

Abstract
Pannexin 1 (PANX1)-mediated ATP release in vascular smooth muscle coordinates α1-adrenergic receptor (α1-AR) vasoconstriction and blood pressure homeostasis. We recently identified amino acids 198–200 (YLK) on the PANX1 intracellular loop that are critical for α1-AR–mediated vasoconstriction and PANX1 channel function. We report herein that the YLK motif is contained within an SRC homology 2 domain and is directly phosphorylated by SRC proto-oncogene, nonreceptor tyrosine kinase (SRC) at Tyr198. We demonstrate that PANX1-mediated ATP release occurs independently of intracellular calcium but is sensitive to SRC family kinase (SFK) inhibition, suggestive of channel regulation by tyrosine phosphorylation. Using a PANX1 Tyr198–specific antibody, SFK inhibitors, SRC knockdown, temperature-dependent SRC cells, and kinase assays, we found that PANX1-mediated ATP release and vasoconstriction involves constitutive phosphorylation of PANX1 Tyr198 by SRC. We specifically detected SRC-mediated Tyr198 phosphorylation at the plasma membrane and observed that it is not enhanced or induced by α1-AR activation. Last, we show that PANX1 immunostaining is enriched in the smooth muscle layer of arteries from hypertensive humans and that Tyr198 phosphorylation is detectable in these samples, indicative of a role for membrane-associated PANX1 in small arteries of hypertensive humans. Our discovery adds insight into the regulation of PANX1 by post-translational modifications and connects a significant purinergic vasoconstriction pathway with a previously identified, yet unexplored, tyrosine kinase–based α1-AR constriction mechanism. This work implicates SRC-mediated PANX1 function in normal vascular hemodynamics and suggests that Tyr198-phosphorylated PANX1 is involved in hypertensive vascular pathology.
Keywords: pannexin 1 (PANX1), SRC, smooth muscle, ATP, ATP release, vascular biology, adrenergic receptor, hypertension, vasoconstriction, SRC family kinase (SFK), kinase signaling, muscle contraction, membrane channel
Introduction
The synchronous and coordinated constriction of vascular smooth muscle cells (VSMCs)2 in resistance arteries is necessary for controlling total peripheral resistance and blood pressure homeostasis (1). VSMCs on resistance arteries are innervated by sympathetic nerves (2), which elicit local constriction events through the co-release of neuronal derived norepinephrine (NE) and presumably ATP (3). Following α1-AR activation, latent NE-mediated VSMC signals (i.e. ATP) promote and coordinate vasoconstriction of neighboring cells, which can be enhanced and propagated to a significant extent by autocrine/paracrine signaling within resistance vessels (4, 5). The regulated release of VSMC-derived ATP has therefore emerged as a predominant signal for controlling hemodynamics.
In the vascular wall, the location of ATP release governs its effect either as a vasodilator (from endothelial cells) or as a potent vasoconstrictor (from VSMCs) (4). This functional dichotomy highlights a unique mechanism for the regulated release of ATP from vascular cells, which has only recently come to light (6). Pannexin 1 (PANX1) channels, the prototypical member of a class of channel-forming transmembrane glycoproteins, have been established as the main conduit by which ATP is released from VSMCs (7) and other cell types (8) under physiological conditions. Recent work from our laboratory (and others) has demonstrated that PANX1-mediated ATP release uniquely couples to α1-AR vasoconstriction in resistance arteries, where VSMC PANX1 is highly expressed (9–11). Moreover, we have identified an important PANX1 intracellular loop motif, residues Tyr198–Lys200 (mouse) and Tyr199–Lys201 (human), that is critical for adrenergic receptor–mediated channel function. In in vitro and in vivo experimental models, pharmacological inhibition and genetic deletion targeting the YLK motif reduced ATP release, inhibited PANX1 current, blunted adrenergic vasoconstriction, and reduced mean arterial pressure (5, 12). Thus, the PANX1 YLK motif functions as an important regulatory site.
The traditional view of α1-AR activation and subsequent VSMC constriction is that they are thought to mechanistically couple heterotrimeric G-protein activation to increased intracellular calcium via the generation of inositol triphosphate. Alternatively, a number of studies have provided evidence for a secondary and, as of yet, unclear tyrosine kinase–mediated component of adrenergic constriction that might co-regulate vasoconstriction events (13–17). Recent evidence in the pannexin literature also suggests a regulatory role for tyrosine kinases in receptor-stimulated PANX1 activity and downstream function (i.e. channel gating and ATP release) responsible for neuronal excitotoxic cell death (18). Similarly, in endothelial cells of peripheral veins, receptor-mediated activation of PANX1 channels and endothelial ATP release were significantly blocked using SRC family kinase (SFK) inhibitors (19). These findings suggest a common tyrosine kinase–based regulatory mechanism for PANX1 channel regulation that, until now, has not been explored in VSMCs of resistance arteries.
Here, we show that SRC kinase, the archetypal SFK, is responsible for the direct phosphorylation of Tyr198 on the intracellular loop of PANX1 in VSCMs and that modulation of SRC activity and phospho-Tyr198 status is critical for supporting proper channel function. Notably, we find that Tyr198 phosphorylation is constitutive in nature and is not induced or further enhanced upon α1-AR stimulation. Moreover, inhibition of SFKs, in particular SRC kinase, and the concomitant loss of constitutive tyrosine phosphorylation at Tyr198 is detrimental to channel opening, ATP release, and adrenergic vasoconstriction. We also find that increased detection of PANX1 Tyr198 phosphorylation in hypertensive human vessel biopsies correlates with PANX1 protein expression in hypertensive but not normotensive arteries. Together, our results suggest that the increased PANX1 at the plasma membrane may contribute to pathological hypertensive responses that occur in resistant hypertension.
Results
SRC family kinases regulate phenylephrine-induced pannexin 1 channel function independent of Ca2+
We have previously shown that α1-AR–stimulated vasoconstriction uniquely couples with PANX1-mediated ATP release from VSMCs of resistance arteries and requires the PANX1 intracellular motif (YLK) (5, 7). To examine whether PANX1-mediated ATP release requires cytosolic calcium (Ca2+), we first sought to determine whether adrenergic stimulated ATP release depended on intracellular Ca2+. Isolated resistance arteries were incubated with BAPTA-AM to chelate intracellular Ca2+. Application of the α1-AR selective agonist phenylephrine (PE; 20 μm) caused a significant increase in extracellular ATP both in the presence and the absence of BAPTA-AM (Fig. 1A). These observations suggest a Ca2+-independent mechanism for α1-AR–mediated ATP release, at least after adrenergic stimulation, and agree with previous observations suggesting that Ca2+ is not always involved in PANX1-mediated ATP release (20–23).
Figure 1.

SRC family kinases regulate phenylephrine-induced pannexin 1 channel function independent of Ca2+. A, PE-stimulated ATP release from intact TDAs treated with the intracellular calcium-chelating agent BAPTA-AM. n = 4 TDAs (from four mice). Data are presented as percentage increase from the unstimulated condition. Student's t test was performed to test for significance. B, mouse pannexin 1 membrane topology denoting SH2 linear motif (red) containing the putative intracellular loop tyrosine 198 (yellow) SRC phosphorylation site and the upstream proline-rich SH3-binding domain (green). C, ex vivo ATP release from TDA stimulated with PE (20 μm) and norepinephrine (10 μm) in the presence and absence of the SFK inhibitors PP2 (10 μm), dasatinib (10 nm), and negative control PP3 (10 μm). The nonadrenergic vasoconstrictors 5-HT (100 nm) and ET-1 (10 nm) were assessed. n = 4 TDA (from four mice) per group. One-way ANOVA was performed for statistical significance. #, significant increase from vehicle; p < 0.5. Asterisks, significant reduction from PE-stimulated; *, p < 0.05; **, p < 0.01; ***, p < 0.001. D–G, contractile responses to increasing concentration of adrenergic agonists (PE or norepinephrine) in the presence of SFK inhibitors PP2 (10 μm; purple line), inactive control PP3 (10 μm; green line), and dasatinib (10 nm; red line). n = 4–8 TDA (from 4–8 mice). Data are presented as mean ± S.E. (error bars) and were assessed for significance using two-way ANOVA with Bonferroni post hoc test of multiple comparisons. *, p < 0.05 compared with control response (black line).
Next, we scanned short eukaryotic linear motif databases to analyze regions of PANX1 that might confer modifications to the channel independently of Ca2+, specifically near the intracellular loop motif (YLK). We identified a putative SRC homology 2 recognition site in both mouse and human amino acid sequences, which was contained within the YLK sequence. In the mouse sequence, this site localized near an SRC homology 3 (proline-rich) SRC-binding region, indicative of regulation by SFKs (Fig. 1B). To confirm a possible role for SFK activity in adrenergic mediated ATP release, we measured ATP release from human embryonic kidney 293T (HEK293T) cells co-expressing PANX1 and the α1-AR in the presence of the SFK inhibitor PP2 and the inactive analog PP3 (Fig. S1). PE-stimulated ATP release was significantly blunted by PP2 (10 μm) treatment, but not with PP3 (10 μm). We performed similar experiments using isolated murine resistance arteries in the presence of PP2, PP3, and the more potent tyrosine kinase inhibitor dasatinib (Fig. 1C). Application of PP2 (10 μm) significantly blunted PE- and NE-induced ATP release as compared with control arteries or arteries treated with the inactive analog PP3 (10 μm). The same inhibitory effect was observed using dasatinib (10 nm). No increase in ATP release was observed when vessels were stimulated with nonadrenergic vasoconstrictors endothelin-1 (ET-1; 10 nm) or serotonin (5-HT; 100 nm), thus confirming the specificity of adrenergic stimuli for PANX1-mediated ATP release, as described previously (5).
In addition to ATP release, we assessed vasoconstriction responses to cumulative doses of adrenergic and nonadrenergic agonists using pressure myography in isolated resistance arteries (Fig. 1, D–G). Pretreatment (15 min) of arteries with PP2 (10 μm) significantly blunted α1-AR constriction as compared with control arteries or arteries treated with the inactive analog PP3 (10 μm) (Fig. 1D). Application of ET-1 or 5-HT produced strong vasoconstriction responses in the presence and absence of the PP2, highlighting again a key pathway between SFK activity and adrenergic mediated vasoconstriction, but not ET-1 or 5-HT (Fig. 1, E and F). Similar inhibitory effects on α1-AR vasoconstriction were observed when vessels were pretreated (15 min) with dasatinib (10 nm) (Fig. 1G).
