

Abstract
Mitochondria are major sites of energy metabolism that influence numerous cellular events, including immunity and cancer development. Previously, we reported that the mitochondrion-specific antioxidant enzyme, manganese-containing superoxide dismutase (MnSOD), has dual roles in early- and late-carcinogenesis stages. However, how defective MnSOD impacts the chain of events that lead to cell transformation in pathologically normal epidermal cells that have been exposed to carcinogens is unknown. Here, we show that UVB radiation causes nitration and inactivation of MnSOD leading to mitochondrial injury and mitophagy. In keratinocytes, exposure to UVB radiation decreased mitochondrial oxidative phosphorylation, increased glycolysis and the expression of autophagy-related genes, and enhanced AKT Ser/Thr kinase (AKT) phosphorylation and cell growth. Interestingly, UVB initiated a prosurvival mitophagy response by mitochondria-mediated reactive oxygen species (ROS) signaling via the mammalian target of the mTOR complex 2 (mTORC2) pathway. Knockdown of rictor but not raptor abrogated UVB-induced mitophagy responses. Furthermore, fractionation and proximity-ligation assays reveal that ROS-mediated mTOC2 activation in mitochondria is necessary for UVB-induced mitophagy. Importantly, pretreatment with the MnSOD mimic MnTnBuOE-2-PyP5+ (MnP) attenuates mTORC2 activation and suppresses UVB-induced mitophagy. UVB radiation exposure also increased cell growth as assessed by soft-agar colony survival and cell growth assays, and pretreatment with MnP or the known autophagy inhibitor 3-methyladenine abrogated UVB-induced cell growth. These results indicate that MnSOD is a major redox regulator that maintains mitochondrial health and show that UVB-mediated MnSOD inactivation promotes mitophagy and thereby prevents accumulation of damaged mitochondria.
Keywords: autophagy, mitophagy, metabolism, cancer, reactive oxygen species (ROS), oxidative stress, MnSOD, mTOR, mTOR complex 2 (mTOR C2), rictor
Introduction
Manganese-containing superoxide dismutase (MnSOD)2 is a nuclear-encoded mitochondrial antioxidant enzyme that maintains normal mitochondrial functions by removing superoxide anion radicals generated in mitochondria. Under physiological conditions, superoxide anion (O2˙̄) is rapidly dismutated by MnSOD to hydrogen peroxide and molecular oxygen (1). However, under stress conditions, such as from UVB, radiation can form peroxynitrite (OONO−) by generating O2˙̄ as well as nitric oxide (NO) and can nitrate tyrosine residues (2, 3). The effect of tyrosine nitration on protein structure and function depends on the location of tyrosine residues in the proteins, for example its location in a loop or a hydrophobic milieu, such as the active site of an enzyme (4). Thus, MnSOD is highly susceptible to OONO− attack and inactivation due to the presence of tyrosine residues in its active site. The specific tyrosine (Tyr-34) residues of MnSOD protein nitrated by peroxynitrite are detected by mass spectroscopic analysis (5).
Previously, we reported changes in expression of MnSOD during cancer initiation and progression in a multistage carcinogenesis model (6). We showed that overexpression of MnSOD reduces radiation-induced neoplastic transformation (7), suppresses tumor incidence and tumor multiplicity in multistage skin carcinogenesis (8, 9), and protects mitochondrial proteins from UVB-induced damage in a mouse skin keratinocyte model (10). However, how inactivation of MnSOD triggers the chain of events in pathologically normal epidermal cells that leads to cell transformation is unknown.
Several lines of evidence demonstrate that oxidative stress provokes organelle defects that activate autophagic responses, leading to either cell survival or cell death (11). Mitophagy occurs when autophagy facilitates the turnover of damaged mitochondria. Thus, mitophagic signals initiated by damaged mitochondria may lead to survival or apoptosis. If the damage is below the threshold level of lethality, defective mitochondria are sequestered by autophagosome. This process can support cell growth and proliferation by removing damaged mitochondria that can contribute to ROS generation and apoptosis. Moreover, mitochondrial defects can compromise oxidative phosphorylation, which leads cells to acquire energy from glycolysis (12). Because MnSOD plays a role in maintaining mitochondrial redox status and is inactivated by UVB exposure, we investigated the impact of UVB radiation on the signaling events associated with mitophagy. Here, we provide the evidence that exposure to UVB radiation causes changes in oxygen consumption rate (OCR) and extracellular acidification rate (ECAR), implying alterations in mitochondrial functions and consequent metabolic activities.
Results
Nitration and inactivation of MnSOD in vitro and in vivo
To directly test the effect of peroxynitrite on MnSOD activity, we exposed purified recombinant MnSOD proteins to various concentrations of peroxynitrite and measured the MnSOD activity. The results (Fig. 1a) show that MnSOD enzyme activity decreases rapidly with increased concentration of peroxynitrite at the low concentration range and continues to decline at the maximum concentration used. Next, we confirmed the formation of peroxynitrite in JB6 cells following UVB treatment (Fig. S1a). Our results are consistent with the published literature (13, 14) and show that UVB increases peroxynitrite formation, which is attenuated by MnSOD mimetics (MnP, 10 μm). To verify that UVB treatment causes MnSOD nitration and inactivation at the cellular level, we performed immunoprecipitation of nitrated MnSOD from primary human keratinocytes (HEKn) with and without exposure to UVB radiation using an antibody to 3-nitrotyrosine to pull down nitrated proteins, followed by Western blotting with an antibody specific to MnSOD. Complementarily, we immunoprecipitated the MnSOD protein using MnSOD antibody after the UVB radiation treatment, and the nitrated MnSOD was detected by Western blotting using 3-nitrotyrosine antibody. The data show that MnSOD is nitrated after UVB exposure (Fig. 1b). We also examined the effect of UVB exposure on MnSOD enzyme activity in HEKn and in established mouse epidermal (JB6) cells. The results show that MnSOD activity decreases after UVB treatment both in primary human keratinocytes as well as in normal mouse epithelial cells (Fig. 1c). Consistent with the in vitro data, MnSOD activity decreases significantly in mouse skin exposed to UVB even 24 h after treatment (Fig. 1d), but MnSOD protein levels do not decrease. They remain unchanged 1 h after UVB treatment and increase 24 h after UVB treatment (Fig. 1e). These in vitro and in vivo data demonstrate that MnSOD is nitrated, and the MnSOD enzyme is inactivated following exposure to UVB.
Figure 1.

MnSOD nitration, activity, and mitochondrial function. a, purified MnSOD protein was incubated with peroxynitrite at various concentrations, and MnSOD activity was determined by activity gel analysis. b, panel i, nitrated MnSOD detected in UVB-treated primary HEKn cells by immunoprecipitation (IP) with 3-nitrotyrosine antibody followed by Western blotting with MnSOD antibody. Panel ii, reverse immunoprecipitation was performed using MnSOD antibody, and the nitrated MnSOD was detected by 3-nitrotyrosine antibody. c, MnSOD activity in HEKn cells in panel i and JB6 cells in panel ii using activity gel and spectrophotometry assays, respectively. d, MnSOD enzyme activity in mouse skin tissues following UVB treatment. e, MnSOD protein level estimated by Western blotting in mouse skin tissues after UVB treatment (5 kJ/m2). f, mitochondrial oxygen consumption was measured as described under “Experimental procedures.” g, glycolysis was measured as described under “Experimental procedures.” In all bar graphs and line graphs, each data point represents the mean ± S.D. of three to four individual samples. Each experiment was repeated at least three times, and statistical analysis was performed using t tests for two groups or one one-way ANOVA analysis followed by Bonferroni's post-test for multiple-group comparisons. Statistical significance is indicated by asterisks: *, p ≤ 0.05, and **, p ≤ 0.01.
