

Viral encephalitis is a significant cause of worldwide morbidity and mortality, and specific treatments are extremely limited. Virus infection of the brain triggers neuroinflammation; however, the role of neuroinflammation in the pathogenesis of viral encephalitis is unclear. Initial neuroinflammatory responses likely contribute to viral clearance, but prolonged exposure to proinflammatory cytokines released during neuroinflammation may be deleterious and contribute to neuronal death and tissue injury. Activation of astrocytes is a hallmark of neuroinflammation. Here, we show that reovirus infection of the brain results in the activation of astrocytes via an IFN-β-mediated process and that these astrocytes later die by Bak-mediated apoptosis. A better understanding of neuroinflammatory responses during viral encephalitis may facilitate the development of new treatment strategies for these diseases.
KEYWORDS: astrocyte, encephalitis, interferon, reovirus
ABSTRACT
Reovirus encephalitis in mice was used as a model system to investigate astrocyte activation (astrogliosis) following viral infection of the brain. Reovirus infection resulted in astrogliosis, as evidenced by increased expression of glial fibrillary acidic protein (GFAP), and the upregulation of genes that have been previously associated with astrocyte activation. Astrocyte activation occurred in regions of the brain that are targeted by reovirus but extended beyond areas of active infection. Astrogliosis also occurred following reovirus infection of ex vivo brain slice cultures (BSCs), demonstrating that factors intrinsic to the brain are sufficient to activate astrocytes and that this process can occur in the absence of any contribution from the peripheral immune response. In agreement with previous reports, reovirus antigen did not colocalize with GFAP in infected brains, suggesting that reovirus does not infect astrocytes. Reovirus-infected neurons produce interferon beta (IFN-β). IFN-β treatment of primary astrocytes resulted in both the upregulation of GFAP and cytokines that are associated with astrocyte activation. In addition, the ability of media from reovirus-infected BSCs to activate primary astrocytes was blocked by anti-IFN-β antibodies. These results suggest that IFN-β, likely released from reovirus-infected neurons, results in the activation of astrocytes during reovirus encephalitis. In areas where infection and injury were pronounced, an absence of GFAP staining was consistent with activation-induced cell death as a mechanism of inflammation control. In support of this, activated Bak and cleaved caspase 3 were detected in astrocytes within reovirus-infected brains, indicating that activated astrocytes undergo apoptosis.
IMPORTANCE Viral encephalitis is a significant cause of worldwide morbidity and mortality, and specific treatments are extremely limited. Virus infection of the brain triggers neuroinflammation; however, the role of neuroinflammation in the pathogenesis of viral encephalitis is unclear. Initial neuroinflammatory responses likely contribute to viral clearance, but prolonged exposure to proinflammatory cytokines released during neuroinflammation may be deleterious and contribute to neuronal death and tissue injury. Activation of astrocytes is a hallmark of neuroinflammation. Here, we show that reovirus infection of the brain results in the activation of astrocytes via an IFN-β-mediated process and that these astrocytes later die by Bak-mediated apoptosis. A better understanding of neuroinflammatory responses during viral encephalitis may facilitate the development of new treatment strategies for these diseases.
INTRODUCTION
Astrocytes are the most abundant cells in the central nervous system (CNS) and are responsible for maintenance of homeostasis and modulation of neuronal activity. Astrocytes act cooperatively with neurons to facilitate a wide variety of functions, including neurotransmitter trafficking and recycling, ion homeostasis, energy metabolism, defense against oxidative stress, and glutamate uptake. This critical dependence of neurons upon astrocyte-derived trophic support confers astrocytes with intrinsic neuroprotective properties (1). Astrocyte reactivity can be triggered by any alteration in brain homeostasis, and they become reactive in response to virtually all pathological conditions in the CNS. Accordingly, astrocytes are equipped with many receptors and intracellular signaling cascades that respond quickly to changes in their environment, such as the presence of viral or bacterial proteins in the extracellular space, increased concentrations of cytokines or chemokines, and even the absence of “normal” signals (growth factors or neurotransmitters) from neighboring cells (2). Astrocyte activation is characterized by increased expression of glial fibrillary acidic protein (GFAP), an intermediate filament protein, as well as cell hypertrophy and proliferation (3, 4). Activated astrocytes act as important regulators of CNS inflammation and release a wide variety of immune mediators, such as cytokines, chemokines, and growth factors that may exert either neuroprotective or neurotoxic effects. Consistent with the neuroprotective role of astrocytes in controlling neuroinflammation, astrocytes participate in the suppression of microglia through negative-feedback loops (5). However, astrocytes are also associated with pathogenesis and selective inhibition of their proinflammatory processes is neuroprotective in some models of CNS injury (6).
