

Abstract
IL-32 is a cytokine involved in proinflammatory immune responses to bacterial and viral infections. However, the role of epigenetic events in the regulation of IL-32 gene expression is understudied. Here we show that IL-32 is repressed by DNA methylation in HEK293 cells. Using ChIP sequencing, locus-specific methylation analysis, CRISPR/Cas9-mediated genome editing, and RT-qPCR (quantitative RT-PCR) and immunoblot assays, we found that short-term treatment (a few hours) with the proinflammatory cytokine tumor necrosis factor α (TNFα) activates IL-32 in a DNA demethylation–independent manner. In contrast, prolonged TNFα treatment (several days) induced DNA demethylation at the promoter and a CpG island in the IL-32 gene in a TET (ten-eleven translocation) family enzyme– and NF-κB–dependent manner. Notably, the hypomethylation status of transcriptional regulatory elements in IL-32 was maintained for a long time (several weeks), causing elevated IL-32 expression even in the absence of TNFα. Considering that IL-32 can, in turn, induce TNFα expression, we speculate that such feedforward events may contribute to the transition from an acute inflammatory response to chronic inflammation.
Keywords: DNA methylation, DNA demethylation, tumor necrosis factor (TNF), gene regulation, NF-κB, inflammation, epigenetics, transcription, gene activation, IL-32, TNFα
Introduction
IL-32 is a proinflammatory cytokine (1–4). The IL-32 gene emerges quite late during evolution and exists only in certain mammals such as humans, chimpanzees, cattle, and horses; however, it does not exist in rodents (5, 6). Moreover, IL-32 shares little sequence identity with other interleukins (1, 5).
Consistent with a role of IL-32 in the inflammatory response, IL-32 expression is induced by TNFα3 in various human cell types, including synovial fibroblasts, intestinal epithelial cell lines, and pancreatic cancer cell lines (7–9). Reciprocally, IL-32 can also induce the expression of TNFα and other cytokines in human THP-1 monocytic cells (1). Interestingly, although mice do not contain the IL-32 gene, ectopic treatment with human IL-32 can induce TNFα expression in mouse Raw macrophage cells (1). Moreover, injection of human IL-32 protein into the knee joints of WT mice, but not into the knee joints of Tnf gene knockout mice, provokes severe inflammation, suggesting that IL-32 exerts direct effects on joint inflammation in a TNFα-dependent manner (2). Functionally, IL-32 promotes the differentiation of monocytes toward macrophage-like cells that display phagocytic activity, further supporting a role of IL-32 in the immune response (10).
IL-32 plays important roles in inflammatory autoimmune diseases (11, 12). IL-32 is highly expressed in rheumatoid arthritis synovial tissue biopsies (2), inflamed mucosa of inflammatory bowel disease (9), and chronic pancreatitis duct cells (8). These reports suggest that IL-32 is likely a cytokine involved in chronic inflammation and that it may serve as a potential therapeutic target.
As a proinflammatory cytokine, the expression of IL-32 is induced during bacterial and viral infections, and its expression improves host immunity in controlling these infections (13). For example, in patients with active Mycobacterium tuberculosis infection, IL-32 expression is induced, and it protects human macrophages and peripheral blood mononuclear cells against M. tuberculosis (14–16). Likewise, the expression of IL-32 is induced during HIV infection and influenza virus infection, as it contributes to the antiviral response (4, 17, 18).
DNA methylation is an important gene silencing mechanism that functions by recruiting corepressor proteins to impede the binding of DNA methylation–sensitive transcription factors (19, 20). DNA demethylation can be achieved by enzyme-mediated active demethylation or by passive DNA demethylation caused by interfering with maintenance DNA methylation (21). TET family methylcytosine dioxygenases catalyze active DNA demethylation through the sequential oxidation of 5mC (5-methylcytosine) to 5hmC (5-hydroxymethylcytosine), 5fC (5-formylcytosine), and 5caC (5-carboxylcytosine) (22–24), followed by TDG (thymine DNA glycosylase)-mediated base excision repair (24).
Gene expression is often regulated by sequence-specific transcription factors and epigenetic regulators. Given that IL-32 expression is regulated during inflammation, understanding whether epigenetic events occur during the induction of IL-32 expression is interesting. Here we report that IL-32 is silenced by DNA methylation and that TNFα induces DNA demethylation–dependent and –independent mechanisms to control IL-32 activation. We also discuss the potential significance of these mechanisms.
Results
IL-32 is silenced by DNA methylation in HEK293 cells
In our previous work, we performed RNA-Seq experiments using HEK293 cells treated with the DNA-demethylating agent 5-aza-2′-deoxycytidine (5-aza-dC) and identified genes silenced by DNA methylation (25, 26). IL-32 was one of the genes strongly activated upon 5-aza-dC treatment (Figs. 1, A and B), suggesting that IL-32 is a gene silenced by DNA methylation in HEK293 cells. Indeed, bisulfite sequencing data revealed that both the promoter and CpG island (CGI) predicted by the Sequence Manipulation Suite (27) of the IL-32 gene (Fig. 1C) are highly methylated (Fig. 1D).
Figure 1.
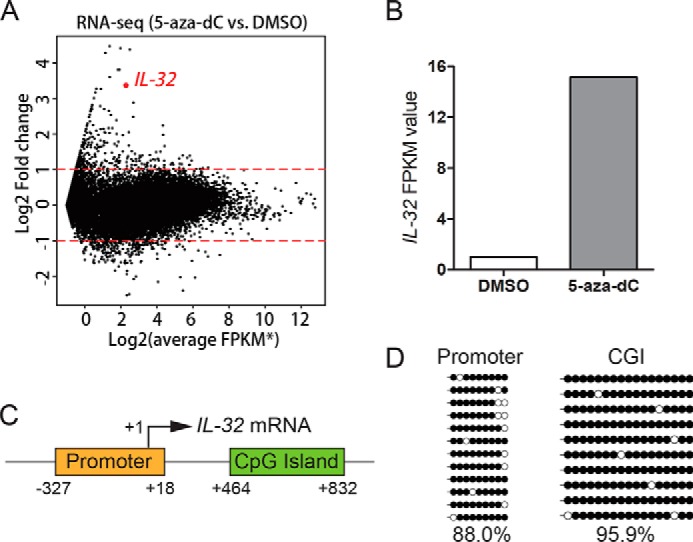
The IL-32 gene is silenced by DNA methylation in HEK293 cells. A, RNA-Seq results showed that 5-aza-dC treatment activates IL-32 expression in HEK293 cells. The asterisk indicates that the FPKM values were added a pseudocount of 0.5 to avoid being divided by zero. B, IL-32 FPKM values in 5-aza-dC- and DMSO-treated samples. C, schematic representation of the IL-32 promoter and CGI. D, locus-specific bisulfite sequencing results revealed that the promoter and CGI of IL-32 are highly methylated in HEK293 cells.