UVB promotes metabolic adaptation
To determine how cells adjust their metabolism in response to UVB treatment, we measured the OCR and ECAR in JB6 cells, using the Agilent Seahorse FX analyzer. The OCR data show that basal respiration, maximum respiration, spare respiratory capacity, and ATP-linked activities are significantly decreased following UVB treatment (Fig. 1f). The ECAR data show increased glycolysis after UVB exposure, compared with control, with significant increases of glycolytic capacity and glycolytic reserve (Fig. 1g). However, nonglycolytic acidification is also significantly increased in UVB-treated murine cells compared with control (Fig. 1g). These results suggest that treatment with UVB radiation shifts the cells from oxidative phosphorylation toward glycolysis.
UVB induces autophagy/mitophagy
As autophagy is critical for removal of damaged organelles, we investigated the effect of UVB exposure on autophagy. Microtubule-associated protein light chain 3 (LC3) is an abundant cytoplasmic protein that is cleaved and lipidated leading to the formation of LC3 II during initiation of autophagy and is incorporated into autophagosomal membranes in a punctate pattern (15). Thus, LC3 II, which participates in autophagosome formation and maturation (16), is considered to be a downstream indicator of the autophagy pathway.
To investigate the role of UVB radiation in autophagy, LC3–GFP expression plasmid vectors were overexpressed in JB6 cells. We then determined the levels of LC3 punctation and LC3 II formation by fluorescence microscopy and Western blotting, respectively. Fig. 2a shows that LC3 punctation significantly increases in UVB-treated cells. Consistent with this finding, the endogenous level of LC3 II also increases in UVB-exposed cells (Fig. 2b). We further assessed the levels of critical upstream autophagy regulators beclin 1, ATG5, and ATG7. Beclin 1 is responsible for initiation of autophagosome formation (17, 18). Consistent with the increased LC3 II formation, beclin 1 also significantly increases in UVB-treated cells compared with control (Fig. 2c). The autophagy initiators ATG5 and ATG7 also significantly increase in UVB-treated cells compared with control (Fig. 2c). In addition, we assessed autophagy flux in UVB-treated cells with or without autophagy inhibitors. We used the manganese superoxide dismutase mimetic MnP as an oxidative stress reducer, 3-methyladenine (3-MA) as a PI3K/mTOR inhibitor, and bafilomycin as a lysosome inhibitor to modulate autophagy. JB6 cells that ectopically express LC3 were pretreated 24 h prior to UVB treatment with the manganese superoxide dismutase mimetic MnP (10 μm), an oxidative stress inhibitor; 3-MA (2.5 mm), a PI3K/mTOR inhibitor; and bafilomycin (50 nm), a lysosome inhibitor. Puncta formation in GFP–LC3-positive cells was considered to be the sign of autophagy. UVB treatment increases the number of punctated cells that were attenuated by pretreatment with MnP or 3-MA (Fig. 2d). Bafilomycin increases LC3 punctation by inhibiting autolysosome formation, resulting in the accumulation of autophagic puncta even in cells not treated with UVB (Fig. 2d). Similarly, Western blotting data (Fig. 2e) show that UVB induces LC3 II formation compared with the untreated control. Pretreatment with MnP or 3-MA significantly reduces UVB-induced LC3 II formation to near that of control levels. However, UVB-induced LC3 II levels are further increased following pretreatment with bafilomycin (Fig. 2e). Because UVB-induced autophagy is associated with inactivation of MnSOD and reduction in mitochondrial respiration, we investigated mitophagy as a result of UVB-induced autophagy. First, we assessed the mitophagy marker, Bcl-2 19-kDa interacting protein 3 (BNIP3), by Western blotting. BNIP3 is a mitochondrially localized Bcl-2 family protein that plays an important role in inducing mitochondrial autophagy. The data show that the BNIP3 level is increased in UVB-treated murine epidermal keratinocytes compared with untreated control (Fig. 2f, panel i). Additionally, we evaluated the presence of mitochondria in the autophagosome by staining the mitochondria with MitoTracker Red and detected its presence within the LC3 puncta in LC3 overexpressed cells. Co-localization of MitoTracker Red with LC3 puncta is increased after UVB treatment (Fig. 2f, panel ii). To further verify mitophagy, we co-stained the lysosome and mitochondria with LysoTrackerTM Green and MitoTracker Red, respectively, to visualize mitochondria and lysosome co-localization. The results (Fig. 2f, panel iii) show that the co-localization of mitochondria and lysosome is increased in UVB-treated cells. Consistent with the in vitro findings, autophagy marker LC3 II and beclin 1 levels were found to be significantly increased in mouse skin tissues as early as 1 h following UVB treatment (Fig. 3).
Figure 2.

UVB induces autophagy/mitophagy. a, JB6 cells were transfected with LC3 expression vector using Lipofectamine transfection protocol. LC3 punctation was detected in UVB-treated cells by a fluorescence microscope. For quantification of autophagic response, 100 GFP-positive cells were counted and compared with control, and the data are presented as fold changes. b and c, Western blot analysis was performed to detect LC3 II, beclin 1, ATG7, and ATG5 proteins in UVB-treated cells and compared with control. Bar graph shows the relative levels of each protein upon UVB treatment compared with control. d, autophagy flux was determined by detecting the puncta formation with or without autophagy inhibitors. The bar graph shows the quantification of punctated cells (100 GFP-positive cells were counted for each cell type). e, autophagy flux was also detected by Western blotting in UVB-treated cells following treatment of autophagy inhibitors (MnP, MnTnBuOE-2-PyP5+, 3-MA, and bafilomycin). The bar graph shows the quantification of LC3 II band intensity normalized to β-actin. f, panel i, BNIP3 proteins are detected by Western blotting in UVB-treated JB6 cells as a marker of mitophagy. The bar graph shows the quantification of BNIP3 band intensity normalized with β-actin. Panel ii, increase of mitophagy was observed by detecting the autophagosome (LC3 II) and mitochondria co-localization. One hundred LC3-positive cells were selected, and the number of LC3 puncta was selected, followed by counting the number of co-localized LC3 puncta with mitochondria. The mitochondria and LC3 puncta were identified and arbitrarily gated. The co-localized area is counted as LC3 and mitochondria co-localization. The mitochondria–LC3 co-localized puncta was normalized with the total number of puncta in each cell. Bar graph shows the relative number of LC3–puncta–mitochondrial co-localization in UVB-treated cells compared with control. Panel iii, mitophagy was also monitored by observing mitochondria–lysosome co-localization. Live cells were stained with MitoTracker Red and LysoTracker Green for mitochondria and lysosomes, respectively; the co-localized mitochondria and lysosomes were estimated by counting the number of arbitrarily selected co-localized areas, panel ii. Each experiment was repeated at least three times, and statistical analysis was performed using t tests for two groups or one-way ANOVA followed by Bonferroni's post-test analysis for multiple-group comparisons. Statistical significance is indicated by asterisks: *, p ≤ 0.05, and **, p ≤ 0.01.
Figure 3.
