

Tens of millions of people are infected with hepatitis C virus (HCV) worldwide. Recently marketed direct-acting antivirals (DAAs) targeting HCV proteins have enhanced the efficacy of treatment. However, due to its high cost, this new therapy is not accessible to the vast majority of infected patients. Furthermore, treatment failures have also been reported due to the appearance of viral resistance. Here, we report on the identification of a new HCV inhibitor, dehydrojuncusol, that targets HCV NS5A and is able to inhibit RNA replication of replicons harboring resistance mutations to anti-NS5A DAAs used in current therapy. Dehydrojuncusol is a natural compound isolated from Juncus maritimus, a halophilic plant species that is very common in coastlines worldwide. This molecule might serve as a lead for the development of a new therapy that is more accessible to hepatitis C patients in the future.
KEYWORDS: antiviral agents, dehydrojuncusol, hepatitis C virus, natural antimicrobial products, phenanthrene, viral replication
ABSTRACT
Recent emergence of direct-acting antivirals (DAAs) targeting hepatitis C virus (HCV) proteins has considerably enhanced the success of antiviral therapy. However, the appearance of DAA-resistant-associated variants is a cause of treatment failure, and the high cost of DAAs renders the therapy not accessible in countries with inadequate medical infrastructures. Therefore, the search for new inhibitors with a lower cost of production should be pursued. In this context, the crude extract of Juncus maritimus Lam. was shown to exhibit high antiviral activity against HCV in cell culture. Bio-guided fractionation allowed the isolation and identification of the active compound, dehydrojuncusol. A time-of-addition assay showed that dehydrojuncusol significantly inhibited HCV infection when added after virus inoculation of HCV genotype 2a (50% effective concentration [EC50] = 1.35 µM). This antiviral activity was confirmed with an HCV subgenomic replicon, and no effect on HCV pseudoparticle entry was observed. Antiviral activity of dehydrojuncusol was also demonstrated in primary human hepatocytes. No in vitro toxicity was observed at active concentrations. Dehydrojuncusol is also efficient on HCV genotype 3a and can be used in combination with sofosbuvir. Interestingly, dehydrojuncusol was able to inhibit RNA replication of two frequent daclatasvir-resistant mutants (L31M or Y93H in NS5A). Finally, mutants resistant to dehydrojuncusol were obtained and showed that the HCV NS5A protein is the target of the molecule. In conclusion, dehydrojuncusol, a natural compound extracted from J. maritimus, inhibits infection of different HCV genotypes by targeting the NS5A protein and is active against resistant HCV variants frequently found in patients with treatment failure.
IMPORTANCE Tens of millions of people are infected with hepatitis C virus (HCV) worldwide. Recently marketed direct-acting antivirals (DAAs) targeting HCV proteins have enhanced the efficacy of treatment. However, due to its high cost, this new therapy is not accessible to the vast majority of infected patients. Furthermore, treatment failures have also been reported due to the appearance of viral resistance. Here, we report on the identification of a new HCV inhibitor, dehydrojuncusol, that targets HCV NS5A and is able to inhibit RNA replication of replicons harboring resistance mutations to anti-NS5A DAAs used in current therapy. Dehydrojuncusol is a natural compound isolated from Juncus maritimus, a halophilic plant species that is very common in coastlines worldwide. This molecule might serve as a lead for the development of a new therapy that is more accessible to hepatitis C patients in the future.
INTRODUCTION
Hepatitis C is a major cause of chronic hepatitis often associated with complications, such as liver cirrhosis and hepatocellular carcinoma (1, 2). A recent report estimates that approximately 71.1 million people are infected with hepatitis C virus (HCV) worldwide (3). To date, no vaccine is available against HCV, mainly due to the diversity of viral isolates (4). In recent decades, various treatments have been established. The first direct-acting antiviral (DAA) agents targeting viral proteins were released in 2011. Since then, many DAAs against NS3/4A, NS5A, and NS5B have been marketed with high rates of sustained viral response against all HCV genotypes and very few side effects (5). However, these treatments are very expensive and not accessible to all people infected (6). Moreover, there is a risk of selecting viral variants resistant to DAAs leading to failure of the therapy, which will require new treatments (7).
HCV, a member of the Flaviviridae family, is an enveloped, single-stranded RNA virus (8) encoding a single polyprotein which is cleaved co- and posttranslationally. Nonstructural proteins are involved in the replication of the viral genome and the production of new infectious particles in infected cells. The HCV life cycle can be divided into three major steps: entry, replication, and assembly/release. At each step, different sets of viral proteins and host factors are involved. The replication of the RNA genome takes place in the “membranous web,” which is composed of endoplasmic reticulum rearranged membranes (9). The RNA replication complex includes viral proteins NS3/4A, NS4B, NS5A, and NS5B.
Current hepatitis C therapy is very efficient but leads to the appearance of resistant-associated substitutions (RASs). Some of the RASs are specific to a DAA. This is the case for NS5B RASs, but some of them appear independently of the DAA used, especially for NS3/4A and NS5A RASs (10). Patients with the resistant NS5A variant are very difficult to treat, and retreatment with a combination of anti-NS5B and anti-NS5A antivirals does not always lead to a highly sustained viral response. Moreover, these mutated viruses are persistent for years in blood serum (7). A lack of alternative therapy might be a problem for these patients in the future.
Natural products from plant species maintain a strong position in drug discovery (11). A number of metabolites found in plants, among which are the phenolic compounds, are being cited as antimicrobials and resistance-modifying agents (12, 13). Natural products often remain a source of inspiration for medicinal chemistry, with semisynthetic modifications or pharmacophore with natural origin (14). Numerous DAAs from natural origins are now described and are able to target different steps of the HCV infectious cycle (15). Crude extracts from plants used in traditional medicine are also promising sources of antiviral molecules. However, only silymarin, a standardized extract of milk thistle (Silybum marianum [L.] Gaertn., Asteraceae), and some of its metabolites, silibinin A and B, a mixture of two diastereoisomers of flavonolignans, are developed in clinical trials to be used in hepatitis C-infected patients (16). Extremophile plants, xerophytes (growing in arid climate) or halophytes (growing in saline soils), are natural reservoirs of rare bioactive compounds. By several molecular mechanisms, extremophile plants can resist abiotic stresses and infections by producing phytoalexins (17). Rare compounds with unique or common chemical structures present in extremophile plants might help to identify new DAAs against HCV with unexpected mechanisms of action.
It is estimated that new DAA therapy against hepatitis C will benefit a very small proportion of patients (18). The use of plant extracts or compounds isolated from plant extracts should render the therapy more accessible to many patients in developing countries. Therefore, the search for such compounds and extracts is still needed to treat the vast majority of patients in the coming decades. In this context, we have screened halophilic and xerophytic plant extracts from Tunisia for their anti-HCV activity. This led us to identify Juncus maritimus rhizome extract for its capacity to inhibit HCV infection (19). Using a bio-guided fractionation approach, dehydrojuncusol, the active compound inhibiting HCV infection, was identified and its mechanism of action against HCV RNA replication was characterized.
(This article was submitted to an online preprint archive [20].)
RESULTS
Dehydrojuncusol present in the methylene chloride partition of J. maritimus rhizome extract is an inhibitor of a postentry step of HCV infection.
We previously screened sixteen plant extracts from eight different Tunisian extremophile plants for the presence of antiviral compounds (19). The strongest antiviral activity was observed for Juncus maritimus rhizome extract, a halophyte belonging to the Juncaceae family. The methylene chloride partition of the J. maritimus rhizome extract was the most active against HCV (19). To go further in the characterization of antiviral activity and identify active compound(s), bio-guided fractionation was performed, leading to the isolation of two major phenanthrene derivatives (compounds 1 and 2). The chemical structures of these two major compounds were determined by comparison of their spectroscopic data (nuclear magnetic resonance [NMR] and mass spectrometry [MS]) with literature values. Compounds 1 and 2 were identified as two known phenanthrene derivatives, respectively, juncusol and dehydrojuncusol (21) (Fig. 1A). The purity of these compounds was checked by liquid chromatography-ultraviolet-diode array detector (LC-UV-DAD) (juncusol, 99.5%; dehydrojuncusol, 98.8%). Their antiviral activity was determined against HCV in cell culture (HCVcc) of genotype 2a. A time-of-addition assay was performed with the purified compounds at 10 µg/ml (corresponding to 37.5 and 37.8 µM for juncusol and dehydrojuncusol, respectively) added either during virus inoculation, postinoculation, or continuously during both steps. Epigallocatechin gallate (EGCG), an inhibitor of HCV entry (22, 23), and boceprevir, an inhibitor of the NS3/4A protease, were used as controls. As shown in Fig. 1B, juncusol and dehydrojuncusol were both active against HCV, with dehydrojuncusol being the most active in the different conditions tested (coaddition, postinoculation, and continuously during infection). Only juncusol significantly decreased the number of cells (Fig. 1C) showing limited cytotoxicity. Juncusol inhibited HCV infection at the postinoculation step, whereas dehydrojuncusol was active both at the inoculation and postinoculation steps. Dehydrojuncusol was the most active at the postinoculation step, with more than 95% inhibition of HCV infection. As this compound was the most active, we decided to further analyze its antiviral activity.