Pannexin 1 tyrosine 198 is constitutively phosphorylated in vascular smooth muscle cells
Our previously published mutagenesis experiments (5) and SFK-dependent vasoconstriction implicate PANX1 Tyr198 in the PANX1 intracellular loop motif (YLK) as a SFK phosphorylation site. To analyze the phosphorylation status of PANX1 Tyr198 in VSMCs after adrenergic stimulation, human coronary smooth muscle cells (hCoSMCs) were differentiated to a contractile phenotype and assessed by Western blotting for PANX1 expression (Fig. 2A). Typical 48-h serum starvation up-regulated smooth muscle contractile proteins, including α-smooth muscle actin and PANX1 (Fig. 2A). We next immunoprecipitated PANX1 from differentiated hCoSMCs following PE (20 μm) stimulation using a total PANX1 antibody and magnetic bead separation. The status of PANX1 Tyr198 phosphorylation was assessed by Western blotting using a phospho-PANX1 (Tyr198) antibody (pPANX1Y198) (Fig. 2B). Our data revealed that PANX1 phosphorylation at Tyr198 was unchanged by PE stimulation. Mutagenesis of tyrosine 198 to phenylalanine abolished detectable phosphorylation, confirming PANX1 Tyr198 antibody specificity in VSMCs (Fig. S2A). We next showed that the PxIL2 peptide (composed of the PANX1 Tyr198–containing SA2 region and a TAT peptide tag (5)) significantly reduced PANX1 Tyr198 phosphorylation detection (Fig. 2C). Again, following adrenergic stimulation, we observed similar levels of PANX1 Tyr198 phosphorylation both in vehicle-treated and PE-stimulated samples, indicative of constitutive phosphorylation at Tyr198. We confirmed observations from immunoprecipitation experiments by probing for constitutive Tyr198 phosphorylation in differentiated hCoSMCs and tested Tyr198 phospho-status after treatment of cells with pharmacological SFK inhibitors (or vehicle control). As with our other assay, PE did not enhance PANX1 Tyr198 phosphorylation (Fig. 2D). However, treatment with PP2 (10 μm) and dasatinib (10 nm) significantly reduced PANX1 Tyr198 levels. No reduction in phosphorylation was observed with the inactive control PP3 (10 μm). These results were recapitulated in HEK293 cells expressing PANX1 (Fig. S2B). As an additional control to confirm the detection of Tyr198 as a phosphorylation residue, cell lysates were treated with λ-phosphatase, which completely removes phosphate groups (Fig. 2D). As expected, λ-phosphatase completely reduced pPANX1Y198 levels. These data suggest that SFK activity constitutively phosphorylates Tyr198 to support PANX1 channel function.
Figure 2.

Pannexin 1 tyrosine 198 is constitutively phosphorylated in vascular smooth muscle cells. A, confirmation of protein expression of PANX1 and SRC kinase in smooth muscle cells following differentiation to a contractile phenotype with 48-h serum starvation (0.2% FBS). Data are presented as mean ± S.E. (error bars), n = 4 independent experiments. Student's t test was performed to test for statistical significance. *, p < 0.05; **, p < 0.01 compared with high FBS level. B, immunoprecipitation of PANX1 from differentiated hCoSMCs stimulated with PE (20 μm) and immunoblotted for pannexin 1 (Tyr198) phosphorylation. C, the PxIL2 peptide was used at 10 mm to inhibit pPANX1Y198 phosphorylation after adrenergic stimulation. λ-Phosphatase was used to eliminate all tyrosine phosphorylation of PANX1. D, representative Western blotting and quantification of phosphorylation status of pannexin 1 Tyr198 in hCoSMCs treated with SFK inhibitors dasatinib (10 nm; red), PP2 (10 μm; purple), and negative control PP3 (10 μm; green). λ-Phosphatase–treated lysates were used as a negative control; n = 6 independent experiments. Data quantification is presented as mean ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001 compared with unstimulated control using one-way ANOVA.
SRC kinase activity modulates phosphorylation of pannexin 1 at tyrosine 198
Due to the significant inhibitory effects of SFK-modulating agents on adrenergic mediated vasoconstriction, ATP release, and PANX1 Tyr198 phosphorylation in VSMCs, we set out to specifically address whether SRC kinase modulates PANX1 Tyr198 phosphorylation, as was observed in other cell types (18, 19). We performed knockdown of SRC kinase using SRC-specific siRNA in hCoSMCs. Similar to the effects observed using SFK inhibitors, PANX1 Tyr198 phosphorylation was equally abundant in control and PE-stimulated conditions yet significantly reduced after SRC knockdown. No reduction in PANX1 Tyr198 was observed using negative control siRNA (scramble siRNA) (Fig. 3A). To further validate the specificity of SRC kinase to phosphorylate PANX1 Tyr198, HEK cells were transfected with expression vectors for the α1-AR, PANX1, and mutant isoforms of SRC kinase that were constitutively active (cSRC) or harbored a kinase-inactivating mutation (SRC-KD) (Fig. 3B). Western blotting detection for SRC and the pSFK(Y416) autophosphorylation activation loop residue revealed an increase in total expression of SRC kinase following transfection. pSFK(Y416) was moderately increased in SRC-KD, possibly due to overexpression in a cell type containing native SRC kinase. In the presence of constitutively active cSRC, an additional increase in pSFK(Y416) was observed. Under basal transfection conditions (only empty vector, α1-AR, and PANX1), PANX1 Tyr198 phosphorylation was not different between cells treated with PE or vehicle control. However, co-transfection with the cSRC isoform, but not the SRC-KD isoform, generated intense hyperphosphorylation of PANX1 Tyr198. Thus, PANX1 Tyr198 phosphorylation is regulated by SRC kinase activity.
Figure 3.

SRC kinase activity modulates pannexin 1 (Tyr198) phosphorylation. A, representative Western blotting and quantification of SRC kinase and pPANX1Y198 following siRNA-mediated knockdown of SRC in hCoSMCs stimulated with PE (20 μm). Data quantification is presented as -fold change ± S.E. (error bars). Statistical analyses were performed using one-way ANOVA. *, p < 0.05; ***, p < 0.001. B, in vitro analysis of adrenergic stimulated pannexin 1 (Tyr198) phosphorylation in HEK293T cells co-expressing the α1-AR and pannexin 1. Overexpression of the cSRC or SRC-KD isoform influenced pPANX1Y198 signal. C, time course for SRC kinase activation (35 °C) and inhibition (40 °C) using mutant temperature-sensitive LA-25 cells. SRC activity was validated using specific antibodies for the SFK activation residue (Tyr416) and an SRC kinase–specific phosphorylation site (Tyr118) on the focal adhesion protein paxillin. D, Western blot analysis of SRC kinase activity on pPANX1Y198 in LA-25 cells transfected with a combination of pannexin 1, α1-AR, empty vector, or cSRC. The SFK inhibitor PP2 (10 μm) was used to inhibit the endogenous temperature-active SRC kinase. WT cSRC expression rescued temperature-inactive SRC activity.
Next, we performed a temperature-dependent SRC kinase assay using LA-25 cells transfected with α1-AR and PANX1 (Fig. 3C). LA-25 cells harbor a temperature-sensitive SRC mutation (24), which at the permissive temperature (35 °C) constitutively activates SRC kinase but at the nonpermissive temperature (40 °C) inactivates kinase activity (25). After a 30-min incubation of LA-25 cells at 35 °C, SRC kinase was activated as indicated by enhanced pSFK(Y416) autophosphorylation, as well as the phosphorylation of the SRC substrate paxillin (Tyr118). pSFK(Y416) or paxillin (Tyr118) phosphorylation was not observed after 30 min at 40 °C. Analysis of PANX1 in vehicle and PE-stimulated conditions revealed constitutive phosphorylation of PANX1 (Tyr198) at 35 °C and reduced signal at 40 °C. Incubating cells with PP2 (10 μm) at the permissive temperature was also sufficient to obstruct constitutive SRC kinase activity and reduce detection of PANX1 Tyr198. Importantly, PANX1 Tyr198 signal could be rescued at the nonpermissive temperature when LA-25 cells were supplemented with the constitutive cSRC isoform. These data support the notion that SRC kinase activity modulates constitutive PANX1 Tyr198 phosphorylation.
SRC kinase directly phosphorylates pannexin 1 at Tyr198
To determine whether phosphorylation of PANX1 Tyr198 is directly mediated by SRC kinase, we performed an in vitro kinase assay using recombinant PANX1 protein isolated from Sf9 cells (26) and recombinant/active SRC kinase (Fig. 4, A and B). It was first necessary to dephosphorylate purified recombinant PANX1 protein using λ-phosphatase. Immunoblotting with antibodies for pan-phosphotyrosine or pPANX1Y198 antibodies revealed basally phosphorylated substrates that could be completely dephosphorylated using λ-phosphatase (Fig. 4A). Following dephosphorylation, PANX1 protein (lane 4) was subsequently incubated with constitutively active SRC-kinase (Fig. 4B). The addition of SRC restored detection of PANX1 Tyr198 phosphorylation (lane 5). As a confirmation of SRC activity, the SRC-dependent PANX1 Tyr308 phosphosite was also detectable, as described previously (18). The presence of doublet patterns in our kinase assay was attributed to a glycosylation modification made in Sf9 cells during recombinant protein production. To ensure that doublets were due to glycosylation, PANX1 was treated with N-glycosidase, which resulted in a single PANX1 band (Fig. S3), as reported previously (27). Based on these results, we propose that SRC kinase associates with PANX1, allowing for the direct phosphorylation of PANX1 at Tyr198.
Figure 4.

SRC kinase directly phosphorylates pannexin 1 (Tyr198). A, in vitro phosphotyrosine analysis using pan-tyrosine phosphorylation antibody of recombinant PANX1 protein treated with λ-phosphatase. Pannexin 1 (Tyr198) phosphorylation immunoblotted using pPANX1Y198. B, in vitro SRC kinase assay using sequentially dephosphorylated recombinant pannexin 1 with λ-phosphatase and rephosphorylation of Tyr198 by recombinant active SRC kinase. Pannexin 1 (Tyr308) was assessed to validate SRC activity (using pPANX1Y308 antibody).
PANX1 Tyr198 phosphorylation occurs at the plasma membrane, where it associates with SRC
Functional PANX1 channels localize to the plasma membrane of VSMCs, where they mediate cellular ATP release. To determine whether phosphorylation of PANX1 Tyr198 corresponds with plasma membrane localization, we performed cell-surface biotinylation and immunoprecipitation (IP) experiments in hCoSMCs (Fig. 5, A and B). Successful membrane biotinylation was confirmed using streptavidin-conjugated antibodies, which detected biotin only in IP fractions and not in cytoplasmic fractions (Fig. 5A). PANX1 protein was resolved in all VSMC fractions, with a proportion of low-molecular weight cytoplasm-associated PANX1 fractions (endoplasmic reticulum, Golgi apparatus, or vesicles (28)) detected in whole-cell lysates and not IP fractions. A 50 kDa PANX1 band was enriched in IP fractions. Moreover, a pool of IP-associated SRC kinase was also resolved in membrane fractions, indicative of its presence and interaction with plasma membrane proteins (29). In support of PANX1 Tyr198 membrane-specific association, a single pPANX1Y198 band (50 kDa) was highly enriched in immunoprecipitated fractions compared with whole-cell lysates or cytoplasmic fractions from cells stimulated with phenylephrine (Fig. 5B). There was no significant difference in pPANX1Y198 between stimulated and control conditions. Due to direct phosphorylation of PANX1 Tyr198 by SRC kinase observed in our in vitro preparations and the membrane-associated pool of SRC detected in our membrane biotinylation IP, we tested whether PANX1 and SRC kinase associate with each other in smooth muscle cells. Using co-immunoprecipitation from hCoSMCs, we observed an association of PANX1 with SRC kinase that was independent of adrenergic stimulation (Fig. 5C). Based on these results, we propose that SRC kinase associates with PANX1 at the plasma membrane and phosphorylates PANX1 Tyr198 to support adrenergic mediated channel activation.