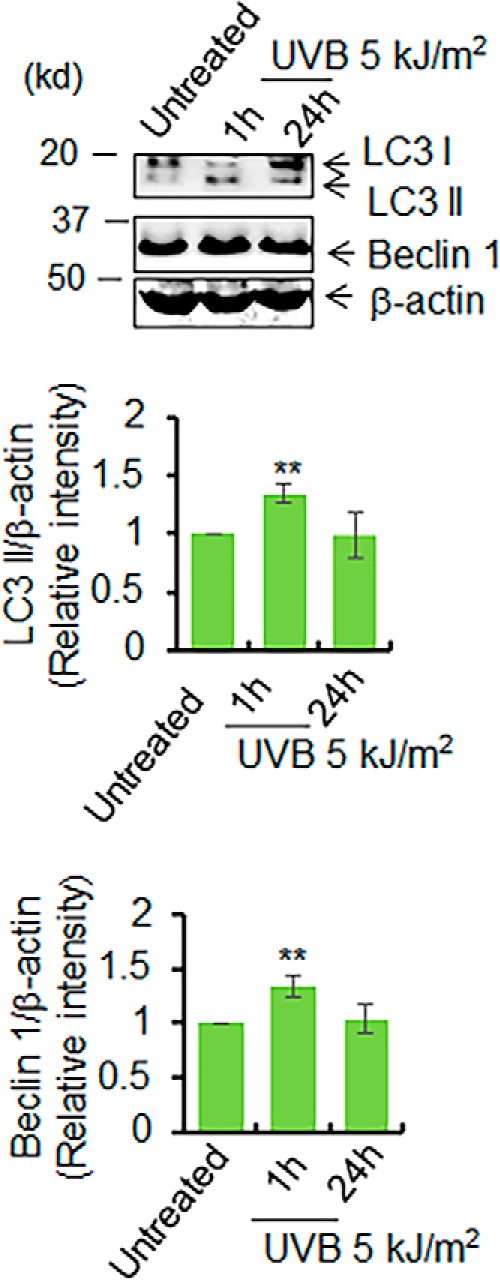
UVB induces autophagy in vivo. The levels of LC3 II formation and beclin 1 were examined in mouse skin tissue lysates after UVB treatment by Western blotting. The bar graph shows the quantification of the band intensity of LC3 II and beclin 1 after normalization with β-actin. Each experiment was repeated at least three times, and statistical analysis was performed using one-way ANOVA followed by Bonferroni's post-test analysis for multiple-group comparisons. Statistical significance is indicated by asterisks: *, p ≤ 0.05, and **, p ≤ 0.01.
UVB activates mTOR pathways
The mTOR pathway is implicated as a regulator of autophagy. However, how UVB-induced activation of the mTOR pathway is linked to the induction of mitophagy is not clear. We hypothesized that UVB exposure, which causes aberrant mitochondrial function and ROS production, could activate stress signals that affect the mTOR signaling pathway in the mitochondria. To test this hypothesis, we first measured the mTOR components in JB6 cells following UVB exposure. The results (Fig. 4) show that rictor (a component of the mTORC2 complex) is significantly increased in JB6 cells following UVB treatment; however, raptor, a component of the mTORC1 complex, does not change after UVB treatment (Fig. 4, a and b). Consistently, mTOR phosphorylation at serine 2481, which represents mTORC2 activation, is increased in UVB-exposed cells compared with control. In contrast, mTOR phosphorylation at serine 2448, which represents mTORC1 activation, is not altered in UVB-treated cells compared with control (Fig. 4, a and b). In agreement with these findings, AKT phosphorylation at serine 473 is increased following UVB treatment; however, there is no significant alteration of AKT phosphorylation at threonine 308 in UVB-treated cells as compared with untreated control. (Fig. 4, a and b). PI3K does not change following UVB treatment compared with control (Fig. 4, a and b). These data confirm that the mTORC2 complex is activated following UVB treatment, which in turn activates AKT phosphorylation at serine 473. We cannot rule out the contribution of PI3K activation and AKT phosphorylation at serine 473. However, in the context of UVB treatment, PI3K remains unchanged, which further supports the role of mTORC2 in AKT phosphorylation at serine 473. To further assess the presence of increased mTORC2 complex after UVB treatment, we overexpressed the FLAG-tagged mTOR expression vector in JB6 cells. The ectopically expressed mTOR was immunoprecipitated with FLAG antibody followed by Western blotting with rictor and raptor antibodies. The results show increased interaction of rictor with mTOR (FLAG) in UVB-treated cells compared with the untreated control; however, the raptor interaction with mTOR (FLAG) remains unchanged (Fig. 4c). Consistent with immunoprecipitation data, mTOR and rictor co-localization is significantly increased after UVB treatment, but the co-localization of mTOR and raptor remains unchanged in UVB-treated cells compared with untreated control (Fig. 4d). These results confirm the activation of mTORC2 pathways following UVB treatment.
Figure 4.

Activation of AKT/mTORC2 pathway after UVB treatment. a, increased levels of mTOR phosphorylation at Ser-2481, AKT phosphorylation at Ser-473, AKT phosphorylation at Thr-308, and rictor were detected by Western blotting in JB6 cells after UVB treatment. b, quantification of protein band intensity by densitometric scanning; results are normalized with corresponding β-actin band intensity. c and d, co-localization of mTOR with raptor and rictor was detected by immunoprecipitation (IP) with mTOR antibody followed by Western blot analysis (c). Immunocytochemistry was performed using mTOR, raptor, or rictor antibody after UVB treatment. The bar graph represents the relative level of mTOR and raptor co-localization or mTOR and rictor co-localization. For quantification, 100 cells were randomly selected, and the density of the merged image was quantified by densitometric scaning using ImageJ software (d). Each experiment was repeated at least three times, and statistical analysis of the quantifications was performed using t test. Statistical significance is indicated by asterisks: *, p ≤ 0.05, and **, p ≤ 0.01.
UVB induces mTOR activation in mitochondria
To investigate the role of mTOR signaling in mitochondria, we performed proximity-ligation assay (PLA) targeting mitochondria by using the mitochondria resident protein DNA-polymerase γ and mTOR (Ser-2481) antibodies. Our PLA data show that p-mTOR (Ser-2481) and DNA-polymerase γ are co-localized and interact with each other and that this interaction is increased following UVB treatment compared with untreated control (Fig. 5a). This result confirms the presence of activated mTOR in the mitochondria. To further demonstrate the presence of activated mTORC2 in mitochondria, we purified the mitochondrial fraction and examined the rictor expression in the mitochondria by Western blotting. The purity of the mitochondrial fraction was assessed by the presence of an enriched level of MnSOD in mitochondria. In agreement with the results from the proximity-ligation assay, the data show that the rictor level is increased in the mitochondrial fraction following UVB treatment, and it is attenuated by MnP treatment (Fig. 5, b and c). Consistent with the rictor level, phosphorylated mTOR (Ser-2481) and phosphorylated AKT (Ser-473) are increased in mitochondrial fraction after UVB treatment and are correspondingly reduced by pretreatment with MnP (Fig. 5, b and c). These results reveal activation of the mTORC2 signaling pathway in mitochondria.
Figure 5.
