

Abstract
Aims/Introduction
Diabetes is an important risk factor for atherosclerotic disease. The initiating factor of atherosclerosis is local endothelial cell injury. The arachidonic acid metabolite, 12(S)‐hydroxyeicosatetraenoic acid (12[S]‐HETE), might be involved in this process. In recent years, some studies have discussed the effect of 12(S)‐HETE on vascular endothelial cell function. In the present study, we investigated the effect of 12(S)‐HETE on vascular endothelial cell function in high‐glucose conditions and the mechanisms involved.
Materials and Methods
Human umbilical vein endothelial cells were cultured in conventional M199 medium and high‐glucose M199 medium. Human umbilical vein endothelial cells were stimulated with 12(S)‐HETE and cinnamyl‐3,4‐dihydroxy‐α‐cyanocinnamate (a 12/15‐lipoxygenases inhibitor). A type 1 diabetes mellitus model was established in C57BL/6 or 12/15‐lipoxygenases knockout mice with streptozotocin. Aortic tissue was harvested for subsequent testing. The transmembrane transport of dextran and human acute monocytic leukaemia cell line (THP‐1) cells was measured. The adherens junction protein, IkBα, nuclear factor kappa Bp65 (P65), intercellular adhesion molecule 1 and vascular cell adhesion protein 1 expression and phosphorylation, and the binding/dissociation of endothelial cell components were observed.
Results
Transendothelial migration of dextran and THP‐1 cells was significantly increased by stimulation of human umbilical vein endothelial cell monolayers with high glucose and 12(S)‐HETE (P < 0.05). High glucose and 12(S)‐HETE altered the vascular endothelial cadherin and β‐catenin phosphorylation level, and promoted the dissociation of β‐catenin and vascular endothelial cadherin. Expression levels of P‐Ikbα, P‐P65, intercellular adhesion molecule 1 and vascular cell adhesion protein 1 were elevated in high glucose and 12(S)‐HETE treated cells and diabetic mice compared with controls (P < 0.05).
Conclusions
The lipoxygenases metabolite, 12(S)‐HETE, can impair vascular endothelial permeability by altering adherens junction phosphorylation levels, and affecting the binding and dissociation of its components in high‐glucose conditions.
Keywords: 12(S)‐hydroxyeicosatetraenoic acid, Endothelial permeability, High glucose
Introduction
Diabetes is one of the most important risk factors for atherosclerotic disease (equivalent to coronary heart disease). The risk of atherosclerotic disease in patients with diabetes mellitus is two‐ to threefold that of non‐diabetes patients1. Although atherosclerosis is a complex process, excessive inflammation and lipid accumulation are at its essence2. The primary initiating factor is local endothelial cell injury, and the integrity of vascular endothelial function primarily depends on three aspects: (i) actin–myosin‐based contraction machinery; (ii) adherens junctions; and (iii) tight junctions3.
Adherens junctions are involved in physical connections between cells and structures underneath cells. The normal structure of adherens junctions is necessary for proper structure of tight junctions4. Simultaneously, adherens junctions also play an important regulatory role in the expression and function of tight junction proteins5, 6. Each adherens junction is composed of a transmembrane component (vascular endothelial cadherin [VE‐cadherin]) and intracellular components (β‐catenin, P120‐catenin and α‐catenin)3. VE‐cadherin can directly bind to β‐catenin, which connects to the actin cytoskeleton and an array of signaling proteins, thereby stabilizing VE‐cadherin‐mediated cell adhesion and related signal transmission. As such, the VE‐cadherin–β‐catenin complex plays an important role in maintaining the integrity of endothelial cells, angiogenesis, vascularization and stabilization of the vascular environment7, 8. Therefore, studies of VE‐cadherin and β‐catenin expression have important implications for our understanding of endothelial cell permeability and integrity.
Lipoxygenases (LO) are a group of oxygenases that do not contain heme iron, making it possible to insert oxygen molecules into polyunsaturated fatty acids represented by arachidonic acid and linoleic acid9. Thus, LO are important components of three major metabolic pathways for unsaturated fatty acids, together with cyclooxygenase and cytochrome P‐45010. Depending on the location of carbon atoms onto which oxygen molecules are inserted, LO are classified into 5‐LO, 8‐LO, 12‐LO and 15‐LO. As human and rabbit 12‐LO and 15‐LO are highly homologous, and can both decompose substrates to produce 12(S)‐hydroxyeicosatetraenoic acid (HETE) and 15(S)‐HETE, they are collectively called 12/15‐LO11.
Under normal circumstances, the content of free arachidonic acid within the body is very low. However, in some pathological conditions, such as hyperglycemia, hyperlipidemia, hypertension and hypoxia, arachidonic acid can be released from cell membranes and converted into biologically active metabolites under the action of 12/15‐LO12, 13. 12‐HETE and 15‐HETE, the major metabolites of 12/15‐LO, have important physiological functions and are important pathological factors. Furthermore, 12(S)‐HETE and 15(S)‐HETE have been shown to participate in vasoactive, inflammatory and proliferative responses, and regulate ion transport, and kidney and respiratory functions14, 15, 16. Some studies have also shown that 12(S)‐HETE and 15(S)‐HETE have important roles in promoting proliferation and inflammatory factors, especially in vascular endothelial and kidney cells15, 16, 17. 12(S)‐HETE is increased in diabetes patients with incipient and early renal disease18, and is increased in diabetes patients with coronary artery disease19. In recent years, some reports have shown the effect of 12(S)‐HETE on vascular endothelial cell function. Studies showed that 12‐ and 15‐HETE increased retinal endothelial cell permeability during diabetic retinopathy through nicotinamide adenine dinucleotide phosphate oxidase or nicotinamide adenine dinucleotide phosphate oxidase‐dependent mechanism20, 21, 22. 12/15‐LO and its AA metabolite, 15(S)‐HETE, play an important role in high‐fat diet‐induced endothelial tight junction disruption and macrophage adhesion, which are crucial events underlying the pathogenesis of atherosclerosis23.