Short-term TNFα treatment induces IL-32 expression in a DNA demethylation–independent manner
Given that the IL-32 gene is induced by TNFα treatment (7–9) and repressed by DNA methylation in HEK293 cells, we wondered whether TNFα treatment is sufficient to overcome DNA methylation–mediated silencing. Thus, we treated HEK293 cells with 50 ng/ml TNFα and analyzed IL-32 expression at various time points. IL-32 expression began to be induced as early as 1 h post-TNFα treatment and was potently activated after 3 h of TNFα treatment (Fig. 2A).
Figure 2.
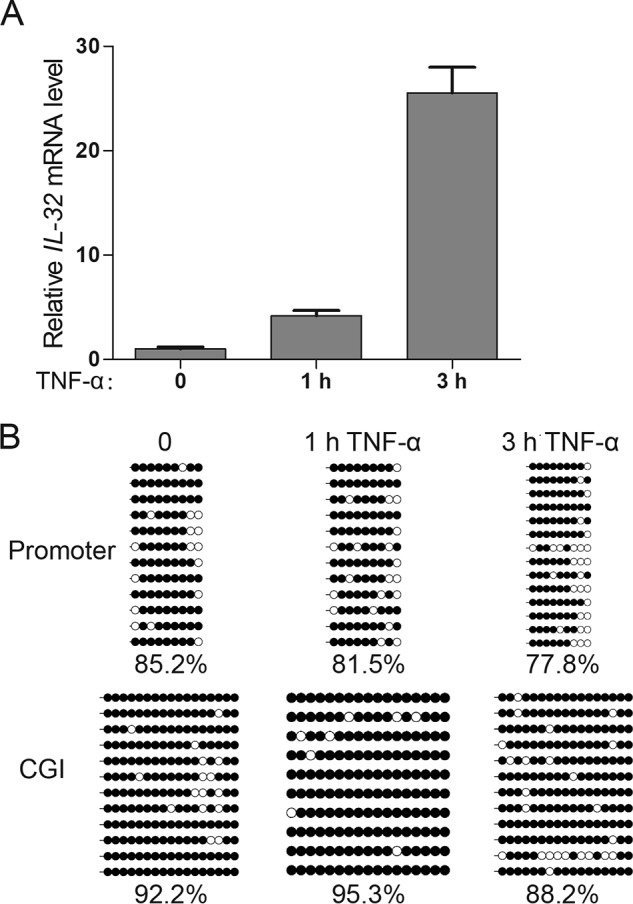
TNFα treatment overcomes DNA methylation–mediated silencing and activates IL-32 expression. A, RT-qPCR results showed that IL-32 expression is quickly activated upon TNFα treatment. Averages from three independent experiments are shown, and error bars represent standard deviation. B, locus-specific bisulfite sequencing data showed that the IL-32 transcriptional regulatory regions remain largely methylated upon 1-h TNFα treatment and that the IL-32 promoter is slightly demethylated after 3-h TNFα treatment.
We next wanted to find out whether IL-32 activation was accompanied by DNA demethylation. Interestingly, despite the apparent transcriptional activation, no substantial DNA demethylation at the promoter or CGI of the IL-32 gene was observed after 1 h of TNFα treatment (Fig. 2, A and B). These results indicate that TNFα treatment could activate IL-32 gene expression in a DNA demethylation–independent manner. We then examined THP-1 cells, a human monocyte-like cell line (28), and HAP1 cells, a human leukemia cell line (29) and also observed DNA demethylation–independent activation of IL-32 expression upon short-term TNFα treatment in these cells (Fig. S1).
Long-term TNFα treatment induces significant DNA demethylation of the IL-32 transcriptional regulatory region
We noticed a slight decrease in DNA methylation at the IL-32 promoter after 3 h of TNFα treatment (Fig. 2B). This finding prompted us to perform longer TNFα treatments with measurements of IL-32 expression and DNA methylation at various time points. As we anticipated, long-term TNFα treatment (12 days) resulted in clear DNA demethylation of the IL-32 transcriptional regulatory regions; furthermore, the accumulation of DNA demethylation was accompanied by IL-32 induction (Figs. 3, A–C).
Figure 3.

IL-32 transcriptional regulatory regions undergo DNA demethylation during 12 days of TNFα treatment. A, RT-qPCR analysis showed that the IL-32 mRNA level can be more efficiently activated via long-term TNFα treatment. Averages from three independent experiments are shown, and error bars represent standard deviation. B, Western blot results showed that the IL-32 protein level can be induced with 12 h and 12 days of TNFα treatment. C, locus-specific bisulfite sequencing results revealed that the promoter and CGI of the IL-32 gene are gradually demethylated during long-term TNFα treatment. Filled circles indicate methylated CpG sites, and open circles indicate unmethylated CpG sites. CpG site methylation percentages are shown.
Hypomethylation triggered by long-term TNFα treatment leads to elevated IL-32 expression after the removal of TNFα
DNA methylation is a relatively stable epigenetic mark; therefore, we wanted to find out whether the methylation status of the IL-32 transcriptional regulatory regions could be stably maintained after TNFα treatment. We treated HEK293 cells with TNFα for 12 h or 12 days and then cultured the cells in TNFα-free medium for an additional 10-day period. Bisulfite sequencing data revealed that the promoter and CGI of the IL-32 gene remained largely hypomethylated in the cells that underwent 12 days of TNFα treatment and 10 days of withdrawal (Fig. 4A), indicating that TNFα-induced DNA demethylation could be maintained for a considerable period of time.
Figure 4.

IL-32 basal expression is up-regulated after long-term TNFα treatment and is accompanied by sustained hypomethylation at the promoter and CGI. A, locus-specific bisulfite sequencing data showed that the hypomethylation status of the IL-32 promoter and CGI can be maintained after 10 days of TNFα withdrawal. B, a time course experiment revealed that the IL-32 basal expression level is up-regulated after long-term TNFα treatment and TNFα withdrawal. Averages from three independent experiments are shown, and error bars represent standard deviation in the RT-qPCR results. d, day. C, Western blot results showed that cells treated long-term with TNFα display a higher basal protein expression level of IL-32. D, RT-qPCR results revealed that the up-regulation of IL-32 expression can be maintained for at least 30 days after TNFα withdrawal. Averages from three independent experiments are shown, and error bars represent standard deviation. E, bisulfite sequencing data revealed that cells subjected to 12 days of TNFα treatment maintained relatively low methylation levels at the promoter and CGI of the IL-32 gene even after 30 days of TNFα withdrawal.
