

Abstract
Mutations of the hepatocyte nuclear factor 4α (HNF4α) gene give rise to maturity‐onset diabetes of the young type 1. Although many such mutations have been identified in affected individuals, part of these mutations has been characterized with regard to their pathological relevance. We here identified a missense mutation (c.773G>A, p.R258H) of HNF4A in a mother and daughter with early‐onset diabetes and impaired insulin secretion. In silico simulation and in vitro luciferase reporter analyses showed that the mutation impairs the stability of self‐dimerization and the transactivation activity of HNF4α. Although arginine‐258 does not appear to participate directly in dimerization, its mutation alters the electrostatic surface potential of the dimer interface. Our results thus suggest that this mutation impairs the function of HNF4α and thereby contributes to the pathogenesis of maturity‐onset diabetes of the young type 1.
Keywords: Hepatocyte nuclear factor 4α, Insulin secretion, Maturity‐onset diabetes of the young type 1
Introduction
Maturity‐onset diabetes of the young (MODY) is a monogenic form of diabetes characterized by autosomal dominant inheritance and a defect in pancreatic β‐cell function1. Mutations in the gene for the transcription factor hepatocyte nuclear factor 4α (HNF4α) give rise to MODY1, which accounts for ~5% of all MODY cases1, 2. Although >100 mutations of HNF4A have been identified in individuals clinically diagnosed with MODY1, part of these mutations has been characterized with regard to their pathological relevance3.
Here, we report two cases of MODY1 in a mother and daughter with a missense mutation (c.773G>A, p.R258H) of HNF4A. Although this mutation has been previously identified in siblings diagnosed with MODY14, functional analysis of the mutant protein has not been carried out. We therefore investigated the pathological relevance of this mutation with in silico simulation analysis and in vitro experiments.
Methods
Gene Sequencing
All exons of HNF4A were amplified by polymerase chain reaction from genomic deoxyribonucleic acid (DNA) isolated from peripheral blood cells and with primers listed in Table S1. The polymerase chain reaction products were directly sequenced with the use of a BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Carlsbad, CA, USA) and a 3730 DNA analyzer.
All the data of the proband and her mother presented in this study, including the results of genetic analyses, were collected for the diagnosis and the treatment of their disease. The proband and her mother provided written consent to publish their clinical data.
In silico simulation analysis
The three‐dimensional structure and electrostatic surface potential of mutant forms of HNF4α were analyzed as described previously5 with the use of three‐dimensional structure information for wild‐type human HNF4α obtained from Protein Data Bank (PDB ID: 3FS1). The binding stability of dimerization was evaluated with the ZDOCK score calculated with ZDOCK software6 as described in Data S1.
Luciferase Reporter Assay
The promoter region of the ABCC6 gene (nucleotides −332 to +72 [relative to the transcription start site]), a target of HNF4α7, was subcloned into the pGL3 Basic Vector (Promega, Madison, WI, USA) to yield pABCC6‐luc. COS‐7 cells were transfected with pABCC6‐luc, pcDNA3.1 encoding wild‐type or mutant forms of HNF4α2 (or empty pcDNA3.1) and a plasmid encoding β‐galactosidase. Luciferase and β‐galactosidase activities in cell lysates were measured 48 h after transfection, and the former was normalized by the latter.
Results
Case
The proband, a Japanese woman born with birthweight of 2,554 g at 36 weeks‐of‐gestation was diagnosed with diabetes at 10 years of age, and was admitted to Kobe University Hospital, Kobe, Japan, for evaluation and management of her disease at age 16 years. She was treated with basal–bolus insulin therapy. On admission, her hemoglobin A1c level was 14.5%, and antibodies to glutamic acid decarboxylase and to insulinoma‐associated protein‐2 were negative (Table S2). She had no severe hypoglycemic episode and apparent diabetic microvascular or macrovascular complications. Her mother was the only family member also known to have diabetes (Figure S1), with which she had been diagnosed at age 14 years.