Based on the above, we hypothesized that 12(S)‐HETE was directly implicated in endothelial injury in diabetes by stimulating factors that alter protein phosphorylation levels to damage cell–cell junctions and increase vascular permeability. The present study investigated the mechanisms of vascular endothelial injury induced by 12(S)‐HETE in high‐glucose conditions, and provides information that will be helpful for understanding the process of arterial injury in diabetes patients and might open up new targets for therapeutic development.
Methods
Cell vulture
Human umbilical vein endothelial cells (HUVECs; ATCC, Rockefeller, MD, USA) were cultured in M199 (Gibco, Grand Island, NY, USA) medium supplemented with low‐serum growth supplement (Gibco), 10 mg/L gentamicin, and 0.25 mg/L amphotericin in a 5% CO2 incubator at 37°C and 95% humidity. Cells at passages 6–8 were harvested for experiments. Before testing, HUVECs were seeded in 100‐mm Petri dishes. At 100% confluence, the cells were incubated with M199 medium without growth factors to promote them entering a stationary phase for the following tests. High‐glucose medium was M199 medium containing d‐glucose for a final concentration of 25 mmol/L (high‐glucose M199). Control group cells were cultured in M199 medium containing a normal concentration of glucose (5.6 mmol/L). HUVECs were also incubated with 12(S)‐HETE (0.1 μmol/L), cinnamyl‐3,4‐dihydroxy‐α‐cyanocinnamate (CDC; a 12/15‐Lipoxygenases inhibitor, 10 μmol/L) and mannitol (19.4 mmol/L). CDC can inhibit 12‐ and 15‐lipoxygenaseactivity, so this compound has been used as a pharmacological tool to identify 12/15‐LO‐mediated action24.
The experiment contained six groups: (i) control; (ii) high‐glucose; (iii) 12(S)‐HETE (0.1 μmol/L, normal glucose concentration; Biomol, Plymouth Meeting, PA, USA); (vi) high glucose + 12(S)‐HETE (0.1 μmol/L); (v) high glucose + CDC (10 μmol/L; Biomol); and (vi) normal glucose + mannitol (19.4 mmol/L).
Animal research
An animal model of type 1 diabetes mellitus was established by small‐dose multiple intraperitoneal injection of streptozotocin (Sigma, St. Louis, MO, USA). C57BL/6 (Beijing, China) or 12/15‐LO knockout (12/15‐LOKO) mice of C57BL/6 background were fasted for 12 h and then intraperitoneally injected with streptozotocin 50 mg/kg daily for five consecutive days25. Mice not included in the diabetes control group were intraperitoneally injected with an equal volume of sodium citrate buffer, for five consecutive days. Blood was obtained from the caudal vein. Blood glucose was measured with an Accu‐Chek ActiveX blood glucose meter (Roche Diagnostics GmbH, Mannheim, Germany). More than 30 mmol/L indicated successful model induction 5 days after streptozotocin administration. The experiment had four groups: control, diabetes, 12/15‐LOKO and 12/15‐LOKO + diabetes. There were 10 mice in each group. After 16 weeks, mice were killed and aortic tissue was harvested for subsequent testing.
Ethics approval of the study protocol
All animal protocols for the present study conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health. All experimental procedures were approved by the Committee of Animal Care and Use at Zhengzhou University (Zhengzhou, China).
Fluorescein isothiocyanate‐labeled transendothelial dextran flux
HUVECs were seeded into a Transwell system. At 100% confluence, a monolayer endothelial cell barrier was formed. A total of 100 μg/mL fluorescein isothiocyanate (FITC)‐labeled dextran (MW 70,000; Seebio, Shanghai, China) was added to the lower chamber. According to group assignment, high glucose, mannitol, 12(S)‐HETE (0.1 μmol/L) or high glucose + CDC (10 μmol/L) were added to the lower and upper chambers, and incubated for 2 h26. From this 100 μL of medium was obtained from the lower and upper chambers, separately. Fluorescence intensity was measured by a fluorescence spectrometer (F98, Shanghai Lengguang Technology Co., Ltd., Shanghai, China) and the transendothelial dextran flux rate (% dextran/h/cm2) was calculated.
Transmembrane migration of THP‐1 cells
Transmembrane migration was measured with a Transwell system with 8.0‐μm pores (Millipore, St. Louis, MO, USA)27. Matrigel (50%) was painted onto the back of the Transwell, and HUVECs were seeded onto the other side and cultured until 100% confluence and formation of an endothelial monolayer barrier. THP‐1 cells (ATCC) in the stationary phase were labeled with 2′,7′‐bis‐(2‐carboxyethyl)5‐(and‐6)‐carboxyfluorescein acetoxymethyl ester (Fanbo, Beijing, China; 1 × 105 cells/well), seeded onto the surface of HUVECs and incubated with high glucose, mannitol, 12(S)‐HETE (0.1 μmol/L) and CDC (10 μmol/L) for 6 h. Non‐migrating THP‐1 cells were wiped off with a cotton swab. The Matrigel was cut, fixed and placed on the slide, and the number of migrating cells was observed with an inverted microscope (CX31; Olympus, Tokyo, Japan).