FIG 1.

Anti-HCV activity of compounds isolated from J. maritimus rhizome extract. (A) Chemical structure of juncusol (1) and dehydrojuncusol (2). (B) Infection was separated in different steps. Huh-7 cells were inoculated with HCVcc for 2 h either in the presence (+) or absence (−) of isolated compounds at 10 µg/ml (corresponding to 37.5 µM and 37.8 µM for juncusol and dehydrojuncusol, respectively), 50 µM EGCG, or 1 µM boceprevir. DMSO (0.0001%) was used as a control. Cells were further incubated in medium containing (+) or not containing (−) the compounds for 28 h before fixation. (C) Infectivity was measured by the use of immunofluorescence labeling of the HCV E1 envelope protein and by calculating the number of infected cells. Data are expressed as a percentage of values measured with DMSO. (D) Nuclei were stained with DAPI to quantify the number of cells. Data are means of values obtained in 3 independent experiments performed in triplicate. Error bars represent standard errors of the mean (SEM). Significantly different from the control (DMSO): *, P < 0.05; **, P < 0.01.
The antiviral effect of dehydrojuncusol was examined by measuring HCV infectious titers. A dose-dependent decrease of the infectious titer was observed, with a 1-log10 decrease at 2.2 µM confirming its high anti-HCV activity (Fig. 2A). A time-of-addition assay of dehydrojuncusol was performed and confirmed that this compound was significantly more active during a postinoculation step (Fig. 2B). No major effect of dehydrojuncusol was observed when cells were preincubated with the compound 2 h before inoculation or when it was coadded with the virus. To measure the half maximal effective concentration (EC50), a dose-response inhibition experiment was then performed with Huh-7 cells incubated with increasing concentrations of dehydrojuncusol at different steps of the infection, during inoculation, postinoculation, or continuously (Fig. 2C). The results show that the EC50 of dehydrojuncusol was 1.35 µM when added continuously, 8.21 µM when added during inoculation, and 1.53 µM when added postinoculation, confirming the major effect of the molecule at the postinoculation step. The toxicity of the compound on Huh-7 cells was also tested in parallel at different time points (24 h, 48 h, and 72 h). The results showed that the 50% cytotoxic concentration (CC50) of dehydrojuncusol was approximately 75.6 µM (Fig. 2D), which was much higher than the active dose, yielding a selective index of 56.
FIG 2.

Dehydrojuncusol inhibits HCV infection in a dose-dependent manner. (A) Huh-7 cells were infected with HCVcc in the presence of dehydrojuncusol at different concentrations. Supernatants were collected 48 h postinoculation, and infectious titers (ffu/ml) were measured by serial dilutions of the supernatants and reinfection of naive Huh-7 cells for 48 h. (B) Dehydrojuncusol at 3.8 µM, delphinidin at 50 µM, or boceprevir at 1 µM was added at different time points as indicated (+) and is represented on the right of the panel. Cells were either preincubated with the compounds for 2 h, or compounds were added during HCVcc inoculation (2 h) or postinoculation (28 h). Infectivity was measured by the use of immunofluorescence labeling of the HCV E1 envelope protein and by calculating the number of infected cells. (C) Huh-7 cells were inoculated with HCVcc in the presence of increasing concentrations of dehydrojuncusol either during the 2-h inoculation, 28 h postinoculation, or continuously. Infectivity was measured by the use of immunofluorescence labeling of the HCV E1 envelope protein and by calculating the number of infected cells. (D) Huh-7 cells were cultured in the presence of given concentrations of dehydrojuncusol. The viability was monitored using an MTS-based viability assay after 24 h, 48 h, and 72 h. Data are expressed as a ratio to control, i.e., the condition without extracts. Data are means of values obtained in 3 independent experiments performed in triplicate. Error bars represent SEM. Significantly different from the control (DMSO): *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Taken together, these results show that dehydrojuncusol inhibits predominantly a postentry step of the HCV infectious cycle, most likely RNA replication, and, to a lesser extent, an early step of infection, potentially the entry step.
Dehydrojuncusol inhibits RNA replication of HCV but not entry.
As shown above, dehydrojuncusol inhibits different steps of HCV infection. To determine if the molecule is able to inhibit HCV entry, HCV-pseudotyped retroviral particles (HCVpp) expressing E1E2 envelope glycoprotein of genotype 2a at their surface were produced and used to infect Huh-7 cells in the presence of dehydrojuncusol at different concentrations. Delphinidin at 50 µM, an inhibitor of HCV entry, was added as a control (24). A strong inhibition of HCVpp infection was observed with delphinidin. In contrast, no effect of dehydrojuncusol on HCVpp entry was observed, even at concentrations up to 94.7 µM (Fig. 3A). This result shows that dehydrojuncusol is not an inhibitor of HCV entry.
FIG 3.

Dehydrojuncusol inhibits HCV RNA replication. (A) HCVpp were used to inoculate Huh-7 cells in the presence of dehydrojuncusol at indicated concentrations. Delphinidin at 50 µM was used as a control. Cells were lysed 48 h postinoculation and luciferase activity quantified. Data are expressed as a ratio to the DMSO (0.0001%) control (100%). (B) Huh-7 cells stably expressing the SGR-JFH1-NS5AGFP replicon were incubated for the indicated time with either 0.5 nM daclatasvir, 1 µM boceprevir, or 10 µg/ml of the methylene chloride partition of J. maritimus (J. mar. MeCl). DMSO (0.0001%) was used as a control. Cells were lysed and lysates subjected to SDS-PAGE followed by a Western blot revelation of GFP and tubulin. The quantification of band intensity is shown in the graph. (C) HCVcc-infected Huh-7 cells were maintained in the presence of dehydrojuncusol or daclatasvir at the indicated concentrations for 48 h. Cells were lysed, RNA extracted, and HCV genomic RNA quantified by qRT-PCR. Data are representative of three experiments. Error bars represent standard deviation (SD). (D) Huh-7 cells were inoculated with HCVcc of genotype 2a (strain JFH1), genotype 1a (strain TN), and genotype 3a (DBN3a) for 2 h. Inoculum was removed and replaced with a medium containing an increasing concentration of dehydrojuncusol. Cells were fixed 30 h postinfection and subjected to immunofluorescence staining with monoclonal anti-NS5A antibody (9E10). Data are means of values obtained in 3 independent experiments performed in triplicate. Error bars represent SEM. (E) Huh-7 cells were inoculated with HCVcc or YFV and MDBK cells with BVDV in the presence (+) or absence (−) of 10 µg/ml of the methylene chloride partition of J. maritimus. Inoculum was removed and replaced with medium with (+) or without (−) 10 µg/ml of the methylene chloride partition of J. maritimus. Cells were fixed and infection detected by immunofluorescence labeling of viral proteins. Data are means of values obtained in 3 independent experiments performed in triplicate. Error bars represent SEM. *, P < 0.05; ***, P < 0.001; ****, P < 0.0001.