Figure 5.

Pannexin 1 (Tyr198) phosphorylation occurs at the plasma membrane in VSMCs. A, isolation of plasma membrane proteins using membrane biotin immunoprecipitation in hCoSMCs. Phosphorylation of pannexin 1 Tyr198 was assessed by Western blotting (pPANX1Y198) from whole-cell lysate, membrane fraction, cytoplasmic fractions, or attached to isolation beads. Streptavidin-conjugated antibodies were used to validate immunoprecipitation of membrane fractions. B, Western blot analysis of membrane-associated and cytoplasm-associated pannexin 1 Tyr198 phosphorylation following phenylephrine stimulation (20 μm) in hCoSMCs. C, co-immunoprecipitation of PANX1 and SRC kinase from human coronary smooth muscle cells under basal and phenylephrine (100 μm)–stimulated conditions. L, ladder; IP, low-pH elution; WCL, whole-cell lysate; Cyto, cytoplasmic fraction.
To further validate the finding that SRC-mediated phosphorylation of PANX1 Tyr198 occurs at the plasma membrane, we transfected HeLa cells with a PANX1-GFP expression vector and assessed PANX1 Tyr198 phosphorylation at the plasma membrane. PANX1 Tyr198 resolved predominantly at cell borders, where it co-localized with plasma membrane–localized PANX1-GFP (Fig. 6, A–C). From these findings, we treated cells with PP2 to inhibit SFK activity and found a loss of pPANX1Y198 signal at the plasma membrane (Fig. 6B). From these observations, we reasoned that the phosphorylation state at Tyr198 was a specific marker for the pool of activatable plasma membrane–associated PANX1. Because VSMC PANX1 regulates α1-AR vasoconstriction in resistance arteries and because hypertensive pathologies often involve enhanced sympathetic nerve activity and increased α1-AR–mediated constriction, we tested human vascular biopsies from normotensive and hypertensive (treatment-resistant) volunteers for PANX1 and phosphorylated PANX1 Tyr198. In hypertensive vessels, phosphorylated PANX1 Tyr198 was more prominently detected in the VSMC (SMα-actin–positive) layer of arteries compared with normotensive vessels (Fig. 6, D–E). No signal was detected in isotype controls (Fig. 6F). Thus, phosphorylation of PANX1 Tyr198 is likely an important marker for plasma membrane PANX1 and could be utilized to identify the level of activatable PANX1 channels that associates with vascular pathologies found in sympathetic nerve-mediated hyperstimulation commonly observed in hypertension.
Figure 6.
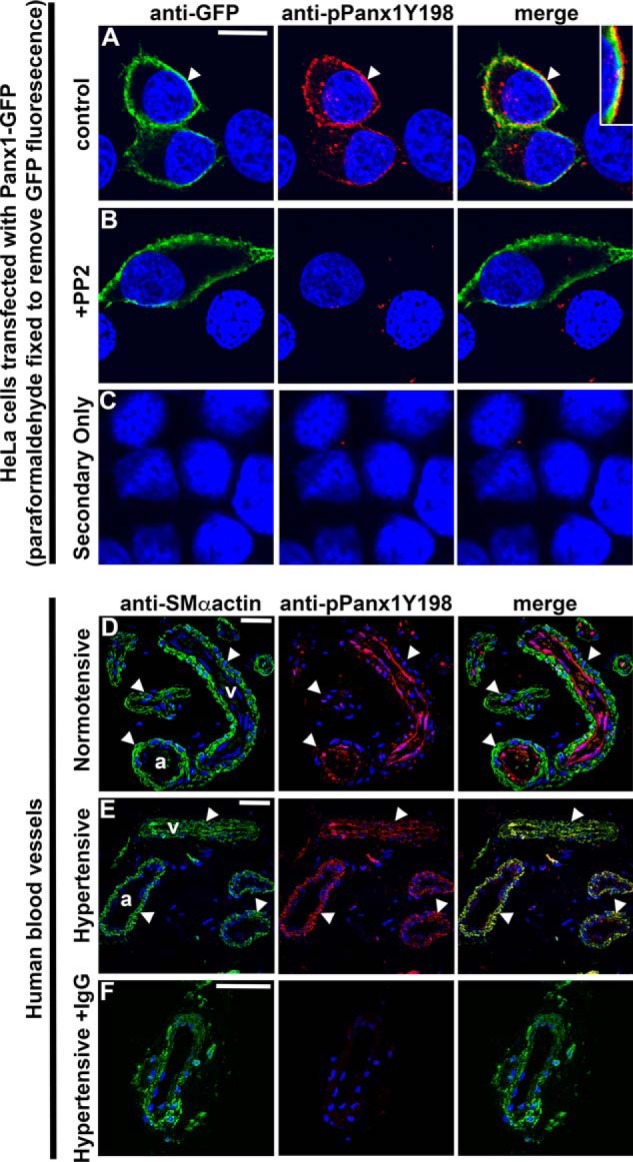
SRC-dependent pannexin 1 Tyr198 phosphorylation is enhanced in resistance arteries of hypertensive patients. In A–C, HeLa cells were transfected with PANX1-GFP. Transfected cells were observed under control conditions (A) or treated with PP2 (10 μm) to inhibit SFK activity (B) or with secondary antibodies only (C). Green, anti-GFP; red, anti-pPANX1Y198; blue, nuclei. Scale bar, 10 μm. In D–F, pPANX1Y198 was detected in small vessels of human gluteal biopsies from normotensive (D) or hypertensive (E) patients. The hypertensive biopsies were also incubated with IgG to determine the specificity of the anti-pPANX1Y198 (F). Smooth muscle cells were detected with smooth muscle (SM) α-actin (green), pPANX1Y198 (red), and nuclei (blue). Scale bar, 20 μm; a, artery; v, vein.
Discussion
The present study demonstrates the direct phosphorylation of a critical amino acid residue (Tyr198) on the PANX1 intracellular loop by SRC kinase in VSMCs and the sufficiency of SFK activity to modulate PANX1 channel function and adrenergic mediated vasoconstriction. Moreover, we find that phosphorylation of the PANX1 Tyr198 residue by SFK activity is constitutive in nature and likely supports the initiation of purinergic signaling cascades (i.e. regulated ATP release) at the plasma membrane. We also demonstrate that enhanced VSMC PANX1 expression and PANX1 Tyr198 immunodetection correlates with a hypertensive vascular phenotype. Thus, our study connects two observations from the pannexin field and the vascular biology field. The first is the modulation of PANX1 function by SFK activity in VSMCs. The second is a possible connection between PANX1-mediated ATP release and a tyrosine kinase–based α1-AR vasoconstriction mechanism that was previously identified to influence hemodynamics in the resistance vasculature (14–16, 18, 30–34).
Currently, there are two main mechanisms ascribed to the activation of PANX1 channels following physiologic stimuli. The first mechanism is a nonreversible cleavage event of PANX1 C-terminal tails, which results in constitutive release of ATP and subsequent cell apoptosis (35, 36). The second mechanism is a reversible receptor-mediated activation of PANX1 channels by ligand-gated signaling (7, 19, 21, 37–42). Previous work from our laboratory has established a significant role for receptor-mediated PANX1 channel function in the vascular wall (5, 7, 19), where PANX1 function specifically couples with adrenergic stimulated events in VSMCs, but not other vasoconstriction pathways (5). We hypothesized that purinergic signaling facilitates autocrine/paracrine coupling of VSMCs during tonic constriction in small resistance arteries where PANX1 is highly expressed (43).
At first, it was unclear whether intracellular calcium was necessary for adrenergic stimulated ATP release from VSMCs. Under physiologic conditions, VSMCs require a rise in intracellular calcium to activate calcium-sensitive kinases and to allow contractile proteins to engage with each other, thus producing a force constriction (44). Our results show that ATP release was unaffected by the membrane-permeable calcium-chelating agent BAPTA-AM, indicating that VSMC PANX1 does not require intracellular calcium to function in this context and likely occurs in the early stages of adrenergic signaling events. We cannot exclude the possibility that calcium sensitive kinases, such as those involved in the canonical RhoA/ROCK sensitization pathway (45), may further potentiate PANX1 channel opening after initial activation, although this remains to be specifically tested. In the literature, evidence of PANX1 channel gating by intracellular calcium is unclear. A handful of studies link PANX1 channel function to direct increases in intracellular calcium using ionophores (46) or, indirectly, through serotonin receptor agonism (47) or thrombin receptor activation (37, 38). From these studies, it remains to be shown whether changes in intracellular calcium directly influence PANX1 gating or if another calcium-dependent process facilitates channel activation. In particular, the requirement for calcium during thrombin-induced ATP release from lung epithelial cells was necessary, but not sufficient on their own to elicit a response (37). A similar observation was made in vein endothelial cells, in which BAPTA-AM treatment blunted thrombin-induced ATP release in a similar fashion to responses from PANX1 shRNA–treated cells (38). Thus, a definitive calcium requirement for PANX1 channel gating has yet to be clearly delineated.
On the other hand, a number of published studies have demonstrated calcium-independent activation of PANX1 channels either by directly manipulating intracellular and extracellular calcium (22) or through activation of P2X7 receptors (23, 48) or NMDA receptors (18, 21). Our data in VSMCs and in resistance arteries support findings in these latter studies, which demonstrated a calcium-independent function of PANX1 channels. Interestingly, the studies that provide evidence of calcium-independent PANX1 activation also demonstrate mechanistic regulation of PANX1 by tyrosine kinase activity (18, 21). Findings from this analysis support a role for SRC kinase, which can be activated by calcium-independent signaling pathways (49, 50). Thus, the necessity for increased calcium during PANX1 activation may reflect either cell type–specific or receptor-specific characteristics that, as of yet, remain to be shown.