Adherens junction protein expression, phosphorylation and component binding/dissociation
Cultured HUVECs were treated with high glucose, 12(S)‐HETE, mannitol or high glucose + CDC for 30 min. Proteins were extracted from HUVECs and aortic tissue, and lysates were used for western blot assay and co‐immunoprecipitation. A total of 50 μg of protein was separated by sodium dodecyl sulfate‐polyacrylamide gel electrophoresis. Western blot analysis was then carried out as described28 to analyze total VE‐cadherin protein, total β‐catenin protein and phosphorylation of VE‐cadherin (anti‐phospho‐VE‐cadherin (Y731) as primary antibody, 1:500; Cell Signaling, Boston, MA, USA). The protein was transferred from the gel to polyvinylidene fluoride membranes, which were incubated with antibody for 1 h at room temperature and then at 4°C overnight. After washing with Tris‐buffered saline Tween‐20, the membranes were incubated with secondary antibody and signals detected by chemiluminescence. Quantification of the bands was carried out using densitometric analysis software (Bio‐Rad, Hercules, CA, USA). Phosphorylation of β‐catenin protein was determined by co‐immunoprecipitation with anti‐serine/threonine phosphorylated antibody (pSer/Thr antibody, 1:500; Cell Signaling). β‐Catenin protein‐specific antibody was used for hybridization to determine its phosphorylation levels. Detection of adherens junction (β‐catenin and VE‐cadherin) binding/dissociation was carried out using co‐immunoprecipitation. A total of 300 μg of protein harvested from the control and treatment groups was incubated with VE‐cadherin antibody (Cell Signaling) overnight, then mixed with 80 μL of protein‐A/G beads at 4°C, shaking for 1 h. The beads were washed three times with cell lysate, and boiled with 20 μL of 2× loading buffer at 100°C for 5 min. After centrifugation, the supernatant was used for western blot assay. β‐Catenin antibody was utilized for hybridization to determine their binding/dissociation with VE‐cadherin protein.
Ikbα, P65, intercellular adhesion molecule 1 and vascular cell adhesion protein 1 protein expression and phosphorylation
The levels of Ikbα, P65, intercellular adhesion molecule 1 (ICAM‐1) and vascular cell adhesion protein 1 (VCAM‐1), and phosphorylated Ikbα and P65 (Cell Signaling) were all measured by western blotting, in a similar way to the method described above. For these samples, the loading amount was 50 μg, the concentration of primary antibody was 1:500 and the concentration of the secondary antibody was 1:3,000.
Histology
Tissue samples were fixed by being immersed in 10% formaldehyde solution at 4°C for 8 h, and were then embedded in paraffin. Sections of 5‐μm slice thickness were prepared, and they underwent hematoxylin–eosin staining by standard techniques, before being observed under a light microscope to visualize vascular endothelial and wall damage.
12(S)‐HETE protein measurement
The level of 12(S)‐HETE protein was measured by enzyme‐linked immunosorbent assay (ELISA). The aorta was lysed in tissue protein lysis solution. Then, protein was extracted after ultrasonic fragmentation, and then total protein concentration was measured. The ELISA was carried out according to the ELISA kit procedure from the manufacturer. Absorbance (A) was measured at 450 nm using a microplate reader. Sample concentration was calculated based on the prepared standard curve. The total protein concentration of the sample was divided by the measured sample concentration to calculate the content of 12(S)‐HETE protein for per unit mass in the sample.
Serum measurements
Mouse blood was taken from the tail vein, and a fasting blood glucose meter (Roche) was used for determination of blood glucose. Insulin was determined by radioimmunoassay, and the ELISA mouse glycosylated hemoglobin A1c test kit (Teco Diagnostics, Anaheim, CA, USA) was used to determine glycosylated hemoglobin A1c.
Statistical analysis
All data were analyzed using SPSS 13.0 (SPSS Inc., Chicago, IL, USA) software. All data were expressed as mean ± standard deviation, and significant differences among groups were evaluated using one‐way analysis of variance and followed by least significant difference t‐tests for post‐hoc comparisons. A value of P < 0.05 was considered statistically significant.
Results
Effects of 12(S)‐HETE on endothelial barrier permeability
FITC‐labeled transendothelial dextran flux and leukocyte transmigration showed that high glucose and 12(S)‐HETE could destroy endothelial cell barriers. High glucose or 12(S)‐HETE remarkably increased transendothelial dextran transport, and in combination it was increased further. Addition of the 12/15‐LO inhibitor, CDC, partially suppressed dextran transport. Similar observations were made for leukocyte transmigration. High glucose or 12(S)‐HETE treatment increased transendothelial transport of THP‐1 cells by threefold, and in combination 12(S)‐HETE and high glucose effects were increased by approximately fivefold (Figure 1).
Figure 1.
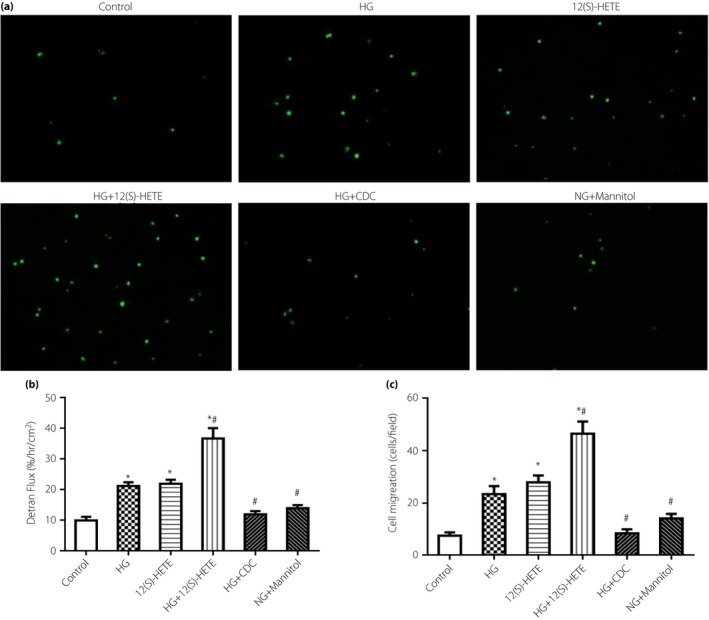
Transendothelial (a,c) cell migration and (b) dextran transport induced by high glucose (HG) and 12(S)‐hydroxyeicosatetraenoic acid (12[S]‐HETE). Compared with the control group, transendothelial transport of human acute monocytic leukaemia cell line (THP‐1) cells was increased after high glucose and 12(S)‐HETE treatment. Cinnamyl‐3,4‐dihydroxy‐α‐cyanocinnamate (CDC) partially inhibited this effect. The addition of mannitol was similar to the control. *P < 0.05, vs the control group; # P < 0.05, vs the 12(S)‐HETE group.