To test the activity of dehydrojuncusol on RNA replication independently of entry, Huh-7 cells stably expressing an HCV replicon were used. This replicon, SGR-JFH1-NS5AGFP, expresses green fluorescent protein (GFP)-tagged NS5A. Due to some difficulties in purifying dehydrojuncusol in sufficient quantity from J. maritimus extract to perform all of the tests, the methylene chloride partition containing dehydrojuncusol was used. The replicon cells were incubated with methylene chloride partition and lysed at different time points. Boceprevir and daclatasvir, two inhibitors of the NS3/4A protease and NS5A protein, respectively, were used as controls. The activity of the molecules against HCV RNA replication was determined by quantifying the expression of NS5A-GFP by Western blotting. As shown in Fig. 3B, the expression of NS5A-GFP was strongly decreased in replicon cells incubated with boceprevir, daclatasvir, and the methylene chloride partition of J. maritimus, showing an inhibitory effect of dehydrojuncusol on HCV RNA replication. Inhibition was observed at all time points (24 h, 48 h, and 72 h of treatment), showing a rapid antiviral activity of the methylene chloride partition. To confirm the antiviral activity of dehydrojuncusol on the RNA replication step, viral RNA quantification was performed in HCV-infected Huh-7 cells treated for 48 h with dehydrojuncusol or daclatasvir at different concentrations. A decrease of more than 2 × log10 of the HCV genomic RNA level was observed with 10 µM dehydrojuncusol, similar to the decrease observed with daclatasvir, an NS5A inhibitor, at 0.1 nM (Fig. 3C). Finally, to determine the effect of dehydrojuncusol on other genotypes, its antiviral activity was tested against two strains of genotype 1a and 3a. As shown in Fig. 3D, dehydrojuncusol is able to inhibit HCVcc of genotype 3a with an EC50 of 9.91 µM, but HCVcc of genotype 1a is resistant to dehydrojuncusol. Taken together, these results show that dehydrojuncusol is an inhibitor of HCV RNA replication with an activity that is not limited to HCV genotype 2a.
Dehydrojuncusol has no effect on bovine viral diarrhea virus and yellow fever virus infections.
To determine if the antiviral activity of dehydrojuncusol is specific to HCV, we tested the antiviral effect of the methylene chloride partition of J. maritimus against two other members of the Flaviviridae family, yellow fever virus (YFV) and bovine viral diarrhea virus (BVDV). As shown in Fig. 3E, no significant decrease in infectivity was observed for YFV or for BVDV after treatment with 10 µg/ml of J. maritimus methylene chloride partition. At this concentration, a strong inhibition of HCV infection was observed. This result shows that dehydrojuncusol is not active against all of the members of the Flaviviridae family, suggesting that it could target a HCV protein(s) directly or a cellular factor specifically involved in HCV RNA replication.
Dehydrojuncusol inhibits HCV infection in PHH.
Primary human hepatocytes (PHH) are a more relevant human preclinical model available to test the activity of an inhibitor on HCV infection. A dose-response experiment was performed in PHH inoculated with JFH1 and treated with dehydrojuncusol for 28 h after inoculation. The infectious titer of the supernatants, negative-strand HCV RNA genome copy number, and cytotoxicity of treatments were assayed 72 h postinfection. As shown in Fig. 4A, a dose-dependent decrease of HCV titer was observed with dehydrojuncusol, showing that the molecule is able to inhibit HCV infection in PHH. Very interestingly, the EC50 of dehydrojuncusol in PHH was 1.14 µM, similar to the EC50 calculated in Huh-7 cells (1.35 µM). The results were confirmed by the quantification of the negative-strand HCV RNA genome (Fig. 4B) that shows a similar decrease in the presence of dehydrojuncusol. The results presented in Fig. 4C show that dehydrojuncusol is not cytotoxic in PHH at active concentrations.
FIG 4.

Dehydrojuncusol inhibits HCV infection in PHH. PHH were inoculated with HCVcc and incubated for 28 h after inoculation in the presence of the indicated concentrations of dehydrojuncusol. Supernatants were harvested 72 h postinoculation for determination of infectivity (A) and cytotoxicity (C). Cells were lysed, RNA extracted, and negative-strand HCV RNA genome copy number was quantified (B). Data are expressed as a ratio to carrier DMSO (0.0001%) control. Data are means (±SEM) of 3 independent experiments performed in triplicate.
Dehydrojuncusol inhibits RNA replication of HCV mutants resistant to NS5A inhibitors.
Some patients treated with the newly approved DAA molecules targeting NS5A or NS5B become resistant to the therapy due to the appearance of resistant HCV mutants. Once present in patients, these resistant mutants are very difficult to eliminate, particularly the resistant mutants selected during treatment with anti-NS5A molecules, like daclatasvir, ledipasvir, or ombitasvir. To determine if dehydrojuncusol could be active against resistant HCV mutants, we generated mutated replicons harboring point mutation L31M or Y93H in NS5A. These two mutations conferred resistance to NS5A-targeting drugs, including daclatasvir, ledipasvir, and ombitasvir, in patients infected with HCV of different genotypes and are often associated with treatment failure (7, 10). Huh-7 cells harboring mutant replicons were incubated with the methylene chloride partition of J. maritimus, containing dehydrojuncusol, and lysed after 72 h of treatment. Daclatasvir and boceprevir were used as controls. As shown in Fig. 5A, in the L31M and Y93H replicon cell lysates, the NS5A-GFP protein was detected after 72 h of treatment, with daclatasvir showing that these replicons are resistant to daclatasvir at 0.5 nM. When treated with boceprevir, NS5A-GFP was not detected, showing that the mutants are still sensitive to boceprevir as expected. In cells expressing mutated replicons treated with the methylene chloride partition at 10 µg/ml, NS5A-GFP was not detected with the L31M mutant, showing that dehydrojuncusol is able to inhibit the RNA replication of a daclatasvir-resistant mutant. However, it was still detectable at a lower level with the Y93H daclatasvir-resistant mutant, showing that this mutant is less sensitive to dehydrojuncusol than the L31M mutant. To quantify this antiviral activity, a dose-response experiment was performed on replicon cells treated with either daclatasvir or dehydrojuncusol, and the number of cells expressing NS5A-GFP after 72 h of treatment was quantified. As shown in Fig. 5B, the L31M mutant was very sensitive to dehydrojuncusol with an EC50 of 0.22 µM, which is similar to the EC50 of the wild type at 0.40 µM, whereas the Y93H daclatasvir-resistant mutant was less sensitive to dehydrojuncusol than the wild-type replicon (EC50 of 3.62 µM), correlating with the results obtained by Western blotting. However, the EC50 fold change between the wild type and Y93H replicon was only 9, much lower than the EC50 fold change observed with daclatasvir (Fig. 5C), which was calculated to be over 100 for both mutants (25).
FIG 5.

Dehydrojuncusol inhibits HCV RNA replication of daclatasvir NS5A resistance mutants. (A) Huh-7 cells stably expressing subgenomic replicon SGR-JFH1-NS5AGFP wild type or harboring the L31M or Y93H mutation in NS5A that confer resistance to daclatasvir were incubated for 72 h with 0.5 nM daclatasvir, 1 µM boceprevir, or 10 µg/ml of the methylene chloride partition of J. maritimus (J. mar. MeCl). DMSO (0.0001%) was used as a control. Cells were lysed and lysates subjected to SDS-PAGE followed by Western blot revelation of GFP and tubulin. (B and C) Huh-7 cells stably expressing subgenomic replicon SGR-JFH1-NS5AGFP wild type or harboring the L31M or Y93H mutation in NS5A that confer resistance to daclatasvir were incubated for 72 h with increasing concentrations of dehydrojuncusol (B) or daclatasvir (C). Cells were fixed, and the percentage of GFP-positive cells was quantified as a measure of RNA replication. Replication of subgenomic replicon was expressed as a ratio to carrier control DMSO (0.0001%). Data are means of values obtained in 3 independent experiments performed in triplicate. Error bars represent SEM.
The target of dehydrojuncusol is NS5A.
In order to identify the viral target of dehydrojuncusol, JFH1 resistance mutants were generated by infecting Huh-7 cells with JFH1, followed by treatment with 5 × EC50 of dehydrojuncusol for 3 weeks. Daclatasvir as well as dimethyl sulfoxide (DMSO) were used as controls. Viral RNA was extracted every 3 to 4 days to follow the appearance of resistant viruses. Infectious titers of the viruses collected at the different time points were quantified (Fig. 6A). As expected, a strong decrease in viral titers was observed after 3 days of treatment of HCV-infected Huh-7 cells with dehydrojuncusol or daclatasvir. However, a rapid increase in viral titers was observed after 7 days, corresponding to the appearance of resistant viruses. The viral titers remained constant in cells treated with DMSO only. Viral RNAs were extracted at day 21, and the nonstructural genes, NS2 to NS5B, were sequenced. No mutation was observed in control DMSO-treated cells, whereas the two frequent mutations of daclatasvir-resistant viruses, NS5A L31M and Y93H, were detected in daclatasvir-treated cells. Importantly, three mutations at the same amino acid position of NS5A were detected, F28I, F28L, and F28V, in viruses produced in dehydrojuncusol-treated cells. These mutations were individually inserted in the JFH1 genome by reverse genetics. The three mutants exhibited a strong resistance to dehydrojuncusol, showing that the amino acid residue F28 is required for the anti-HCV activity of dehydrojuncusol (Fig. 6B). In parallel, we also tested the resistance of these mutants toward daclatasvir. However, mutants F28I, F28L, and F28V remained sensitive to daclatasvir (Fig. 6C). Since another mutation at the position F28 has been described as a resistant variant of HCV JFH1 to daclatasvir (25) and ombitasvir (26), we inserted this mutation in the HCV JFH1 genome to determine the sensitivity of this F28S mutant to dehydrojuncusol. Interestingly, the JFH1 NS5A F28S mutant was sensitive to dehydrojuncusol (Fig. 6B) and, as expected, resistant to daclatasvir (Fig. 6C), showing that the binding sites of daclatasvir and dehydrojuncusol on NS5A might be slightly different.