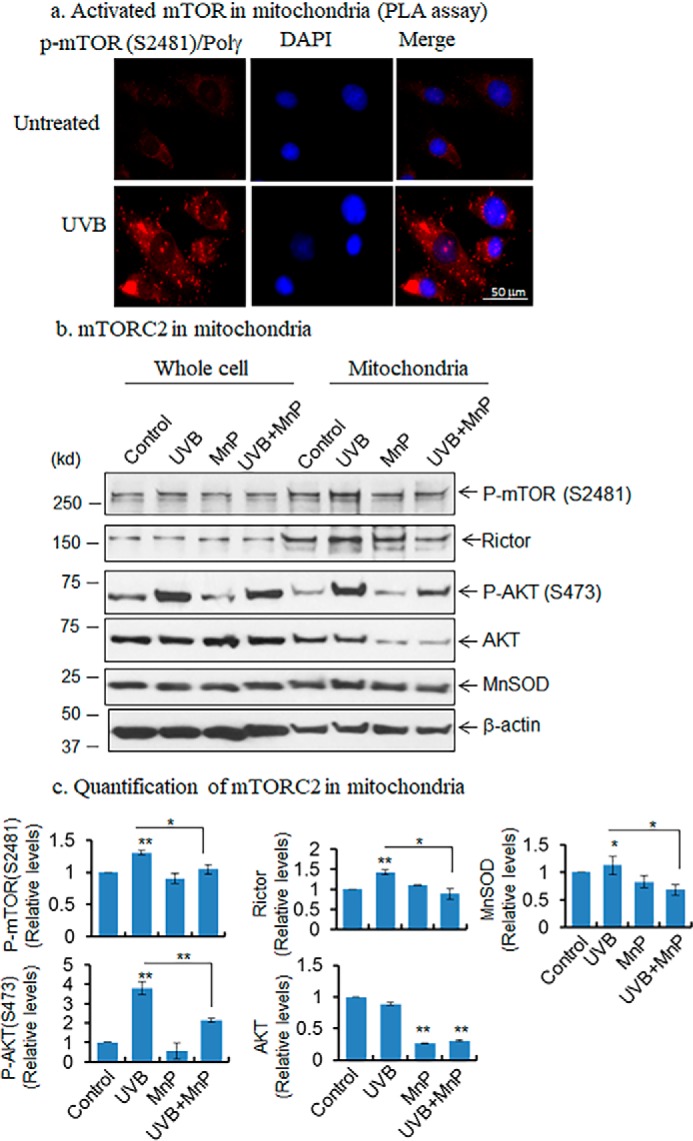
Detection of mTOR in mitochondria. a, presence of activated mTOR in mitochondria was detected by proximity ligation assay using phosphorylated mTOR and DNA polymerase γ antibodies. Briefly, the cells were treated with UVB radiation, and after 1 h cells were fixed with 4% formalin, then treated with two specific antibodies with different origins, and then probed with Duo-linked (PLA kit) secondary antibody according to the manufacturer's instructions. b, presence of mTORC2 in mitochondria was detected in purified mitochondria. First, mitochondria were isolated using mitochondria purification kit and subjected to Western blotting to identify the mTOR component in purified mitochondrial fraction. c, increase in activated mTOR was detected in UVB-treated mitochondria compared with control. Pretreatment with MnP inhibits mTOR phosphorylation (Ser-2481), AKT phosphorylation (Ser-473), and rictor levels in mitochondria. Statistical significance is indicated by asterisks: *, p ≤ 0.05, and **, p ≤ 0.01.
Different roles of rictor and raptor in UVB-induced autophagy
To directly demonstrate the differential role of rictor and raptor on UVB-induced autophagy, we used siRNA to suppress rictor and raptor to determine their effect on LC3 II formation. Suppression of mTOR and rictor by siRNA leads to reduced formation of mTORC2, as measured by mTOR phosphorylation at Ser-2481 and AKT phosphorylation at Ser-473, compared with control siRNA-treated cells. Consistent with these data, the level of LC3 II is significantly suppressed following simultaneous suppression of mTOR and rictor by siRNAs. However, the suppression of mTOR and raptor, which is achieved by treating the cells with mTOR and raptor siRNA, does not reduce the level of LC3 II (Fig. 6, a and b). Treatment of UVB increases mTOR (Ser-2481) and AKT (Ser-473) phosphorylation, which is attenuated by the combined mTOR and rictor siRNA (Fig. 6a). However, the combined treatment of mTOR and raptor siRNA suppresses UVB-induced LC3 II formation less than what occurs in rictor siRNA and mTOR siRNA-mediated reduction of LC3 II following UVB treatment. These results suggest that the mTORC2 complex is playing a dominant role in UVB-induced autophagy.
Figure 6.

Role of mTOR complex in autophagy. a, to demonstrate the role of a specific mTOR complex associated with UVB-induced autophagy, JB6 cells were overexpressed with a combination of mTOR siRNA and raptor siRNA or mTOR siRNA and rictor siRNA to suppress mTORC1 and mTORC2 complexes, respectively. Suppression of mTOR and rictor attenuates LC3 II formation (lanes 2 and 5), but suppression of mTOR and raptor has no effect on LC3 II formation (lanes 3 and 6) with or without UVB treatment. b, effects of the suppression of mTOR complexes on autophagic marker LC3 II formation were quantified and are presented as a bar graph. c, activation of mTORC2 and autophagy/mitophagy was also determined in HEKn cells with both UVB treatment and by suppressing MnSOD levels using MnSOD siRNA. UVB and MnSOD siRNA both increase mTOR and AKT phosphorylation. UVB and MnSOD siRNA also enhance LC3 II formation and BNIP3 expression. d, quantification of Western blotting data are presented as bar graphs. Each experiment was repeated at least three times, and statistical analysis was performed using one-way ANOVA followed by Bonferroni's post-test analysis for multiple-group comparisons. Statistical significance is indicated by asterisks: *, p ≤ 0.05, and **, p ≤ 0.01.
UVB-induced autophagy/mitophagy is MnSOD-dependent
We asked whether UVB-induced mitophagy is altered by MnSOD in mitochondria. It has been shown that UVB increases MnSOD expression as an adaptive response and that the suppression of MnSOD increases the cellular level of superoxide formation. Consistent with these findings, our data show that MnSOD is significantly increased after UVB exposure compared with untreated controls (Fig. 6, c and d). MnSOD siRNA significantly suppresses the endogenous level of MnSOD and increases the levels of LC3 II and BNIP3, with a concomitant increase of mTOR (Ser-2481) and AKT (Ser-473) phosphorylation following UVB treatment (Fig. 6, c and d). Suppression of MnSOD leading to an increase in LC3 II formation was also observed in stably expressed MnSOD shRNA in human keratinocytes (Fig. S3). These results support the connection of UVB-induced mitophagy, mitochondrial ROS, and mTORC2 activation.
Autophagy contributes to UVB-induced cell growth
Our previous studies have demonstrated a prosurvival role of mitochondrial injury (19). Because autophagy can lead to cell survival or death, we asked whether low-dose UVB radiation enhances cell survival. The level of the dose was determined by observing the dose-dependent effects of UVB radiation on autophagy without apoptosis (Fig. S4). We used the BrdU uptake and soft-agar colony formation assays to assess cell growth. The data show that the BrdU uptake is significantly increased after UVB treatment compared with untreated control. Pretreatment of MnP or 3-MA to inhibit autophagy significantly suppresses UVB-induced BrdU incorporation (Fig. 7a). Consistent with the DNA synthesis-based assay, UVB significantly increases cell survival, which is attenuated by the pretreatment with MnP or 3-MA (Fig. 7b). In addition, the number of colonies formed on soft agar also increases compared with untreated control (Fig. 7, c and d). These results provide evidence that autophagy may contribute to the observed UVB-induced cell growth.
Figure 7.