Effects of 12(S)‐HETE on adherens junction protein expression, phosphorylation levels and component binding/dissociation in endothelial cells
Western blot assay and co‐immunoprecipitation were carried out to analyze the phosphorylation levels and total protein changes of VE‐cadherin and β‐catenin. The results showed little change in total protein levels for each group, but some changes to the phosphorylation levels. VE‐cadherin phosphorylation levels increased, but β‐catenin phosphorylation levels decreased in the high glucose and 12(S)‐HETE groups, and the effect was increased with the two conditions combined. The 12/15‐LO inhibitor, CDC, partially reversed this trend. These findings suggested high glucose and 12(S)‐HETE might regulate endothelial barrier permeability by altering phosphorylation of adherens junction proteins (Figure 2a,b). With regard to binding and dissociation of VE‐cadherin and β‐catenin proteins, reduced β‐catenin expression was observed in the high glucose and 12(S)‐HETE groups, suggesting these factors induce the dissociation of β‐catenin from VE‐cadherin (Figure 2c).
Figure 2.
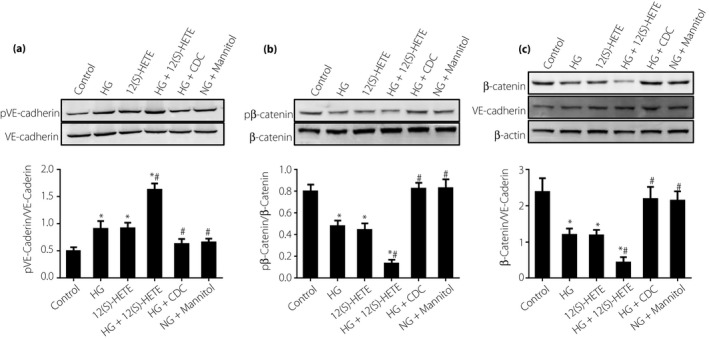
(a) Effects of 12(S)‐hydroxyeicosatetraenoic acid (12[S]‐HETE) on vascular endothelial cadherin (VE‐caderin) expression and phosphorylation levels in endothelial cells. 12(S)‐HETE and high glucose (HG) increased VE‐caderin phosphorylation levels. (b) Effects of 12(S)‐HETE on β‐catenin expression and phosphorylation levels in endothelial cells. (c) Effects of 12(S)‐HETE on adherens junction component binding/dissociation in endothelial cells. 12(S)‐HETE and high glucose promoted the dissociation of β‐catenin and VE‐cadherin. *P < 0.05, vs the control group; # P < 0.05, vs the 12(S)‐HETE group. CDC, cinnamyl‐3,4‐dihydroxy‐α‐cyanocinnamate, NG, normal glucose.
Effects of 12(S)‐HETE on Ikbα, P65, ICAM‐1 and VCAM‐1 protein expression and phosphorylation levels in endothelial cells
The levels of P‐Ikbα, P‐P65, ICAM‐1 and VCAM‐1 were all elevated in the high glucose group and 12(S)‐HETE group, and were highest in the high glucose and 12(S)‐HETE combined group, whereas the expression levels in the high glucose + CDC group and the mannitol group were almost equivalent to that of the normal group (Figure 3).
Figure 3.
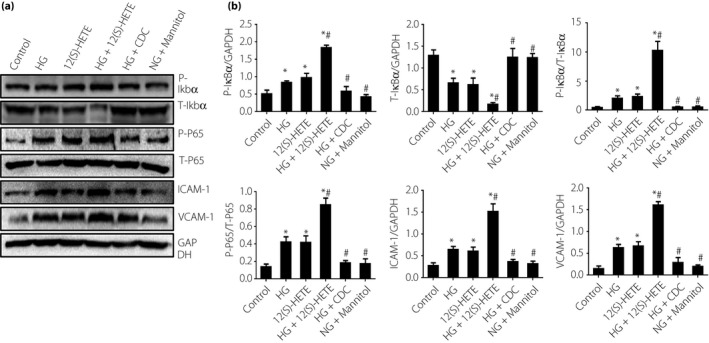
(a, b) Effects of 12(S)‐hydroxyeicosatetraenoic acid (12[S]‐HETE), high glucose (HG), cinnamyl‐3,4‐dihydroxy‐α‐cyanocinnamate (CDC) and mannitol on levels of P‐Ikbα, T‐Ikbα, P‐nuclear factor kappa Bp65 (P‐P65), T‐P65, intercellular adhesion molecule 1 (ICAM‐1) and vascular cell adhesion protein 1 (VCAM‐1) in endothelial cells. 12(S)‐HETE and high glucose increased P‐Ikbα, P‐P65, ICAM‐1 and VCAM‐1 levels, but decreased T‐Ikbα levels compared with control cells. *P < 0.05, vs the control group; # P < 0.05, vs the 12(S)‐HETE group. GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; NG, normal glucose.
T‐Ikbα expression and P‐Ikbα expression showed opposite trends; when T‐Ikbα expression was high in the normal group, it was decreased in the high glucose and 12(S)‐HETE groups, and they were at their lowest in the combination of high glucose and 12(S)‐HETE group. T‐Ikbα expression levels in the high glucose + CDC group and the mannitol group were almost equivalent to that of the normal group (Figure 3).
Changes of T‐P65 expression were not significant in any of the various groups studied (Figure 3).
Effects of 12(S)‐HETE on adherens junction protein expression, phosphorylation levels and component binding/dissociation in diabetic model mice
Table 1 shows the results of establishing the type 1 diabetes mellitus model in the two mice groups. There were significant decreases in bodyweight in the animals treated with streptozotocin compared with the sham groups, and significantly increased serum glucose and glycosylated hemoglobin A1c, with decreased insulin levels (all P < 0.05).
Table 1.