Since ATP release was preserved after calcium chelation in our ex vivo arterial system, we proposed that post-translational modifications might regulate PANX1 function. A large body of evidence points to regulation of PANX1 channels by phosphorylation, predominantly tyrosine phosphorylation by SRC family kinases (18, 19, 21, 30, 51). Moreover, in resistance arteries, a tyrosine kinase–based adrenergic signaling mechanism exists, which significantly modulates tonic and phasic constriction responses to both phenylephrine and norepinephrine in different animal models (13–16, 17, 31–34). Inhibition of adrenergic constriction by tyrosine kinase inhibitors is reversible and has no direct effect on calcium-induced constriction, protein kinase A activity, or myosin light chain kinase activity (13, 14). Thus, there is a strong correlation between tyrosine kinase–regulated adrenergic vasoconstriction and SFK-dependent PANX1 channel activity. In line with these studies, our laboratory mapped and identified a regulatory region on the intracellular loop of PANX1 using mimetic peptides, VSMC-specific PANX knockout mice, and mutated PANX1 expression vectors (5). In this current study, we performed eukaryotic linear motif scanning on human (Tyr199–Lys201) and mouse (Tyr198–Lys200) PANX1 sequences (52). We detected a putative SRC homology 2 (SH2) domain within the same PANX1 regulatory motif (YLK) from our previous investigation, as well as an SH3 proline-rich motif upstream of this regulatory motif (Fig. 1B). SH2 and SH3 domains regulate intra- and intermolecular interactions necessary for SFK catalytic activity, localization, and recruitment of substrates (53–59). The presence of these sequences on the PANX1 intracellular loop points to a role for SFK activity in channel function.
To test if PANX1-mediated ATP release and vasoconstriction depends on SFK activity, we stimulated ex vivo arteries with PE or NE in the presence of the SFK inhibitor PP2 and the Food and Drug Administration–approved SRC/ABL kinase inhibitor dasatinib. Our results show that PE- and NE-stimulated ATP release depends on SFK activity in our model and similarly influences vasoconstriction responses in our pressure myography experiments (Fig. 1, D–G) (5, 7, 60). Our findings confirm observations by Di Salvo et al. and others (5, 13–16, 17, 31–34), who discovered and elaborated on a unique tyrosine kinase–mediated VSMC pathway that regulates adrenergic constriction. Although our mechanistic understanding of receptor-mediated PANX1 activation remains limited, published reports evidence the potential for GPCRs to directly activate tyrosine kinases through guanine nucleotide exchange factor interactions (61–63). These signals could also transactivate growth/proliferation pathways through SRC kinase (17, 64). Thus, a secondary pathway might exist in VSMCs that can regulate adrenergic mediated responses in a nontraditional way. Pathologically, this alternative pathway may contribute to early inward eutrophic vascular remodeling associated with human neurogenic/resistant hypertension (65, 66). For example, norepinephrine-induced signaling has been shown to contribute to VSMC hypertrophy through transactivation of SRC kinase/STAT3/and mitogen-activated protein kinase pathways (64). In line with these observations, we found an important change in pPANX1Y198 immunostaining from EC to VSMC of small vessels from treatment-resistant hypertensive patients (Fig. 6).
The GPCR-mediated tyrosine kinase activity may be a common mechanism for regulating cellular ATP release by PANX1. Although our VSMC models demonstrate a unique role for α-adrenergic receptors, but not serotonin or endothelin receptors in PANX1 function, we do not exclude the possibility that other cell types may couple PANX1 to these GPCRs in unique ways. Serotonin receptors, which can also activate SFKs (e.g. FYN) (67), have been reported to activate PANX1 channels in carotid body cells (47). Evidence for angiotensin II type 1 receptor (AT1R) activation of PANX1 has also been implicated in carotid body cells (68) and in renal mesangial cells (69). In the vasculature, AT1R stimulation results in VSMC constriction, which can directly influence SRC kinase activity, leading to focal adhesion remodeling (70), growth (71), and VSMC proliferation (72). At this time, it is unknown whether AT1R activation can induce PANX1 channel activity in VSMCs or whether the AT1R and the α1-AR share a common tyrosine kinase–mediated mechanism, but it would be interesting if Gαq signaling proteins were directly involved in this process.
Having established the involvement of tyrosine kinases in PE- and NE-induced ATP release and vasoconstriction, we hypothesized that phosphorylation of the PANX1 tyrosine residue (Tyr198), which resides in a SH2 domain, is likely mediated by SFKs. To resolve phosphorylated PANX1 Tyr198, we created a phospho-antibody (pPANX1Y198), which generated a PANX1-specific band at a molecular weight corresponding to a highly glycosylated and plasma membrane–associated form of PANX1 (28). Moreover, the pPANX1Y198 signal was sensitive to phosphatase treatment and could not be detected in cells expressing the PANX1 point-mutant (Y198F). Using differentiated hCoSMCs, which express PANX1 and SRC kinase, we found that PANX1 Tyr198 was constitutively phosphorylated under basal and stimulated conditions. Surprisingly, the addition of an adrenergic stimulus did not enhance PANX1 Tyr198 phosphorylation, but did require SFK activity (Fig. 2C and Fig. S2B). These results thus suggest that whereas adrenergic induced PANX1 activation requires constitutive phosphorylation at Tyr198, another site is likely responsible for direct channel gating. In hippocampal neurons, the PANX1 Tyr308 residue on the C-terminal tail was required for receptor-mediated PANX1 activation by NMDA receptor agonism, and that phosphorylation of Tyr308 and activation of PANX1 currents were similarly sensitive to SFK inhibition (18, 21). Thus, phosphorylation of PANX1 Tyr198 might facilitate selective binding of signaling proteins necessary for post-translational modification of C-terminal residues, which subsequently regulate PANX1 channel activity. In the literature, a quantized mechanism for C-terminal channel regulation has emerged, which supports this concept, whereby individual regulation of C-terminal tails on oligomerized PANX1 subunits can differentially influence both receptor-independent and receptor-dependent functionality (26).
Due to the constitutive nature of PANX1 Tyr198 phosphorylation and the necessity for tyrosine phosphorylation during adrenergic vasoconstriction, we next determined whether the archetypal SRC tyrosine kinase was responsible for PANX1 Tyr198 phosphorylation. The SRC family of tyrosine kinases includes nine isoforms that are expressed in mammalian cells, with each kinase composed of a catalytic domain, an SH2 domain, an SH3 proline-rich domain, and a unique domain for which each isoform is named (73–75). This large kinase family can be divided into two subfamilies; the isoforms SRC, FYN, and Yes are ubiquitously expressed, whereas the isoforms BLK, FGR, HCK, LCK, and LYN are primarily expressed in hemopoietic cells (75). In VSMCs, SRC, FYN, and YES activity has been directly linked to focal adhesion dynamics (15, 76, 77) and VSMC growth/proliferation signaling (71, 78). We selected SRC kinase because of its causal link in regulating PANX1 channel function, but also because SRC activity is linked to constriction of VSMC in a number of contexts (13–16, 17, 31–34). To test the contribution of SRC kinase to phosphorylate PANX1 Tyr198, we utilized SRC kinase-specific siRNA in VSMCs, catalytically dominant or kinase-dead SRC isoforms in HEK cells, and a temperature-sensitive SRC kinase cell line (LA-25) to specifically modulate kinase activity during adrenergic stimulation (Fig. 3, A–C). Consistent with experimental outcomes using pharmacological inhibitors, we show that constitutive phosphorylation of PANX1 Tyr198 is directly mediated by SRC activity. Moreover, direct phosphorylation of PANX1 Tyr198 by SRC kinase was observed using recombinant PANX1 protein, constitutively active recombinant SRC kinase, and in vitro kinase assays (Fig. 4, A and B). In these assays, we found that Sf9-derived recombinant human PANX1 was basally phosphorylated and glycosylated, but not to the same degree as observed in mammalian cells (79–81). Thus, we were able to remove basally phosphorylated protein levels using phosphatase treatment and sequentially restore PANX1 phosphorylation in an SRC-specific manner. Furthermore, we found in human vascular smooth muscle cells that PANX1 and SRC kinase associate with each other, thus supporting findings from our kinase assay that these two proteins interact. Our results are consistent with published findings, in which NMDA receptor agonism leads to the formation of a metabotropic signaling complex in neurons between PANX1, SRC kinase, and the GluN1 subunit of the NMDA receptor, resulting in phosphorylation of tyrosine 308 on the PANX1 C terminus and channel activation (18). These findings demonstrated the presence of SRC kinase under basal and stimulated conditions, suggesting its presence at the plasma membrane with PANX1 to facilitate channel phosphorylation. In support of this, previous studies have demonstrated the myristoylation of SRC kinase and its targeting to the plasma membrane, where it supports a number of cell functions (29, 34, 58, 59, 62, 82). We conclude that PANX1 Tyr198 is a direct post-translational modification target of SRC kinase and may facilitate dual phosphorylation, kinase binding, and channel activity, although this remains to be directly tested.
Last, we investigated whether the phosphorylated PANX1 Tyr198 isoform localizes to the plasma membrane. Western blot analysis of PANX1 Tyr198 revealed the presence of a specific high-molecular weight (∼50 kDa) PANX1 band, indicative of plasma membrane association (83). In our membrane biotinylation experiments, PANX1 Tyr198 was enriched in membrane fractions, but not low-molecular weight species, in both unstimulated and adrenergic stimulated conditions (Fig. 5B). These results were confirmed in cells expressing PANX1-GFP, in which membrane-associated PANX1 co-localized with PANX1 Tyr198 immunostaining (Fig. 6). Because activation of α1-ARs and ATP release occur at the plasma membrane, we presumed that a regulatory role for PANX1 Tyr198 would spatially correlate with SFK effectors. A distinct pool of membrane-associated SRC kinase also localizes to the plasma membrane by both myristoylation at the N terminus (29) and molecular interactions with the caveolae scaffold protein, caveolin-1 (34). Recent evidence from our laboratory demonstrated an important interaction between PANX1 and caveolin-1 at the plasma membrane, which influenced channel function, adrenergic mediated ATP release, and mean arterial pressure (12). In this investigation, we report a pool of SRC kinase that associates at the plasma membrane of vascular smooth muscle cells and with PANX1 that is not dependent on adrenergic stimulation. These data then suggest that interactions between PANX1 and membrane-associated kinases (e.g. SRC), which aggregate at unique plasma membrane domains, support receptor-mediated channel activation to place VSMC effector molecules in close apposition with nerve terminals that release NE. Thus, phosphorylated PANX1 Tyr198 defines the distinct pool of PANX1 channels that are available to be activated by either SRC kinase itself or by an as of yet unidentified effector molecule.