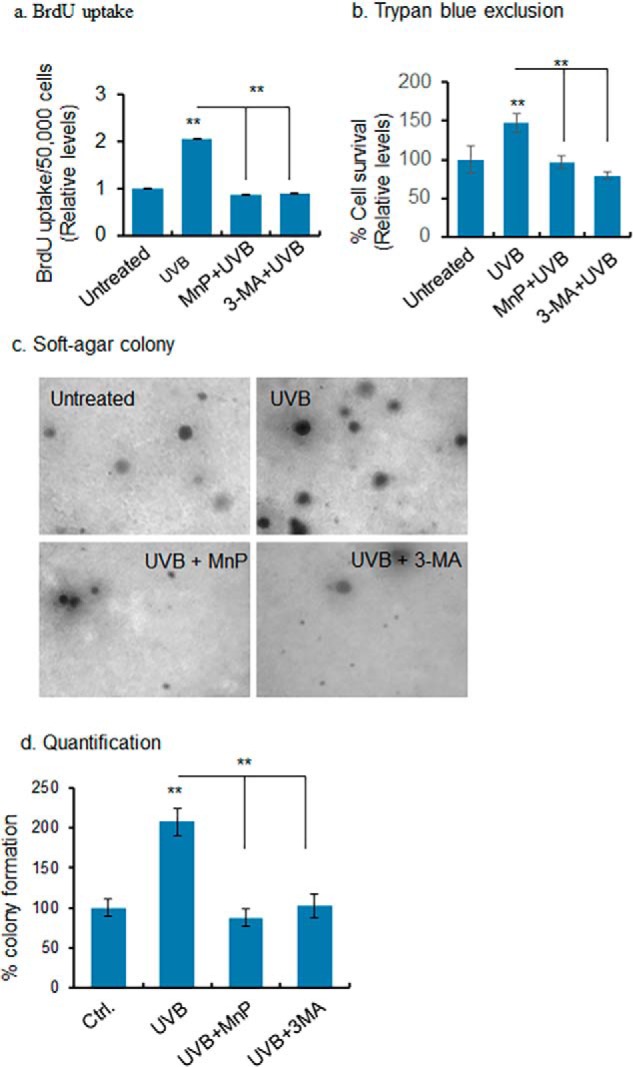
Autophagy-dependent UVB-induced cell proliferation. a, BrdU uptake was determined as a measure of cell growth in UVB-treated cells with or without ROS or autophagy inhibitors. b, cell survival was assessed by counting the cells following UVB treatment using trypan blue exclusion assay. c, soft agar colony formation assay assessed cell growth after UVB treatment with or without ROS and autophagy inhibitor. For each treatment, 6–10 dishes were used, and microscopic images of transformed colonies were taken from six randomly selected fields in each dish (6–10 dishes/group). A representative microscopic view is shown. d, number of transformed colonies was counted and presented as a bar graph. Each experiment was repeated at least three times, and statistical analysis was performed using one-way ANOVA followed by Bonferroni's post-test analysis for multiple-group comparisons. Statistical significance is indicated by asterisks: *, p ≤, 0.05 and **, p ≤ 0.01.
Discussion
Oxidative stress plays a pivotal role in the pathophysiology of various cellular and sub-cellular conditions where MnSOD, a major mitochondrial antioxidant enzyme, imparts defense mechanisms against oxidative stress during cancer initiation and progression. Several studies have reported UVB-induced carcinogenesis in keratinocytes (5–7). However, very little is known about the link between UVB-induced oxidatively modified MnSOD and mitochondrial metabolism in a pre-carcinogenesis stage. The mTOR consists of two distinct complexes, mTORC1 and mTORC2, which play an important role in regulating autophagy and cell growth (20). The role of mTORC1 complexes is well-documented; however, very little is known about the role of mTORC2 in autophagy and UVB-induced cancer. Our data demonstrate that defective mitochondrial function is due, in part, to UVB-induced MnSOD inactivation, which triggers prosurvival mitophagy response via activation of the rictor-mediated mTORC2 signaling pathway.
Our data also suggest that UVB treatment increases MnSOD nitration via a peroxynitrite-mediated mechanism that leads to MnSOD inactivation and consequently alters mitochondrial function (Fig. 1). Although we could not completely rule out the possibility that other oxidants cause oxidative damage to SOD, our data show significant increases in superoxide and nitric oxide but not hydrogen peroxide (Fig. S1, b–d, respectively). These results are supportive of the role of peroxynitrite-mediated MnSOD nitration and inactivation of MnSOD activity. It has been shown that both nitric oxide and the superoxide radical generated by UVB radiation can produce peroxynitrite, leading to nitration of proteins (13, 14). Direct nitration of specific tyrosine residues in MnSOD protein has been established, and peroxynitrite-mediated inactivation of MnSOD has also been investigated (21). Those reports are consistent with our findings that treatment of UVB radiation increases the nitration of MnSOD protein in keratinocytes (Fig. 1) and inactivates the enzymatic activity both in vitro and in vivo (Fig. 1). The loss of MnSOD activity leads to defects in mitochondrial antioxidant response, of which MnSOD is a major component, and explains the decreased level of ATP-linked oxygen consumption, increased glycolysis, higher ROS production, and higher mitophagy markers (Fig. 1f and Fig. S1). These results suggest that UVB-induced suppression of MnSOD enzyme activity is associated with mitophagy.
Our data demonstrate the increase in mitophagy marker BNIP3 (Fig. 2f, panel i). BNIP3 is a member of the BH3-only protein family that forms stable homodimers and localizes to the outer membrane of mitochondria after cellular stress (22). Thus, the increased autophagosome formation suggests a mitophagy process. Although no clear consensus exists as to what constitutes the most critical markers of mitophagy, induction of BNIP3 expression during an increase in survival autophagy could be a signature of mitophagy. Additionally, the activation of Parkin and Pink1 are thought to be biomarkers of mitophagy. However, the Parkin and Pink1 expression is very tissue-specific. For an example, neuronal cells express a high level of Parkin, but skin tissue possess no detectable level of Parkin (23) (www.proteinatlas.org/EnSG00000185345-PARK2/tissue3 (41)). Our experimental investigation found no detectable levels of Parkin or Pink1 in skin keratinocytes (data not shown). It has been demonstrated that mitophagy occurs in mouse skin tissues independent of Pink1 and Parkin interaction (24). Using this information, organelle-specific autophagy can be investigated by monitoring the increased levels of autophagosome containing mitochondria in cases of mitophagy or by monitoring the mitochondria in lysosomes (25). Following these guidelines, we observed increased levels of LC3 II and MitoTracker Red co-localization following UVB treatment (Fig. 2f, panel ii). We also observed increased co-localization of lysosome and mitochondria following UVB treatment compared with untreated control (Fig. 2f, panel iii). These results provide the evidence of UVB-induced mitophagy upon mitochondrial stress.
The mechanism by which inactivation of MnSOD following UVB treatment initiates prosurvival mitophagy is unclear. Our data suggest that UVB targets mitochondria, which leads to metabolic reprogramming, which then generates the ROS that activate mTORC2 pathways. Activation of mTOR was observed to stimulate cell growth in various cell types. For example, it has been reported that it is necessary that mTOR be activated in immune cells (26) during proliferation. Furthermore, endotoxin-induced autophagy is a survival mechanism that drives proliferation of liver cells (27) and cardiomyocytes (28). The central components of the cell survival pathway, including AKT and mTOR activation, sense cellular metabolic alterations and trigger cell survival (29). Our results demonstrate an increase in the phosphorylation of mTOR (Ser-2481) and AKT (Ser-473) in UVB-treated cells (Fig. 4), supporting previous findings that AKT–mTOR regulates autophagy and cell survival. Although it has been demonstrated that the PI3K–AKT–mTOR pathway mediates anti-autophagic signaling, inhibition of mTOR by rapamycin induces autophagic cell death (30). Recent studies, however, have shown that activation of the AKT–mTOR pathways also increases autophagy (31). For example, activation of AKT, which is acquired by phosphorylation at serine 473 residues, increases autophagy by forming a complex with lysosomal protein (32). Serine–threonine kinase AKT and mTOR are interconnected in their signaling pathway, where mTORC2 is known to phosphorylate AKT at Ser-473. Our data show an increase in AKT phosphorylation at Ser-473 as well as a preferential formation of mTORC2 over mTORC1 following UVB treatment. Consistent with AKT phosphorylation, mTOR phosphorylation at serine 2481 (which is also an indication of mTORC2 complex formation and activation) increases after UVB treatment. This is again manifested by an increased level of mTOR and rictor interaction, as detected by co-immunoprecipitation and cellular localization following UVB treatment (Fig. 4). Importantly, the activation of mTORC2 occurs in the mitochondria as demonstrated by proximity ligation assay and the presence of increased mTORC2 components in the mitochondria as compared with control (Fig. 5). Furthermore, our strategy of modulating the mTOR complex by suppressing an individual component of the complex, using an siRNA approach, demonstrates the role of the rictor-mediated mTORC2 pathway in autophagy/mitophagy in UVB-treated cells (Fig. 6). Consistent with our previous findings that rictor plays a critical role in MEF cells during autophagy (19), we now have found that UVB treatment of rictor-deficient cells is unable to induce mitophagy (Fig. S2), suggesting a common mechanism in mitophagy. These data confirm that activation of mTORC2 following UVB treatment is a mitochondrial stress event and that UVB-induced mitophagy is regulated by mTORC2 pathways.