Moreover, we noticed that prior exposure to long-term TNFα treatment led to elevated basal IL-32 expression, even after 10 days of TNFα withdrawal (Fig. 4, B and C). These results indicated that long-term TNFα treatment not only caused a stable epigenetic change but also led to a sustained change in basal expression of the IL-32 gene. Similarly, long-term TNFα treatment caused DNA demethylation and elevated basal expression of the IL-32 gene in HAP1 cells (Fig. S2).
To determine whether the above effect could be maintained for an even longer period, we treated HEK293 cells with TNFα for 12 days and then cultured them in TNFα-free medium for 10, 18, or 30 days. RT-qPCR results showed that the up-regulation of IL-32 was maintained after 10, 18, and 30 days, although the up-regulated level became more moderate after 30 days (Fig. 4D). Consistently, the DNA methylation level of the IL-32 promoter and CpG island began to increase after 30 days of TNFα withdrawal (Fig. 4E). Taken together, these results suggest that long-term TNFα treatment can induce heritable hypomethylation at the promoter and CpG island of the IL-32 gene, causing long-term transcriptional alteration.
TET enzymes mediate IL-32 demethylation during long-term TNFα treatment
DNA demethylation can be achieved by passive demethylation, TET enzyme–mediated active oxidation and demethylation, or both (21, 30). To find out whether passive demethylation was involved in TNFα induced demethylation, we attempted to arrest the cells at S phase and simultaneously treated the cells with TNFα. Unfortunately, these cells suffered from severe cell death, and we were unable to draw a clear conclusion about whether there was any involvement of passive demethylation.
To determine whether DNA demethylation at the promoter and CpG island of the IL-32 gene was mediated by TET enzymes, we generated TET1 KO, TET2 KO, TET3 KO, and TET1/2/3 triple knockout (TKO) cells using the CRISPR-Cas9 system. In these cells, frameshift mutations were introduced at the C terminus of the TET family proteins to abrogate their catalytic activity (Figs. S3 and S4, A and B).
We then performed bisulfite sequencing, and the results revealed that the DNA demethylation induced by TNFα treatment at the IL-32 gene promoter and CGI was largely abrogated in TET TKO cells, with the single knockouts each displaying varied partial defects (Fig. 5A). These results suggested that the TET enzymes function together to promote TNFα-induced IL-32 gene demethylation. We also confirmed that there was no up-regulation of DNMT genes in TET TKO cells by RNA-Seq experiments (Fig. S4C).
Figure 5.

TET enzymes mediate DNA demethylation, leading to up-regulated IL-32 basal expression upon long-term TNFα treatment. A, locus-specific bisulfite sequencing results showed that TET enzymes are responsible for the DNA demethylation events during long-term TNFα treatment. B, RT-qPCR results showed that the up-regulated IL-32 expression that occurred after long-term TNFα treatment depends on TET enzymes. Averages from three independent experiments are shown, and error bars represent standard deviation. d, day.
We next wanted to find out whether IL-32 gene demethylation mediated by TET enzymes was responsible for the elevated IL-32 expression levels in cells recovered from long-term TNFα treatment. Although IL-32 expression was induced by 12 h or 12 days of TNFα treatment in all of the above cells (Fig. S5), elevated IL-32 basal expression was not observed in TET TKO cells withdrawn from long-term TNFα treatment (Fig. 5B). These results are consistent with the methylation states of the promoter and CGI of the IL-32 gene in these cells and support that long-term TNFα treatment induces DNA demethylation at the transcriptional regulatory regions of the IL-32 gene, elevating its basal expression level.
NF-κB–dependent transcriptional activation contributes to IL-32 gene demethylation and long-term elevation of its basal expression
TNFα activates the NF-κB signaling pathway and induces nuclear translocation of the canonical p50/p65 heterodimer (31–36). Interestingly, a p65 binding site (κB site) is located in the promoter of the IL-32 gene (Fig. 6A), and its presence was confirmed by our p65 ChIP-Seq results (Fig. 6B). Therefore, we knocked out the RELA gene, which encodes p65, in HEK293 cells using the CRISPR-Cas9 system (Figs. S6, A and B, and Table S4) and verified the cells using sequencing (Fig. S6C) and Western blotting (Fig. 6C). RT-qPCR data revealed that TNFα-mediated IL-32 activation was significantly impaired in RELA KO cells (Fig. 6D), indicating that p65 is the predominant transcription factor mediating IL-32 induction in response to TNFα. Moreover, in RELA KO cells treated with TNFα for 12 days, the levels of DNA demethylation at the transcriptional regulatory regions of IL-32 were reduced, especially at the CGI of the IL-32 gene (Fig. 6E). The impaired DNA demethylation at the CGI of the IL-32 gene was accompanied by less elevated basal expression of IL-32 in long-term TNFα-treated RELA KO cells (Fig. 6F). These data collectively support that TNFα-induced NF-κB signaling pathway activation leads to DNA demethylation–independent short-term activation and DNA demethylation–dependent elevation of IL-32 basal transcription in the absence of initial TNFα treatment.
Figure 6.

NF-κB–dependent transcriptional activation promotes DNA demethylation and results in IL-32 up-regulation after long-term TNFα treatment. A, schematic representation of a κB site (GGGAGTTTCC) in the IL-32 promoter. B, p65 ChIP-Seq results showed that p65 is enriched at the κB site of the IL-32 promoter after 12 h of TNFα treatment. C, Western blot data validating the RELA KO cell line. D, RT-qPCR results revealed impaired IL-32 induction in RELA KO cells. Averages from three independent experiments are shown, and error bars represent standard deviation. E, locus-specific bisulfite sequencing results showed that the IL-32 CpG island DNA demethylation reaction that occurs during long-term TNFα stimulation is impaired in RELA KO cells. F, RT-qPCR results showed that up-regulation of IL-32 transcription after long-term TNFα treatment is impaired in RELA KO cells. Averages from three independent experiments are shown, and error bars represent standard deviation. d, day.