FIG 6.

Dehydrojuncusol targets NS5A. (A) Huh-7 cells infected with the HCVcc JFH1 strain were treated with 5 × EC50 of dehydrojuncusol and daclatasvir or remained untreated for 21 days. Viral infectious titers were measured in the cell supernatants collected at different time points during the selection of resistant viruses. Huh-7 cells were inoculated with wild-type HCVcc or HCVcc harboring an F28I, F28L, F28V, or F28S mutation in the presence of given concentrations of dehydrojuncusol (B) or daclatasvir (C). Cells were fixed 30 h postinoculation, and infectivity was measured by the use of immunofluorescence labeling of the HCV E1 envelope protein and by calculating the number of infected cells. Data are expressed as a percentage of values measured with 0.0001% DMSO. Data are means of values obtained in 3 independent experiments performed in triplicate. Error bars represent SEM.
Dehydrojuncusol can be used in combination with sofosbuvir.
The results presented here show that dehydrojuncusol encompasses many characteristics of a DAA to be used in therapy. In order to determine the potential use of dehydrojuncusol in combination with the known DAA, a combination study of dehydrojuncusol and sofosbuvir, an inhibitor of HCV polymerase NS5B (27), was performed. Dehydrojuncusol was added at different concentrations along with sofosbuvir at fixed concentrations, 400 and 600 nM, during infection of Huh-7 cells with HCVcc. The result shows that Sofosbuvir can increase the antiviral activity of dehydrojuncusol in an additive manner (Fig. 7). The EC50 of dehydrojuncusol is decreased down to 1.10 nM in the presence of sofosbuvir at 600 nM, demonstrating that dehydrojuncusol could be used in combination with DAAs used in hepatitis C therapy.
FIG 7.
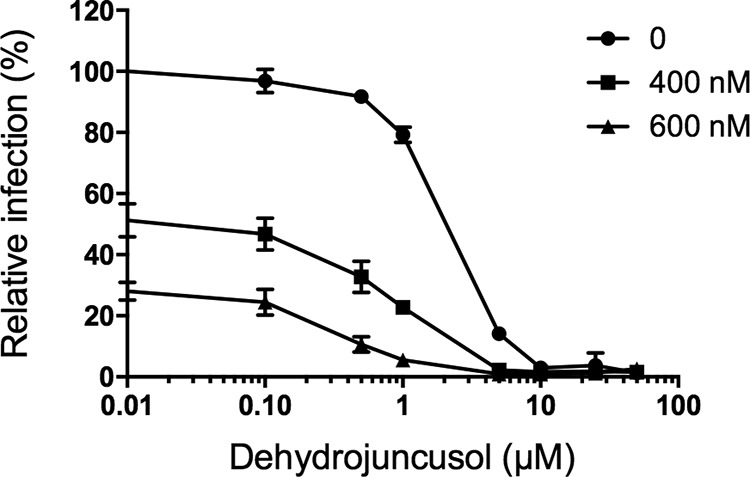
Dehydrojuncusol can be used in combination with sofosbuvir. Huh-7 cells were inoculated with HCVcc. Inoculum was removed and replaced by a medium containing dehydrojuncusol at given concentrations combined with sofosbuvir at 400 or 600 nM. Cells were fixed 30 h postinoculation, and infectivity was measured by the use of immunofluorescence labeling of the HCV E1 envelope protein and by calculating the number of infected cells. Data are means of triplicate values obtained in one experiment representative of 3 independent experiments. Error bars represent SD.
DISCUSSION
Plants are known to be an important source of numerous bioactive compounds (14). Here, we highlighted the strong inhibition of HCV infection obtained in an HCV in cell culture (HCVcc) system by a bioactive molecule, dehydrojuncusol, isolated from a J. maritimus rhizomes extract. More interestingly, we also showed that this natural product strongly inhibits the virus in PHH, which is one of the most relevant human preclinical models for HCV infection. Many different drugs are now available to cure hepatitis C, but in some patients, the appearance of viral variants resistant to the treatment makes them difficult to cure, especially for NS5A resistant mutants. Unfortunately, the sets of marketed molecules available are not efficient to cure such resistant viruses (7). In this work, we demonstrate that dehydrojuncusol has the capacity to inhibit two NS5A resistant mutants appearing in patients treated with anti-NS5A inhibitors, L31M and Y93H, which are two major resistant variants appearing after treatment with daclatasvir, ledipasvir, or ombitasvir (7, 10). These mutants are often associated with treatment failure. Therefore, dehydrojuncusol or molecules derived from dehydrojuncusol might be of great interest in the future to cure patients with resistant variants and treatment failure. Here, we demonstrate that dehydrojuncusol anti-HCV activity is not genotype specific since this molecule is able to inhibit HCV genotype 3a in addition to genotype 2a, even if HCV genotype 1a is resistant to the compound. Furthermore, we also show that dehydrojuncusol can be used in combination with sofosbuvir, the most widely used DAA that targets the NS5B polymerase.
Even if dehydrojuncusol is able to inhibit the RNA replication of HCV replicons harboring point mutations in NS5A (L31M and Y93H), it seems likely that the viral target of this molecule is NS5A. Dehydrojuncusol-resistant HCV mutants were obtained, and we demonstrated that a single mutation of NS5A F28 is responsible for resistance to dehydrojuncusol. This amino acid residue was mutated into three different amino acids, isoleucine, leucine, and valine, and each of them confers resistance to dehydrojuncusol. Phenylalanine 28 is one of the first amino acid residues of NS5A domain 1. This domain is the target of daclatasvir, ombitasvir, and ledipasvir (28). Resistance mutations in NS5A at position 28 already have been described for genotype 1 (M28) and confer resistance to daclatasvir, ombitasvir, or ledipasvir (7, 10, 29, 30). For JFH1 genotype 2a, an F28S resistance mutation has been observed for daclatasvir (25) and ombitasvir (26). Here, we show that HCV JFH1 NS5A F28S is sensitive to dehydrojuncusol. Taken together, these data suggest that dehydrojuncusol and daclatasvir interact with NS5A domain 1 but in different ways.
Unexpectedly for an RNA replication inhibitor, dehydrojuncusol also inhibits HCV infection when it is added to the cells during virus contact and removed later on (Fig. 2B and C). This ability to inhibit an early step of HCVcc infection is probably not due to an inhibition of HCV entry, as this molecule is not active on HCVpp (Fig. 3A). It is likely that this effect results from dehydrojuncusol molecules that enter cells during viral inoculation, remain trapped inside cells, and are able to inhibit HCV RNA replication afterwards.
The EC50 of dehydrojuncusol is close to 1 µM in the replicon, in the HCVcc system, and in PHH, a concentration relatively low for a natural product. This molecule might serve as a starting point for a study of structure activity relationships and drug design to increase its antiviral capacity and reduce its EC50. It is rare that a compound is as active in Huh-7 as in PHH. The EC50 of dehydrojuncusol could even be decreased by 3 × log10 when it was combined with sofosbuvir at 600 nM. The toxicity of dehydrojuncusol in Huh-7 cells is relatively low with a CC50 of approximately 75 µM, leading to a selective index of 56. Similarly, no toxicity was observed in PHH even at high concentrations.