General characteristics of diabetic mice
| Sham | Type 1 diabetes mellitus | 12/15‐LOKO | Type 1 diabetes mellitus + 12/15‐LOKO | |
|---|---|---|---|---|
| Bodyweight (g) | 21.4 ± 2.1 | 16.7 ± 1.3* | 19.1 ± 3.0** | 18.8 ± 2.6** |
| Serum glucose (mmol/L) | 5.6 ± 1.1 | 38.7 ± 7.9* | 5.5 ± 0.9** | 6.5 ± 1.6** |
| HbA1c (%) | 5.5 ± 0.4 | 11.3 ± 0.8* | 5.9 ± 0.6** | 7.0 ± 0.9** |
| Insulin (ng/mL) | 1.51 ± 0.06 | 0.57 ± 0.03* | 1.49 ± 0.05** | 1.33 ± 0.06** |
Values are expressed as means ± standard deviation from 10 mice for each experimental condition. *P < 0.05, vs the sham group; **P < 0.05, vs the type 1 diabetes mellitus group. 12/15‐LOKO, 12/15‐lipoxygenases knockout mice; HbA1c, glycosylated hemoglobin A1c.
hematoxylin–eosin staining showed obvious vascular endothelial wall damage in the type 1 diabetes mellitus mouse group compared with the sham mice, but the damage was less obvious in the type 1 diabetes mellitus + 12/15‐LOKO mice (Figure 4a). The type 1 diabetes mellitus mice showed significantly increased 12(S)‐HETE levels than the sham mice in aortic tissue (Figure 4b).
Figure 4.
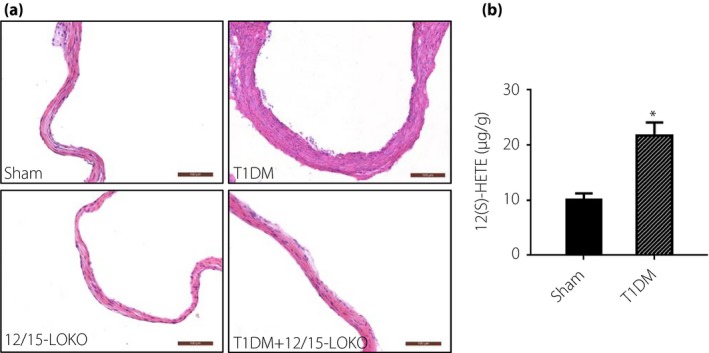
(a) Hematoxylin–eosin staining of mouse vascular endothelial samples showing obvious wall damage in the type 1 diabetes mellitus mouse group. The damage was less obvious in the type 1 diabetes mellitus + 12/15‐lipoxygenases knockout mice (12/15‐LOKO) group with the 12(S)‐hydroxyeicosatetraenoic acid (12[S]‐HETE) knockout mice group. (b) 12(S)‐HETE levels in the mouse samples. *P < 0.05, vs the sham group.
VE‐cadherin phosphorylation levels were obviously increased in diabetic mice. VE‐cadherin phosphorylation levels were higher in diabetic mice than in 12/15‐LOKO mice. Changes in β‐catenin phosphorylation levels showed the opposite trend. β‐Catenin phosphorylation levels were minimal in diabetic mice, but maximal in 12/15‐LOKO mice (Figure 5). Regarding the VE‐cadherin–β‐catenin complex, the level of β‐catenin was decreased in the type 1 diabetes mellitus group. However, the levels were almost equivalent in the sham and 12/15‐LOKO groups. These results showed that high glucose and 12(S)‐HETE promoted the dissociation of β‐catenin and VE‐cadherin, consistent with the cell research results (Figure 5c).
Figure 5.
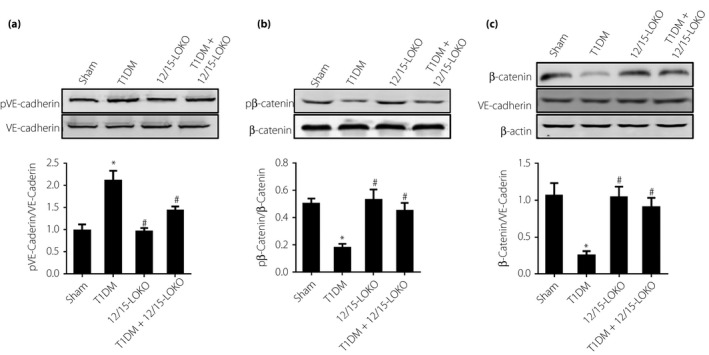
(a) The effects of 12(S)‐hydroxyeicosatetraenoic acid (12[S]‐HETE) on vascular endothelial cadherin (VE‐caderin) expression and phosphorylation levels in diabetic model mice. VE‐cadherin phosphorylation levels were obviously increased in diabetic mice. (b) The effects of 12(S)‐HETE on β‐catenin expression and phosphorylation levels in diabetic model mice. β‐Catenin phosphorylation levels were minimal in diabetic mice. (c) Effects of 12(S)‐HETE on adherens junction component binding/dissociation in aortic endothelium. 12(S)‐HETE promoted the dissociation of β‐catenin and VE‐cadherin. *P < 0.05, vs the sham group. # P < 0.05, vs the type 1 diabetes mellitus group. 12/15‐LOKO, 12/15‐lipoxygenases knockout mice.
Effects of 12(S)‐HETE on Ikbα, P65, ICAM‐1 and VCAM‐1 protein expression and phosphorylation levels in diabetic model mice
Expression levels of P‐Ikbα, P‐P65, ICAM‐1 and VCAM‐1 were elevated significantly in diabetic mice. They were almost equivalent in the 12/15‐LOKO mice and normal mice. The expression levels were significantly decreased in the type 1 diabetes mellitus + 12/15‐LOKO mice compared with diabetic mice (P < 0.05), but were higher than those of normal mice (P > 0.05; Figure 6).
Figure 6.
