

Abstract
Aims/Introduction
To evaluate the contribution of pancreatic α‐cell function to the dawn phenomenon, insulin sensitivity, hepatic glucose uptake and glycemic variability in patients with type 1 diabetes.
Materials and Methods
In 40 patients with type 1 diabetes, arginine stimulation tests were carried out, and the area under the curve (AUC) of glucagon was measured using radioimmunoassays (AUC glc RIA) and enzyme‐linked immunosorbent assays (AUC glc ELISA). The ratio of the insulin dose delivered by an artificial pancreas to maintain euglycemia between 04.00 and 08.00 hours or between 00.00 and 04.00 hours was measured as the dawn index. The glucose infusion rate and hepatic glucose uptake were measured using hyperinsulinemic euglycemic clamp and clamp oral glucose loading tests. Glycemic variability in 96 h was measured by continuous glucose monitoring.
Results
The median dawn index (1.7, interquartile range 1.0–2.8) was not correlated with AUC glc RIA (R 2 = 0.03, P = 0.39) or AUC glc ELISA (R 2 = 0.04, P = 0.32). The median glucose infusion rate (7.3 mg/kg/min, interquartile range 6.4–9.2 mg/kg/min) was significantly correlated with AUC glc RIA (R 2 = 0.20, P = 0.02) and AUC glc ELISA (R 2 = 0.21, P = 0.02). The median hepatic glucose uptake (65.3%, interquartile range 40.0–87.3%) was not correlated with AUC glc RIA (R 2 = 0.07, P = 0.26) or AUC glc ELISA (R 2 = 0.26, P = 0.79). The standard deviation of glucose levels measured by continuous glucose monitoring was significantly correlated with AUC glc RIA (R 2 = 0.11, P = 0.049), but not with AUC glc ELISA (R 2 = 0.01, P = 0.75).
Conclusions
Pancreatic α‐cell function contributed to insulin sensitivity in patients with type 1 diabetes.
Keywords: Insulin sensitivity, Pancreatic α‐cell, Type 1 diabetes
Introduction
Type 1 diabetes mellitus is characterized by insulin deficiency caused by pancreatic β‐cell destruction1. Glucagon inappropriately secretes from pancreatic α‐cells and can exacerbate hyperglycemia due to paradoxical hyperglucagonemia or lead to severe hypoglycemia as a result of failed counter‐regulation in patients with type 1 diabetes2. Notably, patients with type 1 diabetes typically show glucose metabolism mechanisms that are different from those of healthy individuals, including reduced insulin sensitivity, acutely increased blood glucose levels between 05.00 and 09.00 hours (called the “dawn phenomenon”), and impaired hepatic glucose uptake (HGU). These characteristics adversely affect glucose homeostasis and can cause micro‐ or macrovascular complications.
Despite extensive studies, the role of glucagon as a pathophysiological factor remains unclear. In previous studies, glucagon levels were typically measured using conventional radioimmunoassay (RIA) kits. Quantitative assays known as sandwich enzyme‐linked immunosorbent assays (ELISAs) have recently been developed. Thus, it is necessary to compare the results of different assay kits in order to confirm the specific contribution of glucagon to glucose metabolism.
Accordingly, in the present study, we aimed to determine the contribution of pancreatic α‐ cell function evaluated with RIA or ELISA kits to insulin sensitivity, HGU and glycemic variability, including the dawn phenomenon, in patients with type 1 diabetes.
Methods
Study design and patients
The present observational study was carried out at the National Center for Global Health and Medicine in Tokyo, Japan. We examined patients diagnosed with type 1 diabetes who were admitted to our hospital, and met the inclusion criteria and did not meet the exclusion criteria. Inclusion criteria were as follows: patients who were previously diagnosed with type 1 diabetes according to World Health Organization criteria3 and were aged ≥20 years. Exclusion criteria were as follows: current treatment with steroid hormones or immunosuppressants, pregnant or breastfeeding, estimated glomerular filtration rate (GFR) of <45 mL/min/1.73 m2, current infection and refusal to participate in the study. The estimated GFR was calculated using the following formula4: estimated GFR (mL/min/1.73 m2) = 194 × (serum creatinine level, mg/dL)−1.094 × (age, years)−0.287 (×0.739 if the patient was female). Baseline characteristic information was collected from patient medical records. Measurements as baseline characteristics were as follows: age, sex, body mass index (BMI; calculated as weight in kilograms divided by height in meters squared), diabetes duration, glycated hemoglobin, fasting levels of serum C‐peptide, estimated GFR, insulin treatment regimen (multiple daily injection or continuous subcutaneous insulin infusion), total daily insulin dose and basal/bolus ratio. All patients provided informed and written consent. This study conformed to the provisions of the Declaration of Helsinki, and was approved by the institutional review board of the National Center for Global Health and Medicine.