Unfortunately, little is known about the bioavailability of this compound in animal models or in humans. More generally, few/no pharmacokinetics studies of phenanthrene derivatives in vivo or in humans were reported. Natural phenanthrenes are an uncommon class of aromatic metabolites, which in a biosynthetic point of view can originate from stilbene precursors and more rarely from diterpenoid precursors. These compounds are biosynthesized by a limited number of botanical families. Orchidaceae and Juncaceae are the main botanical families known to produce this kind of natural product (31, 32). Phenanthrenes and 9,10-dihydrophenanthrenes showed a large panel of biological activities, including, among others, cytotoxic, antimicrobial, spasmolytic, and anti-inflammatory activities. However, this potential is not sufficiently investigated and not really exploited (32). Concerning the antiviral activities of phenanthrene derivatives, a limited number of studies exist in the literature. Some phenanthrene derivatives isolated from Tamus communis (Dioscoreaceae) showed antiviral activity against two RNA viruses, vesicular stomatitis virus (VSV) and human rhinovirus serotype 1B (33). Other phenanthrenes isolated from Juncus compressus Jacq. showed antiviral activity against human herpes simplex virus 2 (34). So far, no antiviral activity was highlighted for dehydrojuncusol. Only moderate anti-inflammatory activity has been shown for this natural product (35).
J. maritimus is a perennial plant which thrives in particular on saline soils. In the framework of our study, this halophyte was collected in Tunisia, but J. maritimus is present worldwide on the coastline of many different areas, including several European countries, such as France. Few traditional uses are mentioned for this plant in Tunisia, with the exception of its use in making baskets and curtains. Its usage as an antiviral agent is a new opportunity for economic valorization of this plant. One of the disadvantages of our study is that the part of the plant that is used is not renewable (rhizomes), so the chemical synthesis of dehydrojuncusol or a more active derivative could be an alternative. It also would be interesting to search for dehydrojuncusol in other Juncus species and especially in parts of the plant that are renewable, such as aerial parts. Interestingly, this phenanthrene derivative has been previously isolated from the aerial parts of Juncus species, such as Juncus acutus (36) and Juncus effusus (37), increasing its potential for use in low income countries.
In conclusion, we showed that dehydrojuncusol, an active compound present in J. maritimus rhizomes, is a new DAA that targets HCV NS5A and inhibits RNA replication of resistant viruses that appear in patients treated with anti-NS5A inhibitors and are responsible for treatment failure. Plants should be considered more, in the future, as sources of antiviral agents, and dehydrojuncusol could constitute an alternative treatment for patients resistant to currently available DAA.
MATERIALS AND METHODS
Chemicals.
Dulbecco’s modified Eagle’s medium (DMEM), Opti-MEM, phosphate-buffered saline (PBS), GlutaMax-I, and fetal bovine serum were purchased from Invitrogen (Carlsbad, CA). 4’,6-diamidino-2-phenylindole (DAPI) was from Molecular Probes (Thermo Fisher Scientific, Waltham, MA, USA). EGCG was from Calbiochem (Merck Chemicals, Darmstadt, Germany) and was >95% pure. Delphinidin chloride was from Extrasynthèse (Lyon, France) and was >96% pure. Daclatasvir (BMS-790052) and sofosbuvir (PSI-7977, GS-7977) were from Selleckchem (France). Stocks of EGCG and delphinidin were resuspended in dimethyl sulfoxide (DMSO) at 0.5 M. Plant extracts were resuspended in DMSO at 25 mg/ml. Boceprevir was kindly provided by Philippe Halfon (Hôpital Européen, Laboratoire Alphabio, Marseille, France). Other chemicals were from Sigma (St. Louis, MO).
Antibodies.
The anti-E1 monoclonal antibody (A4) (38), anti-YFV envelope protein 2D12 antibody (ATCC CRL-1689), and anti-BVDV NS3 protein OSC-23 antibody (39) were produced in vitro by using the miniPerm apparatus (Heraeus, Hanau, Germany). The anti-NS5A, 2F6/G11, and 9E10 were from Austral Biologicals (San Ramon, CA, USA) or kindly provided by C. M. Rice (Rockefeller University, NY). The mouse monoclonal anti-GFP antibody was from Roche. The mouse anti-β tubulin monoclonal antibody (TUB 2.1) was from Sigma. Cy3-conjugated goat anti-mouse IgG and peroxidase-conjugated goat anti-mouse IgG antibodies were from Jackson ImmunoResearch (West Grove, PA, USA).
Cells and culture conditions.
Huh-7 and HEK 293T (ATCC number CRL-11268) were grown in DMEM supplemented with GlutaMax-I and 10% fetal bovine serum (complete culture medium) and Madin-Darby bovine kidney (MDBK) (ATCC CCL-22) in DMEM supplemented with GlutaMax-I and 10% horse serum in an incubator at 37°C with 5% CO2. The primary human hepatocytes were from Biopredic International (Saint-Grégoire, France) and maintained in primary culture as described previously (24, 40).
Collection of plant, extraction, and purification of natural products.
Juncus maritimus rhizomes were collected in October 2013 from a coastal region in Northeast Tunisia (Soliman), and a voucher specimen (184) was deposited at the Herbarium of the Laboratory of Extremophile Plants at the Biotechnology Centre (Technopark of Borj Cédria). Dried and powdered rhizomes were soaked in methanol (15 ml/g; 1 × 24 h; 2 × 48 h) to afford 300.0 g of a crude methanolic extract. A part of the extract (215.0 g) was dissolved in water and then partitioned with methylene chloride (CH2Cl2) to afford a CH2Cl2 partition (15.0 g). Preparative centrifugal partition chromatography (CPC) (Armen Instrument) was carried out on the CH2Cl2 partition (1.45 g) using a quaternary biphasic solvent system Arizona X (n-Hept/EtOAc/MeOH/H2O, 9:1:9:1 [vol/vol/vol/vol]) in ascending mode for 30 min. Fifteen fractions (MCX1 to MCX15) were pooled according to thin-layer chromatography (TLC) and UV analysis. Compounds 1 (22.2 mg) and 2 (25.9 mg) were isolated from MCX15 (200 mg) by preparative high-performance liquid chromatography (HPLC) (Shimadzu Instruments) using the following elution program (EP1): 10% to 38% acetonitrile (B) (0 to 2 min), 38% to 45% B (2 to 8 min), 45% to 51% B (8 to 25 min), 51% to 96% B (25 to 26 min), 96% to 96.1% B (26 to 29 min), and 100% B (29 to 35 min).
HCVcc.
We used a modified JFH1 strain (Japanese fulminant hepatitis 1, genotype 2a) containing cell culture adaptive mutations (41, 42), the HCV strain TN of genotype 1a (43), and the HCV strain DBN3a of genotype 3a (44). JFH1 was kindly provided by T. Wakita (National Institute of Infectious Diseases, Tokyo, Japan), and TN and DBN3a were kindly provided by Jens Bukh (University of Copenhagen, Denmark). The viral stocks were produced in Huh-7 cells. Huh-7 cells were infected with a prestock of HCVcc in flasks. After 24 h, 48 h, and 72 h, the supernatants of flasks were collected. The titers of the stocks were 5 × 106, 4 × 104, and 5 × 105 focus forming units (ffu)/ml for JFH1, TN, and DBN3a, respectively. For infection assay, Huh-7 cells (6,000/well) seeded in 96-well plates were inoculated with HCVcc at a multiplicity of infection (MOI) of 0.8 for 2 h at 37°C. Then, the inoculum was removed and cells were incubated in complete culture medium for 28 h at 37°C. Compounds were added to cells either for 2 h at 37°C before infection (preincubation condition), for 2 h at 37°C in the presence of the virus (inoculation condition), for 28 h at 37°C after virus removal (postinoculation condition), or both during the 2 h inoculation and 28 h postinoculation periods (inoculation and postinoculation condition). Cells were fixed with ice-cold methanol and subjected to immunofluorescent detection of the viral E1 envelope protein.
HCV grown in primary culture.
For HCV infection, PHH were inoculated 3 days postseeding at an MOI of 2 with a nonmodified JFH1 virus (HCVcc) as previously described (40). After 2 h of incubation at 37°C, the inoculum was removed, and PHH were incubated in complete culture medium for 28 h at 37°C at the indicated concentrations of dehydrojuncusol. The medium was then renewed without treatment. Supernatants containing infectious HCV grown in primary culture (HCVpc) were harvested 3 days postinoculation, and infectious titers were evaluated by focus-forming assay on Huh-7 cells as previously described (45). Intracellular levels of negative-strand HCV RNA were quantified by a strand-specific reverse transcription real-time PCR technique described previously (threshold of detection, 25 copies/reaction) (46). The potential cytotoxicity of dehydrojuncusol was assessed by measurement of the activity of lactate dehydrogenase (LDH) released into culture supernatants as previously described (24).
BVDV and YFV.