Transcription factor–induced DNA demethylation has been widely reported (37–50). In certain cases, these transcription factors can associate with TET enzymes (42–47, 49). In some other cases, no direct evidence supporting the association between transcription factors and TET enzymes is provided (41, 50). We expressed FLAG-TET1, FLAG-TET2, or FLAG-TET3 in HEK293 cells and stimulated the cells for 12 h with TNFα. Then we performed immunoprecipitation experiments with p65 and the TET enzymes, but we did not observe any robust interaction. On the other hand, increased chromatin accessibility has been reported to facilitate DNA demethylation mediated by TET enzymes (51–54). We measured chromatin accessibility at the IL-32 promoter by formaldehyde-assisted isolation of regulatory elements assay (55) and observed increased chromatin accessibility in response to 12-h TNFα treatment (Fig. S6D and Table S5). Thus, we speculate that p65-induced chromatin opening contributes to DNA demethylation mediated by TET enzymes.
CREB and the cAMP response element (CRE) at the IL-32 promoter are not required for elevated IL-32 basal expression upon long-term TNFα treatment
The CpG site within the CRE of the IL-32 promoter has been reported to be demethylated during influenza A virus infection, which increases transcription factor CREB binding (4). We wondered whether this CpG site within the CRE was also a target for TNFα-induced demethylation, playing a role in long-term activation of the IL-32 gene. Therefore, we examined the CRE in the IL-32 promoter (Fig. S7A) and confirmed its demethylation by TNFα treatment (Figs. 2B, 3C, 4A, and 5A). We next asked whether this CRE mediates the up-regulation of IL-32 transcription through long-term TNFα stimulation. Frameshift mutations were introduced in both alleles of the CREB1 gene to disrupt CREB binding to the CRE (Fig. S7B). However, the RT-qPCR results revealed normal IL-32 activation by TNFα in CREB1 KO cells (Fig. S7, C and D).
In addition, we also mutated this CRE within the IL-32 promoter from TGACGTCA to TTTCGTCA (Fig. S7E). Again, RT-qPCR revealed a largely normal elevation of IL-32 basal expression after long-term TNFα treatment (Fig. S7F). Collectively, these data suggest that the long-term effect of TNFα treatment is not solely dependent on DNA demethylation of the CpG site within the CRE of the IL-32 promoter.
Discussion
Signaling events triggered by environmental cues are well known for their roles in transcriptional regulation. In most cases, the majority of transcriptional changes triggered by signals are reset, and target gene expression returns to its initial basal level upon withdrawal of the environmental cues that initiated the signaling events (56, 57). However, sometimes signaling events can also trigger lasting epigenetic changes that facilitate a long-term effect (56–60), which is an interesting field termed “signal to chromatin” (61–63).
DNA methylation is certainly one of the most stable epigenetic marks that can mediate a lasting effect. In recent years, increasing evidence has supported the role of transcription factor binding in facilitating DNA demethylation in neighboring regions (37–50) as well as the role of signaling events in stimulating DNA demethylation (64). However, reports of a full axis from signal to transcription factor to DNA demethylation to a lasting transcriptional change in the absence of the initiating signal are still limited (58). Here we report one such case: an axis involving a TNFα signal, NF-κB pathway activation and association of p65 at the IL-32 promoter, TET enzyme–mediated IL-32 gene demethylation, and long-term activation of IL-32 expression (Fig. 7).
Figure 7.

A model for DNA demethylation–dependent and –independent activation of IL-32 expression upon TNFα treatment.
In addition to the abovementioned case, the discovery of DNA demethylation–dependent and –independent mechanisms involved in activating IL-32 expression may have additional significance worthy of further investigation. As a TNFα target, IL-32 has been reported to reciprocally induce the expression of TNFα in certain cell types (1). We suspect that, under certain in vivo situations, a strong acute inflammation event or the cumulative effect of several acute inflammation events may lead to demethylation of the IL-32 gene and a lasting elevation of IL-32 basal expression, which may, in turn, stimulate TNFα expression in these cells or neighboring cells. Such a self-reinforcing feedforward loop may well contribute to the conversion from acute inflammation to chronic inflammation. Understanding the potential mechanisms governing the conversion from acute inflammation to chronic inflammation is highly important because of its relevance to human health. Although this study does not offer a clear answer for this important question, it provides an interesting direction for future exploration. One obvious difficulty in following up this study is the lack of a mouse model. The IL-32 gene does not exist in rodents (11), and follow-up studies will likely focus on human diseases. Therefore, one key question is what kind of pathological conditions may be relevant to our observations. We reason that chronic inflammatory diseases and autoimmune diseases are potential candidates on which to focus.
TNFα antagonists, including soluble receptors and antibodies, have excellent efficacy for treatment of chronic inflammatory diseases (e.g. rheumatoid arthritis and inflammatory bowel disease) (65, 66). Establishing a connection between TNFα-induced demethylation and long-term activation of proinflammatory genes, including but not limited to IL-32, in any of the above diseases would be highly interesting.
To offer a mechanistic answer for TNFα-induced long-term gene activation in the absence of TNFα, our model is missing one piece. We reason that the long-term effect of TNFα was due to DNA demethylation that facilitated the association of transcription factor(s) sensitive to DNA methylation. However, in this case, we do not yet know the identity of such transcription factor(s). The CREB binding site in the CRE of the IL-32 promoter and its association with CREB provided an ideal candidate, particularly because this site was found to be demethylated in A549 cells infected with influenza virus (4), and the association of CREB with the CRE is DNA methylation–sensitive (67, 68). However, in our case, this site does not appear to be the sole answer because neither mutation of the CREB gene nor mutation of the CRE site in the IL-32 promoter caused sufficient changes (Fig. S7). Future studies in this direction are of great interest.
We also performed HPLC-MRM (multiple reaction monitoring) MS/MS experiments at various time points following TNFα treatment and observed a gradual subtle decline of the global 5mC level (Fig. S8). Obviously, TNFα treatment–induced DNA demethylation is not restricted to the IL-32 gene. The identification of other potential targets and their biological significance are interesting topics for future investigation.
Experimental procedures
Cell culture
HEK293 cells were cultured in DMEM/high glucose (HyClone, catalog no. SH30022.01) supplemented with 10% fetal bovine serum (Biological Industries, catalog no. 04-010-1ACS) and a penicillin–streptomycin solution (BBI Life Sciences, catalog no. E607011-0100). Recombinant human TNFα (Peprotech, catalog no. 300-01A) was used at a final concentration of 50 ng/ml. For long-term TNFα stimulation, TNFα was added to the culture medium immediately after each passage.
Antibodies
Antibodies against IL-32 (Abcam, catalog no. ab172339), p65 (Santa Cruz Biotechnology, catalog no. sc-372), and histone H3 (Abcam, catalog no. ab1791) are commercially available.