Arginine stimulation test
On admission, each patient underwent arginine stimulation tests to evaluate their pancreatic α‐cell function. To exclude the effects of exogenous insulin, the typical basal insulin regimen within 24 h before the arginine stimulation test was replaced with continuous intravenous insulin injection and was stopped 1 h before the arginine stimulation test if the patient was treated with multiple daily injections. If the patient was treated with continuous subcutaneous insulin infusion, treatment with an insulin pump was continued as usual and stopped 1 h before the arginine stimulation test. Patients were asked to rest for 30 min after overnight fasting, and 30 g arginine was intravenously administered as 10% l‐arginine hydrochloride over 30 min. Blood samples were collected before, and 15, 30, 60, 90 and 120 min after arginine loading. The levels of plasma glucose, serum C‐peptide and plasma glucagon were measured at each time‐point. The levels of plasma glucose were measured using a glucose oxidase‐immobilized membrane‐H2O2 electrode (glucose analyzer GA‐1172; Arkray, Kyoto, Japan; the intra‐ and interassay coefficients of variation were <2.0%). The levels of serum C‐peptide were measured by electrochemiluminescence immunoassays (Roche Diagnostics, Mannheim, Germany; the intra‐ and interassay coefficients of variation were 1.9 and 2.3%, respectively). The levels of plasma glucagon were measured by RIA (Sceti Medical Labo, Tokyo, Japan; the intra‐ and interassay coefficients of variation were <20 and <15%, respectively) and sandwich ELISA (Mercodia AB, Uppsala, Sweden; the intra‐ and interassay coefficients of variation were 7.3–9.4% and 7.5–8.5%, respectively). The area under the concentration‐time curve (AUC) of plasma glucagon between 0 and 120 min was calculated using the trapezoidal rule. The AUC of plasma glucagon measured by RIA kits was defined as AUCglcRIA, and that measured by ELISA kits was defined as AUCglcELISA. A peak glucagon level measured by RIA during arginine stimulation tests of ≥300 pg/mL was evaluated as glucagon hyperreactivity, whereas that of <300 pg/mL was evaluated as glucagon hyporeactivity, as previously reported5.
Evaluation of changes in insulin requirements between night and morning as the “dawn phenomenon”
After the arginine stimulation test, we evaluated changes in insulin requirements between night and morning as the “dawn phenomenon.” Continuous intravenous or subcutaneous insulin infusion resumed after arginine stimulation tests and stopped at 19.00 hours. At 20.00 hours, two cannulas were placed in a forearm vein (for infusion of glucose and insulin) and in a heated contralateral forearm vein (for arterialized venous blood sampling), and then connected to an artificial pancreas (STG55; Nikkiso Co., Shizuoka, Japan). The artificial pancreas automatically primed insulin (Humulin R, 250 U in 500 mL saline; Eli Lily and Company, Indianapolis, IN, USA) in accordance with an algorism to maintain blood glucose levels within the range of 80–110 mg/dL throughout the test. Blood was continuously sampled, and glucose levels were measured with a glucose sensor electrode and glucose oxidase membrane every minute. The pump delivering insulin and the glucose sensor electrode each had an accuracy of ±5% according to a previous report6. We evaluated changes in insulin requirements from 00.00 to 08.00 hours as the “dawn phenomenon” using this artificial pancreas. The ratio of the delivered insulin dose average between 04.00 and 08.00 hours to that between 00.00 and 04.00 hours was calculated as the dawn index (Figure S1). To support the relationship between the dawn phenomenon and glucose‐related hormones, we also measured levels of the following hormones after patients were kept at rest for 30 min after overnight fasting: growth hormone (GH; Elecsys immunoassay; Roche Diagnostics), insulin‐like growth factor‐1 (IGF‐1; immunoradiometric assay; Fujirebio, Tokyo, Japan), adrenocorticotropic hormone (Elecsys immunoassay; Roche Diagnostics), cortisol (chemiluminescent immunoassay; Siemens Medical Solutions Diagnostics, Los Angeles, CA, USA), active glucagon‐related protein‐1 (GLP‐1; ELISA; IBL, Hamburg, Germany) and somatostatin (enzyme immunoassay; R&D Systems, Minneapolis, MN, USA).
Hyperinsulinemic euglycemic clamp test
Hyperinsulinemic euglycemic clamp tests were applied to determine insulin sensitivity using the modified technique described by DeFronzo et al 7. At 08.00 hours after evaluating the dawn index, a primed‐constant infusion of insulin was given at a rate of 2.58 mU/kg/min by the artificial pancreas to achieve a desired steady‐state plasma insulin concentration (200 μU/mL). Splanchnic glucose uptake was decreased when the peripheral insulin concentration was raised to such a level8. Subsequently, exogenous glucose infusion was initiated to maintain blood glucose levels within the euglycemic range (95 mg/dL) throughout the study. The blood glucose level was measured every minute, and the exogenous glucose infusion rate (GIR; mg/kg/min) was adjusted by the artificial pancreas. Blood samples to measure levels of serum insulin were taken from a heated superficial hand vein 90 min after achieving steady state. The average of GIR during the last 90 min after achieving a steady state was calculated as an indicator of the insulin sensitivity of peripheral tissue.
Clamp oral glucose loading test
After hyperinsulinemic euglycemic clamp tests, clamp oral glucose loading tests were carried out to evaluate HGU as previously described9. Briefly, 90 min after the blood glucose concentration monitored by the artificial pancreas reached a steady‐state level, a fixed amount of glucose (0.2 g/kg) was orally administered. The glucose infusion rate then started to decrease, because some of the ingested glucose that was not extracted by the splanchnic tissues entered the systemic circulation and reduced the GIR required to maintain euglycemia. After an oral glucose load, in addition to the ingested glucose, recirculating glucose from the systemic circulation was presented to the liver (the HGU). The GIR required to maintain euglycemia then returned to a normal level (approximately 120 min after oral glucose administration). We calculated HGU (%) using the following formula: HGU (%) = ([oral glucose load] – [GIR decrements]) / (oral glucose load). If the GIR decreased to zero after glucose loading, the results were excluded from analysis. To support the relationship between HGU and glucose‐related hormones, we also analyzed the correlation between HGU and fasting levels of GH, IGF‐1, adrenocorticotropic hormone, cortisol, active GLP‐1 and somatostatin.
Assessment of glycemic variability
A total of 24 h after completion of tests using the artificial pancreas, each patient underwent continuous glucose monitoring (CGM; ipro2; Medtronic Minimed, CA, USA) for 96 h. The averages of the following variables over 3 days were calculated using the CGM data: mean blood glucose level, standard deviation (SD), M‐value10, mean amplitude of glycemic excursions11, hyperglycemic time and hypoglycemic time. Hyperglycemic and hypoglycemic times were defined as the average number of minutes during which the patient's glucose levels were >180 or <70 mg/dL in 1 day, respectively.
