% of energy from fat and 1.3 g cholesterol/100 g diet19, and the MCD provides 21.2% of energy from fat, but lacks methionine and choline. At the end of the experimental period, plasma and tissue samples were collected from animals in the ad libitum‐fed state and stored at −80°C.
Biochemical and histological analyses
Blood glucose levels were measured using a GLUCOCARD G+ Meter (Arkray, Kyoto, Japan). Plasma insulin concentrations were determined using a mouse insulin enzyme‐linked immunosorbent assay kit (Wako, Saitama, Japan). Plasma aspartate aminotransferase/alanine aminotransferase (AST/ALT) levels were measured using the Transaminase CII‐Test‐Wako kit (Wako). Liver triglyceride concentrations were measured by using the TG E‐Test‐Wako (Wako), as described previously20. Liver tissues were fixed in 4% paraformaldehyde/phosphate‐buffered saline (Wako), and the sections were stained with Sirius red.
Quantitative polymerase chain reaction
Quantitative polymerase chain reaction was carried out using the SYBR Select Master Mix kit (Thermo Fisher Scientific, Waltham, MA, USA), as described previously21. Quantitative polymerase chain reaction results were analyzed using the Rplp0 gene as an internal control and plotted in arbitrary units as the mean ± standard error. Primer sequences for Rplp0, Srebf1, Fasn, Scd1, Ppara, Cpt1a, Il6, Tnf, Ccl2, Acta2, Col1a1 and Tgfb1 are as described previously19, 22, 23. Primer sequences for Mttp, Itgax, Mrc1 and Cd163 are described in Table S1.
Statistical analysis
Data are represented as the mean ± standard error. Statistical analysis was carried out using Student's t‐test and one‐way anova followed by post‐hoc tests, and differences were considered significant at P < 0.05.
Results
Animals fed an AD are reported to develop NASH accompanied by insulin resistance, hepatic inflammation and liver fibrosis2, 17. In the present study, AD induced a significant increase in body weight, blood glucose levels, plasma insulin and ALT levels, and hepatic triglyceride content (Figure 1a–e). We therefore investigated the effect of α7nAchR deficiency on insulin resistance and liver injury induced by the AD. α7KO mice fed an AD had significantly higher plasma ALT levels and, although insignificant, the levels of plasma AST were also increased (Figure 1d). However, body weights, and blood glucose and plasma insulin levels did not differ significantly between α7KO and control mice (Figure 1a–c). Hepatic triglyceride content was significantly increased in α7KO mice fed an AD (Figure 1e). Hepatic gene expression of Srebf1, a master regulator of hepatic lipogenesis, was higher in α7KO mice fed an AD than in their control (Figure S1a). However, there were no differences in the hepatic expression of lipogenic enzyme genes (Fasn and Scd1), lipid oxidation enzyme genes (Ppara and Cpt1a) or a VLDL secretion‐associated gene (Mttp) between α7KO mice and their control (Figure S1a,b).
Figure 1.
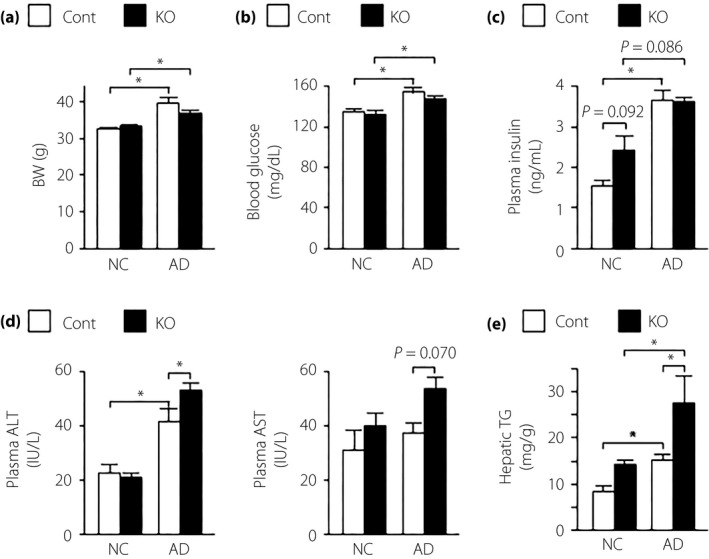
(a) Body weight (BW), (b) blood glucose, (c) plasma insulin, (d) plasma aspartate aminotransferase/alanine aminotransferase (ALT/AST) and (e) hepatic triglyceride (TG) content in mice fed normal chow (NC) or an atherogenic high‐fat diet (AD) for 32 weeks. Values are the mean ± standard error of the mean (NC‐control [Cont], n = 4; NC‐knockout [KO], n = 5; AD‐Cont, n = 6; AD‐KO, n = 10). *P < 0.05.