Insulin secretion was evaluated in the proband after her glycemia had been lowered sufficiently. The average of the sum of serum C‐peptide immunoreactivity (ΣCPR) during an oral glucose tolerance test (8.6 ng/mL) was markedly lower than that previously reported for individuals with diabetes (~20 ng/mL; Table 1)8. The secretion of C‐peptide (peak CPR 2.77 ng/mL), but not that of glucagon, in response to an arginine challenge test was lower than that described for type 2 diabetes patients (peak CPR ~4 ng/mL; Table 1)9. The secretion of C‐peptide during a glucagon challenge test (ΔCPR 1.2 ng/mL) was also lower than that previously reported for type 2 diabetes patients (average ΔCPR 2.0 ng/mL; Table 1)10. A glucagon challenge test carried out with the proband's mother showed that the C‐peptide response was even lower than that of the proband (Table 1). These results were indicative of impaired β‐cell function in both the proband and her mother. Concomitantly, insulin therapy was considered to be necessary for this patient.
Table 1.
Evaluation of insulin secretion in the proband and her mother
| Proband | Mother | |
|---|---|---|
| Oral glucose tolerance test | ||
| C‐peptide (ng/mL) | ||
| Pre | 0.85 | |
| 30 min | 2.53 | |
| 60 min | 2.77 | |
| 120 min | 1.79 | |
| Glucose (mg/dL) | ||
| Pre | 121 | |
| 30 min | 199 | |
| 60 min | 321 | |
| 120 min | 323 | |
| ΣCPR (ng/mL) | 8.6 | |
| Arginine challenge test | ||
| C‐peptide (ng/mL) | ||
| Pre | 0.85 | |
| 30 min | 2.53 | |
| 60 min | 2.77 | |
| 120 min | 1.79 | |
| Glucagon (pg/mL) | ||
| Pre | 113 | |
| 30 min | 521 | |
| 60 min | 365 | |
| 120 min | 134 | |
| Glucagon challenge test | ||
| Glucose (mg/dL) | ||
| Pre | 98 | 94 |
| Post | 131 | 105 |
| C‐peptide (ng/mL) | ||
| Pre | 1.0 | 1.8 |
| Post | 2.2 | 2.6 |
Plasma glucose or glucagon as well as serum C‐peptide concentrations before and 30, 60 or 120 min after the oral administration of glucose (75 g) or intravenous administration of arginine (0.5 g/kg) were measured. Plasma glucagon was measured with a radioimmunoassay (BML, Tokyo, Japan). For the glucagon challenge test, glucagon (1 mg) was injected intravenously after the participants had been deprived of food overnight, with plasma glucose and serum C‐peptide being measured before (pre) and 6 min after (post) the injection. ΣCPR, sum of serum C‐peptide immunoreactivity.
Mutation Analysis of HNF4A
Mutational screening of genes known to be responsible for MODY showed that both the proband and her mother harbored a heterozygous mutation (c.773G>A) of HNF4A that alters the encoded amino acid sequence (p.R258H; Figure S2). HNF4α forms a homodimer through its ligand‐binding domain that is essential for its transactivation activity11. The crystal structure of a complex of HNF4α with DNA and a coactivator11 shows that Arg258 (R258) is located near the dimer interface (Figure S3).
In Silico Simulation Analysis
With the use of in silico simulation, we constructed the three‐dimensional structures of dimers formed by wild‐type HNF4α or by MODY1‐associated mutants (E276Q, R258H, V255M) that harbor amino acid substitutions near the dimer interface, and we calculated the ZDOCK score as a measure of dimer stability. The transactivation activity of the E276Q mutant was previously shown to be impaired in vitro 12. The ZDOCK scores of heterodimers formed by the E276Q or R258H mutants with the wild‐type protein were lower than that of the wild‐type homodimer, and the scores of the mutant homodimers were lower than those of the corresponding heterodimers, suggestive of a pathological role for these mutations (Table 2). In contrast, the ZDOCK score of the heterodimer formed by the wild‐type protein and the V255M mutant, which manifests normal transactivation activity12, was similar to that of the wild‐type homodimer, suggesting that this mutation is of little pathogenic relevance (Table 2).
Table 2.