Statistical analysis
Mann–Whitney U‐tests were used to examine continuous variables, whereas Fisher's exact tests were used for two categorical variables. Pearson correlation analysis was carried out to analyze the correlations among measurements. Multiple regression analysis was carried out to examine the relationships between GIR during hyperinsulinemic euglycemic clamp assays as the dependent variable and the following independent variables: model 1 included age, sex, BMI and AUCglcRIA; and model 2 included age, sex, BMI and AUCglcELISA. Results with P‐values of <0.05 were considered statistically significant. All analyses were carried out using STATA software, version 14.2 (StataCorp, College Station, TX, USA).
Results
Demographics
In total, 40 Japanese patients with type 1 diabetes who met the inclusion criteria and did not meet the exclusion criteria participated in the present study. Table 1 shows the patients’ characteristics. Briefly, the diabetes duration was short, the patients were not obese and the median fasting level of serum C‐peptide was <1.0 ng/mL, suggesting that their β‐cell function was severely impaired.
Table 1.
Clinical characteristics of the patients included in the study
| n = 40 | |
|---|---|
| Age (years) | 43 (31–56) |
| Female | 21 (52.5%) |
| BMI (kg/m2) | 20.5 (19.0–21.7) |
| Diabetes duration (years) | 2.6 (0.08–10.3) |
| HbA1c (%) | 8.2 (7.4–10.3) |
| (mmol/mol) | 66 (57–89) |
| Fasting serum C‐peptide (ng/mL) | 0.32 (0.00–0.94) |
| eGFR (mL/min/1.73 m2) | 111.1 (83.3–124.1) |
| Insulin treatment | |
| MDI/CSII | 32/8 |
| Total daily insulin dose per weight (units/day/kg) | 0.50 (0.33–0.75) |
| Basal/bolus ratio | 0.42 (0.30–0.61) |
Data are presented as n, n (%) or median (interquartile range). BMI, body mass index calculated by weight in kilograms divided by height in meters squared; CSII, continuous subcutaneous insulin infusion; eGFR, estimated glomerular filtration rate calculated using the following formula4: estimated GFR (mL/min/1.73 m2) = 194 × (serum creatinine level, mg/dL)−1.094 × (age, years)−0.287 (×0.739 if the patient was female); HbA1c, glycated hemoglobin; MDI, multiple daily injection.
Glucagon response to arginine stimulation measured by RIA or ELISA
Figure 1a,b show plasma glucose, serum C‐peptide and plasma glucagon levels measured by RIA or ELISA curves in response to arginine stimulation. The levels of plasma glucose were increased in response to arginine stimulation. The response of serum C‐peptide in almost all patients was abolished, although a slight response was observed in some patients (Figure 1a). The median (interquartile range) plasma glucagon levels at preloading and peak, as measured by RIA and the AUCglcRIA, were 133.5 pg/mL (117.0–151.5 pg/mL), 413.0 pg/mL (272.5–507.0 pg/mL) and 3.7 × 104 pg/mL·min (2.6–4.6 × 104 pg/mL·min), respectively, and those measured by ELISA were 2.5 pg/mL (0–7.0 pg/mL), 32.8 pg/mL (10.7–61.2 pg/mL) and 2.0 × 103 pg/mL·min (0.8–4.5 × 103 pg/mL·min), respectively. Trends in the glucagon response to arginine stimulation, as measured by RIA or ELISA, were similar (Figure 1b). Correlations in the levels of plasma glucagon measured by RIA and ELISA at preloading and peak, and those between logarithm‐transformed AUCglcRIA and AUCglcELISA were statistically significant (R 2 = 0.42, 0.25 and 0.20, and P = 0.001, 0.001 and 0.004, respectively; Figure 1c–e). However, the levels of glucagon at preloading were undetectable by ELISA, even if those measured by RIA were detected in 17 of 40 (42.5%) patients. The peak levels and logarithm‐transformed AUC levels of glucagon measured by RIA and ELISA were also decreased in some patients.
Figure 1.
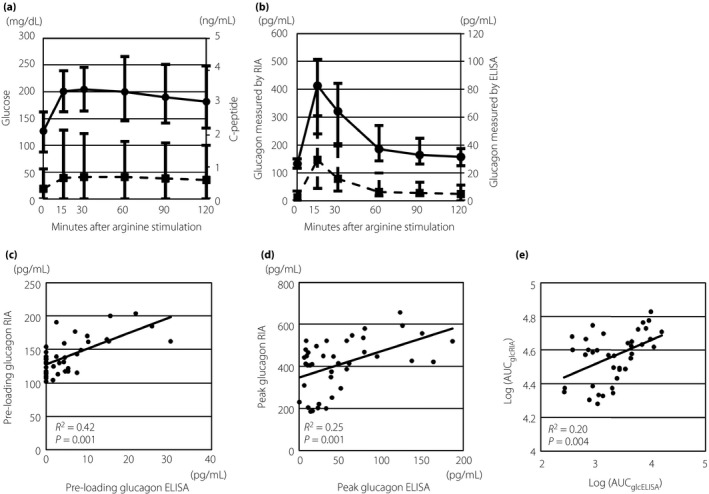
Trends in responses to arginine stimulation and coefficients of plasma glucagon measurements by radioimmunoassay (RIA) and enzyme‐linked immunosorbent assays (ELISA). (a) Trends in plasma glucose (solid line with circles) and serum C‐peptide (dashed line with squares) responses to arginine stimulation. (b) Trends in plasma glucagon responses to arginine stimulation measured by RIA (solid line with circles) and ELISA (dashed line with squares). (c) Scatterplot of plasma glucagon levels at preloading measured by RIA and ELISA. (d) Scatter plot of peak levels of plasma glucagon measured by RIA and ELISA. (e) Scatter plot of logarithm‐transformed area under the curve (AUC) of glucagon measured using radioimmunoassays (log[AUC glc RIA]) and logarithm‐transformed AUC of glucagon measured using enzyme‐linked immunosorbent assays (log[AUC glc ELISA]). Solid lines in (c–e) show approximate lines between each measurement.