We also investigated the effect of α7nAchR deficiency on NASH‐related inflammation and fibrosis. Hepatic gene expression analysis showed significant messenger ribonucleic acid upregulation of the pro‐inflammatory mediators Tnf, Il6 and Ccl2 in mice fed an AD (Figure 2a). The expression of the Ccl2 gene, but not the Tnf and Il6 genes, was significantly higher in α7KO mice than in control mice (Figure 2a). There were no differences in the hepatic expression of the Itgax, encoding the M1 marker CD11c, Mrc1, encoding the M2 marker CD206, and CD163 genes between the groups (Figure 1c). Mice fed an AD showed significant upregulation of the Acta2 and Col1a1 genes, which are associated with fibrosis, and a tendency toward an increase in the Tgfb1 gene, another fibrosis‐related gene (Figure 2b). In α7KO mice, expression of the Col1a1 gene, but not the Acta2 and Tgfb1 genes, was significantly higher than in control mice (Figure 2b). Histological analysis with Sirius red, which stains collagen fibers, clearly showed the exacerbation of liver fibrosis in α7KO mice compared with control mice (Figure 3).
Figure 2.
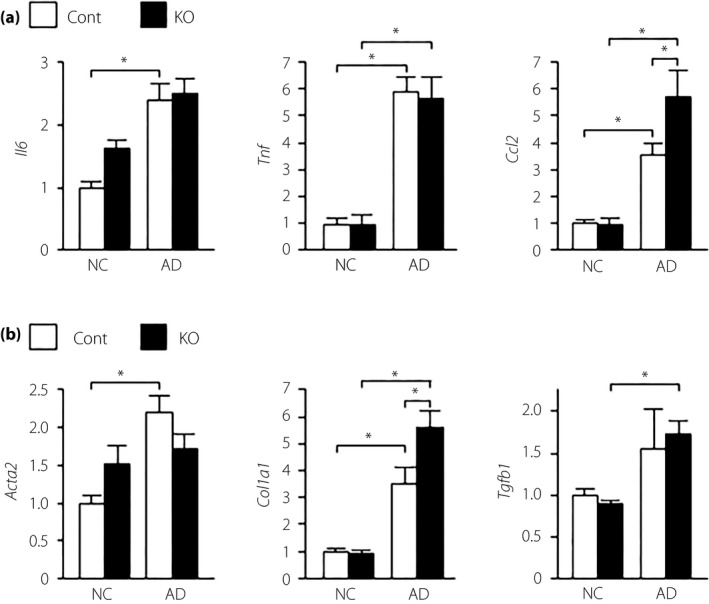
Hepatic messenger ribonucleic acid expression levels of genes related to (a) inflammation and (b) fibrosis in mice fed normal chow (NC) or an atherogenic high‐fat diet (AD) for 32 weeks. Values are the mean ± standard error of the mean (NC‐control [Cont], n = 4; NC‐knockout [KO], n = 5; AD‐Cont, n = 6; AD‐KO, n = 10). *P < 0.05.
Figure 3.
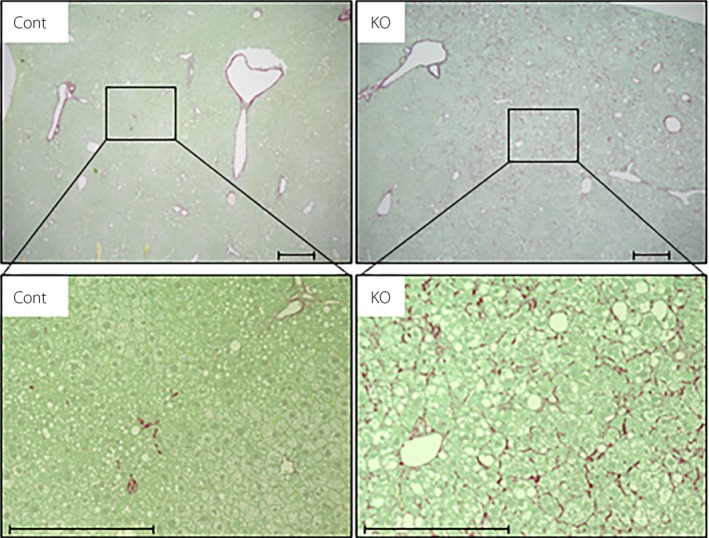
Hepatic fibrosis in mice fed an atherogenic high‐fat diet for 32 weeks, evaluated using representative Sirius red‐stained histological sections of the liver. Scale bar, 500 μm. Cont, control; KO, knockout.