ChIP-Seq
ChIP experiments were performed with HEK293 cells using procedures described previously (69). ChIP-Seq libraries were constructed with a Kapa Hyper Prep Kit (Kapa Biosystems, catalog no. KK8504) and NEBNext multiplex oligos for Illumina (index primer set 1, New England Biolabs, catalog no. E7335). Libraries were sequenced via NovaSeq using the 150-bp paired-end mode.
Bioinformatics
50-bp single-end reads were generated by BGISEQ-500 platforms for mRNA sequencing experiments (BGI, Shenzhen, China). Sequencing quality was evaluated with FastQC software and aligned to human genome hg38 using STAR Aligner. FPKM values were quantified using Cuffdiff (v2.0.2). FPKM values were added to a pseudovalue of 0.5 to avoid being divided by zero. ChIP-Seq reads were generated by Illumina NovaSeq-6000 platforms (paired end, 150 bp). Adaptors were removed by Trim_galore software and then aligned to hg38 genome sequences (<2-bp mismatches allowed) with Bowtie2. Uniquely mapped reads were kept and then extended to the average fragment size. Genome profile files were generated with IGV (integrative genomics viewer) tools and linearly normalized to the same depth of 10 million reads.
IL-32 locus-specific methylation analysis
To perform IL-32 promoter and CpG island (Table S1) locus-specific methylation analysis, purified genomic DNA was treated with an EpiTect Bisulfite Kit (Qiagen, catalog no. 59104), and the converted DNA was amplified using locus-specific nested PCR primers (Table S2). Purified PCR products were cloned, sequenced, and then analyzed using a BiQ Analyzer (70).
Genome editing using the CRISPR-Cas9 system
To generate RELA knockout, CREB1 frameshift mutant, IL-32 promoter CRE mutant, and TET frameshift mutant cell lines, guide RNA sequences (Table S3) were designed and cloned into lentiCRISPR v2 vectors (Addgene, 52961) (71). Individual clones were verified by genotyping PCR and Sanger sequencing.
Primers for RT-qPCR
The sequences of primers used for RT-qPCR included the following: IL-32 forward, TGGCGGCTTATTATGAGGAGC; IL-32 reverse, CTCGGCACCGTAATCCATCTC; GAPDH forward, CTGGGCTACACTGAGCACC; GAPDH reverse, AAGTGGTCGTTGAGGGCAATG.
Author contributions
Z. Zhao, G. L., Z. Zhang, and B. Z. conceptualization; Z. Zhao, Z. Zhang, and B. Z. data curation; Z. Zhao and Z. Zhang software; Z. Zhao, Z. Zhang, and B. Z. formal analysis; Z. Zhao validation; Z. Zhao, Z. Zhang, and B. Z. investigation; Z. Zhao and Z. Zhang visualization; Z. Zhao, B. L., and Z. Zhang methodology; Z. Zhao, Z. Zhang, and B. Z. writing-original draft; Z. Zhao, M. L., J. L., Q. D., X. L., B. L., and Z. Zhang project administration; Q. D. and X. L. resources; G. L., H. W., Z. Zhang, and B. Z. supervision; G. L., Z. Zhang, and B. Z. funding acquisition; G. L., Z. Zhang, and B. Z. writing-review and editing.
Supplementary Material
This work was supported by the NSFC-FDCT Joint Grants 31761163001 (to B. Z.) and 033/2017/AFJ (to G. L.). This work was also supported by Ministry of Science and Technology of the People's Republic of China Grants 2016YFA0100400, 2015CB856200, and 2017YFA0504200; National Natural Science Foundation of China Grants 31521002, 31425013, 31730047, and 31571344; and Chinese Academy of Sciences Grants XDB08010103, XDBP10, and QYZDY-SSW-SMC031). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S8 and Tables S1–S5.
All high-throughput sequencing data have been deposited in the GEO under accession number GSE121361.
- TNFα
- tumor necrosis factor α
- 5-aza-dC
- 5-aza-2′-deoxycytidine
- CGI
- CpG island
- TKO
- triple knockout
- CREB
- cAMP response element–binding protein
- CRE
- cAMP response element
- RT-qPCR
- quantitative RT-PCR
- TET
- ten-eleven translocation
- FPKM
- fragments per kilobase per million mapped fragments.
References
- 1. Kim S. H., Han S. Y., Azam T., Yoon D. Y., and Dinarello C. A. (2005) Interleukin-32: a cytokine and inducer of TNFα. Immunity 22, 131–142 10.1016/j.immuni.2004.12.003 [DOI] [PubMed] [Google Scholar]
- 2. Joosten L. A., Netea M. G., Kim S. H., Yoon D. Y., Oppers-Walgreen B., Radstake T. R., Barrera P., van de Loo F. A., Dinarello C. A., and van den Berg W. B. (2006) IL-32, a proinflammatory cytokine in rheumatoid arthritis. Proc. Natl. Acad. Sci. U.S.A. 103, 3298–3303 10.1073/pnas.0511233103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li W., Liu Y., Mukhtar M. M., Gong R., Pan Y., Rasool S. T., Gao Y., Kang L., Hao Q., Peng G., Chen Y., Chen X., Wu J., and Zhu Y. (2008) Activation of interleukin-32 pro-inflammatory pathway in response to influenza A virus infection. PLoS ONE 3, e1985 10.1371/journal.pone.0001985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li W., Sun W., Liu L., Yang F., Li Y., Chen Y., Fang J., Zhang W., Wu J., and Zhu Y. (2010) IL-32: a host proinflammatory factor against influenza viral replication is upregulated by aberrant epigenetic modifications during influenza A virus infection. J. Immunol. 185, 5056–5065 10.4049/jimmunol.0902667 [DOI] [PubMed] [Google Scholar]
- 5. Joosten L. A., Heinhuis B., Netea M. G., and Dinarello C. A. (2013) Novel insights into the biology of interleukin-32. Cell. Mol. Life Sci. 70, 3883–3892 10.1007/s00018-013-1301-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bai X., Shang S., Henao-Tamayo M., Basaraba R. J., Ovrutsky A. R., Matsuda J. L., Takeda K., Chan M. M., Dakhama A., Kinney W. H., Trostel J., Bai A., Honda J. R., Achcar R., Hartney J., et al. (2015) Human IL-32 expression protects mice against a hypervirulent strain of Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 112, 5111–5116 10.1073/pnas.1424302112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Heinhuis B., Koenders M. I., van Riel P. L., van de Loo F. A., Dinarello C. A., Netea M. G., van den Berg W. B., and Joosten L. A. (2011) Tumour necrosis factor α-driven IL-32 expression in rheumatoid arthritis synovial tissue amplifies an inflammatory cascade. Ann. Rheum. Dis. 70, 660–667 10.1136/ard.2010.139196 [DOI] [PubMed] [Google Scholar]