Associations between AUCglcRIA or AUCglcELISA and the dawn index
Of 40 patients who underwent arginine stimulation tests, four patients could not have a cannula placed in the forearm, and six patients had to discontinue the test during evaluation of the dawn index because of problems with blood collection and were excluded from the analysis. The median (interquartile range) dawn index was 1.7 (1.0–2.8), and was not significantly correlated with AUCglcRIA or AUCglcELISA (R 2 = 0.03, P = 0.39 and R 2 = 0.04, P = 0.32, respectively; Figure 2a,b). We also analyzed the correlations between the dawn index and fasting levels of glucose‐related hormones (i.e., GH, IGF‐1, adrenocorticotropic hormone, cortisol, GLP‐1 and somatostatin). There were no significant correlations among these parameters (Figure S2).
Figure 2.
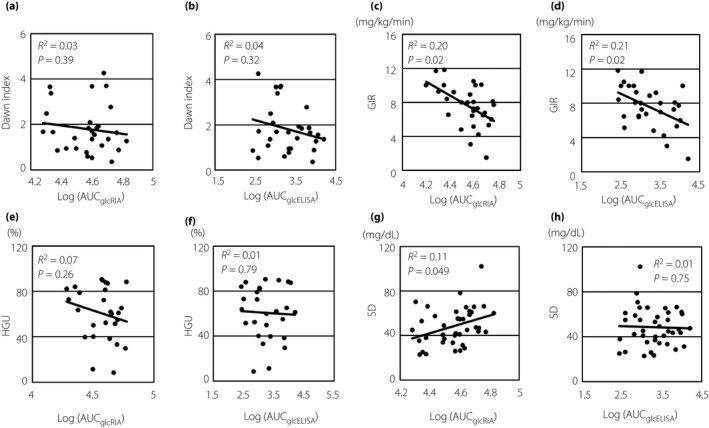
Scatter plots between measurements of glucose metabolism or glycemic variability and log(AUC glc RIA) or log(AUC glc ELISA). (a) Scatter plot between the dawn index and log(AUC glc RIA). (b) Scatter plot between the dawn index and log(AUC glc ELISA). (c) Scatter plot between GIR and log(AUC glc RIA). (d) Scatter plot between GIR and log(AUC glc ELISA). (e) Scatter plot between HGU and log(AUC glc RIA). (f) Scatter plot between HGU and log(AUC glc ELISA). (g) Scatter plot between SD and log(AUC glc RIA). (h) Scatter plot between SD and log(AUC glc ELISA). Solid lines show approximate lines for each measurement. The dawn index was defined as the ratio of the average insulin dose delivered to maintain euglycemia (80–110 mg/dL) with an artificial pancreas between 04:00 and 08:00 to that between 00:00 and 04:00. AUC, area under the curve; ELISA, enzyme‐linked immunosorbent assay; GIR, glucose infusion rate during hyperinsulinemic euglycemic clamp tests; HGU, hepatic glucose uptake evaluated by clamp oral glucose loading tests, as previously described 9 ; Log(AUC glc ELISA), logarithm‐transformed AUC glc ELISA; Log(AUC glc RIA), logarithm‐transformed AUC glc RIA; RIA, radioimmunoassay; SD, standard deviation of glucose levels in 96 h, as evaluated by continuous glucose monitoring.
AUCglcRIA and AUCglcELISA were associated with GIR, but not HGU, as evaluated by hyperinsulinemic euglycemic clamp and clamp oral glucose loading tests
During clamp oral glucose loading tests, GIR in two patients reached zero after glucose loading, and their HGU values were then excluded from analysis. The median (interquartile range) GIR during hyperinsulinemic euglycemic clamp assays and HGU evaluated by clamp oral glucose loading tests were 7.3 mg/kg/min (6.4–9.2 mg/kg/min) and 65.3% (40.0–87.3%), respectively. The AUCglcRIA and AUCglcELISA were significantly negatively correlated with GIR (R 2 = 0.20, P = 0.02 and R 2 = 0.21, P = 0.02, respectively; Figure 2c,d), but not with HGU (R 2 = 0.07, P = 0.26 and R 2 = 0.01, P = 0.79, respectively; Figure 2e,f). Considering confounding variables, multiple regression analysis showed that none of the variables (age, sex, BMI and AUCglcRIA) were significant predictors of GIR in model 1. However, age and AUCglcELISA were significant predictors of GIR in model 2 (Table S1).
We also compared GIR, HGU and baseline characteristics between patients with glucagon hypo‐ or hyperreactivity who could be evaluated for GIR and HGU. Only GIR was significantly higher in patients with glucagon hyporeactivity than those with glucagon hyperreactivity (Table 2).
Table 2.