Bovine viral diarrhea virus (BVDV) (strain NADL) was produced in MDCK cells as previously described (47). Yellow fever virus (YFV) (strain 17D) was produced in SW13 cells. Infection assays were performed with MDBK cells (BVDV) or Huh-7 cells (YFV) seeded in 96-well plates at an MOI of 1.5 or 1, respectively. Cells were infected for 1 h at 37°C. The viral inoculum was removed, and the cells were further cultured for either 15 h (BVDV) or 23 h (YFV). Cells were fixed with paraformaldehyde 3% in PBS and subjected to immunofluorescent detection of NS3 (BVDV) or E (YFV).
HCVpp.
Retroviral pseudoparticles expressing HCV envelope glycoproteins E1E2 of genotype 2a (HCVpp) were produced in HEK 293T cells as described (48). Huh-7 cells were seeded in 48-well plates and inoculated with HCVpp in the presence of compounds for 2 h. Inoculum was removed and replaced with fresh medium without compound. Cells were lysed at 48 h postinoculation in 50 µl of 1× luciferase lysis buffer (Promega, Madison, WI, USA), and luciferase activity was quantified in a TriStar LB 941 luminometer (Berthold Technologies, Bad Wildbad, Germany) using the luciferase assay system (Promega) as recommended by the manufacturer.
Immunofluorescent detection assay.
HCVcc-, YFV-, and BVDV-infected cells grown in 96-well plates were processed for immunofluorescence detection of viral proteins as previously described (49). Nuclei were stained with 1 µg/ml of DAPI, and infected cells were detected by immunofluorescent labeling of the E1 envelope glycoprotein (HCV), E envelope protein (YFV), and NS3 protein (BVDV) followed by Cy3-conjugated anti-mouse secondary antibody. For BVDV and YFV infection assays, the plates were observed with an Axiophot microscope equipped with ×10 magnification objective (Carl Zeiss AG, Oberkochen, Germany). Fluorescent signals were collected with a CoolSnap ES camera (Photometrix, Kew, Australia). Images of infected cells (Cy3 channel) and of nuclei (DAPI channel) present in 10 randomly picked areas from each well were recorded. The numbers of total cells and infected cells were quantified using ImageJ software. For quantification of HCVcc infection, confocal images were recorded with an automated confocal microscope IN Cell Analyzer 6000 (GE Healthcare Life Sciences) using a ×20 objective with exposure parameters 405/450 nm and 561/610 nm. Six fields per well were recorded. Each image was then processed using the Columbus image analysis software (PerkinElmer). Nuclei were first segmented, and the cytoplasm region was extrapolated based on DAPI staining. Objects with a specific and predefined size were defined as cells. The ratio of infected cells over total cells represents the infection rate. For each virus, the number of cells per well and MOI were determined in order to have 30% to 40% of infected cells in control experiments with no inhibitor. Infection rates in DMSO controls were expressed as 100%.
Viral RNA quantification.
Huh-7 cells seeded in 12-well plates were inoculated with HCVcc for 2 h. The inoculum was removed and replaced with culture medium. Forty-eight hours after inoculation, cells were lysed in lysis buffer from the NucleoSpin RNA plus kit (Macherey-Nagel) and total RNA extracted following the manufacturer’s instructions, eluted in a final volume of 60 µl of H2O and quantified. Ten microliters of RNA was used for cDNA synthesis using a high-capacity cDNA reverse transcription kit (Applied Biosystems). Five microliters of cDNA was used for a reverse transcription-quantitative PCR (RT-qPCR) assay using TaqMan probes as described previously (50).
Viability assay.
Huh-7 cells were plated in 96-well plates at a density of 6,000 cells/well and were then incubated the next day in 100 μl of culture medium containing increasing concentrations of dehydrojuncusol for either 24 h, 48 h, or 72 h. An MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium]-based viability assay (CellTiter 96 aqueous nonradioactive cell proliferation assay, Promega) was performed as recommended by the manufacturer. The absorbance of formazan at 490 nm is detected using an enzyme-linked immunosorbent assay (ELISA) plate reader (ELx808, BioTek Instruments, Inc.). Each measure was performed in triplicate.
HCV replicon.
The plasmid pSGR-JFH1 encoding a subgenomic replicon of the JFH1 strain was obtained from T. Wakita (51). BglII and an NsiI restriction sites were inserted between codons Pro419 and Leu420 of NS5A, and the coding sequence of EGFP was then inserted between these two sites. This position was previously shown to accept a GFP insertion in a subgenomic replicon of the Con1 strain (52). Mutations Leu31Met (L31M) and Tyr93His (Y93H) were inserted independently by PCR. Plasmid pSGR-JFH1-NS5AGFP and PCR-amplified DNA containing mutated sequences were digested with NsiI and BglII restriction enzymes, and the PCR fragment was inserted by ligation into the plasmid. All constructs were verified by sequencing. These plasmids were transcribed in vitro before electroporation into Huh-7 cells. Cells that express wild-type and mutated replicons were selected by using 500 µg/ml of Geneticin for 15 days and cultured in a medium containing 250 µg/ml of Geneticin.
Replication assay.
Huh-7 cells stably expressing wild-type or mutated replicons were seeded in 24-well plates and incubated with the different compounds for 24 h, 48 h, and 72 h. They were lysed in ice cold lysis buffer (Tris HCl, 50 mM; NaCl, 100 mM; EDTA, 2 mM; Triton X-100, 1%; SDS, 0.1%) on ice for 20 min. Lysates were collected, and 20 µg of proteins was analyzed by Western blotting using anti-NS5A and anti-β tubulin antibodies. Peroxidase-conjugated goat anti-mouse secondary antibody (Jackson ImmunoResearch) was used for the revelation using enhanced chemiluminescence (ECL) Western blotting substrate (Thermo Fisher Scientific). The intensity of the bands was quantified using ImageJ software.
Wild-type, L31M, and Y93H replicon cell lines were seeded in 96-well plates and incubated with boceprevir, daclatasvir, or methylene chloride partition at the given concentration for 72 h. Cells were fixed with paraformaldehyde (PFA), 3%, for 30 min. The cells were counted by labeling of nuclei with DAPI (0.5 µg/ml). The number of GFP-positive cells was quantified with a Zeiss Axiophot 2 microscope equipped with a ×40/1.3 numerical aperture lens. Fluorescence signals were analyzed as described previously.
Selection of a dehydrojuncusol-resistant virus and identification of resistance mutations.
Huh-7 cells were infected with JFH1 followed by treatment with 5 × EC50 of dehydrojuncusol (6.55 µM) or daclatasvir (0.05 nM) or were untreated. Cells were split every 3 to 4 days in the presence of dehydrojuncusol or daclatasvir at 6.55 µM or 0.05 nM, respectively. Supernatants were collected every 3 to 4 days, and viral infectious titers were measured and viral RNA was quantified in parallel to follow the appearance of resistant viruses. At day 21, viral RNA was extracted, and the genomic sequence of HCV from NS2 to NS5B was amplified by reverse transcription-PCR (RT-PCR) and sequenced. Amino acid changes that arose during inhibitor selection were identified by analysis of the DNA sequence compared to the initial and control passages in the absence of drug. The identified mutations were reintroduced into the JFH1 plasmid by PCR mutagenesis, and the plasmids were sequenced.
Statistical analysis.
The results were presented as means ± standard error of the mean (SEM) of three independent experiments performed in triplicate. The statistical test used is a Kruskal-Wallis nonparametric test followed by Dunn’s multicomparison post hoc test with a confidence interval of 95% to identify individual differences between treatments. P values of <0.05 were considered to be significantly different from the control. The data were analyzed using GraphPad Prism (version 5.0b) by comparisons between each treated group and untreated group (DMSO control).
ACKNOWLEDGMENTS
This work was supported by the French National Agency for Research on AIDS and Viral Hepatitis (ANRS-18208), the European Community (ERC-STG INTRACELLTB grant 260901), the Agence Nationale de la Recherche (ANR-10-EQPX-04-01), the Fonds Européen de Développement Régional (Feder) (12001407 [D-AL] EquipEx ImagInEx BioMed), and the Région Nord-Pas-de-Calais (convention 12000080).
We thank platforms of CUMA (University of Lille 2, J. F. Goossens) and LARMN (University of Lille 2, N. Azaroual) for access to equipment. We are grateful to J. Bukh, C. M. Rice, and T. Wakita for providing essential reagents. We are also grateful to Abderrazak Smaoui (Biotechnology Centre of Borj-Cédria) concerning botanical identification and to Thibaut Vausselin for useful discussions.
M.-E.S. is a recipient of a Ph.D. Fellowship provided by the French Government.