Although the outcome of autophagosome formation and its consequence in cell survival and cell death depends on cell type and context (33, 34), our current investigation shows that organelle-specific autophagy (mitophagy) serves a critical role in deciding cell survival in a pre-neoplastic stage. These results add critical insights into the mechanism of mitophagy-associated cell survival and have broad implications for understanding mitochondrial pathology in the development of cancer and other metabolism-related disorders.
Experimental procedures
Reagents
Anti-LC3A/B (catalog no. 1274), anti-Beclin 1 (catalog no. 3495), and ATG7 (catalog no. 8558) were purchased from Cell Signaling Technology (Danvers, MA). Anti-β-actin (catalog no. SAB2100037), anti-mTOR (catalog no. PLA0114), and anti-FLAG (F3165) antibodies were purchased from Sigma. Anti-raptor (catalog no. 05-1470) and anti-rictor (catalog no. 05-1471) antibodies were purchased from Millipore (Temecula, CA). 3-MA (catalog no. tlrl-3ma) and bafilomycin A1 (catalog no. tlrl-baf1) were purchased from InvivoGen (San Diego). The mTOR, rictor, and raptor siRNAs were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), and MnSOD siRNA was purchased from Dharmacon (Lafayette, CO). All other chemicals were purchased from Sigma unless otherwise specified. MnTnBuOE-2-PyP5+ is synthesized as described by Rajic et al. (35). Its therapeutic actions and the most recent insight into its mechanism of action have been recently reviewed (36).
Cell culture and treatments
The JB6 mouse epidermal cell line was originally obtained from Dr. Nancy H. Colburn of the NCI (National Institutes of Health), Rockville, MD, and was maintained as described previously (37, 38). The primary human epidermal keratinocyte cells (HEKn, catalog no. C-001-5C) were purchased from Invitrogen. Both WT and rictor-knockout MEFs were originally obtained from David Sabatini (Whitehead Institute, Cambridge, MA). All cells were grown in a 5% CO2 incubator at 37 °C in media consisting of either MEM supplemented with 10% fetal bovine serum (Hyclone Inc., Logan, UT), 1% (w/v) l-glutamine (Invitrogen), and 1% penicillin/streptomycin antibiotics (Invitrogen), or Epilife medium supplemented with S7 (Invitrogen) for primary cells. Cells were exposed to a single dose of UV radiation (50 mJ/cm2) using UVB lamps as described previously (9). Cells were also treated with 10 μm MnP, 2.5 mm 3-MA, and 50 nm bafilomycin A1 for 24 h.
Cell transfection
Cells were grown for 24 h with no antibiotics to obtain 70–80% confluency. The cells were then transfected with plasmids following a transfection protocol using Lipofectamine® as directed by the manufacturer. JB6 cells are easily transfectable, and we obtained 50–70% transfection efficiency using the Lipofectamine transfection protocol. Cells were transfected with 1–2 μg of plasmid DNA containing LC3–GFP (equilibrated to the same amount of DNA by adding control vector) or control vector alone. Twenty four hours after transfection, the cells were washed twice with PBS and incubated in fresh medium for another 24 h with or without treatment for an indicated time. Cells were then processed for whole-cell lysate preparation or fluorescence microscopy. Similarly, mTOR, rictor, and raptor siRNAs were transfected using Transfectin® reagent (Santa Cruz Biotechnology). MnSOD siRNA was transfected in cells using the Dharmafect® transfection reagent, which was obtained from Dharmacon (Lafayette, CO), according to the manufacturer's protocol. Cells were exposed to the siRNA for 72 h.
Animals and UVB exposure studies
WT and heterozygous MnSOD knockdown mice (MnSOD+/−) with a C57BL/6 background were used in this study. The animal experimental procedures carried out in this study were approved by the Institutional Animal Care and Use Committee of the University of Kentucky. Six- to 8-week-old mice in resting stage of hair cycle were exposed to UV irradiation in a Plexiglas cabinet (Plastic Design Corp., Scottsdale, AZ). A single dose of 5 kJ/m2 was delivered by UVB lamp (black light blue lamp, Sankyo Denki Ltd., Japan). The UV emittance was measured using a UVB photometer radiometer (International Light Technologies, Peabody, MA) equipped with a UVB measuring head.
Measurement of oxygen consumption rate and extracellular acidification rate
XF extracellular flux assays (Seahorse-Bioscience, North Billerica, MA) were utilized to measure the OCR and the ECAR. The OCR experiments were performed by sequentially adding to cells the substrate oligomycin, carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP) and antimycin/rotenone, and the ECAR measurements were performed by using glucose, oligomycin, and 2-deoxyglucose as substrates.
MnSOD activity assay
Both cultured cells and mouse skin tissues were harvested and homogenized. MnSOD activity was measured by nitro blue tetrazolium reduction using xanthine and xanthine oxidase for superoxide generation as described previously (39).
In-gel MnSOD activity assay
Human embryonic keratinocyte (HEKn) cells were collected, homogenized and then sonicated on ice for 30 s. Protein concentrations were estimated by the Bradford protein assay. One hundred micrograms of the total protein were loaded onto each lane of the gel, and the SOD activity was visualized by the nitro blue tetrazolium method (40).
Western blotting
Proteins were analyzed by Western blotting. Briefly, cell extracts were subjected to 10 or 12% SDS-PAGE and transferred to a nitrocellulose membrane. Following blocking with 5% BSA, membranes were then probed with specific primary antibody (by diluting at a range from 500 to 5,000) followed by secondary antibody (dilution range from 2,000 to 6,000) to detect specific proteins. Protein bands were detected using the enhanced chemiluminescence detection system (ECL®, Amersham Biosciences). Densitometric analysis was performed for quantification of proteins using ImageJ software (National Institutes of Health).
Fluorescence microscopy
Green fluorescent LC3 puncta formation was detected in cultured cells by a fluorescence microscope (model IX71, Tokyo, Japan) following transient transfection of pEGFP–LC3 expression vector.
Immunoprecipitation
Immunoprecipitation studies were carried out, as described previously (14), with whole-cell extracts, using specific antibodies in a binding buffer (9.1 mm Na2HPO4, 1.7 mm NaH2PO4, 150 mm NaCl (pH 7.4), 0.1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS) containing 10 μg/ml phenylmethylsulfonyl fluoride and 1 μg/ml aprotinin as protease inhibitor. Rabbit anti-3-nitrotyrosine antibodies were used for immunoprecipitation. One mg of protein from whole-cell lysate was incubated overnight with 2 μg of antibody at 4 °C with continuous rotation, at which point 20 μl of protein A/G beads (Santa Cruz Biotechnology) were added to the reaction mixture, and the rotation continued for another 2 h at 4 °C. Immunoprecipitates were collected by centrifugation at 2,500 × g for 5 min followed by washing with binding buffers. After the final wash, all the adhering liquids were removed from the beads. Samples were then suspended in 1× Laemmli buffer and subjected to SDS-PAGE, and MnSOD proteins were detected by Western blotting.