Clinical characteristics, glucose infusion rate and hepatic glucose uptake in patients with glucagon hyporeactivity or hyperreactivity
| Glucagon hyporeactivity (n = 11) | Glucagon hyperreactivity (n = 17) | P | |
|---|---|---|---|
| Age (years) | 46 (31–66) | 62 (44–72) | 0.28 |
| Female | 4 (36.3%) | 9 (52.9%) | 0.48 |
| BMI (kg/m2) | 22.3 (19.8–24.0) | 21.0 (19.3–22.5) | 0.64 |
| Diabetes duration (years) | 1.9 (0.5–11.6) | 2.6 (0.1–8.2) | 0.80 |
| HbA1c (%) | 8.6 (7.5–14.8) | 8.2 (7.2–9.2) | 0.19 |
| (mmol/mol) | 70 (58–138) | 66 (55–77) | 0.19 |
| Fasting serum C‐peptide (ng/mL) | 0.33 (0–1.08) | 0.17 (0–0.94) | 0.71 |
| eGFR (mL/min/1.73 m2) | 92.7 (80.0–120.9) | 85.9 (69.6–102.9) | 0.20 |
| Insulin treatment | |||
| MDI/CSII | 9/2 | 14/3 | 0.67 |
| Total daily insulin dose per weight (units/day/kg) | 0.65 (0.35–0.78) | 0.49 (0.31–0.73) | 0.40 |
| Basal/bolus ratio | 0.53 (0.36–1.27) | 0.40 (0.28–0.60) | 0.12 |
| GIR (mg/min/kg) | 9.24 (7.02–11.67) | 6.75 (5.14–8.08) | 0.03 |
| HGU (%) | 82.5 (40.0–84.0) | 62.9 (42.4–84.8) | 0.22 |
Data are presented as n, n (%) or median (interquartile range). A peak level of glucagon evaluated by radioimmunoassay during arginine stimulation tests of ≥300 pg/mL was defined as glucagon hyperreactivity, and that of <300 pg/mL was defined as glucagon hyporeactivity5. BMI, body mass index calculated by weight in kilograms divided by height in meters squared; CSII, continuous subcutaneous insulin infusion; eGFR, estimated glomerular filtration rate calculated using the following formula4: estimated GFR (mL/min/1.73 m2) = 194 × (serum creatinine level, mg/dL)−1.094 × (age, years)−0.287 (×0.739 if the patient was female); GIR, glucose infusion rate during hyperinsulinemic euglycemic clamp; HbA1c, glycated hemoglobin; HGU, hepatic glucose uptake evaluated by clamp oral glucose loading tests, as previously described9; MDI, multiple daily injection.
We also analyzed correlations between HGU and fasting levels of glucose‐related hormones. Hepatic glucose uptake was significantly correlated with fasting cortisol levels (R 2 = 0.28, P = 0.003), and was not correlated with any other glucose‐related hormones (Figure S3).
AUCglcRIA, but not AUCglcELISA, was associated with glycemic variability evaluated by CGM
The median (interquartile) values for the average, SD, mean amplitude of glycemic excursions, M‐value, hyperglycemic time and hypoglycemic time of glucose levels, as evaluated by CGM, within 96 h were 148.4 mg/dL (126.1–175.9 mg/dL), 46.7 mg/dL (35.1–60.1 mg/dL), 111.4 (90–132.2), 18.8 mg/dL (11.8–48.0 mg/dL), 465.0 min/day (216.7–893.3 min/day) and 15.0 min/day (0–120.0 min/day), respectively. Of these measurements, SD was significantly correlated with logarithm‐transformed AUCglcRIA positively (R 2 = 0.11, P = 0.049), but not with logarithm‐transformed AUCglcELISA (R 2 = 0.01, P = 0.75; Figure 2g,h). Other measurements of glycemic variability were not significantly correlated with AUCglcRIA or AUCglcELISA (Figure S4).
Discussion
To the best of our knowledge, this is the first study to report the associations between glucagon response to arginine stimulation measured by RIA or ELISA, and dawn phenomenon, insulin sensitivity, HGU and measurement of glycemic variability in patients with type 1 diabetes. The glucagon response to arginine stimulation involves the reproducible and complementary pancreatic endocrinological functions of both α‐ and β‐cells12, 13. In the present study, trends in the glucagon response to arginine stimulation measured by RIA or ELISA were generally similar, and glucagon levels at preloading, peak and logarithm‐transformed AUC measured by RIA or ELISA were significantly correlated. However, these measurements varied in some patients, as shown in Figure 1. Glucagon (1–29) is produced through processing of proglucagon by proglucagon convertase14. In this process, other proglucagon fragments (e.g., oxyntomodulin, glicentin and GLP‐1) were also produced. Measurement of glucagon with the RIA kit uses polyclonal antibodies against the glucagon C‐terminal region, and these antibodies crossreact with other proglucagon fragments that also contain the C‐terminal region. In contrast, double‐sandwich ELISA kits use monoclonal antibodies against both the C‐ and N‐terminal regions of glucagon and measure glucagon concentrations with much lower cross‐reactivity against proglucagon fragments other than glucagon (1–29)15. In a previous report, secretion of GLP‐1 was also stimulated by arginine loading16. The discrepancy between AUCglcRIA and AUCglcELISA in some patients suggested that the differences between responses of glucagon (1–29) to arginine stimulation and those of other proglucagon fragments. We did not measure the levels of other proglucagon fragments in the present study. Further studies are required to evaluate the responses of other proglucagon fragments.
In terms of associations between glucagon and insulin sensitivity, previous studies have shown that increased fasting levels of glucagon or glucagon responses to arginine stimulation can contribute to worsening insulin sensitivity in healthy individuals or patients with impaired glucose tolerance17, 18. The mechanisms through which α‐cells adapt to insulin sensitivity remain unclear. The primary mechanism is thought to be “paracrinopathy,” which designates the loss of tonic restraint normally exerted by a high local concentration of insulin on α‐ cells2. In the present study, GIR during hyperinsulinemic euglycemic clamp tests was significantly negatively correlated with AUCglcRIA and AUCglcELISA. Furthermore, age and AUCglcELISA were independent variables for GIR in multiple regression analysis. These results suggested that pancreatic α‐cell function independently contributed to insulin sensitivity, even in patients with type 1 diabetes, whose β‐cell function is diminished. Potential adaptive mediators, such as nutrients (branched amino acid and free fatty acids)19, incretin hormones and adipocytokines, can be considered. Indeed, clinical data show that GLP‐1 improves insulin sensitivity20, 21. The stress effects of obesity might also involve α‐cell function22. However, we showed that AUCglcELISA was an independent variable of GIR in multiple regression analysis, suggesting that α‐cell function independently contributes to insulin sensitivity.