REFERENCES
- 1.Levrero M. 2006. Viral hepatitis and liver cancer: the case of hepatitis C. Oncogene 25:3834–3847. doi: 10.1038/sj.onc.1209562. [DOI] [PubMed] [Google Scholar]
- 2.Messina JP, Humphreys I, Flaxman A, Brown A, Cooke GS, Pybus OG, Barnes E. 2015. Global distribution and prevalence of hepatitis C virus genotypes. Hepatology 61:77–87. doi: 10.1002/hep.27259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Polaris Observatory HCV Collaborators. 2017. Global prevalence and genotype distribution of hepatitis C virus infection in 2015: a modelling study. Lancet Gastroenterol Hepatol 2:161–176. doi: 10.1016/S2468-1253(16)30181-9. [DOI] [PubMed] [Google Scholar]
- 4.Tarr AW, Khera T, Hueging K, Sheldon J, Steinmann E, Pietschmann T, Brown RJP. 2015. Genetic diversity underlying the envelope glycoproteins of hepatitis C virus: structural and functional consequences and the implications for vaccine design. Viruses 7:3995–4046. doi: 10.3390/v7072809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pawlotsky J. 2014. New hepatitis C therapies: the toolbox, strategies, and challenges. Gastroenterology 146:1176–1192. doi: 10.1053/j.gastro.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 6.Graham CS, Swan T. 2015. A path to eradication of hepatitis C in low- and middle-income countries. Antiviral Res 119:89–96. doi: 10.1016/j.antiviral.2015.01.004. [DOI] [PubMed] [Google Scholar]
- 7.Pawlotsky J-M. 2016. Hepatitis C virus resistance to direct-acting antiviral drugs in interferon-free regimens. Gastroenterology 151:70–86. doi: 10.1053/j.gastro.2016.04.003. [DOI] [PubMed] [Google Scholar]
- 8.Lindenbach BD, Thiel H-J, Rice CM. 2007. Flaviviridae: the viruses and their replication, p 1101–1152. In Knipe DM, Howley PM (ed), Fields virology, 5th ed Lippincott, William & Wilkins, Philadelphia, PA. [Google Scholar]
- 9.Egger D, Wölk B, Gosert R, Bianchi L, Blum HE, Moradpour D, Bienz K. 2002. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J Virol 76:5974–5984. doi: 10.1128/JVI.76.12.5974-5984.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sarrazin C. 2016. The importance of resistance to direct antiviral drugs in HCV infection in clinical practice. J Hepatol 64:486–504. doi: 10.1016/j.jhep.2015.09.011. [DOI] [PubMed] [Google Scholar]
- 11.Newman DJ, Cragg GM. 2016. Natural products as sources of new drugs from 1981 to 2014. J Nat Prod 79:629–661. doi: 10.1021/acs.jnatprod.5b01055. [DOI] [PubMed] [Google Scholar]
- 12.Abreu AC, McBain AJ, Simões M. 2012. Plants as sources of new antimicrobials and resistance-modifying agents. Nat Prod Rep 29:1007–1021. doi: 10.1039/c2np20035j. [DOI] [PubMed] [Google Scholar]
- 13.Gibbons S. 2004. Anti-staphylococcal plant natural products. Nat Prod Rep 21:263–277. doi: 10.1039/b212695h. [DOI] [PubMed] [Google Scholar]
- 14.Newman DJ, Cragg GM. 2012. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat Prod 75:311–335. doi: 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Calland N, Dubuisson J, Rouillé Y, Séron K. 2012. Hepatitis C virus and natural compounds: a new antiviral approach? Viruses 4:2197–2217. doi: 10.3390/v4102197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Polyak SJ, Oberlies NH, Pécheur E-I, Dahari H, Ferenci P, Pawlotsky J-M. 2013. Silymarin for HCV infection. Antivir Ther 18:141–147. doi: 10.3851/IMP2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ksouri R, Ksouri WM, Jallali I, Debez A, Magné C, Hiroko I, Abdelly C. 2012. Medicinal halophytes: potent source of health promoting biomolecules with medical, nutraceutical and food applications. Crit Rev Biotechnol 32:289–326. doi: 10.3109/07388551.2011.630647. [DOI] [PubMed] [Google Scholar]
- 18.Hajarizadeh B, Grebely J, Dore GJ. 2013. Epidemiology and natural history of HCV infection. Nat Rev Gastroenterol Hepatol 10:553–562. doi: 10.1038/nrgastro.2013.107. [DOI] [PubMed] [Google Scholar]
- 19.Sahli R, Rivière C, Neut C, Bero J, Sahuc M-E, Smaoui A, Beaufay C, Roumy V, Hennebelle T, Rouillé Y, Quetin-Leclercq J, Séron K, Ksouri R, Sahpaz S. 2017. An ecological approach to discover new bioactive extracts and products: the case of extremophile plants. J Pharm Pharmacol 69:1041–1055. doi: 10.1111/jphp.12728. [DOI] [PubMed] [Google Scholar]
- 20.Sahuc M-E, Sahli R, Rivière C, Pène V, Lavie M, Vandeputte A, Brodin P, Rosenberg A, Dubuisson JD, Ksouri R, Rouillé Y, Sahpaz S, Séron K. 2018. Dehydrojuncusol, a natural phenanthrene compound extracted from Juncus maritimus is a new inhibitor of hepatitis C virus replication. bioRxiv 10.1101/469361. [DOI] [PMC free article] [PubMed]
- 21.Sarkar H, Zerezghi M, Bhattacharyya J. 1988. Dehydrojuncusol, a constituent of the roots of Juncus roemerianus. Phytochemistry 27:3006–3008. doi: 10.1016/0031-9422(88)80714-3. [DOI] [Google Scholar]
- 22.Ciesek S, Von Hahn T, Colpitts CC, Schang LM, Friesland M, Steinmann J, Manns MP, Ott M, Wedemeyer H, Meuleman P, Pietschmann T, Steinmann E. 2011. The green tea polyphenol, epigallocatechin-3-gallate, inhibits hepatitis C virus entry. Hepatology 54:1947–1955. doi: 10.1002/hep.24610. [DOI] [PubMed] [Google Scholar]
- 23.Calland N, Albecka A, Belouzard S, Wychowski C, Duverlie G, Descamps V, Hober D, Dubuisson J, Rouillé Y, Séron K. 2012. (–)-Epigallocatechin-3-gallate is a new inhibitor of hepatitis C virus entry. Hepatology 55:720–729. doi: 10.1002/hep.24803. [DOI] [PubMed] [Google Scholar]
- 24.Calland N, Sahuc M-E, Belouzard S, Pène V, Bonnafous P, Mesalam AA, Deloison G, Descamps V, Sahpaz S, Wychowski C, Lambert O, Brodin P, Duverlie G, Meuleman P, Rosenberg AR, Dubuisson J, Rouillé Y, Séron K. 2015. Polyphenols inhibit hepatitis C virus entry by a new mechanism of action. J Virol 89:10053–10063. doi: 10.1128/JVI.01473-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fridell RA, Qiu D, Valera L, Wang C, Rose RE, Gao M. 2011. Distinct functions of NS5A in hepatitis C virus RNA replication uncovered by studies with the NS5A inhibitor BMS-790052. J Virol 85:7312–7320. doi: 10.1128/JVI.00253-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krishnan P, Beyer J, Mistry N, Koev G, Reisch T, DeGoey D, Kati W, Campbell A, Williams L, Xie W, Setze C, Molla A, Collins C, Pilot-Matias T. 2015. In vitro and in vivo antiviral activity and resistance profile of ombitasvir, an inhibitor of hepatitis C virus NS5A. Antimicrob Agents Chemother 59:979–987. doi: 10.1128/AAC.04226-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lam AM, Murakami E, Espiritu C, Steuer HMM, Niu C, Keilman M, Bao H, Zennou V, Bourne N, Julander JG, Morrey JD, Smee DF, Frick DN, Heck JA, Wang P, Nagarathnam D, Ross BS, Sofia MJ, Otto MJ, Furman PA. 2010. PSI-7851, a pronucleotide of beta-D-2’-deoxy-2’-fluoro-2’-C-methyluridine monophosphate, is a potent and pan-genotype inhibitor of hepatitis C virus replication. Antimicrob Agents Chemother 54:3187–3196. doi: 10.1128/AAC.00399-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ascher DB, Wielens J, Nero TL, Doughty L, Morton CJ, Parker MW. 2014. Potent hepatitis C inhibitors bind directly to NS5A and reduce its affinity for RNA. Sci Rep 4:4765. doi: 10.1038/srep04765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lontok E, Harrington P, Howe A, Kieffer T, Lennerstrand J, Lenz O, McPhee F, Mo H, Parkin N, Pilot-Matias T, Miller V. 2015. Hepatitis C virus drug resistance-associated substitutions: state of the art summary. Hepatology 62:1623–1632. doi: 10.1002/hep.27934. [DOI] [PubMed] [Google Scholar]