- 8. Nishida A., Andoh A., Inatomi O., and Fujiyama Y. (2009) Interleukin-32 expression in the pancreas. J. Biol. Chem. 284, 17868–17876 10.1074/jbc.M900368200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shioya M., Nishida A., Yagi Y., Ogawa A., Tsujikawa T., Kim-Mitsuyama S., Takayanagi A., Shimizu N., Fujiyama Y., and Andoh A. (2007) Epithelial overexpression of interleukin-32α in inflammatory bowel disease. Clin. Exp. Immunol. 149, 480–486 10.1111/j.1365-2249.2007.03439.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Netea M. G., Lewis E. C., Azam T., Joosten L. A., Jaekal J., Bae S. Y., Dinarello C. A., and Kim S. H. (2008) Interleukin-32 induces the differentiation of monocytes into macrophage-like cells. Proc. Natl. Acad. Sci. U.S.A. 105, 3515–3520 10.1073/pnas.0712381105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim S. (2014) Interleukin-32 in inflammatory autoimmune diseases. Immune Network 14, 123–127 10.4110/in.2014.14.3.123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hong J. T., Son D. J., Lee C. K., Yoon D. Y., Lee D. H., and Park M. H. (2017) Interleukin 32, inflammation and cancer. Pharmacol. Ther. 174, 127–137 10.1016/j.pharmthera.2017.02.025 [DOI] [PubMed] [Google Scholar]
- 13. Ribeiro-Dias F., Saar Gomes R., de Lima Silva L. L., Dos Santos J. C., and Joosten L. A. (2017) Interleukin 32: a novel player in the control of infectious diseases. J. Leukoc. Biol. 101, 39–52 10.1189/jlb.4RU0416-175RR [DOI] [PubMed] [Google Scholar]
- 14. Netea M. G., Azam T., Lewis E. C., Joosten L. A., Wang M., Langenberg D., Meng X., Chan E. D., Yoon D. Y., Ottenhoff T., Kim S. H., and Dinarello C. A. (2006) Mycobacterium tuberculosis induces interleukin-32 production through a caspase-1/IL-18/interferon-γ-dependent mechanism. PLoS Med. 3, e277 10.1371/journal.pmed.0030277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bai X., Kim S. H., Azam T., McGibney M. T., Huang H., Dinarello C. A., and Chan E. D. (2010) IL-32 is a host protective cytokine against Mycobacterium tuberculosis in differentiated THP-1 human macrophages. J. Immunol. 184, 3830–3840 10.4049/jimmunol.0901913 [DOI] [PubMed] [Google Scholar]
- 16. Montoya D., Inkeles M. S., Liu P. T., Realegeno S., Teles R. M., Vaidya P., Munoz M. A., Schenk M., Swindell W. R., Chun R., Zavala K., Hewison M., Adams J. S., Horvath S., Pellegrini M., et al. (2014) IL-32 is a molecular marker of a host defense network in human tuberculosis. Sci. Transl. Med. 6, 250ra114 10.1126/scitranslmed.3009546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nold M. F., Nold-Petry C. A., Pott G. B., Zepp J. A., Saavedra M. T., Kim S. H., and Dinarello C. A. (2008) Endogenous IL-32 controls cytokine and HIV-1 production. J. Immunol. 181, 557–565 10.4049/jimmunol.181.1.557 [DOI] [PubMed] [Google Scholar]
- 18. Rasool S. T., Tang H., Wu J., Li W., Mukhtar M. M., Zhang J., Mu Y., Xing H. X., Wu J., and Zhu Y. (2008) Increased level of IL-32 during human immunodeficiency virus infection suppresses HIV replication. Immunol. Lett. 117, 161–167 10.1016/j.imlet.2008.01.007 [DOI] [PubMed] [Google Scholar]
- 19. Li E., and Zhang Y. (2014) DNA methylation in mammals. Cold Spring Harb. Perspect. Biol. 6, a019133 10.1101/cshperspect.a019133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang C., Zhu B., and Xiong J. (2018) Recruitment and reinforcement: maintaining epigenetic silencing. Sci. China Life Sci. 61, 515–522 10.1007/s11427-018-9276-7 [DOI] [PubMed] [Google Scholar]
- 21. Wu X., and Zhang Y. (2017) TET-mediated active DNA demethylation: mechanism, function and beyond. Nat. Rev. Genet. 18, 517–534 10.1038/nrg.2017.33,10.1038/nrm.2017.35 [DOI] [PubMed] [Google Scholar]
- 22. Tahiliani M., Koh K. P., Shen Y., Pastor W. A., Bandukwala H., Brudno Y., Agarwal S., Iyer L. M., Liu D. R., Aravind L., and Rao A. (2009) Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935 10.1126/science.1170116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ito S., Shen L., Dai Q., Wu S. C., Collins L. B., Swenberg J. A., He C., and Zhang Y. (2011) Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333, 1300–1303 10.1126/science.1210597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. He Y. F., Li B. Z., Li Z., Liu P., Wang Y., Tang Q., Ding J., Jia Y., Chen Z., Li L., Sun Y., Li X., Dai Q., Song C. X., Zhang K., et al. (2011) Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333, 1303–1307 10.1126/science.1210944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li X., Shang E., Dong Q., Li Y., Zhang J., Xu S., Zhao Z., Shao W., Lv C., Zheng Y., Wang H., Lei X., Zhu B., and Zhang Z. (2018) Small molecules capable of activating DNA methylation-repressed genes targeted by the p38 mitogen-activated protein kinase pathway. J. Biol. Chem. 293, 7423–7436 10.1074/jbc.RA117.000757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dong Q., Li X., Wang C. Z., Xu S., Yuan G., Shao W., Liu B., Zheng Y., Wang H., Lei X., Zhang Z., and Zhu B. (2018) Roles of the CSE1L-mediated nuclear import pathway in epigenetic silencing. Proc. Natl. Acad. Sci. U.S.A. 115, E4013–E4022 10.1073/pnas.1800505115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stothard P. (2000) The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques 28, 1102, 1104 10.2144/00286ir01 [DOI] [PubMed] [Google Scholar]
- 28. Bosshart H., and Heinzelmann M. (2016) THP-1 cells as a model for human monocytes. Ann. Transl. Med. 4, 438 10.21037/atm.2016.08.53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kotecki M., Reddy P. S., and Cochran B. H. (1999) Isolation and characterization of a near-haploid human cell line. Exp. Cell Res. 252, 273–280 10.1006/excr.1999.4656 [DOI] [PubMed] [Google Scholar]