Next, we investigated the effect of α7nAchR deficiency on MCD‐induced NASH, and found no significant difference in bodyweight and blood glucose and plasma insulin levels between control and α7KO mice (Figure 4a–c). In α7KO mice fed an MCD, plasma AST levels were significantly increased, whereas plasma ALT levels tended to increase (Figure 4d). Although increased, hepatic triglyceride content did not significantly differ between control and A7KO mice (Figure 4e). Hepatic gene expression levels of lipogenesis, lipid oxidation and VLDL secretion did not differ between control and α7KO mice (Figure S2a,b). The expression levels of Tnf, Ccl2, Acta2, Col1a1, Tgfb1 and Itgax genes were upregulated in the liver of α7KO mice fed an MCD compared with α7KO mice fed a control diet, whereas there was no change in the hepatic expression of M2 marker genes between the groups (Figure 5a,b and Figure S2c). α7KO mice showed higher expression of Tnf and Tgfb1 genes, and exacerbation of liver fibrosis on hepatic histology with Sirius red stain compared with control mice (Figure 5a–c).
Figure 4.
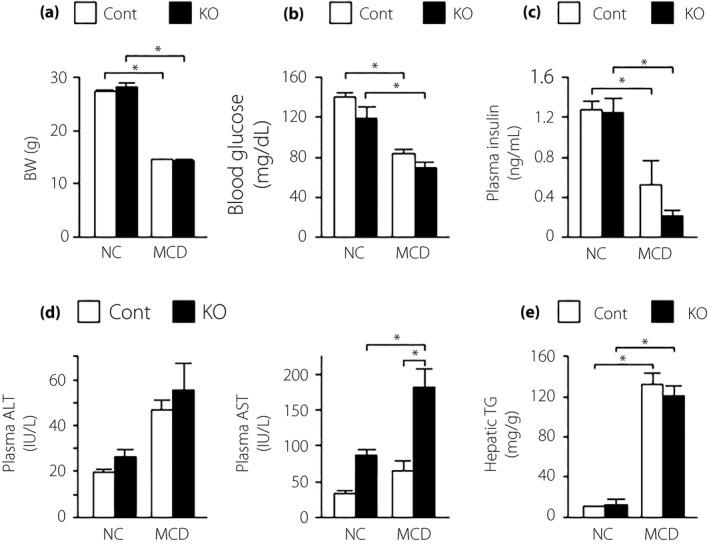
(a) Body weight (BW), (b) blood glucose, (c) plasma insulin, (d) plasma aspartate aminotransferase/alanine aminotransferase (ALT/AST) and (e) hepatic triglyceride (TG) content in mice fed normal chow (NC) or a methionine/choline‐deficient diet (MCD) for 6 weeks. Values are the mean ± standard error of the mean (NC‐control [Cont], n = 4; NC‐knockout [KO], n = 4; MCD‐Cont, n = 6; MCD‐KO, n = 8). *P < 0.05.
Figure 5.