Quantification of ROS levels by flow cytometry
MitoSox Red (ThermoFisher Scientific, catalog no. M36008), a highly selective mitochondrial superoxide indicator of live cells, was used to measure ROS levels. Rotenone (200 nm, Sigma), known to be a mitochondrial superoxide inducer, was used as a positive control. To account for superoxide-specific fluorescence, the cells were pretreated with 100 units/ml PEG–SOD (Sigma) or 10 μm MnP for 24 h prior to measurement. Briefly, cells were loaded with 5 μm MitoSox Red for 10 min at 37 °C and rinsed three times with 1× PBS, and then the intensity of MitoSox Red fluorescence was measured in live cells by a fluorometer (SpectraMax Gemini, Molecular Devices) at an excitation/emission maxima of 510/580 nm. Similarly, for hydrogen peroxide measurements, the MitoPY1 (Tocris Bioscience, catalog no. 4428), a selective mitochondrial hydrogen peroxide indicator in live cells, was used to measure hydrogen peroxide levels. Catalase was used to confirm the presence of hydrogen peroxide.
BrdU incorporation and detection
Cell proliferation was assessed based on 5-bromo-2′-deoxyuridine (BrdU) incorporation followed by ELISA according to the manufacturer's protocol (Cell Biolabs, Inc.). Briefly, 30,000 cells were grown in a 96-well plate for 24 h at 37 °C in a 5% CO2 incubator. BrdU solutions were added to the well to achieve 1 nm final concentration and incubated for 6 h. Cells were then washed, fixed, and denatured at 37 °C for 30 min. Following successive washings with PBS, cellular BrdU uptake was detected by colorimetric ELISA (450 nm) using anti-BrdU antibody. Data were normalized to the protein concentration of the corresponding sample.
Soft-agar colony formation assay
Mouse epidermal cells (5,000 cells) were mixed with 0.33% agar in MEM and overlaid on top of the 5% agar medium. After solidification, 2–3 ml of culture media were added (on a 60-mm dish) over the top of a soft agar layer and incubated at 37 °C in a 5% CO2-humidified incubator for 14 days. Colonies consisting of at least 50 cells were counted under a light microscope.
Statistical analysis
Data were analyzed using one-way analysis of variance for multiple-group comparisons, and Student's t test for two-group comparisons. For multiple-group statistics, Bonferroni's post-test for multiple comparisons was used to determine statistical significance.
Author contributions
S. K. D. and D. K. S. C. conceptualization; S. K. D. and D. K. S. C. data curation; S. K. D. formal analysis; S. K. D. validation; S. K. D. investigation; S. K. D. writing-original draft; I. B.-H. resources; I. B.-H. visualization; D. K. S. C. supervision; D. K. S. C. funding acquisition.
Supplementary Material
Acknowledgments
We thank Dr. Mihail I. Mitov of the Redox Metabolism Shared Resource Facility (RMSRF) for outstanding technical help in acquiring data from Seahorse experiments. We acknowledge with gratitude the assistance of the RMSRF of the Markey Cancer Center, which is the recipient of National Institutes of Health Cancer Center Support Grant 2P30 CA177558 from NCI.
This work was supported by National Institutes of Health Grant CA 214638. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S4.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- MnSOD
- manganese-containing superoxide dismutase
- mTOR
- mammalian target of rapamycin
- ROS
- reactive oxygen species
- MnP
- MnTnBuOE-2PyP5+
- 3-MA
- 3-methyladenine
- MEF
- mouse embryo fibroblast
- MEM
- minimum essential medium
- OCR
- oxygen consumption rate
- ECAR
- extracellular acidification rate
- ANOVA
- analysis of variance
- PI3K
- phosphatidylinositol 3-kinase
- PLA
- proximity-ligation assay.
References
- 1. Gregory E. M., and Fridovich I. (1973) Oxygen toxicity and the superoxide dismutase. J. Bacteriol. 114, 1193–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aitken G. R., Henderson J. R., Chang S. C., McNeil C. J., and Birch-Machin M. A. (2007) Direct monitoring of UV-induced free radical generation in HaCaT keratinocytes. Clin. Exp. Dermatol. 32, 722–727 10.1111/j.1365-2230.2007.02474.x [DOI] [PubMed] [Google Scholar]
- 3. Moldogazieva N. T., Lutsenko S. V., and Terentiev A. A. (2018) Reactive oxygen and nitrogen species-induced protein modifications: implication in carcinogenesis and anticancer therapy. Cancer Res. 78, 6040–6047 10.1158/0008-5472.CAN-18-0980 [DOI] [PubMed] [Google Scholar]
- 4. Alvarez B., and Radi R. (2003) Peroxynitrite reactivity with amino acids and proteins. Amino Acids 25, 295–311 10.1007/s00726-003-0018-8 [DOI] [PubMed] [Google Scholar]
- 5. Justilien V., Pang J.-J., Renganathan K., Zhan X., Crabb J. W., Kim S. R., Sparrow J. R., Hauswirth W. W., and Lewin A. S. (2007) SOD2 knockdown mouse model of early AMD. Invest. Ophthalmol. Vis. Sci. 48, 4407–4420 10.1167/iovs.07-0432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dhar S. K., Tangpong J., Chaiswing L., Oberley T. D., and St Clair D. K. (2011) Manganese superoxide dismutase is a p53-regulated gene that switches cancers between early and advanced stages. Cancer Res. 71, 6684–6695 10.1158/0008-5472.CAN-11-1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. St Clair D. K., Wan X. S., Oberley T. D., Muse K. E., and St Clair W. H. (1992) Suppression of radiation-induced neoplastic transformation by overexpression of mitochondrial superoxide dismutase. Mol. Carcinog. 6, 238–242 10.1002/mc.2940060404 [DOI] [PubMed] [Google Scholar]
- 8. Zhao Y., Oberley T. D., Chaiswing L., Lin S. M., Epstein C. J., Huang T. T., and St Clair D. (2002) Manganese superoxide dismutase deficiency enhances cell turnover via tumor promoter-induced alterations in AP-1 and p53-mediated pathways in a skin cancer model. Oncogene 21, 3836–3846 10.1038/sj.onc.1205477 [DOI] [PubMed] [Google Scholar]
- 9. St Clair D., Zhao Y., Chaiswing L., and Oberley T. (2005) Modulation of skin tumorigenesis by SOD. Biomed. Pharmacother. 59, 209–214 10.1016/j.biopha.2005.03.004 [DOI] [PubMed] [Google Scholar]
- 10. Bakthavatchalu V., Dey S., Xu Y., Noel T., Jungsuwadee P., Holley A. K., Dhar S. K., Batinic-Haberle I., and St Clair D. K. (2012) Manganese superoxide dismutase is a mitochondrial fidelity protein that protects polγ against UV-induced inactivation. Oncogene 31, 2129–2139 10.1038/onc.2011.407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rodriguez-Rocha H., Garcia-Garcia A., Panayiotidis M. I., and Franco R. (2011) DNA damage and autophagy. Mutat. Res. 711, 158–166 10.1016/j.mrfmmm.2011.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vander Heiden M. G., Cantley L. C., and Thompson C. B. (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 10.1126/science.1160809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gonzalez Maglio D. H., Paz M. L., Ferrari A., Weill F. S., Czerniczyniec A., Leoni J., and Bustamante J. (2005) Skin damage and mitochondrial dysfunction after acute ultraviolet B irradiation: relationship with nitric oxide production. Photodermatol. Photoimmunol. Photomed. 21, 311–317 10.1111/j.1600-0781.2005.00185.x [DOI] [PubMed] [Google Scholar]