In silico simulation analysis of hepatocyte nuclear factor 4α dimer stability
| ZDOCK score | Obtained dimers | P‐value 1 | P‐value 2 | |
|---|---|---|---|---|
| Wild type | ||||
| Homodimer | 133.31 ± 20.35 | 24 | – | – |
| R258H | ||||
| Heterodimer | 95.64 ± 7.66 | 15 | <0.001 | – |
| Homodimer | 81.90 ± 9.48 | 35 | <0.001 | <0.001 |
| V255M | ||||
| Heterodimer | 120.64 ± 33.66 | 47 | NS | – |
| Homodimer | 108.35 ± 33.55 | 62 | <0.05 | NS |
| E276Q | ||||
| Heterodimer | 113.25 ± 33.09 | 25 | <0.01 | – |
| Homodimer | 93.27 ± 16.80 | 54 | <0.001 | <0.01 |
ZDOCK scores are mean ± standard deviation. Obtained dimers derived from 2,000 results of docking analyses were chosen on the basis of similar structure of mouse hepatocyte nuclear factor 4α docking. P‐values 1 and 2 are for comparison with the wild‐type homodimer or with the corresponding heterodimer formed with the wild‐type protein. NS, not significant (P ≥ 0.05).
We also analyzed the electrostatic surface potential of the dimer interface for wild type and R258H. Although the electrostatic potential at position 258 is similar, the surface of helix 10/11 (H10/11), which plays an important role in dimerization11, is acidic in the wild type, but neutral in the R258H (Figure 1). Collectively, these results suggest that the R258H mutation alters the function of HNF4α by affecting protein structure adjacent to its position.
Figure 1.
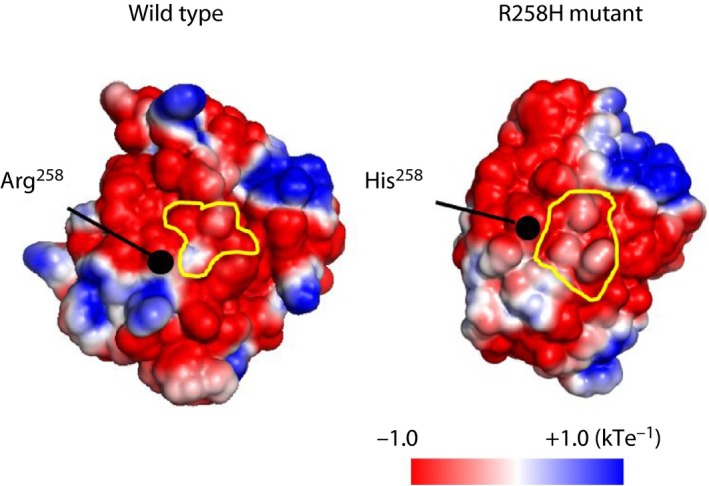
Electrostatic surface potential of wild‐type and R258H mutant forms of hepatocyte nuclear factor 4α. Potentials from negative to positive are shown as red to blue, respectively. Dimer interfaces are outlined in yellow. [Colour figure can be viewed at wileyonlinelibrary.com]
Luciferase Reporter Analysis
Finally, we measured the transactivation activity of the R258H mutant in vitro. Forced expression of wild‐type HNF4α in COS‐7 cells resulted in a marked increase in luciferase expression driven by the ABCC6 promoter (Figure 2). The luciferase activity in cells expressing the R258H or E276Q mutants was significantly lower than that in those expressing the wild‐type protein (Figure 2), providing further support for the pathological relevance of the R258H mutation.
Figure 2.