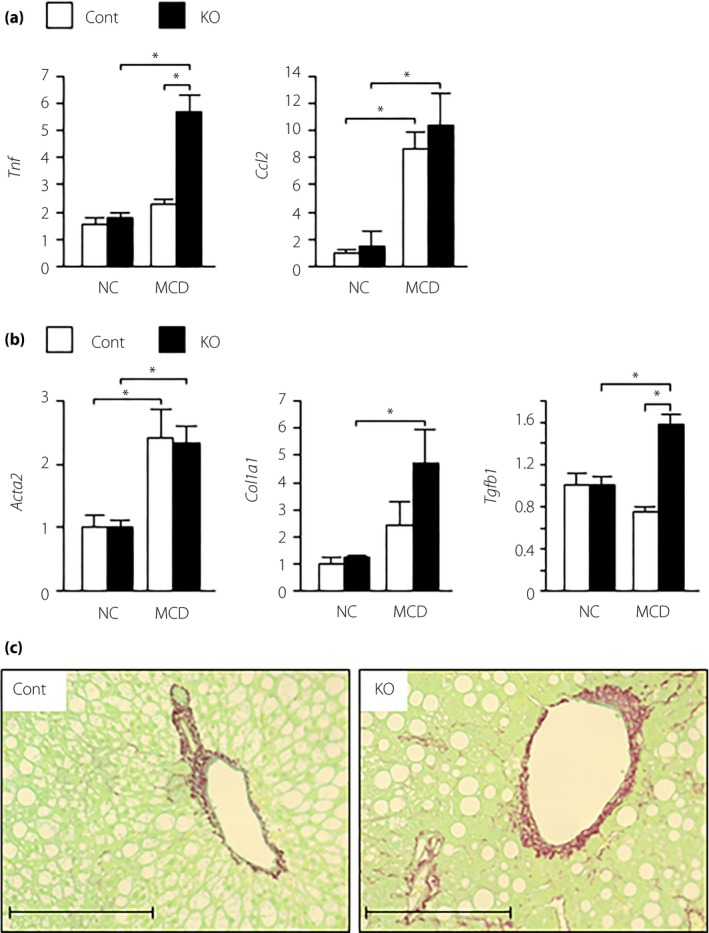
Hepatic expression of genes related to (a) inflammation and (b) fibrosis, and (c) liver histological sections of Sirius red staining in mice fed normal chow (NC) or a methionine/choline‐deficient diet (MCD) for 6 weeks. Values are the mean ± standard error of the mean (NC‐control [Cont], n = 4; NC‐knockout [KO], n = 4; MCD‐Cont, n = 6; MCD‐KO, n = 8). *P < 0.05. Scale bar, 500 μm.
Discussion
Along with hepatic fat accumulation as a result of insulin resistance, hepatic inflammation is deeply involved in the onset and progression of NASH1, 7. α7nAchR in the vagus nerve plays an important role in the regulation of hepatic inflammation, but this vagal regulation is impaired in individuals with insulin resistance12. Although previous studies have reported that acute or chronic α7nAchR impairment exacerbates hepatic inflammation in NAFLD13, 24, 25, no previous study has elucidated the effect of α7nAchR impairment on the progression of NASH; that is, the exacerbation of not only inflammation, but also fibrosis. In the present study, using α7KO mice and diet‐induced animal models of NASH, we found that α7nAchR impairment leads to both inflammation and fibrosis related to the exacerbation of NASH. In AD‐ and MCD‐fed mice, α7nAchR deficiency induced elevation of plasma transaminase levels, markers of liver injury; upregulation of genes associated with inflammation and fibrosis; and an increase in fibrosing lesions on liver histology.
In the present study, we used AD and MCD to establish two diet‐induced NASH models with distinct mechanisms, and showed that α7nAchR deficiency exacerbates both NASH‐related inflammation and fibrosis. In particular, we found that α7nAchR deficiency resulted in hepatic pericellular fibrosis in collagen fiber staining, in addition to the increase in the expression of inflammation‐ and fibrosis‐associated genes. Previous studies have reported the association of α7nAchR deficiency with the exacerbation of hepatic inflammation caused by bacterial endotoxins13 and of inflammation induced by HFD or short‐term MCD loading24, 25. However, HFD or short‐term MCD loading is insufficient to induce liver fibrosis, making it difficult to clarify the role of α7nAchR impairment in liver fibrosis, although liver fibrosis, together with hepatic inflammation, is a major manifestation of NASH26. The present study, by examining liver fibrosis in tissue sections stained with Sirius red after the administration of AD and MCD for 32 and 6 weeks, respectively, has shown that α7nAchR deficiency exacerbates liver fibrosis in NASH. AD and MCD are widely used to establish a diet‐induced animal model of NASH2, 17. The AD model is thought to closely represent the pathology of NASH in humans, because the diet induces and promotes insulin resistance and hepatic fat accumulation, eventually triggering inflammation and fibrosis in the liver2, 17. In contrast, the MCD model triggers NASH through hepatic fat accumulation caused largely by impaired release of hepatic triglycerides as a result of the lower synthesis of VLDL2, 17. Indeed, animals fed an MCD have substantial weight loss. and reduced blood glucose and plasma insulin levels. The MCD model showed no difference in the hepatic triglyceride concentration between α7KO mice and their control, whereas AD loading exacerbated hepatic steatosis in α7KO mice. This phenotypic difference between MCD and AD might depend on the difference in the effect on plasma insulin levels. Scherer et al. reported that central insulin action regulates VLDL secretion and reduces hepatic triglyceride content, although the mechanism underlying the central insulin regulation of VLDL secretion remains unclear.27 α7nAchR plays an important role in brain–liver interaction through the vagus nerve12. If α7nAchR also affects central insulin‐mediated regulation of VLDL secretion, α7nAchR deficiency could increase the hepatic triglyceride concentration.