- 30.Fridell RA, Wang C, Sun J-H, O'Boyle DR, Nower P, Valera L, Qiu D, Roberts S, Huang X, Kienzle B, Bifano M, Nettles RE, Gao M. 2011. Genotypic and phenotypic analysis of variants resistant to hepatitis C virus nonstructural protein 5A replication complex inhibitor BMS-790052 in humans: in vitro and in vivo correlations. Hepatology 54:1924–1935. doi: 10.1002/hep.24594. [DOI] [PubMed] [Google Scholar]
- 31.El-Shamy AI, Abdel-Razek AF, Nassar MI. 2015. Phytochemical review of Juncus L. genus (Fam. Juncaceae). Arab J Chem 8:614–623. doi: 10.1016/j.arabjc.2012.07.007. [DOI] [Google Scholar]
- 32.Kovács A, Vasas A, Hohmann J. 2008. Natural phenanthrenes and their biological activity. Phytochemistry 69:1084–1110. doi: 10.1016/j.phytochem.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 33.Aquino R, Conti C, De Simone F, Orsi N, Pizza C, Stein ML. 1991. Antiviral activity of constituents of Tamus communis. J Chemother 3:305–309. doi: 10.1080/1120009X.1991.11739110. [DOI] [PubMed] [Google Scholar]
- 34.Bús C, Kúsz N, Jakab G, Senobar Tahaei SA, Zupkó I, Endrész V, Bogdanov A, Burián K, Csupor-Löffler B, Hohmann J, Vasas A. 2018. Phenanthrenes from Juncus Compressus Jacq. with promising antiproliferative and anti-HSV-2 activities. Molecules 23:E2085. doi: 10.3390/molecules23082085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Behery FAA, Naeem ZEM, Maatooq GT, Amer MMA, Wen Z-H, Sheu J-H, Ahmed AF. 2007. Phenanthrenoids from Juncus acutus L., new natural lipopolysaccharide-inducible nitric oxide synthase inhibitors. Chem Pharm Bull 55:1264–1266. doi: 10.1248/cpb.55.1264. [DOI] [PubMed] [Google Scholar]
- 36.Della Greca M, Fiorentino A, Isidori M, Lavorgna M, Monaco P, Previtera L, Zarrelli A. 2002. Phenanthrenoids from the wetland Juncus acutus. Phytochemistry 60:633–638. doi: 10.1016/S0031-9422(02)00152-8. [DOI] [PubMed] [Google Scholar]
- 37.Wang Y-G, Wang Y-L, Zhai H-F, Liao Y-J, Zhang B, Huang J-M. 2012. Phenanthrenes from Juncus effusus with anxiolytic and sedative activities. Nat Prod Res 26:1234–1239. doi: 10.1080/14786419.2011.561491. [DOI] [PubMed] [Google Scholar]
- 38.Dubuisson J, Hsu HH, Cheung RC, Greenberg HB, Russell DG, Rice CM. 1994. Formation and intracellular localization of hepatitis C virus envelope glycoprotein complexes expressed by recombinant vaccinia and Sindbis viruses. J Virol 68:6147–6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boulanger D, Waxweiler S, Karelle L, Loncar M, Mignon B, Dubuisson J, Thiry E, Pastoret PP. 1991. Characterization of monoclonal antibodies to bovine viral diarrhoea virus: evidence of a neutralizing activity against gp48 in the presence of goat anti-mouse immunoglobulin serum. J Gen Virol 72:1195–1198. doi: 10.1099/0022-1317-72-5-1195. [DOI] [PubMed] [Google Scholar]
- 40.Podevin P, Carpentier A, Pène V, Aoudjehane L, Carrière M, Zaïdi S, Hernandez C, Calle V, Méritet J, Scatton O, Dreux M, Cosset F, Wakita T, Bartenschlager R, Demignot S, Conti F, Rosenberg AR, Calmus Y. 2010. Production of infectious hepatitis C virus in primary cultures of human adult hepatocytes. Gastroenterology 139:1355–1364. doi: 10.1053/j.gastro.2010.06.058. [DOI] [PubMed] [Google Scholar]
- 41.Delgrange D, Pillez A, Castelain S, Cocquerel L, Rouillé Y, Dubuisson J, Wakita T, Duverlie G, Wychowski C. 2007. Robust production of infectious viral particles in Huh-7 cells by introducing mutations in hepatitis C virus structural proteins. J Gen Virol 88:2495–2503. doi: 10.1099/vir.0.82872-0. [DOI] [PubMed] [Google Scholar]
- 42.Goueslain L, Alsaleh K, Horellou P, Roingeard P, Descamps V, Duverlie G, Ciczora Y, Wychowski C, Dubuisson J, Rouillé Y. 2010. Identification of GBF1 as a cellular factor required for hepatitis C virus RNA replication. J Virol 84:773–787. doi: 10.1128/JVI.01190-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Y-P, Ramirez S, Jensen SB, Purcell RH, Gottwein JM, Bukh J. 2012. Highly efficient full-length hepatitis C virus genotype 1 (strain TN) infectious culture system. Proc Natl Acad Sci U S A 109:19757–19762. doi: 10.1073/pnas.1218260109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ramirez S, Mikkelsen LS, Gottwein JM, Bukh J. 2016. Robust HCV genotype 3a infectious cell culture system permits identification of escape variants with resistance to sofosbuvir. Gastroenterology 151:973–985. doi: 10.1053/j.gastro.2016.07.013. [DOI] [PubMed] [Google Scholar]
- 45.Pène V, Hernandez C, Vauloup-Fellous C, Garaud-Aunis J, Rosenberg AR. 2009. Sequential processing of hepatitis C virus core protein by host cell signal peptidase and signal peptide peptidase: a reassessment. J Viral Hepat 16:705–715. doi: 10.1111/j.1365-2893.2009.01118.x. [DOI] [PubMed] [Google Scholar]
- 46.Carrière M, Pène V, Breiman A, Conti F, Chouzenoux S, Meurs E, Andrieu M, Jaffray P, Grira L, Soubrane O, Sogni P, Calmus Y, Chaussade S, Rosenberg AR, Podevin P. 2007. A novel, sensitive, and specific RT-PCR technique for quantitation of hepatitis C virus replication. J Med Virol 79:155–160. doi: 10.1002/jmv.20773. [DOI] [PubMed] [Google Scholar]
- 47.Lecot S, Belouzard S, Dubuisson J, Rouillé Y. 2005. Bovine viral diarrhea virus entry is dependent on clathrin-mediated endocytosis. J Virol 79:10826–10829. doi: 10.1128/JVI.79.16.10826-10829.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bartosch B, Dubuisson J, Cosset F-L. 2003. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J Exp Med 197:633–642. doi: 10.1084/jem.20021756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rouillé Y, Helle F, Delgrange D, Roingeard P, Voisset C, Blanchard E, Belouzard S, McKeating J, Patel AH, Maertens G, Wakita T, Wychowski C, Dubuisson J. 2006. Subcellular localization of hepatitis C virus structural proteins in a cell culture system that efficiently replicates the virus. J Virol 80:2832–2841. doi: 10.1128/JVI.80.6.2832-2841.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Castelain S, Descamps V, Thibault V, François C, Bonte D, Morel V, Izopet J, Capron D, Zawadzki P, Duverlie G. 2004. TaqMan amplification system with an internal positive control for HCV RNA quantitation. J Clin Virol 31:227–234. doi: 10.1016/j.jcv.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 51.Kato T, Date T, Miyamoto M, Furusaka A, Tokushige K, Mizokami M, Wakita T. 2003. Efficient replication of the genotype 2a hepatitis C virus subgenomic replicon. Gastroenterology 125:1808–1817. doi: 10.1053/j.gastro.2003.09.023. [DOI] [PubMed] [Google Scholar]
- 52.Moradpour D, Evans MJ, Gosert R, Yuan Z, Blum HE, Goff SP, Lindenbach BD, Rice CM. 2004. Insertion of green fluorescent protein into nonstructural protein 5A allows direct visualization of functional hepatitis C virus replication complexes. J Virol 78:7400–7409. doi: 10.1128/JVI.78.14.7400-7409.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]