- 14. Wu S., Wang L., Jacoby A. M., Jasinski K., Kubant R., and Malinski T. (2010) Ultraviolet B light-induced nitric oxide/peroxynitrite imbalance in keratinocytes–implications for apoptosis and necrosis. Photochem. Photobiol. 86, 389–396 10.1111/j.1751-1097.2009.00682.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tanida I., Ueno T., and Kominami E. (2004) LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol. 36, 2503–2518 10.1016/j.biocel.2004.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Aoki H., Kondo Y., Aldape K., Yamamoto A., Iwado E., Yokoyama T., Hollingsworth E. F., Kobayashi R., Hess K., Shinojima N., Shingu T., Tamada Y., Zhang L., Conrad C., Bogler O., et al. (2008) Monitoring autophagy in glioblastoma with antibody against isoform B of human microtubule-associated protein 1 light chain 3. Autophagy 4, 467–475 10.4161/auto.5668 [DOI] [PubMed] [Google Scholar]
- 17. He C., and Klionsky D. J. (2009) Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 43, 67–93 10.1146/annurev-genet-102808-114910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Levine B., and Kroemer G. (2008) Autophagy in the pathogenesis of disease. Cell 132, 27–42 10.1016/j.cell.2007.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dhar S. K., Bakthavatchalu V., Dhar B., Chen J., Tadahide I., Zhu H., Gao T., and St Clair D. K. (2018) DNA polymerase γ (polγ) deficiency triggers a selective mTORC2 prosurvival autophagy response via mitochondria-mediated ROS signaling. Oncogene 37, 6225–6242 10.1038/s41388-018-0404-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sabatini D. M. (2006) mTOR and cancer: insights into a complex relationship. Nat. Rev. Cancer 6, 729–734 10.1038/nrc1974 [DOI] [PubMed] [Google Scholar]
- 21. MacMillan-Crow L. A., and Thompson J. A. (1999) Tyrosine modifications and inactivation of active site manganese superoxide dismutase mutant (Y34F) by peroxynitrite. Arch. Biochem. Biophys. 366, 82–88 10.1006/abbi.1999.1202 [DOI] [PubMed] [Google Scholar]
- 22. Chinnadurai G., Vijayalingam S., and Gibson S. B. (2008) BNIP3 subfamily BH3-only proteins: mitochondrial stress sensors in normal and pathological functions. Oncogene 27, Suppl. 1, S114–S127 10.1038/onc.2009.49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lippolis R., Siciliano R. A., Pacelli C., Ferretta A., Mazzeo M. F., Scacco S., Papa F., Gaballo A., Dell'Aquila C., De Mari M., Papa S., and Cocco T. (2015) Altered protein expression pattern in skin fibroblasts from parkin-mutant early-onset Parkinson's disease patients. Biochim. Biophys. Acta 1852, 1960–1970 10.1016/j.bbadis.2015.06.015 [DOI] [PubMed] [Google Scholar]
- 24. McWilliams T. G., Prescott A. R., Montava-Garriga L., Ball G., Singh F., Barini E., Muqit M. M. K., Brooks S. P., and Ganley I. G. (2018) Basal mitophagy occurs independently of PINK1 in mouse tissues of high metabolic demand. Cell Metab. 27, 439–449.e5 10.1016/j.cmet.2017.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ding W. X., and Yin X. M. (2012) Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol. Chem. 393, 547–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Matsuzawa Y., Oshima S., Takahara M., Maeyashiki C., Nemoto Y., Kobayashi M., Nibe Y., Nozaki K., Nagaishi T., Okamoto R., Tsuchiya K., Nakamura T., Ma A., and Watanabe M. (2015) TNFAIP3 promotes survival of CD4 T cells by restricting mTOR and promoting autophagy. Autophagy 11, 1052–1062 10.1080/15548627.2015.1055439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dangi A., Huang C., Tandon A., Stolz D., Wu T., and Gandhi C. R. (2016) Endotoxin-stimulated rat hepatic stellate cells induce autophagy in hepatocytes as a survival mechanism. J. Cell. Physiol. 231, 94–105 10.1002/jcp.25055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Duan H., Li Y., Yan L., Yang H., Wu J., Qian P., Li B., and Wang S. (2015) Rcan1–1L overexpression induces mitochondrial autophagy and improves cell survival in angiotensin II-exposed cardiomyocytes. Exp. Cell Res. 335, 99–106 10.1016/j.yexcr.2015.05.003 [DOI] [PubMed] [Google Scholar]
- 29. Laplante M., and Sabatini D. M. (2012) mTOR signaling in growth control and disease. Cell 149, 274–293 10.1016/j.cell.2012.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jung C. H., Ro S. H., Cao J., Otto N. M., and Kim D. H. (2010) mTOR regulation of autophagy. FEBS Lett. 584, 1287–1295 10.1016/j.febslet.2010.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang P., Guo Q. S., Wang Z. W., and Qian H. X. (2013) HBx induces HepG-2 cells autophagy through PI3K/Akt-mTOR pathway. Mol. Cell. Biochem. 372, 161–168 10.1007/s11010-012-1457-x [DOI] [PubMed] [Google Scholar]
- 32. Matsuda-Lennikov M., Suizu F., Hirata N., Hashimoto M., Kimura K., Nagamine T., Fujioka Y., Ohba Y., Iwanaga T., and Noguchi M. (2014) Lysosomal interaction of Akt with Phafin2: a critical step in the induction of autophagy. PLoS ONE 9, e79795 10.1371/journal.pone.0079795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pua H. H., Guo J., Komatsu M., and He Y. W. (2009) Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J. Immunol. 182, 4046–4055 10.4049/jimmunol.0801143 [DOI] [PubMed] [Google Scholar]
- 34. Fan Q. W., and Weiss W. A. (2011) Autophagy and Akt promote survival in glioma. Autophagy 7, 536–538 10.4161/auto.7.5.14779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rajic Z., Tovmasyan A., Spasojevic I., Sheng H., Lu M., Li A. M., Gralla E. B., Warner D. S., Benov L., and Batinic-Haberle I. (2012) A new SOD mimic, Mn(III) ortho N-butoxyethylpyridylporphyrin, combines superb potency and lipophilicity with low toxicity. Free Radic. Biol. Med. 52, 1828–1834 10.1016/j.freeradbiomed.2012.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Batinic-Haberle I., Tovmasyan A., and Spasojevic I. (2018) Mn porphyrin-based redox-active drugs: differential effects as cancer therapeutics and protectors of normal tissue against oxidative injury. Antioxid. Redox Signal. 29, 1691–1724 10.1089/ars.2017.7453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Colburn N. H., Former B. F., Nelson K. A., and Yuspa S. H. (1979) Tumour promoter induces anchorage independence irreversibly. Nature 281, 589–591 10.1038/281589a0 [DOI] [PubMed] [Google Scholar]
- 38. Dhar S. K., Xu Y., and St Clair D. K. (2010) Nuclear factor κB- and specificity protein 1-dependent p53-mediated bi-directional regulation of the human manganese superoxide dismutase gene. J. Biol. Chem. 285, 9835–9846 10.1074/jbc.M109.060715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Spitz D. R., and Oberley L. W. (2001) Measurement of MnSOD and CuZnSOD activity in mammalian tissue homogenates. Curr. Protoc. Toxicol. Chapter 7, Unit 7.5 10.1002/0471140856.tx0705s08 [DOI] [PubMed] [Google Scholar]
- 40. Beauchamp C., and Fridovich I. (1971) Superoxide dismutase improved assays and an assay applicable to polyacrylamide gel. Anal. Biochem. 44, 276–287 10.1016/0003-2697(71)90370-8 [DOI] [PubMed] [Google Scholar]
- 41. Uhlen M., Fagerberg L., Hallstrom B. M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson A., Kampf C., Sjostedt E., Asplund A., Olsson I., Edlund K., Lundberg E., Navani S., Szigyarto C. A., et al. (2015) Proteomics: Tissue-based map of the human proteome. Science 347, 1260419 10.1126/science.1260419 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.