After 18‐week administration of HFD, α7KO mice have impaired glucose tolerance accompanied by insulin resistance and high fasting blood glucose levels14. The HFD model induces severe insulin resistance and hepatic inflammation, although the animals lack clear signs of liver fibrosis2, 17. In the present study, even though blood glucose and plasma insulin levels increased after AD administration, no significant difference was observed between control and α7KO mice, suggesting that the mechanism underlying the exacerbation of inflammation/fibrosis as a result of α7nAchR deficiency is different from the mechanism underlying the impairment of glucose tolerance and the exacerbation of insulin resistance. Insulin resistance induced by the AD is reportedly milder than that induced by the HFD2, and this might explain why AD‐fed α7KO mice did not have clear signs of hyperglycemia or hyperinsulinemia in the present study.
According to the Mouse Expression Database, α7nAchR is expressed in the nervous system and hemolymphoid system28. Our previous study also showed the expression of α7nAchR in the central nervous system and Kupffer cells12. In α7KO mice, exacerbation of diet‐induced hepatic inflammation is likely mediated by α7nAchR in inflammatory cells in the liver, such as Kupffer cells. We have reported that α7KO mice show an increase in hepatic inflammatory responses, and that bone marrow‐specific restoration of α7nAChR in α7KO mice results in amelioration of the increased hepatic inflammatory response12. Furthermore, hepatic inflammation induced by short‐term administration of MCD is exacerbated in bone marrow‐derived cell‐specific α7nAChR knockout mice generated by transplantation of α7KO bone marrow to wild‐type mice24.
The exacerbation of diet‐induced liver fibrosis in α7KO mice might be attributable to persistent severe hepatic inflammation, instead of being mediated by α7nAchR in stellate cells, the major player in liver fibrosis. This is because an immunohistological study has shown that α7nAchR is less abundantly expressed in hepatic stellate cells29. However, a future challenge would necessarily be to elucidate the functional role of α7nAchR in stellate cells and macrophage cells in hepatic fibrosis in NASH by using stellate cell‐ and macrophage‐specific α7nAchR knockout mice. AD administration increased Col1a1 gene expression in α7KO mice, but not Tgfb1 and Acta2 gene expression. Meanwhile, MCD administration increased Tgfb1 gene expression, although Col1a1 gene expression showed a tendency for an increase in α7KO mice. The difference in the fibrosis‐associated gene expression pattern might be explained by the timing of the liver sample harvest, because both AD and MCD models showed hepatic fibrosis in collagen fiber staining.
The present study shows that the functional deficit of α7nAchR exacerbates both inflammation and fibrosis attributable to hepatic fat accumulation. Given that insulin resistance impedes vagal regulation of inflammation, insulin resistance might develop and exacerbate NASH through the impairment of vagal α7nAchR activity. α7nAchR agonists alleviate acute inflammatory diseases in the liver, such as toxic liver damage and ischemic liver injury30, 31, and improve chronic hepatic inflammation induced by HFD25. α7nAchR might be a novel drug candidate for the prevention and treatment of NASH, but further study is required to verify the utility of α7nAchR agonists for liver fibrosis in NASH. Because α7nAchR is abundant in the central nervous system, α7nAchR agonists might be promising candidate therapeutic targets for cognitive disorders, such as schizophrenia and Alzheimer's disease32, 33. The application of activated α7nAchR in the prevention and treatment of NASH requires further investigation of α7nAchR action in organs other than the liver, and the development of a liver‐specific drug delivery system for α7nAchR agonists.
Disclosure
The authors declare no conflict of interest.
Supporting information
Figure S1¦ Hepatic messenger ribonucleic acid expression levels of genes related to (a) lipogenesis, (b) β‐oxidation and very high‐density lipoprotein (VLDL) secretion, and macrophage polarization markers in mice fed a normal chow (NC) or an atherogenic high‐fat diet (AD)for 32 weeks.