Collectively, our data confirm previous observations in the vasculature, in which SFKs specifically regulate adrenergic mediated vasoconstriction and regulate cellular ATP release by PANX1. We demonstrate that phosphorylation of the PANX1 regulatory motif at Tyr198, by SRC kinase, is both constitutive and required for activation by adrenergic receptors in VSMCs. The phosphorylation of Tyr198 likely supports activation of PANX1 channels by other kinases, which can utilize Tyr198 phosphorylation to localize and bind to the intracellular loop of PANX1. These data link the longstanding observation of tyrosine kinase–based adrenergic constriction with adrenergic stimulated ATP release through the supportive activation of PANX1 channels. In the pathological context of human resistant hypertension, a PANX1-mediated constriction pathway may contribute to dysfunctional hemodynamics. The phosphorylation state of PANX1 Tyr198 is therefore an important marker for the distinct pool of membrane-associated PANX1 that is activated by receptor-dependent stimuli and might be useful as a quantitative biomarker for vascular pathologies associated with hypertensive phenotypes.
Experimental procedures
Animals
All mice were male, 10–15 weeks of age, on a C57Bl/6 genetic background, and were cared for under the provisions of the University of Virginia Animal Care and Use Committee and followed the National Institutes of Health guidelines for the care and use of laboratory animals. WT C57Bl/6 male mice were purchased from Taconic. All experiments were performed on a minimum of three mice.
Cell culture
Primary human coronary smooth muscle cells (hCoSMCs) were purchased from Lonza (catalog no. CC-2583). All cells were maintained under standard cell culture conditions (5% CO2 at 37 °C) in smooth muscle growth medium (Lonza, catalog no. CC-3181) supplemented with growth factors (Lonza, catalog no. CC-3182) and 10% fetal bovine serum (Lonza, catalog no. CC-4102D). Prior to use, primary cells were transfected with plasmids using Lipofectamine 3000 (Invitrogen, catalog no. L300015) or siRNA (RNAiMAX Invitrogen, catalog no. 13778075) where noted and incubated in low-serum medium conditions (0.2% fetal bovine serum) for 48 h to differentiate smooth muscle cells. For siRNA-mediated knockdown of SRC kinase, hCoSMCs were plated in 6-well plates and grown to 70–80% confluence. Nontargeting control siRNAs or siRNAs targeting the human SRC gene (Invitrogen Silencer Select, s13414) were transfected into cells using Lipofectamine RNAiMAX reagent (Invitrogen) according to the manufacturer's instructions, and knockdown efficiency was assessed via Western blotting following a 48-h incubation. Vasoconstrictive agonists and pharmacological SFK inhibitors were added after dedifferentiation in low-serum medium. hCoSMCs were used for experimentation up to passage 10. LA-25 cells (normal rat kidney epithelial cells containing temperature-sensitive v-SRC) (24, 84, 85) were cultured in Dulbecco's minimal essential medium (Life Technologies, Inc.) supplemented with 10% fetal bovine serum and antibiotics under standard conditions and were used to specifically manipulate SRC tyrosine kinase activity at the permissive (35 °C) or nonpermissive (40 °C) temperatures. LA-25 cells were co-transfected with laboratory-generated PANX1 (pEBB vector) and α1D-adrenergic receptor (OriGene, catalog no. sc119760) expression plasmids, using electroporation (Lonza Nucleofector Kit T, catalog no. VCA-1002) according to the manufacturer's instructions. WT SRC kinase expression plasmids were a gift from Joan Brugge and Peter Howley (Addgene plasmid 13663). All cells were used between passages 3 and 10. HEK293T and HeLa cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% FBS, 1% penicillin/streptomycin, and 1% l-glutamine. Cells were transfected using Lipofectamine 2000 (Invitrogen, catalog no. L11668-019) according to the manufacturer's instructions. Cells were maintained under standard conditions and used for experimentation under passage 20. Anti-GFP antibody (Abcam, catalog no. 13970; 1:500 dilution) was used to visualize PANX1.
Human vascular biopsies
Adipose tissue biopsy samples (∼1.5 g) were obtained from the gluteal region under aseptic conditions under the supervision of Dr. Eugene Barrett at the University of Virginia outpatient clinic (institutional review board approval 20408), which abides by the Declaration of Helsinki principles. Biopsies were obtained for research purposes from (nonobese) adult consented patients (40–60 years old) that were normotensive (mean arterial pressure <130/80 mm Hg systolic/diastolic) or hypertensive (mean arterial pressure >140/90 mm Hg) and not being treated with adrenergic blocking medications. Biopsies were fixed in 4% paraformaldehyde, thin-sectioned (5 μm), and immunostained as described previously (12). Antibodies directed toward phospho-PANX1 (Tyr198) (Millipore, ABN1681; 1:500 dilution), α-Smactin (Acta2; Sigma, catalog no. A2547; 1:500 dilution), and the C terminus of pannexin 1 (80, 86) were used for immunostaining.
Extracellular ATP measurements
For the measurement of extracellular ATP, intact thoracodorsal arteries (TDAs) were singly placed in a well of a 96-well plate and incubated in Krebs-HEPES physiologic solution alone or containing the SFK inhibitors PP2 (10 μm) and dasatinib (10 nm) for 1 h or 30 min, respectively. The ecto-nucleotidase inhibitor ARL 67156 (Tocris, catalog no. 1283) was added 30 min prior to treatment with contractile agonists as described previously (16). The medium surrounding the vessel was collected before stimulation and immediately after stimulation, placed into prechilled 1.5-ml Eppendorf tubes on ice, and centrifuged at 10,000 × g for 5 min. To chelate intracellular calcium, BAPTA-AM (10 μm) (Sigma, catalog no. A1076) was added to TDA incubation medium (Krebs-HEPES, 2 mm Ca2+) for 30 min prior to stimulation with vasoconstrictor compounds, including PE (20 μm), NE (10 μm), 5-HT (100 nm), and ET-1 (10 nm) (Sigma). The amount of ATP in the medium was quantified using ATP bioluminescence assay kit HSII (Roche Applied Science, catalog no. 11699709001) and a FluoStar Omega luminometer. Extracellular ATP measurements for each sample were tested in triplicate and calculated using an ATP standard curve for all experiments. Data are presented as percentage change of ATP from each sample's baseline (prestimulation) level. Data are expressed as mean ± S.E.
Pressure myography
Pressure myography was performed on thoracodorsal arteries of C57Bl/6 as described previously (15, 32, 33). Briefly, mice were sacrificed using CO2 asphyxia. TDAs were microdissected, cannulated on glass pipettes in a pressure arteriography chamber, and pressurized to 80 mm Hg. After a 30-min equilibration period in Krebs-HEPES with 2 mm Ca2+, vessels were preincubated with SFK inhibitors as described above for ATP measurements and treated with cumulative doses of PE (10−10 to 10−3 m), 5-HT (10−11 to 10−5 m), and ET-1 (10−14 to 10−7 m) applied to the bath. The luminal diameter was analyzed using digital calipers using the DMT Vessel Acquisition Software (Danish MyoTechnology). Endothelial and smooth muscle cell viability was assessed using KCl or a variety of vasodilators, which included endothelia-dependent and -independent vasodilatory agents, as described previously (15, 32, 33).
Cell membrane biotinylation
Differentiated hCoSMCs were washed in PBS and then incubated in Krebs buffer (118.4 mm NaCl, 4.7 mm KCl, 1.2 mm MgSO4, 4 mm NaHCO3, 1.2 mm KH2PO4, 10 mm Hepes, 6 mm glucose) containing 2 mmol/liter CaCl2 and 10 μmol/liter carbenoxalone (Sigma). Cells were then biotinylated using the EZ-Link NHS-Biotin kit (Thermo Fisher Scientific, catalog no. 20217) according to the manufacturer's instructions for 30 min at 4 °C. Subsequently, cells were washed in Krebs buffer, quenched with glycine (20 mm) for 15 min, washed again in Krebs buffer, and lysed in ice-cold radioimmune precipitation assay buffer. Membrane fractions were isolated by centrifugation at 13,000 rpm, and biotinylated proteins were immunoprecipitated for 16 h using prewashed agarose-avidin beads. Beads were collected by centrifugation at 5000 × g for 1 min, washed three times in lysis buffer, and reduced in 5× SDS reducing buffer for Western blot analysis.
Western blotting
Samples were subjected to SDS-gel electrophoresis using 4–12% BisTris gels (Invitrogen) and transferred to nitrocellulose membrane for immunoblotting. Membranes were blocked for 1 h at room temperature in a solution containing 3% BSA in TBS and then incubated overnight at 4 °C with primary antibodies against pannexin 1 (Cell Signaling Technology; D9M1C mAb 91137; 1:1000), caveolin-1 (BD Biosciences, catalog no. 610059; 1:1000), SRC (Cell Signaling Technology, L4A1 mAb 2110; 1:1000), phospho-SRC family (Tyr416) (Cell Signaling Technology, D49G4 mAb 6943; 1:1000), phospho-paxillin (Tyr118) (Cell Signaling Technology, catalog no. 2541; 1:1000), phospho-PANX1 (Tyr198) (Millipore, ABN1681; 1:1000), phospho-PANX1 (Tyr308) (Millipore, ABN1680; 1:1000), β-tubulin (Invitrogen, catalog no. MA5–16308-BTIN; 1:5000), α-actin (Sigma, mAb A2547; 1:2000), and GAPDH (Sigma, G8795; 1:10,000) or an antibody against the C terminus of pannexin 1 (87). Membranes were washed and incubated in LI-COR IR dye secondary antibodies (1:15,000) for 1 h and viewed/quantified using the LI-COR Odyssey imager with Image Studio software. Representative Western blotting images have been cropped for presentation. Data are presented as mean ± S.E.
Co-immunoprecipitation
Differentiated hCoSMCs and HEK293T cells were grown to 70–80% confluence in either SMC growth medium or Dulbecco's modified Eagle's medium, respectively. HEK293T cells were transfected with epitope-tagged human pannexin 1-pcDNA3.1-FLAG expression plasmids (WT or mutated to Y198F) using Lipofectamine 3000 (Invitrogen, catalog no. L3000-015) according to the manufacturer's instructions. Plasmid mutagenesis was performed using the Q5 site-directed mutagenesis kit (New England Biolabs, E0554S) with conventional PCR and pannexin 1–specific primers designed using NEBaseChanger analytical tools (New England Biolabs). Cells were equilibrated for 30 min in Krebs-HEPES supplemented with 2 mm Ca2+ and stimulated with 20 μm phenylephrine for 2 min. Cells were lysed in ice-cold co-immunoprecipitation buffer, 20 mm Tris-HCl, pH 8, 120 mm NaCl, 1% Nonidet P-40, 2 mm EDTA, 1 mm sodium orthovanadate, and 20 mm NaF supplemented with protease and phosphatase inhibitors (Sigma), and Dounce-homogenized (10 strokes) on ice. Protein extracts were incubated overnight at 4 °C in 20 μl of anti-FLAG–conjugated magnetic beads (Clontech) on a tube rotator. The beads were magnetically separated and washed three times in ice-cold co-immunoprecipitation buffer. Next, the beads were pulled down, eluted using a low-pH elution buffer, and allowed to incubate at room temperature for 10 min to remove bound proteins. The eluent and beads were separated, and the eluent was incubated in Laemmli buffer for analysis by SDS-gel electrophoresis. Immunoprecipitation from hCoSMCs was similarly performed as indicated above. Pannexin 1 was detected using antibody against the C terminus of pannexin 1 (80, 86) and phospho-PANX1 (Tyr198) (Millipore, ABN1681; 1:1000), whereas SRC kinase was detected using SRC (36D10) (Cell Signaling Technology, mAb 2109; 1:1000).