- 30. Pastor W. A., Aravind L., and Rao A. (2013) TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat. Rev. Mol. Cell Biol. 14, 341–356 10.1038/nrm3589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Duh E. J., Maury W. J., Folks T. M., Fauci A. S., and Rabson A. B. (1989) Tumor necrosis factor α activates human immunodeficiency virus type 1 through induction of nuclear factor binding to the NF-κB sites in the long terminal repeat. Proc. Natl. Acad. Sci. U.S.A. 86, 5974–5978 10.1073/pnas.86.15.5974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lowenthal J. W., Ballard D. W., Böhnlein E., and Greene W. C. (1989) Tumor necrosis factor α induces proteins that bind specifically to κB-like enhancer elements and regulate interleukin 2 receptor α-chain gene expression in primary human T lymphocytes. Proc. Natl. Acad. Sci. U.S.A. 86, 2331–2335 10.1073/pnas.86.7.2331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Osborn L., Kunkel S., and Nabel G. J. (1989) Tumor necrosis factor α and interleukin 1 stimulate the human immunodeficiency virus enhancer by activation of the nuclear factor κB. Proc. Natl. Acad. Sci. U.S.A. 86, 2336–2340 10.1073/pnas.86.7.2336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hayden M. S., and Ghosh S. (2008) Shared principles in NF-κB signaling. Cell 132, 344–362 10.1016/j.cell.2008.01.020 [DOI] [PubMed] [Google Scholar]
- 35. Hayden M. S., and Ghosh S. (2012) NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 26, 203–234 10.1101/gad.183434.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang Q., Lenardo M. J., and Baltimore D. (2017) 30 Years of NF-κB: a blossoming of relevance to human pathobiology. Cell 168, 37–57 10.1016/j.cell.2016.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Brandeis M., Frank D., Keshet I., Siegfried Z., Mendelsohn M., Nemes A., Temper V., Razin A., and Cedar H. (1994) Sp1 elements protect a CpG island from de novo methylation. Nature 371, 435–438 10.1038/371435a0 [DOI] [PubMed] [Google Scholar]
- 38. Macleod D., Charlton J., Mullins J., and Bird A. P. (1994) Sp1 sites in the mouse aprt gene promoter are required to prevent methylation of the CpG island. Genes Dev. 8, 2282–2292 10.1101/gad.8.19.2282 [DOI] [PubMed] [Google Scholar]
- 39. Silke J., Rother K. I., Georgiev O., Schaffner W., and Matsuo K. (1995) Complex demethylation patterns at Sp1 binding sites in F9 embryonal carcinoma cells. FEBS Lett. 370, 170–174 10.1016/0014-5793(95)00830-3 [DOI] [PubMed] [Google Scholar]
- 40. Kirillov A., Kistler B., Mostoslavsky R., Cedar H., Wirth T., and Bergman Y. (1996) A role for nuclear NF-κB in B-cell-specific demethylation of the Igκ locus. Nat. Genet. 13, 435–441 10.1038/ng0895-435 [DOI] [PubMed] [Google Scholar]
- 41. Sérandour A. A., Avner S., Oger F., Bizot M., Percevault F., Lucchetti-Miganeh C., Palierne G., Gheeraert C., Barloy-Hubler F., Péron C. L., Madigou T., Durand E., Froguel P., Staels B., Lefebvre P., et al. (2012) Dynamic hydroxymethylation of deoxyribonucleic acid marks differentiation-associated enhancers. Nucleic Acids Res. 40, 8255–8265 10.1093/nar/gks595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Costa Y., Ding J., Theunissen T. W., Faiola F., Hore T. A., Shliaha P. V., Fidalgo M., Saunders A., Lawrence M., Dietmann S., Das S., Levasseur D. N., Li Z., Xu M., Reik W., et al. (2013) NANOG-dependent function of TET1 and TET2 in establishment of pluripotency. Nature 495, 370–374 10.1038/nature11925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. de la Rica L., Rodríguez-Ubreva J., García M., Islam A. B., Urquiza J. M., Hernando H., Christensen J., Helin K., Gómez-Vaquero C., and Ballestar E. (2013) PU.1 target genes undergo Tet2-coupled demethylation and DNMT3b-mediated methylation in monocyte-to-osteoclast differentiation. Genome Biol. 14, R99 10.1186/gb-2013-14-9-r99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fujiki K., Shinoda A., Kano F., Sato R., Shirahige K., and Murata M. (2013) PPARγ-induced PARylation promotes local DNA demethylation by production of 5-hydroxymethylcytosine. Nat. Commun. 4, 2262 10.1038/ncomms3262 [DOI] [PubMed] [Google Scholar]
- 45. Dubois-Chevalier J., Oger F., Dehondt H., Firmin F. F., Gheeraert C., Staels B., Lefebvre P., and Eeckhoute J. (2014) A dynamic CTCF chromatin binding landscape promotes DNA hydroxymethylation and transcriptional induction of adipocyte differentiation. Nucleic Acids Res. 42, 10943–10959 10.1093/nar/gku780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rampal R., Alkalin A., Madzo J., Vasanthakumar A., Pronier E., Patel J., Li Y., Ahn J., Abdel-Wahab O., Shih A., Lu C., Ward P. S., Tsai J. J., Hricik T., Tosello V., et al. (2014) DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell Rep. 9, 1841–1855 10.1016/j.celrep.2014.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tsai Y. P., Chen H. F., Chen S. Y., Cheng W. C., Wang H. W., Shen Z. J., Song C., Teng S. C., He C., and Wu K. J. (2014) TET1 regulates hypoxia-induced epithelial-mesenchymal transition by acting as a co-activator. Genome Biol. 15, 513 10.1186/s13059-014-0513-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Perera A., Eisen D., Wagner M., Laube S. K., Künzel A. F., Koch S., Steinbacher J., Schulze E., Splith V., Mittermeier N., Müller M., Biel M., Carell T., and Michalakis S. (2015) TET3 is recruited by REST for context-specific hydroxymethylation and induction of gene expression. Cell Rep. 11, 283–294 10.1016/j.celrep.2015.03.020 [DOI] [PubMed] [Google Scholar]