Figure S2¦ Hepatic messenger ribonucleic acid expression levels of genes related to (a) lipogenesis, (b) β‐oxidation and VLDL secretion, and macrophage polarization markers in mice fed a normal chow (NC) or a methionine/choline‐deficient diet (MCD) for 6 weeks.
Table S1¦ Primer sequences.
Acknowledgments
We thank K Nagamori and C Asahi (Kanazawa University) for providing technical assistance, and ThinkSCIENCE (Tokyo, Japan) for help with the preparation of the manuscript. This work was supported by the Japan Society for the Promotion of Science KAKENHI (grant number 15H05678 to Y Inaba, grant number 16K01815 to K Kimura, and grant number 17H05499 to H Inoue); a research grant from the Naito Foundation (to K Kimura); and a research grant from the Uehara Memorial Foundation and the Takeda Science Foundation (to H Inoue).
J Diabetes Investig 2019; 10: 659–666
References
- 1. Smith BW, Adams LA. Nonalcoholic fatty liver disease and diabetes mellitus: pathogenesis and treatment. Nat Rev Endocrinol 2011; 7: 456–465. [DOI] [PubMed] [Google Scholar]
- 2. Hebbard L, George J. Animal models of nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol 2011; 8: 35–44. [DOI] [PubMed] [Google Scholar]
- 3. Tanaka M, Itoh M, Ogawa Y, et al Molecular mechanism of obesity‐induced ‘metabolic’ tissue remodeling. J Diabetes Investig 2018; 9: 256–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sica A, Invernizzi P, Mantovani A. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology 2014; 59: 2034–2042. [DOI] [PubMed] [Google Scholar]
- 5. Wan J, Benkdane M, Teixeira‐Clerc F, et al M2 Kupffer cells promote M1 Kupffer cell apoptosis: a protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology 2014; 59: 130–142. [DOI] [PubMed] [Google Scholar]
- 6. Kitade H, Chen G, Ni Y, et al Nonalcoholic fatty liver disease and insulin resistance: new insights and potential new treatments. Nutrients 2017; 9: E387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Anstee QM, Targher G, Day CP. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat Rev Gastroenterol Hepatol 2013; 10: 330–344. [DOI] [PubMed] [Google Scholar]
- 8. Polyzos SA, Mantzoros CS. Adiponectin as a target for the treatment of nonalcoholic steatohepatitis with thiazolidinediones: a systematic review. Metabolism 2016; 65: 1297–1306. [DOI] [PubMed] [Google Scholar]
- 9. Papandreou D, Andreou E. Role of diet on non‐alcoholic fatty liver disease: an updated narrative review. World J Hepatol 2015; 7: 575–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pavlov VA, Tracey KJ. The vagus nerve and the inflammatory reflex–linking immunity and metabolism. Nat Rev Endocrinol 2012; 8: 743–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Borovikova LV, Ivanova S, Zhang M, et al Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000; 405: 458–462. [DOI] [PubMed] [Google Scholar]
- 12. Kimura K, Tanida M, Nagata N, et al Central insulin action activates kupffer cells by suppressing hepatic vagal activation via the nicotinic alpha 7 acetylcholine receptor. Cell Rep 2016; 14: 2362–2374. [DOI] [PubMed] [Google Scholar]
- 13. Wang H, Yu M, Ochani M, et al Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003; 421: 384–388. [DOI] [PubMed] [Google Scholar]
- 14. Wang X, Yang Z, Xue B, et al Activation of the cholinergic antiinflammatory pathway ameliorates obesity‐induced inflammation and insulin resistance. Endocrinology 2011; 152: 836–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Inoue H, Ogawa W, Asakawa A, et al Role of hepatic STAT3 in brain‐insulin action on hepatic glucose production. Cell Metab 2006; 3: 267–275. [DOI] [PubMed] [Google Scholar]
- 16. Kimura K, Nakamura Y, Inaba Y, et al Histidine augments the suppression of hepatic glucose production by central insulin action. Diabetes 2013; 62: 2266–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sanches SC, Ramalho LN, Augusto MJ, et al Nonalcoholic steatohepatitis: a search for factual animal models. Biomed Res Int 2015; 2015: 574832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rinella ME, Elias MS, Smolak RR, et al Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline‐deficient diet. J Lipid Res 2008; 49: 1068–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Watanabe H, Inaba Y, Kimura K, et al Dietary mung bean protein reduces hepatic steatosis, fibrosis, and inflammation in male mice with diet‐induced, nonalcoholic fatty liver disease. J Nutr 2017; 147: 52–60. [DOI] [PubMed] [Google Scholar]