In vitro kinase assay
Recombinant mouse pannexin 1 protein was purified as described previously for Sf9 cells (26). To remove endogenous phosphorylation, 3.2 μg of recombinant mouse pannexin 1 protein was incubated with 2000 units of recombinant λ-phosphatase (Cell Signaling Technology, P0753S) in 10× phosphatase buffer supplemented with 1 mm Mn2+ for 16 h at 30 °C with shaking at 500 rpm. The enzyme mix was heat-inactivated at 65 °C for 1 h and supplemented with 1 mm sodium orthovanadate to inhibit phosphatase activity. To phosphorylate pannexin 1 protein, 1.25 μg of dephosphorylated pannexin 1 stock protein was added to a 0.6-ml Eppendorf tube containing kinase assay buffer (25 mm MOPS, pH 7.2, 20 mm MgCl2, 5 mm EGTA, 2 mm EDTA, 0.25 mm DTT) diluted 1:5 with 0.05 mg/ml BSA. The reaction was supplemented with 0.25 mm ATP, 0.5 mm MnCl2, and 0.3 μg/ml recombinant human (active) SRC-GST kinase (PRECISIO© Kinase; Sigma, S1076) in a 25-μl reaction volume. Samples were incubated for 1 h at 30 °C with shaking at 500 rpm. 5× SDS buffer was added to terminate the kinase reaction. Samples were boiled for 5 min at 95 °C and subjected to SDS-gel electrophoresis as described for Western blotting. Phosphorylation was confirmed using phosphotyrosine (P-Tyr-100) antibody (Cell Signaling Technology, mAb 9411; 1:1000), phospho-PANX1 (Tyr198) (Millipore, ABN1681; 1:1000), phospho-PANX1 (Tyr308) (Millipore, ABN1680; 1:1000), and pannexin 1 (Cell Signaling Technology, D9M1C mAb 91137; 1:1000).
Prediction of pannexin membrane topology and phosphorylation sites
A pannexin 1 topology map was generated using PROTTER version 1.0 (88). Analysis of pannexin 1 protein linear motif sequences was performed using the Eukaryotic Linear Motif computational biology resource (52).
Statistics
All data were analyzed using GraphPad Prism version 7.0 software. Briefly D'Agostino–Pearson tests were used to determine normality. Brown–Forsthe/Bartlett tests were used to determine equal variance for ANOVA, and F-test was used to determine equal variance for t test. Data that passed normality tests and equal variance tests were analyzed by t test for two groups or ANOVA for three or more groups. p value < 0.05 was considered significant.
Author contributions
L. J. D. and B. E. I. conceptualization; L. J. D., M. B., C. A. R., S. R. J., J. T. B., A. G. W., X. J., T. S. K., A. S. K., T. R., M. E. G., and L. A. S. data curation; L. J. D., M. B., C. A. R., S. R. J., J. T. B., A. G. W., X. J., A. S. K., T. R., M. E. G., and L. A. S. formal analysis; L. J. D. and B. E. I. funding acquisition; L. J. D., T. S. K., A. W. L., S. P., R. J. T., P. D. L., and M. Y. validation; L. J. D., M. B., C. A. R., S. R. J., J. T. B., A. G. W., X. J., A. S. K., T. R., A. K. B., and R. J. T. investigation; L. J. D., M. B., C. A. R., S. R. J., J. T. B., A. G. W., X. J., A. S. K., T. R., A. K. B., A. W. L., S. P., R. J. T., and P. D. L. methodology; L. J. D., and M. E. G. writing-original draft; T. S. K., M. E. G., A. W. L., L. A. S., S. P., R. J. T., P. D. L., M. Y., and B. E. I. writing-review and editing; A. K. B. and B. E. I. supervision; A. W. L., S. P., M. Y. Y., and B. E. I. project administration; M. Y. visualization.
Supplementary Material
Acknowledgment
We thank the University of Virginia Medical Histology Core.
This work was supported by NIGMS, National Institutes of Health, Grant F31 HL137270 (to L. J. D.), National Institutes of Health Grant T32 GM00813 (to L. J. D.), and NHLBI, National Institutes of Health, Grant P01 HL120840 (to B. E. I.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S3.
- VSMC
- vascular smooth muscle cell
- NMDA
- N-methyl-d-aspartate
- NE
- norepinephrine
- SFK
- SRC family kinase
- BAPTA-AM
- 1,2-bis(o-aminophenoxy)ethane-N,N,N,N-tetraacetic acid
- PE
- phenylephrine
- ET-1
- endothelin-1
- 5-HT
- serotonin
- hCoSMC
- human coronary smooth muscle cell
- cSRC
- constitutively active SRC
- SRC-KD
- kinase-dead SRC
- IP
- immunoprecipitation
- SH2 and SH3
- Src homology 2 and 3, respectively
- AT1R
- angiotensin II type 1 receptor
- TDA
- thoracodorsal artery
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- ANOVA
- analysis of variance
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- GPCR
- G protein–coupled receptor
- HEK
- human embryonic kidney
- α1-AR
- α1-adrenergic receptor.
References
- 1. Tanoue A., Nasa Y., Koshimizu T., Shinoura H., Oshikawa S., Kawai T., Sunada S., Takeo S., and Tsujimoto G. (2002) The α1D-adrenergic receptor directly regulates arterial blood pressure via vasoconstriction. J. Clin. Invest. 109, 765–775 10.1172/JCI200214001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fisher S. A. (2010) Vascular smooth muscle phenotypic diversity and function. Physiol. Genomics 42A, 169–187 10.1152/physiolgenomics.00111.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jackson W. F., Boerman E. M., Lange E. J., Lundback S. S., and Cohen K. D. (2008) Smooth muscle α1D-adrenoceptors mediate phenylephrine-induced vasoconstriction and increases in endothelial cell Ca2+ in hamster cremaster arterioles. Br. J. Pharmacol. 155, 514–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Burnstock G. (2010) Control of vascular tone by purines and pyrimidines. Br. J. Pharmacol. 161, 527–529 10.1111/j.1476-5381.2010.00937.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Billaud M., Chiu Y.-H., Lohman A. W., Parpaite T., Butcher J. T., Mutchler S. M., DeLalio L. J., Artamonov M. V., Sandilos J. K., Best A. K., Somlyo A. V., Thompson R. J., Le T. H., Ravichandran K. S., Bayliss D. A., and Isakson B. E. (2015) A molecular signature in the Pannexin 1 intracellular loop confers channel activation by the α1-adrenergic receptor in smooth muscle cells. Sci. Signal. 8, ra17 10.1126/scisignal.2005824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lohman A. W., Billaud M., and Isakson B. E. (2012) Mechanisms of ATP release and signalling in the blood vessel wall. Cardiovasc. Res. 95, 269–280 10.1093/cvr/cvs187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Billaud M., Lohman A. W., Straub A. C., Looft-Wilson R., Johnstone S. R., Araj C. A., Best A. K., Chekeni F. B., Ravichandran K. S., Penuela S., Laird D. W., and Isakson B. E. (2011) Pannexin1 regulates 1-adrenergic receptor-mediated vasoconstriction. Circ. Res. 109, 80–85 10.1161/CIRCRESAHA.110.237594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bond S. R., and Naus C. C. (2014) The pannexins: past and present. Front. Physiol. 5, 58 10.3389/fphys.2014.00058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Billaud M., Lohman A. W., Straub A. C., Parpaite T., Johnstone S. R., and Isakson B. E. (2012) Characterization of the thoracodorsal artery: morphology and reactivity. Microcirculation 19, 360–372 10.1111/j.1549-8719.2012.00172.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nyberg M., Piil P., Kiehn O. T., Maagaard C., Jørgensen T. S., Egelund J., Isakson B. E., Nielsen M. S., Gliemann L., and Hellsten Y. (2018) Probenecid inhibits α-adrenergic receptor–mediated vasoconstriction in the human leg vasculature. Hypertension 71, 151–159 10.1161/HYPERTENSIONAHA.117.10251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Angus J. A., and Wright C. E. (2016) Novel α 1-adrenoceptor antagonism by the fluroquinolone antibiotic trovafloxacin. Eur. J. Pharmacol. 791, 179–184 10.1016/j.ejphar.2016.08.035 [DOI] [PubMed] [Google Scholar]
- 12. DeLalio L. J., Keller A. S., Chen J., Boyce A. K. J., Artamonov M. V., Askew-Page H. R., Keller T. C. S. 4th, Johnstone S. R., Weaver R. B., Good M. E. J. A., Murphy S. A., Best A. K., Mintz E. L., Penuela S., Greenwood I. A., et al. (2018) Interaction between pannexin 1 and caveolin-1 in smooth muscle can regulate blood pressure. Arterioscler. Thromb. Vasc. Biol. 38, 2065–2078 10.1161/ATVBAHA.118.311290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Di Salvo J., Steusloff A., Semenchuk L., Satoh S., Kolquist K., and Pfitzer G. (1993) Tyrosine kinase inhibitors suppress agonist-induced contraction in smooth muscle. Biochem. Biophys. Res. Commun. 190, 968–974 10.1006/bbrc.1993.1144 [DOI] [PubMed] [Google Scholar]
- 14. Di Salvo J., Semenchuk L. A., and Lauer J. (1993) Vanadate-induced contraction of smooth muscle and enhanced protein tyrosine phosphorylation. Arch. Biochem. Biophys. 304, 386–391 10.1006/abbi.1993.1366 [DOI] [PubMed] [Google Scholar]
- 15. Min J., Reznichenko M., Poythress R. H., Gallant C. M., Vetterkind S., Li Y., and Morgan K. G. (2012) Src modulates contractile vascular smooth muscle function via regulation of focal adhesions. J. Cell Physiol. 227, 3585–3592 10.1002/jcp.24062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hughes A. D., and Wijetunge S. (1998) Role of tyrosine phosphorylation in excitation-contraction coupling in vascular smooth muscle. Acta Physiol. Scand. 164, 457–469 10.1046/j.1365-201X.1998.00446.x [DOI] [PubMed] [Google Scholar]
- 17. Ward D. T., Alder A. C., Ohanian J., and Ohanian V. (2002) Noradrenaline-induced paxillin phosphorylation, ERK activation and MEK-regulated contraction in intact rat mesenteric arteries. J. Vasc. Res. 39, 1–11 10.1159/000048988 [DOI] [PubMed] [Google Scholar]
- 18. Weilinger N. L., Lohman A. W., Rakai B. D., Ma E. M. M., Bialecki J., Maslieieva V., Rilea T., Bandet M. V., Ikuta N. T., Scott L., Colicos M. A., Teskey G. C., Winship I. R., and Thompson R. J. (2016) Metabotropic NMDA receptor signaling couples Src family kinases to pannexin-1 during excitotoxicity. Nat. Neurosci. 19, 432–442 10.1038/nn.4236 [DOI] [PubMed] [Google Scholar]