- 49. Wang Y., Xiao M., Chen X., Chen L., Xu Y., Lv L., Wang P., Yang H., Ma S., Lin H., Jiao B., Ren R., Ye D., Guan K. L., and Xiong Y. (2015) WT1 recruits TET2 to regulate its target gene expression and suppress leukemia cell proliferation. Mol. Cell 57, 662–673 10.1016/j.molcel.2014.12.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xiong J., Zhang Z., Chen J., Huang H., Xu Y., Ding X., Zheng Y., Nishinakamura R., Xu G. L., Wang H., Chen S., Gao S., and Zhu B. (2016) Cooperative action between SALL4A and TET proteins in stepwise oxidation of 5-methylcytosine. Mol. Cell 64, 913–925 10.1016/j.molcel.2016.10.013 [DOI] [PubMed] [Google Scholar]
- 51. Shen L., Wu H., Diep D., Yamaguchi S., D'Alessio A. C., Fung H. L., Zhang K., and Zhang Y. (2013) Genome-wide analysis reveals TET- and TDG-dependent 5-methylcytosine oxidation dynamics. Cell 153, 692–706 10.1016/j.cell.2013.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wu H., Wu X., Shen L., and Zhang Y. (2014) Single-base resolution analysis of active DNA demethylation using methylase-assisted bisulfite sequencing. Nat. Biotechnol. 32, 1231–1240 10.1038/nbt.3073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sun Z., Dai N., Borgaro J. G., Quimby A., Sun D., Corrêa I. R. Jr, Zheng Y., Zhu Z., and Guan S. (2015) A sensitive approach to map genome-wide 5-hydroxymethylcytosine and 5-formylcytosine at single-base resolution. Mol. Cell 57, 750–761 10.1016/j.molcel.2014.12.035 [DOI] [PubMed] [Google Scholar]
- 54. Xia B., Han D., Lu X., Sun Z., Zhou A., Yin Q., Zeng H., Liu M., Jiang X., Xie W., He C., and Yi C. (2015) Bisulfite-free, base-resolution analysis of 5-formylcytosine at the genome scale. Nat. Methods 12, 1047–1050 10.1038/nmeth.3569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rodriguez-Gil A., Riedlinger T., Ritter O., Saul V. V., and Schmitz M. L. (2018) Formaldehyde-assisted isolation of regulatory elements to measure chromatin accessibility in mammalian cells. J. Vis. Exp. 134, e57272 10.3791/57272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ding Y., Fromm M., and Avramova Z. (2012) Multiple exposures to drought “train” transcriptional responses in Arabidopsis. Nat. Commun. 3, 740 10.1038/ncomms1732 [DOI] [PubMed] [Google Scholar]
- 57. Naik S., Larsen S. B., Gomez N. C., Alaverdyan K., Sendoel A., Yuan S., Polak L., Kulukian A., Chai S., and Fuchs E. (2017) Inflammatory memory sensitizes skin epithelial stem cells to tissue damage. Nature 550, 475–480 10.1038/nature24271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Thomassin H., Flavin M., Espinás M. L., and Grange T. (2001) Glucocorticoid-induced DNA demethylation and gene memory during development. EMBO J. 20, 1974–1983 10.1093/emboj/20.8.1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Steffen P. A., and Ringrose L. (2014) What are memories made of? How Polycomb and Trithorax proteins mediate epigenetic memory. Nat. Rev. Mol. Cell Biol. 15, 340–356 10.1038/nrm3789 [DOI] [PubMed] [Google Scholar]
- 60. D'Urso A., and Brickner J. H. (2014) Mechanisms of epigenetic memory. Trends Genet. 30, 230–236 10.1016/j.tig.2014.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Badeaux A. I., and Shi Y. (2013) Emerging roles for chromatin as a signal integration and storage platform. Nat. Rev. Mol. Cell Biol. 14, 211–224 10.1038/nrm3545 [DOI] [PubMed] [Google Scholar]
- 62. Johnson D. G., and Dent S. Y. (2013) Chromatin: receiver and quarterback for cellular signals. Cell 152, 685–689 10.1016/j.cell.2013.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Suganuma T., and Workman J. L. (2013) Chromatin and signaling. Curr. Opin. Cell Biol. 25, 322–326 10.1016/j.ceb.2013.02.016 [DOI] [PubMed] [Google Scholar]
- 64. Wu D., Hu D., Chen H., Shi G., Fetahu I. S., Wu F., Rabidou K., Fang R., Tan L., Xu S., Liu H., Argueta C., Zhang L., Mao F., Yan G., et al. (2018) Glucose-regulated phosphorylation of TET2 by AMPK reveals a pathway linking diabetes to cancer. Nature 559, 637–641 10.1038/s41586-018-0350-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lin J., Ziring D., Desai S., Kim S., Wong M., Korin Y., Braun J., Reed E., Gjertson D., and Singh R. R. (2008) TNFα blockade in human diseases: an overview of efficacy and safety. Clin. Immunol. 126, 13–30 10.1016/j.clim.2007.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wong M., Ziring D., Korin Y., Desai S., Kim S., Lin J., Gjertson D., Braun J., Reed E., and Singh R. R. (2008) TNFα blockade in human diseases: mechanisms and future directions. Clin. Immunol. 126, 121–136 10.1016/j.clim.2007.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Iguchi-Ariga S. M., and Schaffner W. (1989) CpG methylation of the cAMP-responsive enhancer/promoter sequence TGACGTCA abolishes specific factor binding as well as transcriptional activation. Genes Dev. 3, 612–619 10.1101/gad.3.5.612 [DOI] [PubMed] [Google Scholar]
- 68. Altarejos J. Y., and Montminy M. (2011) CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat. Rev. Mol. Cell Biol. 12, 141–151 10.1038/nrm3072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kasowski M., Grubert F., Heffelfinger C., Hariharan M., Asabere A., Waszak S. M., Habegger L., Rozowsky J., Shi M., Urban A. E., Hong M. Y., Karczewski K. J., Huber W., Weissman S. M., Gerstein M. B., et al. (2010) Variation in transcription factor binding among humans. Science 328, 232–235 10.1126/science.1183621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bock C., Reither S., Mikeska T., Paulsen M., Walter J., and Lengauer T. (2005) BiQ Analyzer: visualization and quality control for DNA methylation data from bisulfite sequencing. Bioinformatics 21, 4067–4068 10.1093/bioinformatics/bti652 [DOI] [PubMed] [Google Scholar]
- 71. Sanjana N. E., Shalem O., and Zhang F. (2014) Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 10.1038/nmeth.3047 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.