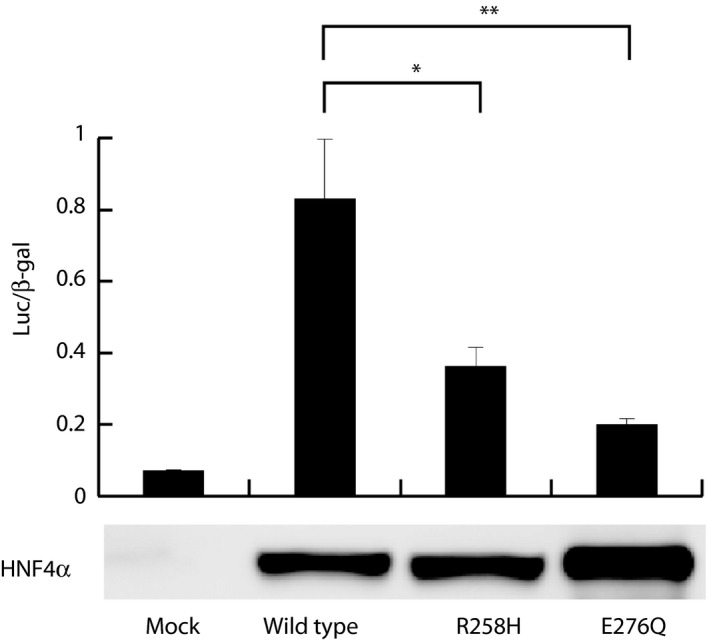
Luciferase reporter assay of transactivation activity for wild‐type and mutant forms of hepatocyte nuclear factor 4α (HNF4α). The luciferase/β‐galactosidase (Luc/β‐gal) activity ratios are shown as mean ± standard error of the mean from three independent experiments. *P < 0.05 (Student's t‐test). The cell lysates were subjected to immunoblot analysis with anti‐HNF4α antibody (sc‐374229; Santa Cruz Biotechnology, Santa Cruz, CA, USA), **P < 0.01
Discussion
We have characterized the pathological relevance of a mutation of HNF4A identified in a mother and daughter with early‐onset diabetes and impaired insulin secretion. Both women were thus diagnosed with MODY1. Although the mutation (c.773G>A) was previously identified in a single family4, we now show that it confers a functional defect with the use of in silico simulation analysis and in vitro experiments.
The simulation analysis and luciferase reporter assay showed that the stability of the dimers formed by and the transactivation activity of the R258H mutant are impaired. The crystal structure of HNF4α shows that Arg258 is located near the dimer interface11. Whereas Arg258 does not directly participate in dimerization, it forms an intramolecular salt‐bridge with Glu262, as well as hydrogen bonds with Ser337 and Gln341 (Figure S3). These hydrophilic interactions might stabilize the orientation of H10/11, which plays a pivotal role in dimerization. The electrostatic surface potential of H10/11 differs between the wild‐type and R258H mutant proteins. Instability of dimerization conferred by the R258H mutation might thus result from an allosteric structural change at H10/11.
Disclosure
The authors declare no conflict of interest.
Supporting information
Figure S1 ¦ Pedigree of the proband. Males are shown as squares and females as circles, the proband is indicated by the arrow, and family members known to have diabetes are shaded black. Ages at the time of the present study are indicated in years (y).
Figure S2 ¦ Sequence analysis of hepatocyte nuclear factor 4α (HNF4A). (a) Sequences of the HNF4A gene of the proband and her mother. Partial sequences surrounding the mutation site (arrow) of the proband and her mother, as well as the corresponding sequence for a control individual are shown. (b) Domain structure of HNF4α and partial sequence surrounding the mutation site are shown. The substitution of amino acid is shown in red.
Figure S3 ¦ Mapping of Arg258 to the crystal structure of human hepatocyte nuclear factor 4α (HNF4α). The structure of full‐length HNF4α complexed with deoxyribonucleic acid and a coactivator (PDB ID: 4IQR) is presented. The left panel shows the entire complex, whereas the right panel shows a zoomed‐in image of the dimer interface. Three residues, Glu 262, ser337 and Gln341, involved in the interaction with Arg258 are shown.
Table S1 ¦ Polymerase chain reaction primers used for amplification of hepatocyte nuclear factor 4α exons.
Table S2 ¦ Laboratory data for the proband.
Data S1 ¦ Supporting method.
Acknowledgments
This study was supported by a Grant‐in‐Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan to KS (16K19537) and YH (16K09750).