The dawn phenomenon, first reported by Schmidt et al.23, refers to the concept that the levels of blood glucose rise acutely between 04.00 and 08.00 hours, and is typically observed in patients with type 1 diabetes. A previous report showed that the dawn phenomenon could affect overall glycemic control24, and that we should clarify the etiology of this phenomenon. Circadian variations in counter‐regulatory hormones (e.g., GH, IGF‐1 and cortisol) could affect endogenous glucose production and cause the observed increase in blood glucose levels25, 26. In a study of healthy individuals, endogenous glucose production was found to increase as glucagon concentrations increased in the morning27. However, subsequent studies concluded that there were no associations between the glucagon concentration and the dawn phenomenon26, 28. We also did not find any correlations between the dawn index and AUCglcRIA or AUCglcELISA. Thus, pancreatic α‐cell function appeared not to be related to the dawn phenomenon.
Cortisol has been shown to play a pivotal role in stimulation of HGU29. The significant correlation between HGU evaluated by clamp oral glucose loading tests and fasting levels of cortisol in the present study appeared to show the pathophysiological effects of cortisol on hepatic glucose metabolism. Interestingly, hepatic glucose production is rapidly stimulated by the physiological rise in glucagon, which is entirely attributable to enhancement of glycogenolysis30. Other previous studies in animals have reported that increasing intraportal infusion of glucagon decreases HGU31. Although a study of patients with type 2 diabetes also suggested the association between glucagon and HGU32, another report of patients with insulin‐dependent diabetes could not find any association between glucagon response to oral glucose loading and HGU33. In the present study, we also did not find any significant correlations between HGU and AUCglcRIA or AUCglcELISA. These results indicated that pancreatic α‐cell function was not associated with HGU in patients with type 1 diabetes.
Emerging evidence suggests that glycemic variability contributes to adverse clinical outcomes34. Notably, glycemic instability is caused by deficiency of intrinsic insulin secretion and the paradoxical behaviors of α‐cells during glycemic changes2; that is, a deficient glucagon response to hypoglycemia35 and an inappropriately high glucagon response to hyperglycemia36. A previous report showed a positive correlation between glucagon responses to arginine stimulation and several parameters of glycemic variability evaluated by CGM in patients with type 1 diabetes37. However, the plasma glucagon levels in these previous reports were measured with RIA kits. In the present study, AUCglcRIA was significantly correlated with the SD of glucose levels, similar to the findings of a previous report. In contrast, AUCglcELISA was not correlated with the measurement of glycemic variability. According to a previous report, the trend of glucagon levels measured by an ELISA kit differed from that measured by a RIA kit during the meal tolerance test; the former returned slightly elevated results, whereas the latter produced significantly lower levels38. The accuracy of glucagon levels measured by an ELISA kit was confirmed with novel liquid chromatography‐high resolution mass spectroscopy. Indeed, proglucagon fragments, such as the glicentin and oxyntomodulin, are secreted from the intestine in response to feeding39, 40. The discrepancy between the correlation of glycemic variability with AUCglcRIA and AUCglcELISA in the present study appeared to show that proglucagon fragments other than glucagon (1–29) could contribute to glycemic variability.
The present study had several limitations. First, this was an observational study carried out at a single national center, and the sample size was small. Prospective studies carried out at multiple centers with large sample sizes are required in order to confirm the present results. Second, we evaluated HGU with clamp oral glucose loading tests during hyperinsulinemic euglycemic clamp tests, as described previously9. We chose this method because we could evaluate GIR and HGU continuously during hyperinsulinemic euglycemic clamp test, and because the use of radioactive tracers for human studies is limited in Japan. Although the reliability of this method was confirmed in a previous report41, we should carry out the direct method to more accurately evaluate HGU.
In conclusion, we found that pancreatic α‐cell function contributed to insulin sensitivity, but did not affect HGU and glycemic variability including the dawn phenomenon, in patients with type 1 diabetes. The relationships between pancreatic α‐cell function and glycemic variability could be affected by the purity of glucagon assays. These data provide an important context for the multifactorial role of glucagon in glucose metabolism in patients with type 1 diabetes.
Disclosure
The authors declare no conflict of interest.
Supporting information
Figure S1 | Schematic presentation of changes in insulin requirements between night and morning, as evaluated using an artificial pancreas.
Figure S2 | Scatter plots between the dawn index and fasting levels of glucose‐related hormones.
Figure S3 | Scatter plots between hepatic glucose uptake evaluated by clamp oral glucose loading tests and fasting levels of glucose‐related hormones.
Figure S4 | Scatter plots between measurements of glucose variability, except standard deviation (average, mean amplitude of glycemic excursions, M‐value, hyperglycemic time and hypoglycemic time), as evaluated by continuous glucose monitoring in 96 h and (a–e) logarithm‐transformed area under the curve (AUC) of glucagon measured using radioimmunoassays (log[AUC glc RIA]) or (f–j) logarithm‐transformed AUC of glucagon measured using enzyme‐linked immunosorbent assays (log[AUC glc ELISA]).
Table S1 | Multiple regression analysis of glucose infusion rate during hyperinsulinemic euglycemic clamp tests.
Acknowledgments
This work was supported in part by a Junior Scientist Development Grant supported by Novo Nordisk Pharma Ltd., The Japan Diabetes Society.