- 20. Inaba Y, Furutani T, Kimura K, et al Growth arrest and DNA damage‐inducible 34 regulates liver regeneration in hepatic steatosis in mice. Hepatology 2015; 61: 1343–1356. [DOI] [PubMed] [Google Scholar]
- 21. Inoue H, Ogawa W, Ozaki M, et al Role of STAT‐3 in regulation of hepatic gluconeogenic genes and carbohydrate metabolism in vivo. Nat Med 2004; 10: 168–174. [DOI] [PubMed] [Google Scholar]
- 22. Watanabe H, Inaba Y, Kimura K, et al Sirt2 facilitates hepatic glucose uptake by deacetylating glucokinase regulatory protein. Nat Commun 2018; 9: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kimura K, Yamada T, Matsumoto M, et al Endoplasmic reticulum stress inhibits stat3‐dependent suppression of hepatic gluconeogenesis via dephosphorylation and deacetylation. Diabetes 2012; 61: 61–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nishio T, Taura K, Iwaisako K, et al Hepatic vagus nerve regulates Kupffer cell activation via alpha7 nicotinic acetylcholine receptor in nonalcoholic steatohepatitis. J Gastroenterol 2017; 52: 965–976. [DOI] [PubMed] [Google Scholar]
- 25. Li DJ, Liu J, Hua X, et al Nicotinic acetylcholine receptor alpha7 subunit improves energy homeostasis and inhibits inflammation in nonalcoholic fatty liver disease. Metabolism 2018; 79: 52–63. [DOI] [PubMed] [Google Scholar]
- 26. Younossi ZM, Stepanova M, Rafiq N, et al Pathologic criteria for nonalcoholic steatohepatitis: interprotocol agreement and ability to predict liver‐related mortality. Hepatology 2011; 53: 1874–1882. [DOI] [PubMed] [Google Scholar]
- 27. Scherer T, Lindtner C, O'Hare J, et al Insulin regulates hepatic triglyceride secretion and lipid content via signaling in the brain. Diabetes 2016; 65: 1511–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Finger JH, Smith CM, Hayamizu TF, et al The mouse Gene Expression Database (GXD): 2017 update. Nucleic Acids Res 2017; 45: D730–D736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hajiasgharzadeh K, Tavangar SM, Javan M, et al Does hepatic vagus nerve modulate the progression of biliary fibrosis in rats? Auton Neurosci 2014; 185: 67–75. [DOI] [PubMed] [Google Scholar]
- 30. Hiramoto T, Chida Y, Sonoda J, et al The hepatic vagus nerve attenuates Fas‐induced apoptosis in the mouse liver via alpha7 nicotinic acetylcholine receptor. Gastroenterology 2008; 134: 2122–2131. [DOI] [PubMed] [Google Scholar]
- 31. Li F, Chen Z, Pan Q, et al The protective effect of PNU‐282987, a selective alpha7 nicotinic acetylcholine receptor agonist, on the hepatic ischemia‐reperfusion injury is associated with the inhibition of high‐mobility group box 1 protein expression and nuclear factor kappaB activation in mice. Shock 2013; 39: 197–203. [DOI] [PubMed] [Google Scholar]
- 32. Freedman R. alpha7‐nicotinic acetylcholine receptor agonists for cognitive enhancement in schizophrenia. Annu Rev Med 2014; 65: 245–261. [DOI] [PubMed] [Google Scholar]
- 33. Hernandez CM, Dineley KT. alpha7 nicotinic acetylcholine receptors in Alzheimer's disease: neuroprotective, neurotrophic or both? Curr Drug Targets 2012; 13: 613–622. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1¦ Hepatic messenger ribonucleic acid expression levels of genes related to (a) lipogenesis, (b) β‐oxidation and very high‐density lipoprotein (VLDL) secretion, and macrophage polarization markers in mice fed a normal chow (NC) or an atherogenic high‐fat diet (AD)for 32 weeks.
Figure S2¦ Hepatic messenger ribonucleic acid expression levels of genes related to (a) lipogenesis, (b) β‐oxidation and VLDL secretion, and macrophage polarization markers in mice fed a normal chow (NC) or a methionine/choline‐deficient diet (MCD) for 6 weeks.
Table S1¦ Primer sequences.