J Diabetes Investig 2019; 10: 680–684
References
- 1. Horikawa Y. Maturity‐onset diabetes of the young as a model for elucidating the multifactorial origin of type 2 diabetes mellitus. J Diabetes Investig 2018; 9: 704–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yamagata K, Furuta H, Oda N, et al Mutations in the hepatocyte nuclear factor‐4α gene in maturity‐onset diabetes of the young (MODY1). Nature 1996; 384: 458–460. [DOI] [PubMed] [Google Scholar]
- 3. Colclough K, Bellanne‐Chantelot C, Saint‐Martin C, et al Mutations in the genes encoding the transcription factors hepatocyte nuclear factor 1α and 4α in maturity‐onset diabetes of the young and hyperinsulinemic hypoglycemia. Hum Mutat 2013; 34: 669–685. [DOI] [PubMed] [Google Scholar]
- 4. Motzkau M, Meyer P, Mertens PR, et al Monogenic diabetes in a family with 2 unknown HNF‐4A gene mutations. Exp Clin Endocrinol Diabetes 2012; 120: 89–90. [DOI] [PubMed] [Google Scholar]
- 5. Nakano E, Ono R, Masaki T, et al Differences in clinical phenotype among patients with XP complementation group D: 3D structure and ATP‐docking of XPD in silico . J Invest Dermatol 2014; 134: 1775–1778. [DOI] [PubMed] [Google Scholar]
- 6. Chen R, Li L, Weng Z. ZDOCK: an initial‐stage protein‐docking algorithm. Proteins 2003; 52: 80–87. [DOI] [PubMed] [Google Scholar]
- 7. de Boussac H, Ratajewski M, Sachrajda I, et al The ERK1/2‐hepatocyte nuclear factor 4α axis regulates human ABCC6 gene expression in hepatocytes. J Biol Chem 2010; 285: 22800–22808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Maruyama T, Tanaka S, Shimada A, et al Insulin intervention in slowly progressive insulin‐dependent (type 1) diabetes mellitus. J Clin Endocrinol Metab 2008; 93: 2115–2121. [DOI] [PubMed] [Google Scholar]
- 9. Tsuchiyama N, Takamura T, Ando H, et al Possible role of alpha‐cell insulin resistance in exaggerated glucagon responses to arginine in type 2 diabetes. Diabetes Care 2007; 30: 2583–2587. [DOI] [PubMed] [Google Scholar]
- 10. Fujioka Y, Okura T, Sumi K, et al Normal meal tolerance test is preferable to the glucagon stimulation test in patients with type 2 diabetes that are not in a hyperglycemic state: comparison with the change of C‐peptide immunoreactivity. J Diabetes Investig 2018; 9: 274–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chandra V, Huang P, Potluri N, et al Multidomain integration in the structure of the HNF‐4α nuclear receptor complex. Nature 2013; 495: 394–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Navas MA, Munoz‐Elias EJ, Kim J, et al Functional characterization of the MODY1 gene mutations HNF4(R127W), HNF4(V255M), and HNF4(E276Q). Diabetes 1999; 48: 1459–1465. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 ¦ Pedigree of the proband. Males are shown as squares and females as circles, the proband is indicated by the arrow, and family members known to have diabetes are shaded black. Ages at the time of the present study are indicated in years (y).
Figure S2 ¦ Sequence analysis of hepatocyte nuclear factor 4α (HNF4A). (a) Sequences of the HNF4A gene of the proband and her mother. Partial sequences surrounding the mutation site (arrow) of the proband and her mother, as well as the corresponding sequence for a control individual are shown. (b) Domain structure of HNF4α and partial sequence surrounding the mutation site are shown. The substitution of amino acid is shown in red.
Figure S3 ¦ Mapping of Arg258 to the crystal structure of human hepatocyte nuclear factor 4α (HNF4α). The structure of full‐length HNF4α complexed with deoxyribonucleic acid and a coactivator (PDB ID: 4IQR) is presented. The left panel shows the entire complex, whereas the right panel shows a zoomed‐in image of the dimer interface. Three residues, Glu 262, ser337 and Gln341, involved in the interaction with Arg258 are shown.
Table S1 ¦ Polymerase chain reaction primers used for amplification of hepatocyte nuclear factor 4α exons.
Table S2 ¦ Laboratory data for the proband.
Data S1 ¦ Supporting method.