J Diabetes Investig 2019; 10: 690–698
References
- 1. American Diabetes Association . Report of the expert committee on the diagnosis and classification of diabetes mellitus. Diabetes Care 1997; 20: 1183–1197. [DOI] [PubMed] [Google Scholar]
- 2. Unger RH, Orci L. Paracrinology of islets and the paracrinopathy of diabetes. Proc Natl Acad Sci USA 2010; 107: 16009–16012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. World Health Organization: World Health Organization, International Diabetes Federation , editors. Definition and diagnosis of diabetes mellitus and intermediate hyperglycemia. Report of a WHO/IDF Consultation. Geneva, Switzerland: WHO Press; 2006. [Google Scholar]
- 4. Matsuo S, Imai E, Horio M, et al Revised equations for estimated GFR from serum creatinine in Japan. Am J Kidney Dis 2009; 53: 982–992. [DOI] [PubMed] [Google Scholar]
- 5. Ito N, Yagi K, Kawano M, et al Analysis of pancreatic endocrine function in patients with IgG4‐related diseases, in whom autoimmune pancreatitis was ruled out by diagnostic imaging. Endocr J 2014; 61: 765–772. [DOI] [PubMed] [Google Scholar]
- 6. Mita N, Kawahito S, Soga T, et al Strict blood glucose control by an artificial endocrine pancreas during hepatectomy may prevent postoperative acute kidney injury. J Artif Organs 2017; 20: 76–83. [DOI] [PubMed] [Google Scholar]
- 7. DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol 1979; 237: E214–E223. [DOI] [PubMed] [Google Scholar]
- 8. DeFronzo RA, Ferrannini E, Hendler R, et al Influence of hyperinsulinemia, hyperglycemia, and the route of glucose administration on splanchnic glucose exchange. Proc Natl Acad Sci USA 1978; 75: 5173–5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kawamori R, Morishima T, Ikeda M, et al Effect of strict metabolic control on glucose handling by the liver and peripheral tissues in non‐insulin‐dependent diabetes mellitus. Diabetes Res Clin Pract 1994; 23: 155–161. [DOI] [PubMed] [Google Scholar]
- 10. Schlichtkrull J, Munck O, Jersild M. The M‐value, an index of blood‐sugar control in diabetics. Acta Med Scand 1965; 177: 95–102. [DOI] [PubMed] [Google Scholar]
- 11. Service FJ, Molnar GD, Rosevear JW, et al Mean amplitude of glycemic excursions, a measure of diabetic instability. Diabetes 1970; 19: 644–655. [DOI] [PubMed] [Google Scholar]
- 12. Shankar SS, Vella A, Raymond RH, et al Standardized mixed‐meal tolerance and arginine stimulation tests provide reproducible and complementary measures of beta‐cell function: results from the foundation for the national institutes of health biomarkers consortium investigative series. Diabetes Care 2016; 39: 1602–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tsuchiyama N, Takamura T, Ando H, et al Possible role of alpha‐cell insulin resistance in exaggerated glucagon responses to arginine in type 2 diabetes. Diabetes Care 2007; 30: 2583–2587. [DOI] [PubMed] [Google Scholar]
- 14. Rouille Y, Westermark G, Martin SK, et al Proglucagon is processed to glucagon by prohormone convertase PC2 in alpha TC1‐6 cells. Proc Natl Acad Sci USA 1994; 91: 3242–3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wewer Albrechtsen NJ, Hartmann B, Veedfald S, et al Hyperglucagonaemia analysed by glucagon sandwich ELISA: nonspecific interference or truly elevated levels? Diabetologia 2014; 57: 1919–1926. [DOI] [PubMed] [Google Scholar]
- 16. Hirota M, Shimizu I, Ohboshi C, et al A large molecular form of glucagon‐like peptide‐1 (GLP‐1) immunoreactivity is co‐released with glucagon from pancreas by arginine in normal subjects. Clin Chim Acta 1987; 167: 293–302. [DOI] [PubMed] [Google Scholar]
- 17. Ferrannini E, Muscelli E, Natali A, et al Association of fasting glucagon and proinsulin concentrations with insulin resistance. Diabetologia 2007; 50: 2342–2347. [DOI] [PubMed] [Google Scholar]
- 18. Faerch K, Vistisen D, Pacini G, et al Insulin resistance is accompanied by increased fasting glucagon and delayed glucagon suppression in individuals with normal and impaired glucose regulation. Diabetes 2016; 65: 3473–3481. [DOI] [PubMed] [Google Scholar]
- 19. Groop LC, Ferrannini E. Insulin action and substrate competition. Baillieres Clin Endocrinol Metab 1993; 7: 1007–1032. [DOI] [PubMed] [Google Scholar]
- 20. Santilli F, Simeone PG, Guagnano MT, et al Effects of liraglutide on weight loss, fat distribution, and beta‐cell function in obese subjects with prediabetes or early type 2 diabetes. Diabetes Care 2017; 40: 1556–1564. [DOI] [PubMed] [Google Scholar]
- 21. Schmitt C, Portron A, Jadidi S, et al Pharmacodynamics, pharmacokinetics and safety of multiple ascending doses of the novel dual glucose‐dependent insulinotropic polypeptide/glucagon‐like peptide‐1 agonist RG7697 in people with type 2 diabetes mellitus. Diabetes Obes Metab 2017; 19: 1436–1445. [DOI] [PubMed] [Google Scholar]
- 22. Rhodes CJ, Alarcon C. What beta‐cell defect could lead to hyperproinsulinemia in NIDDM? Some clues from recent advances made in understanding the proinsulin‐processing mechanism. Diabetes 1994; 43: 511–517. [DOI] [PubMed] [Google Scholar]