- 19. Lohman A. W., Leskov I. L., Butcher J. T., Johnstone S. R., Stokes T. A., Begandt D., DeLalio L. J., Best A. K., Penuela S., Leitinger N., Ravichandran K. S., Stokes K. Y., and Isakson B. E. (2015) Pannexin 1 channels regulate leukocyte emigration through the venous endothelium during acute inflammation. Nat. Commun. 6, 7965 10.1038/ncomms8965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thompson R. J., Zhou N., and MacVicar B. A. (2006) Ischemia opens neuronal gap junction hemichannels. Science 312, 924–927 10.1126/science.1126241 [DOI] [PubMed] [Google Scholar]
- 21. Weilinger N. L., Tang P. L., and Thompson R. J. (2012) Anoxia-induced NMDA receptor activation opens pannexin channels via Src family kinases. J. Neurosci. 32, 12579–12588 10.1523/JNEUROSCI.1267-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ma W., Hui H., Pelegrin P., and Surprenant A. (2009) Pharmacological characterization of pannexin-1 currents expressed in mammalian cells. J. Pharmacol. Exp. Ther. 328, 409–418 10.1124/jpet.108.146365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Iglesias R., Locovei S., Roque A., Alberto A. P., Dahl G., Spray D. C., and Scemes E. (2008) P2X7 receptor-Pannexin1 complex: pharmacology and signaling. Am. J. Physiol. Cell Physiol. 295, C752–C760 10.1152/ajpcell.00228.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Solan J. L., and Lampe P. D. (2008) Connexin 43 in LA-25 cells with active v-src is phosphorylated on Y247, Y265, S262, S279/282, and S368 via multiple signaling pathways. Cell Commun. Adhes. 15, 75–84 10.1080/15419060802014016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Atkinson M. M., Menko A. S., Johnson R. G., Sheppard J. R., and Sheridan J. D. (1981) Rapid and reversible reduction of junctional permeability in cells infected with a temperature-sensitive mutant of avian sarcoma virus. J. Cell Biol. 91, 573–578 10.1083/jcb.91.2.573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chiu Y.-H., Jin X., Medina C. B., Leonhardt S. A., Kiessling V., Bennett B. C., Shu S., Tamm L. K., Yeager M., Ravichandran K. S., and Bayliss D. A. (2017) A quantized mechanism for activation of pannexin channels. Nat. Commun. 8, 14324 10.1038/ncomms14324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bargiotas P., Krenz A., Hormuzdi S. G., Ridder D. A., Herb A., Barakat W., Penuela S., von Engelhardt J., Monyer H., and Schwaninger M. (2011) Pannexins in ischemia-induced neurodegeneration. Proc. Natl. Acad. Sci. U.S.A. 108, 20772–20777 10.1073/pnas.1018262108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Penuela S., Bhalla R., Nag K., and Laird D. W. (2009) Glycosylation regulates pannexin intermixing and cellular localization. Mol. Biol. Cell 20, 4313–4323 10.1091/mbc.e09-01-0067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Patwardhan P., and Resh M. D. (2010) Myristoylation and membrane binding regulate c-Src stability and kinase activity. Mol. Cell. Biol. 30, 4094–4107 10.1128/MCB.00246-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Poornima V., Vallabhaneni S., Mukhopadhyay M., and Bera A. K. (2015) Nitric oxide inhibits the pannexin 1 channel through a cGMP–PKG dependent pathway. Nitric Oxide 47, 77–84 10.1016/j.niox.2015.04.005 [DOI] [PubMed] [Google Scholar]
- 31. Di Salvo J., Pfitzer G., and Semenchuk L. A. (1994) Protein tyrosine phosphorylation, cellular Ca2+, and Ca2+ sensitivity for contraction of smooth muscle. Can. J. Physiol. Pharmacol. 72, 1434–1439 10.1139/y94-207 [DOI] [PubMed] [Google Scholar]
- 32. Abebe W., and Agrawal D. K. (1995) Role of tyrosine kinases in norepinephrine-induced contraction of vascular smooth muscle. J. Cardiovasc. Pharmacol. 26, 153–159 10.1097/00005344-199507000-00024 [DOI] [PubMed] [Google Scholar]
- 33. Jin N., Siddiqui R. A., English D., and Rhoades R. A. (1996) Communication between tyrosine kinase pathway and myosin light chain kinase pathway in smooth muscle. Am. J. Physiol. 271, H1348–H1355 10.1152/ajpheart.1996.271.4.H1348 [DOI] [PubMed] [Google Scholar]
- 34. Li S., Couet J., and Lisanti M. P. (1996) Src tyrosine kinases, Gα subunits, and H-Ras share a common membrane-anchored scaffolding protein, caveolin: caveolin binding negatively regulates the auto-activation of Src tyrosine kinases. J. Biol. Chem. 271, 29182–29190 10.1074/jbc.271.46.29182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chekeni F. B., Elliott M. R., Sandilos J. K., Walk S. F., Kinchen J. M., Lazarowski E. R., Armstrong A. J., Penuela S., Laird D. W., Salvesen G. S., Isakson B. E., Bayliss D. A., and Ravichandran K. S. (2010) Pannexin 1 channels mediate “find-me” signal release and membrane permeability during apoptosis. Nature 467, 863–867 10.1038/nature09413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sandilos J. K., Chiu Y.-H., Chekeni F. B., Armstrong A. J., Walk S. F., Ravichandran K. S., and Bayliss D. A. (2012) How the tail of pannexin 1 inhibits the ATP release channel: pannexin 1, an ATP release channel, is activated by caspase cleavage of its pore-associated C-terminal autoinhibitory region. J. Biol. Chem. 287, 11303–11311 10.1074/jbc.M111.323378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Seminario-Vidal L., Kreda S., Jones L., O'Neal W., Trejo J., Boucher R. C., and Lazarowski E. R. (2009) Thrombin promotes release of ATP from lung epithelial cells through coordinated activation of rho- and Ca2+-dependent signaling pathways. J. Biol. Chem. 284, 20638–20648 10.1074/jbc.M109.004762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gödecke S., Roderigo C., Rose C. R., Rauch B. H., Gödecke A., and Schrader J. (2012) Thrombin-induced ATP release from human umbilical vein endothelial cells. Am. J. Physiol. Cell Physiol. 302, C915–C923 10.1152/ajpcell.00283.2010 [DOI] [PubMed] [Google Scholar]
- 39. Zhang M., Piskuric N. A., Vollmer C., and Nurse C. A. (2012) P2Y2 receptor activation opens pannexin-1 channels in rat carotid body type II cells: potential role in amplifying the neurotransmitter ATP. J. Physiol. 590, 4335–4350 10.1113/jphysiol.2012.236265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gulbransen B. D., Bashashati M., Hirota S. A., Gui X., Roberts J. A., MacDonald J. A., Muruve D. A., McKay D. M., Beck P. L., Mawe G. M., Thompson R. J., and Sharkey K. A. (2012) Activation of neuronal P2X7 receptor–pannexin-1 mediates death of enteric neurons during colitis. Nat. Med. 18, 600–604 10.1038/nm.2679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pinheiro A. R., Paramos-de-Carvalho D., Certal M., Costa C., Magalhães Cardoso M. T. M., Ferreirinha F., Costa M. A., and Correia-de-Sá P. (2013) Bradykinin-induced Ca2+ signaling in human subcutaneous fibroblasts involves ATP release via hemichannels leading to P2Y12 receptors activation. Cell Commun. Signal. 11, 70 10.1186/1478-811X-11-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pinheiro A. R., Paramos-de-Carvalho D., Certal M., Costa M. A., Costa C., Magalhães-Cardoso M. T., Ferreirinha F., Sévigny J., and Correia-de-Sá P. (2013) Histamine induces ATP release from human subcutaneous fibroblasts, via pannexin-1 hemichannels, leading to Ca2+ mobilization and cell proliferation. J. Biol. Chem. 288, 27571–27583 10.1074/jbc.M113.460865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lohman A. W., Billaud M., Straub A. C., Johnstone S. R., Best A. K., Lee M., Barr K., Penuela S., Laird D. W., and Isakson B. E. (2012) Expression of pannexin isoforms in the systemic murine arterial network. J. Vasc. Res. 49, 405–416 10.1159/000338758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Somlyo A. P., and Somlyo A. V. (1998) From pharmacomechanical coupling to G-proteins and myosin phosphatase. Acta Physiol. Scand. 164, 437–448 10.1046/j.1365-201X.1998.00454.x [DOI] [PubMed] [Google Scholar]
- 45. Somlyo A. P., and Somlyo A. V. (2003) Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol. Rev. 83, 1325–1358 10.1152/physrev.00023.2003 [DOI] [PubMed] [Google Scholar]
- 46. Locovei S., Wang J., and Dahl G. (2006) Activation of pannexin 1 channels by ATP through P2Y receptors and by cytoplasmic calcium. FEBS Lett. 580, 239–244 10.1016/j.febslet.2005.12.004 [DOI] [PubMed] [Google Scholar]
- 47. Murali S., Zhang M., and Nurse C. A. (2017) Evidence that 5-HT stimulates intracellular Ca2+ signalling and activates pannexin-1 currents in type II cells of the rat carotid body. J. Physiol. 595, 4261–4277 10.1113/JP273473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Boyce A. K. J., and Swayne L. A. (2017) P2X7 receptor cross-talk regulates ATP-induced pannexin 1 internalization. Biochem. J. 474, 2133–2144 10.1042/BCJ20170257 [DOI] [PubMed] [Google Scholar]
- 49. Shah B. H., and Catt K. J. (2002) Calcium-independent activation of extracellularly regulated kinases 1 and 2 by angiotensin II in hepatic C9 cells: roles of protein kinase Cδ, Src/proline-rich tyrosine kinase 2, and epidermal growth factor receptor trans-activation. Mol. Pharmacol. 61, 343–351 10.1124/mol.61.2.343 [DOI] [PubMed] [Google Scholar]
- 50. Vazquez G., Wedel B. J., Kawasaki B. T., Bird G. S. J., and Putney J. W. (2004) Obligatory role of Src kinase in the signaling mechanism for TRPC3 cation channels. J. Biol. Chem. 279, 40521–40528 10.1074/jbc.M405280200 [DOI] [PubMed] [Google Scholar]
- 51. Riquelme M. A., Cea L. A., Vega J. L., Boric M. P., Monyer H., Bennett M. V., Frank M., Willecke K., and Sáez J. C. (2013) The ATP required for potentiation of skeletal muscle contraction is released via pannexin hemichannels. Neuropharmacology 75, 594–603 10.1016/j.neuropharm.2013.03.022 [DOI] [PubMed] [Google Scholar]