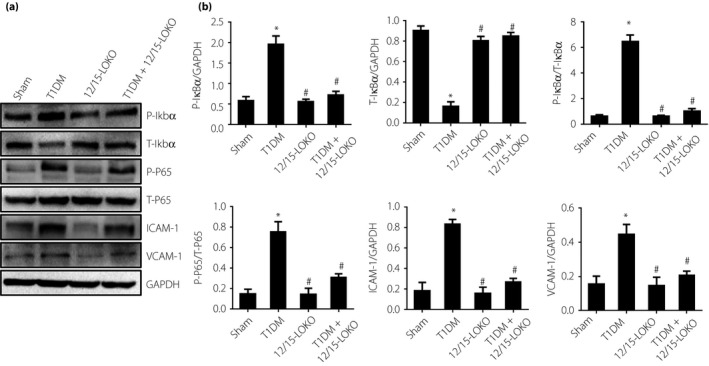
(a,b) Effects of 12(S)‐hydroxyeicosatetraenoic acid (12[S]‐HETE) on levels of P‐Ikbα, T‐Ikbα, P‐nuclear factor kappa Bp65 (P‐P65), T‐P65, intercellular adhesion molecule 1 (ICAM‐1) and vascular cell adhesion protein 1 (VCAM‐1) in diabetic model mice. P‐Ikbα, P‐P65, ICAM‐1 and VCAM‐1 levels were increased in diabetic mice, whereas T‐Ikbα was decreased in diabetic mice and higher in 2/15‐lipoxygenases knockout (12/15‐LOKO) mice. *P < 0.05, vs the sham group. # P < 0.05, vs the type 1 diabetes mellitus group. GAPDH, glyceraldehyde 3‐phosphate dehydrogenase.
T‐Ikbα expression showed an opposite trend to that of P‐Ikbα. T‐Ikbα expression was highest in the normal mice and 12/15‐LOKO mice, but lowest in diabetic mice. T‐Ikbα expression increased significantly in DM + 12/15‐LOKO mice compared with diabetic mice (P < 0.05), but was lower than that of normal mice (P > 0.05; Figure 6).
Changes of T‐P65 expressions were not significant in various groups (Figure 6). These results were consistent with the cell culture experiments.
Discussion
The vascular endothelium controls the transendothelial transport of macromolecules and inflammatory cells. Endothelial dysfunction is a key step in the early stage of atherosclerosis. Currently, the most widely accepted mechanism for vascular endothelial dysfunction involves inflammatory and oxidative damage. However, although high glucose and LO play a clear role in atherosclerosis, how these factors damage the vascular endothelial barrier to commence the process of atherosclerosis is unknown. The present study confirmed that high glucose and the LO metabolite 12(S)‐HETE can impair vascular endothelial permeability by altering adherens junction phosphorylation levels, and affecting the binding and dissociation of its components – the combination of which exerts a strong effect. Thus, it is proposed that 12(S)‐HETE plays an important role in the occurrence and development of atherosclerosis in diabetes patients.
Other studies have also shown that 12(S)‐HETE is likely to have an important role in the development of atherosclerosis in diabetes. A pig model showed that 12‐LO activation plays a key role in accelerated atherosclerosis29, and hyperglycemia causes upregulation of 12‐LO activity30. The increased production of 15(S)‐HETE activates monocyte integrins, which results in enhanced adhesion of monocytes to endothelium, a key event in the early development of atherosclerosis. However, the mechanisms involved require further exploration.
An essential component of the adherens junction, VE‐cadherin exerts a key effect for maintaining vascular integrity31, including cytoskeletal rearrangement32, establishment of cell polarity33 and tight junction remodeling34. Phosphorylation of VE‐cadherin protein regulates its activity, leading to instability of the adherens junction complex and changes in vascular permeability. At present, many soluble molecules are thought to cause VE‐cadherin tyrosine phosphorylation, thereby increasing vascular permeability, including vascular endothelial growth factor, tumor necrosis factor, platelet‐activating factor, thrombin and histamine35. Furthermore, these factors can lead to changes in β‐catenin phosphorylation levels and formation of VE‐cadherin–β‐catenin complexes, which contributes to disruption of intercellular connections. The present study explored the effects of high glucose and 12(S)‐HETE on VE‐cadherin and β‐catenin, and verified that high glucose and 12(S)‐HETE could alter VE‐cadherin and β‐catenin phosphorylation levels, but did not alter total protein expression. However, the 12/15‐LO inhibitor, CDC, antagonized the effect of high glucose on protein phosphorylation to mitigate destruction of the endothelial cell barrier, and the mouse diabetes mellitus model further confirmed these conclusions.
The interaction of VE‐cadherin with β‐catenin and other molecules has been shown to regulate the stability of adherens junctions. β‐Catenin binds to and connects VE‐cadherin with cytoskeletal proteins to allow endothelial cells to form tight attachments to each other, thus forming a selective semipermeable barrier between blood and tissue to control the exchange of liquid and macromolecular material on both sides of the vessel wall36, 37. When free in the cytoplasm, β‐catenin is phosphorylated and ubiquitinated by protein complexes, and becomes inactivated38. The present study observed the effects of high glucose and 12(S)‐HETE on the binding and dissociation of VE‐cadherin and β‐catenin in vascular endothelial cells. Both cell research and animal research found that high glucose and 12(S)‐HETE could induce the dissociation of β‐catenin from VE‐cadherin to affect vascular endothelial permeability.
With regard to the relationship between high glucose and 12(S)‐HETE, another study found that 12/15‐LO expression increased after in vitro cultured renal mesangial cells were treated with high glucose26. Thus, we propose that in diabetes patients, increased 12/15‐LO expression might be an important cause of endothelial function impairment induced by high glucose. Furthermore, the present study showed that the destructive effect of high glucose on vascular endothelium was significantly reduced after the addition of CDC in high‐glucose medium. Animal research also confirmed that adherens junction protein phosphorylation levels were minimal in 12(S)‐HETE knockout mice, and had only a minimal effect on the VE‐cadherin–β‐catenin complex.