- 23. Schmidt MI, Hadji‐Georgopoulos A, Rendell M, et al The dawn phenomenon, an early morning glucose rise: implications for diabetic intraday blood glucose variation. Diabetes Care 1981; 4: 579–585. [DOI] [PubMed] [Google Scholar]
- 24. Monnier L, Colette C, Dejager S, et al The dawn phenomenon in type 2 diabetes: how to assess it in clinical practice? Diabetes Metab 2015; 41: 132–137. [DOI] [PubMed] [Google Scholar]
- 25. Campbell PJ, Bolli GB, Cryer PE, et al Pathogenesis of the dawn phenomenon in patients with insulin‐dependent diabetes mellitus. Accelerated glucose production and impaired glucose utilization due to nocturnal surges in growth hormone secretion. N Engl J Med 1985; 312: 1473–1479. [DOI] [PubMed] [Google Scholar]
- 26. Mallad A, Hinshaw L, Dalla Man C, et al Nocturnal glucose metabolism in type 1 diabetes: a study comparing single versus dual tracer approaches. Diabetes Technol Ther 2015; 17: 587–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Miyoshi H, Shulman GI, Peters EJ, et al Hormonal control of substrate cycling in humans. J Clin Invest 1988; 81: 1545–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Campbell PJ, Bolli GB, Cryer PE, et al Sequence of events during development of the dawn phenomenon in insulin‐dependent diabetes mellitus. Metabolism 1985; 34: 1100–1104. [DOI] [PubMed] [Google Scholar]
- 29. Goldstein RE, Wasserman DH, McGuinness OP, et al Effects of chronic elevation in plasma cortisol on hepatic carbohydrate metabolism. Am J Physiol 1993; 264: E119–E127. [DOI] [PubMed] [Google Scholar]
- 30. Ramnanan CJ, Edgerton DS, Kraft G, et al Physiologic action of glucagon on liver glucose metabolism. Diabetes Obes Metab 2011; 13(Suppl 1): 118–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen SS, Zhang Y, Santomango TS, et al Glucagon chronically impairs hepatic and muscle glucose disposal. Am J Physiol Endocrinol Metab 2007; 292: E928–E935. [DOI] [PubMed] [Google Scholar]
- 32. Muller WA, Faloona GR, Aguilar‐Parada E, et al Abnormal alpha‐cell function in diabetes. Response to carbohydrate and protein ingestion. N Engl J Med 1970; 283: 109–115. [DOI] [PubMed] [Google Scholar]
- 33. Dinneen S, Alzaid A, Turk D, et al Failure of glucagon suppression contributes to postprandial hyperglycaemia in IDDM. Diabetologia 1995; 38: 337–343. [DOI] [PubMed] [Google Scholar]
- 34. Gorst C, Kwok CS, Aslam S, et al Long‐term glycemic variability and risk of adverse outcomes: a systematic review and meta‐analysis. Diabetes Care 2015; 38: 2354–2369. [DOI] [PubMed] [Google Scholar]
- 35. Siafarikas A, Johnston RJ, Bulsara MK, et al Early loss of the glucagon response to hypoglycemia in adolescents with type 1 diabetes. Diabetes Care 2012; 35: 1757–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Porksen S, Nielsen LB, Kaas A, et al Meal‐stimulated glucagon release is associated with postprandial blood glucose level and does not interfere with glycemic control in children and adolescents with new‐onset type 1 diabetes. J Clin Endocrinol Metab 2007; 92: 2910–2916. [DOI] [PubMed] [Google Scholar]
- 37. Bessho M, Murase‐Mishiba Y, Tsutsumi C, et al Glycaemic instability correlates with a hyperglucagonaemic response in patients with type 1 diabetes without residual beta‐cell function. Diabetes Res Clin Pract 2013; 102: e38–e40. [DOI] [PubMed] [Google Scholar]
- 38. Miyachi A, Kobayashi M, Mieno E, et al Accurate analytical method for human plasma glucagon levels using liquid chromatography‐high resolution mass spectrometry: comparison with commercially available immunoassays. Anal Bioanal Chem 2017; 409: 5911–5918. [DOI] [PubMed] [Google Scholar]
- 39. Raffort J, Lareyre F, Massalou D, et al Insights on glicentin, a promising peptide of the proglucagon family. Biochem Med (Zagreb) 2017; 27: 308–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Holst JJ, Albrechtsen NJW, Gabe MBN, et al Oxyntomodulin: actions and role in diabetes. Peptides 2018; 100: 48–53. [DOI] [PubMed] [Google Scholar]
- 41. Matsuhisa M, Shi ZQ, Wan C, et al The effect of pioglitazone on hepatic glucose uptake measured with indirect and direct methods in alloxan‐induced diabetic dogs. Diabetes 1997; 46: 224–231. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 | Schematic presentation of changes in insulin requirements between night and morning, as evaluated using an artificial pancreas.
Figure S2 | Scatter plots between the dawn index and fasting levels of glucose‐related hormones.
Figure S3 | Scatter plots between hepatic glucose uptake evaluated by clamp oral glucose loading tests and fasting levels of glucose‐related hormones.
Figure S4 | Scatter plots between measurements of glucose variability, except standard deviation (average, mean amplitude of glycemic excursions, M‐value, hyperglycemic time and hypoglycemic time), as evaluated by continuous glucose monitoring in 96 h and (a–e) logarithm‐transformed area under the curve (AUC) of glucagon measured using radioimmunoassays (log[AUC glc RIA]) or (f–j) logarithm‐transformed AUC of glucagon measured using enzyme‐linked immunosorbent assays (log[AUC glc ELISA]).
Table S1 | Multiple regression analysis of glucose infusion rate during hyperinsulinemic euglycemic clamp tests.