- 52. Dinkel H., Van Roey K., Michael S., Kumar M., Uyar B., Altenberg B., Milchevskaya V., Schneider M., Kühn H., Behrendt A., Dahl S. L., Damerell V., Diebel S., Kalman S., Klein S., et al. (2016) ELM 2016—data update and new functionality of the eukaryotic linear motif resource. Nucleic Acids Res. 44, D294–D300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yu H., Rosen M. K., Shin T. B., Seidel-Dugan C., Brugge J. S., and Schreiber S. L. (1992) Solution structure of the SH3 domain of Src and identification of its ligand-binding site. Science 258, 1665–1668 10.1126/science.1280858 [DOI] [PubMed] [Google Scholar]
- 54. Nakamoto T., Sakai R., Ozawa K., Yazaki Y., and Hirai H. (1996) Direct binding of C-terminal region of p130 to SH2 and SH3 domains of Src kinase. J. Biol. Chem. 271, 8959–8965 10.1074/jbc.271.15.8959 [DOI] [PubMed] [Google Scholar]
- 55. Ren R., Mayer B. J., Cicchetti P., and Baltimore D. (1993) Identification of a ten-amino acid proline-rich SH3 binding site. Science 259, 1157–1161 10.1126/science.8438166 [DOI] [PubMed] [Google Scholar]
- 56. Shvartsman D. E., Donaldson J. C., Diaz B., Gutman O., Martin G. S., and Henis Y. I. (2007) Src kinase activity and SH2 domain regulate the dynamics of Src association with lipid and protein targets. J. Cell Biol. 178, 675–686 10.1083/jcb.200701133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gottlieb-Abraham E., Shvartsman D. E., Donaldson J. C., Ehrlich M., Gutman O., Martin G. S., and Henis Y. I. (2013) Src-mediated caveolin-1 phosphorylation affects the targeting of active Src to specific membrane sites. Mol. Biol. Cell 24, 3881–3895 10.1091/mbc.e13-03-0163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Xing Z., Chen H. C., Nowlen J. K., Taylor S. J., Shalloway D., and Guan J. L. (1994) Direct interaction of v-Src with the focal adhesion kinase mediated by the Src SH2 domain. Mol. Biol. Cell 5, 413–421 10.1091/mbc.5.4.413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Thomas S. M., and Brugge J. S. (1997) Cellular functions regulated by Src family kinases. Annu. Rev. Cell Dev. Biol. 13, 513–609 10.1146/annurev.cellbio.13.1.513 [DOI] [PubMed] [Google Scholar]
- 60. Sumi Y., Woehrle T., Chen Y., Yao Y., Li A., and Junger W. G. (2010) Adrenergic receptor activation involves ATP release and feedback through purinergic receptors. Am. J. Physiol. Cell Physiol. 299, C1118–C1126 10.1152/ajpcell.00122.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Luttrell D. K., and Luttrell L. M. (2004) Not so strange bedfellows: G-protein-coupled receptors and Src family kinases. Oncogene 23, 7969–7978 10.1038/sj.onc.1208162 [DOI] [PubMed] [Google Scholar]
- 62. McGarrigle D., and Huang X.-Y. (2007) GPCRs signaling directly through Src-family kinases. Sci. STKE 2007, pe35 10.1126/stke.3922007pe35 [DOI] [PubMed] [Google Scholar]
- 63. Ma Y.-C., Huang J., Ali S., Lowry W., and Huang X.-Y. (2000) Src tyrosine kinase is a novel direct effector of G proteins. Cell 102, 635–646 10.1016/S0092-8674(00)00086-6 [DOI] [PubMed] [Google Scholar]
- 64. Han C., Bowen W. C., Michalopoulos G. K., and Wu T. (2008) α-1 adrenergic receptor transactivates signal transducer and activator of transcription-3 (Stat3) through activation of Src and epidermal growth factor receptor (EGFR) in hepatocytes. J. Cell Physiol. 216, 486–497 10.1002/jcp.21420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Grassi G. (2009) Assessment of sympathetic cardiovascular drive in human hypertension: achievements and perspectives. Hypertension 54, 690–697 10.1161/HYPERTENSIONAHA.108.119883 [DOI] [PubMed] [Google Scholar]
- 66. Grassi G., Mark A., and Esler M. (2015) The sympathetic nervous system alterations in human hypertension. Circ. Res. 116, 976–990 10.1161/CIRCRESAHA.116.303604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yun H.-M., Kim S., Kim H.-J., Kostenis E., Kim J. I., Seong J. Y., Baik J.-H., and Rhim H. (2007) The novel cellular mechanism of human 5-HT6 receptor through an interaction with Fyn. J. Biol. Chem. 282, 5496–5505 10.1074/jbc.M606215200 [DOI] [PubMed] [Google Scholar]
- 68. Murali S., Zhang M., and Nurse C. A. (2014) Angiotensin II mobilizes intracellular calcium and activates pannexin-1 channels in rat carotid body type II cells via AT1 receptors. J. Physiol. 592, 4747–4762 10.1113/jphysiol.2014.279299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Gómez G. I., Fernández P., Velarde V., and Sáez J. C. (2018) Angiotensin II-induced mesangial cell damage is preceded by cell membrane permeabilization due to upregulation of non-selective channels. Int. J. Mol. Sci. 19, E957 10.3390/ijms19040957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Weng Y.-I., and Shukla S. D. (2002) Angiotensin II activation of focal adhesion kinase and pp60c-Src in relation to mitogen-activated protein kinases in hepatocytes. Biochim. Biophys. Acta 1589, 285–297 10.1016/S0167-4889(02)00189-1 [DOI] [PubMed] [Google Scholar]
- 71. Bokemeyer D., Schmitz U., and Kramer H. J. (2000) Angiotensin II-induced growth of vascular smooth muscle cells requires an Src-dependent activation of the epidermal growth factor receptor1. Kidney Int. 58, 549–558 10.1046/j.1523-1755.2000.t01-1-00201.x [DOI] [PubMed] [Google Scholar]
- 72. Li L., Zhou Y., Wang C., Zhao Y.-L., Zhang Z.-G., Fan D., Cui X.-B., and Wu L.-L. (2010) Src tyrosine kinase regulates angiotensin II-induced protein kinase Cζ activation and proliferation in vascular smooth muscle cells. Peptides 31, 1159–1164 10.1016/j.peptides.2010.03.014 [DOI] [PubMed] [Google Scholar]
- 73. Parsons S. J., and Parsons J. T. (2004) Src family kinases, key regulators of signal transduction. Oncogene 23, 7906–7909 10.1038/sj.onc.1208160 [DOI] [PubMed] [Google Scholar]
- 74. Roskoski R., Jr. (2004) Src protein–tyrosine kinase structure and regulation. Biochem. Biophys. Res. Commun. 324, 1155–1164 10.1016/j.bbrc.2004.09.171 [DOI] [PubMed] [Google Scholar]
- 75. Bolen J. B., and Brugge J. S. (1997) Leukocyte protein tyrosine kinases: potential targets for drug discovery. Annu. Rev. Immunol. 15, 371–404 10.1146/annurev.immunol.15.1.371 [DOI] [PubMed] [Google Scholar]
- 76. Mitra S. K., and Schlaepfer D. D. (2006) Integrin-regulated FAK–Src signaling in normal and cancer cells. Curr. Opin. Cell Biol. 18, 516–523 10.1016/j.ceb.2006.08.011 [DOI] [PubMed] [Google Scholar]
- 77. Bolós V., Gasent J. M., López-Tarruella S., and Grande E. (2010) The dual kinase complex FAK-Src as a promising therapeutic target in cancer. Onco Targets Ther. 3, 83–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Parsons J. T., and Parsons S. J. (1997) Src family protein tyrosine kinases: cooperating with growth factor and adhesion signaling pathways. Curr. Opin. Cell Biol. 9, 187–192 10.1016/S0955-0674(97)80062-2 [DOI] [PubMed] [Google Scholar]
- 79. Penuela S., Simek J., and Thompson R. J. (2014) Regulation of pannexin channels by post-translational modifications. FEBS Lett. 588, 1411–1415 10.1016/j.febslet.2014.01.028 [DOI] [PubMed] [Google Scholar]
- 80. Penuela S., Bhalla R., Gong X.-Q., Cowan K. N., Celetti S. J., Cowan B. J., Bai D., Shao Q., and Laird D. W. (2007) Pannexin 1 and pannexin 3 are glycoproteins that exhibit many distinct characteristics from the connexin family of gap junction proteins. J. Cell Sci. 120, 3772–3783 10.1242/jcs.009514 [DOI] [PubMed] [Google Scholar]
- 81. Boassa D., Ambrosi C., Qiu F., Dahl G., Gaietta G., and Sosinsky G. (2007) Pannexin1 channels contain a glycosylation site that targets the hexamer to the plasma membrane. J. Biol. Chem. 282, 31733–31743 10.1074/jbc.M702422200 [DOI] [PubMed] [Google Scholar]
- 82. Calalb M. B., Polte T. R., and Hanks S. K. (1995) Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Mol. Cell Biol. 15, 954–963 10.1128/MCB.15.2.954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Penuela S., Lohman A. W., Lai W., Gyenis L., Litchfield D. W., Isakson B. E., and Laird D. W. (2014) Diverse post-translational modifications of the pannexin family of channel-forming proteins. Channels 8, 124–130 10.4161/chan.27422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Di Salvo J., Gifford D., and Kokkinakis A. (1989) ATP- and polyphosphate-mediated stimulation of pp60c-src kinase activity in extracts from vascular smooth muscle. J. Biol. Chem. 264, 10773–10778 [PubMed] [Google Scholar]
- 85. Poste G., and Flood M. K. (1979) Cells transformed by temperature-sensitive mutants of avian sarcoma virus cause tumors in vivo at permissive and nonpermissive temperatures. Cell 17, 789–800 10.1016/0092-8674(79)90319-2 [DOI] [PubMed] [Google Scholar]
- 86. Shao Q., Lindstrom K., Shi R., Kelly J., Schroeder A., Juusola J., Levine K. L., Esseltine J. L., Penuela S., and Jackson M. F. (2016) A germline variant in PANX1 has reduced channel function and is associated with multisystem dysfunction. J. Biol. Chem. 291, 12432–12443 10.1074/jbc.M116.717934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Penuela S., Harland L., Simek J., and Laird D. W. (2014) Pannexin channels and their links to human disease. Biochem. J. 461, 371–381 10.1042/BJ20140447 [DOI] [PubMed] [Google Scholar]
- 88. Omasits U., Ahrens C. H., Müller S., and Wollscheid B. (2014) Protter: interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics 30, 884–886 10.1093/bioinformatics/btt607 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.