To further investigate the role of 12(S)‐HETE in the mechanism of inflammation and the development and promotion of atherosclerosis, we investigated expression levels of Ikbα and P65 and their phosphorylated forms, and the levels of ICAM‐1 and VCAM‐1. Both Ikbα and P65 are important factors in regulating nuclear factor‐kappa B, a transcription factor that has a crucial role in inflammation39. Although ICAM‐1 and VCAM‐1 are cell surface adhesion molecules that are expressed in endothelial cells, and are implicated in the early development and progression of atherosclerosis40, 41. The results showed that in both the cell culture system and the mouse DM model, the levels of P‐Ikbα and P‐P65 increased in the presence of high glucose and 12(S)‐HETE and in the diabetic mice, whereas T‐Ikbα decreased and T‐P65 was relatively unchanged. These results suggest that inflammation via factor‐kappa B might be implicated in the development of atherosclerosis in the present study. This is supported by increased levels of ICAM‐1 and VCAM‐1 in the high glucose and 12(S)‐HETE treated cells, and the diabetic mice.
The present study suggests a potential mechanism by which underlying vascular endothelial injury is associated with diabetes. Full understanding of the mechanism requires further study to continue to explore how altered phosphorylation levels of adherens junction proteins affect downstream molecules to increase permeability. Understanding the theoretical basis for the role of oxidized lipid molecules in the pathogenesis of atherosclerosis might provide novel targets for potential therapeutic development in the future.
Disclosure
The authors declare no conflict of interest.
Acknowledgments
This study was supported by the National Nature Science Foundation of China (81400323) and the Key Scientific Research Project of Henan Province College (15A320029). All authors acknowledged the funding received. We thank Dr Jun‐nan Tang for editing the manuscript.
J Diabetes Investig 2019; 10: 639–649
References
- 1. Chiha M, Njeim M, Chedrawy EG. Diabetes and coronary heart disease: a risk factor for the global epidemic. Int J Hypertens 2012; 2012: 697240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature 1993; 362: 801–809. [DOI] [PubMed] [Google Scholar]
- 3. Dejana E, Orsenigo F, Lampugnani MG. The role of adherens junctions and VE‐cadherin in the control of vascular permeability. J Cell Sci 2008; 121: 2115–2122. [DOI] [PubMed] [Google Scholar]
- 4. Zhu H, Takahashi Y, Xu W, et al Low density lipoprotein receptor‐related protein‐mediated membrane translocation of 12/15‐lipoxygenase is required for oxidation of low density lipoprotein by macrophages. J Biol Chem 2003; 278: 13350–13355. [DOI] [PubMed] [Google Scholar]
- 5. Bazzoni G, Dejana E. Endothelial cell‐to‐cell junctions: molecular organization and role in vascular homeostasis. Physiol Rev 2004; 84: 869–901. [DOI] [PubMed] [Google Scholar]
- 6. Dejana E. Endothelial cell‐cell junctions: happy together. Nat Rev Mol Cell Biol 2004; 5: 261–270. [DOI] [PubMed] [Google Scholar]
- 7. Herwig MC, Muller KM, Muller AM. Endothelial VE‐cadherin expression in human lungs. Pathol Res Pract 2008; 204: 725–730. [DOI] [PubMed] [Google Scholar]
- 8. Panorchan P, Thompson MS, Davis KJ, et al Single‐molecule analysis of cadherin‐mediated cell‐cell adhesion. J Cell Sci 2006; 119: 66–74. [DOI] [PubMed] [Google Scholar]
- 9. Joo YC, Oh DK. Lipoxygenases: potential starting biocatalysts for the synthesis of signaling compounds. Biotechnol Adv 2012; 30: 1524–1532. [DOI] [PubMed] [Google Scholar]
- 10. Vega M, Karboune S, Kermasha S. Stability of immobilized soybean lipoxygenase in selected organic solvent media. Appl Biochem Biotechnol 2005; 127: 29–42. [DOI] [PubMed] [Google Scholar]
- 11. Yamamoto S. Mammalian lipoxygenases: molecular structures and functions. Biochim Biophys Acta 1992; 1128: 117–131. [DOI] [PubMed] [Google Scholar]
- 12. Natarajan R, Nadler JL. Lipid inflammatory mediators in diabetic vascular disease. Arterioscler Thromb Vasc Biol 2004; 24: 1542–1548. [DOI] [PubMed] [Google Scholar]
- 13. Yla‐Herttuala S, Rosenfeld ME, Parthasarathy S, et al Colocalization of 15‐lipoxygenase mRNA and protein with epitopes of oxidized low density lipoprotein in macrophage‐rich areas of atherosclerotic lesions. Proc Natl Acad Sci USA 1990; 87: 6959–6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ma C, Li Y, Ma J, et al Key role of 15‐lipoxygenase/15‐hydroxyeicosatetraenoic acid in pulmonary vascular remodeling and vascular angiogenesis associated with hypoxic pulmonary hypertension. Hypertension 2011; 58: 679–688. [DOI] [PubMed] [Google Scholar]
- 15. Reddy MA, Thimmalapura PR, Lanting L, et al The oxidized lipid and lipoxygenase product 12(S)‐hydroxyeicosatetraenoic acid induces hypertrophy and fibronectin transcription in vascular smooth muscle cells via p38 MAPK and cAMP response element‐binding protein activation. Mediation of angiotensin II effects. J Biol Chem 2002; 277: 9920–9928. [DOI] [PubMed] [Google Scholar]
- 16. Natarajan R, Gonzales N, Lanting L, et al Role of the lipoxygenase pathway in angiotensin II‐induced vascular smooth muscle cell hypertrophy. Hypertension 1994; 23: I142–I1147. [DOI] [PubMed] [Google Scholar]
- 17. Natarajan R, Reddy MA, Malik KU, et al Signaling mechanisms of nuclear factor‐kappab‐mediated activation of inflammatory genes by 13‐hydroperoxyoctadecadienoic acid in cultured vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 2001; 21: 1408–1413. [DOI] [PubMed] [Google Scholar]
- 18. Antonipillai I, Nadler J, Vu EJ, et al A 12‐lipoxygenase product, 12‐hydroxyeicosatetraenoic acid, is increased in diabetics with incipient and early renal disease. J Clin Endocrinol Metab 1996; 81: 1940–1945. [DOI] [PubMed] [Google Scholar]
- 19. Zhang HJ, Sun CH, Kuang HY, et al 12S‐hydroxyeicosatetraenoic acid levels link to coronary artery disease in Type 2 diabetic patients. J Endocrinol Invest 2013; 36: 385–389. [DOI] [PubMed] [Google Scholar]
- 20. Ibrahim AS, Elshafey S, Sellak H, et al A lipidomic screen of hyperglycemia‐treated HRECs links 12/15‐Lipoxygenase to microvascular dysfunction during diabetic retinopathy via NADPH oxidase. J Lipid Res 2015; 56: 599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Othman A, Ahmad S, Megyerdi S, et al 12/15‐Lipoxygenase‐derived lipid metabolites induce retinal endothelial cell barrier dysfunction: contribution of NADPH oxidase. PLoS ONE 2013; 8: e57254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ibrahim AS, Tawfik AM, Hussein KA, et al Pigment epithelium‐derived factor inhibits retinal microvascular dysfunction induced by 12/15‐lipoxygenase‐derived eicosanoids. Biochim Biophys Acta 2015; 1851: 290–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kundumani‐Sridharan V, Dyukova E, Hansen DR, et al 12/15‐Lipoxygenase mediates high‐fat diet‐induced endothelial tight junction disruption and monocyte transmigration: a new role for 15(S)‐hydroxyeicosatetraenoic acid in endothelial cell dysfunction. J Biol Chem 2013; 288: 15830–15842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tornhamre S, Elmqvist A, Lindgren JA. 15‐Lipoxygenation of leukotriene A(4). Studies of 12‐ and 15‐lipoxygenase efficiency to catalyze lipoxin formation. Biochim Biophys Acta 2000; 1484: 298–306. [DOI] [PubMed] [Google Scholar]
- 25. Yuan H, Lanting L, Xu ZG, et al Effects of cholesterol‐tagged small interfering RNAs targeting 12/15‐lipoxygenase on parameters of diabetic nephropathy in a mouse model of type 1 diabetes. Am J Physiol Renal Physiol 2008; 295: F605–F617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xu ZG, Miao LN, Cui YC, et al Angiotensin II type 1 receptor expression is increased via 12‐lipoxygenase in high glucose‐stimulated glomerular cells and type 2 diabetic glomeruli. Nephrol Dial Transplant 2009; 24: 1744–1752. [DOI] [PubMed] [Google Scholar]
- 27. Lin CI, Chen CN, Lin PW, et al Lysophosphatidic acid regulates inflammation‐related genes in human endothelial cells through LPA1 and LPA3. Biochem Biophys Res Commun 2007; 363: 1001–1008. [DOI] [PubMed] [Google Scholar]
- 28. Wang XF, Zhang JY, Li L, et al Metformin improves cardiac function in rats via activation of AMP‐activated protein kinase. Clin Exp Pharmacol Physiol 2011; 38: 94–101. [DOI] [PubMed] [Google Scholar]
- 29. Natarajan R, Gerrity RG, Gu JL, et al Role of 12‐lipoxygenase and oxidant stress in hyperglycaemia‐induced acceleration of atherosclerosis in a diabetic pig model. Diabetologia 2002; 45: 125–133. [DOI] [PubMed] [Google Scholar]
- 30. Hedrick CC, Kim MD, Natarajan RD, et al 12‐Lipoxygenase products increase monocyte:endothelial interactions. Adv Exp Med Biol 1999; 469: 455–460. [DOI] [PubMed] [Google Scholar]
- 31. Giannotta M, Trani M, Dejana E. VE‐cadherin and endothelial adherens junctions: active guardians of vascular integrity. Dev Cell 2013; 26: 441–454. [DOI] [PubMed] [Google Scholar]
- 32. Komarova Y, Malik AB. Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu Rev Physiol 2010; 72: 463–493. [DOI] [PubMed] [Google Scholar]
- 33. Lampugnani MG, Orsenigo F, Rudini N, et al CCM1 regulates vascular‐lumen organization by inducing endothelial polarity. J Cell Sci 2010; 123: 1073–1080. [DOI] [PubMed] [Google Scholar]
- 34. Taddei A, Giampietro C, Conti A, et al Endothelial adherens junctions control tight junctions by VE‐cadherin‐mediated upregulation of claudin‐5. Nat Cell Biol 2008; 10: 923–934. [DOI] [PubMed] [Google Scholar]
- 35. Breslin JW, Sun H, Xu W, et al Involvement of ROCK‐mediated endothelial tension development in neutrophil‐stimulated microvascular leakage. Am J Physiol Heart Circ Physiol 2006; 290: H741–H750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tinsley JH, Wu MH, Ma W, et al Activated neutrophils induce hyperpermeability and phosphorylation of adherens junction proteins in coronary venular endothelial cells. J Biol Chem 1999; 274: 24930–24934. [DOI] [PubMed] [Google Scholar]
- 37. Lampugnani MG, Corada M, Caveda L, et al The molecular organization of endothelial cell to cell junctions: differential association of plakoglobin, beta‐catenin, and alpha‐catenin with vascular endothelial cadherin (VE‐cadherin). J Cell Biol 1995; 129: 203–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Stelzner TJ, Weil JV, O'Brien RF. Role of cyclic adenosine monophosphate in the induction of endothelial barrier properties. J Cell Physiol 1989; 139: 157–166. [DOI] [PubMed] [Google Scholar]
- 39. Viatour P, Merville MP, Bours V, et al Phosphorylation of NF‐kappaB and IkappaB proteins: implications in cancer and inflammation. Trends Biochem Sci 2005; 30: 43–52. [DOI] [PubMed] [Google Scholar]
- 40. Roth FR, Skoura A, Matevossian A, et al Endothelial protein kinase MAP4K4 promotes vascular inflammation and atherosclerosis. Nat Commun 2015; 6: 8995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fotis L, Agrogiannis G, Vlachos IS, et al Intercellular adhesion molecule (ICAM)‐1 and vascular cell adhesion molecule (VCAM)‐1 at the early stages of atherosclerosis in a rat model. Vivo 2012; 26: 243–250. [PubMed] [Google Scholar]