Virus-induced CNS disease is a significant cause of morbidity and mortality and treatment options are severely limited. Autopsy studies have shown that astrocytes are activated following infection of the brain with several human viruses (7–10), and recent studies have shown that astrocytes are important interferon (IFN)-producing cells in various neurotropic viral infections (11–13). It has been suggested that uninfected astrocytes may exist in an antiviral state and respond very quickly to viral infection by upregulating important antiviral IFN-stimulated genes (14). However, many questions remain involving the role of astrocytes in viral pathogenesis within the CNS, including the mechanisms of astrocyte activation and how activated astrocytes are regulated to prevent deleterious effects on neurons that may result from prolonged activation.
Reovirus encephalitis in neonatal mice is a classic model of viral pathogenesis in the CNS. We have previously shown that reovirus infection of the spinal cord of newborn mice results in astrocyte activation, as characterized by changes in astrocyte morphology and increased expression of GFAP (15). We now show that reovirus infection of the brain causes astrocyte activation in regions of the brain that are targeted by reovirus, but that this activation is not limited to areas of active infection.
Reovirus antigen was not detected in astrocytes in the brain, consistent with our previous studies showing that reovirus does not infect astrocytes in infected spinal cords, brain slice cultures (BSCs), or primary glial cultures (15–17). Our prior studies also suggested that activation of astrocytes following reovirus infection is absent in mice deficient in the expression of the IFN-α/β receptor, suggesting that astrocyte activation may be triggered by IFN-α/β released from reovirus-infected neurons (15). We now show that IFN-β treatment of primary astrocytes results in their activation and that anti-IFN-β antibodies block the ability of supernatant from reovirus-infected BSC to activate primary astrocytes. In areas of pronounced reovirus infection and significant injury, there was a loss of astrocytes and associated GFAP staining. In addition, in these areas GFAP staining in residual astrocytes colocalized with proapoptotic Bak-NT and activated caspase 3, suggesting that activated astrocytes undergo apoptotic cell death to curtail excessive inflammation. These data indicate a prominent role for astrocytes in viral pathogenesis within the CNS and suggest that manipulation of astrocyte function may provide a novel treatment strategy for virus-induced CNS disease.
RESULTS
Infection of the brain with reovirus results in astrogliosis.
Astrocyte activation is characterized by increased expression of GFAP. We used Western blot analysis to determine levels of GFAP expression in reovirus-infected brains. Two-day-old Swiss Webster (SW) mice were infected with reovirus by intracerebral (i.c.) inoculation of 100 PFU of reovirus (T3 Dearing/T3D). Brains were harvested from virus- and mock-infected mice at 3, 5, and 7 days postinfection (p.i.). Western blot analysis (Fig. 1A and B) was then used to demonstrate that reovirus (T3D) infection resulted in increased GFAP expression. Increased GFAP expression was apparent as early as day 3 p.i. in reovirus-infected brains, compared to mock-infected controls, and continued to rise through the course of infection with increases in GFAP expression of around 10-fold seen in reovirus-infected brains at 7 days p.i., compared to mock-infected controls. Our prior microarray analysis (18) also showed increased GFAP expression in T3D-infected brains, compared to mock-infected controls at the mRNA level (Table 1). This microarray analysis also identified additional genes associated with astrocyte activation that were upregulated in the brain following reovirus infection (Table 1), including vimentin (Vim), lipocalin 2 (Lcn2), tumor necrosis factor alpha (TNF-α)-induced protein 9 (TNFAIP9), sphingosine 1-phosphate receptor-3 (S1pr3), tissue inhibitor of metalloproteinases 1 (Timp1), C-X-C motif chemokine 10 (Cxcl10), and oncostatin M receptor (Osmr). These genes represent a core set of genes that is upregulated in reactive astrocytes (19). Chemokine (C-C motif) ligand 5 (CCL5) and interleukin-6 (IL-6) that are upregulated in cultured astrocytes in response to viral infection and may be important for viral CNS pathogenesis (20–25) were also upregulated in reovirus-infected brains compared to mock-infected controls (Table 1). Taken together, these data indicate that markers of astrocyte activation are increased in the brain following reovirus infection.
FIG 1.

Reovirus infection of the brain results in increased expression of GFAP. Two-day-old Swiss Webster (SW) mice were infected with reovirus by i.c. inoculation of 100 PFU of reovirus (T3 Dearing/T3D). (A) Western blot analysis (see representative blot) shows increased GFAP levels at 3, 5, and 7 days p.i. (DPI) in reovirus (Reo)-infected brains compared to mock-infected controls. Increased reovirus antigen (σ3) was also seen at 3, 5, and 7 days p.i. Actin levels were used to demonstrate equivalent protein loading between samples. (B) Densitometric analysis of three blots. The graph shows the mean intensity of GFAP bands. Error bars represent the standard errors of the mean (SEM). **, P < 0.01; ****, P < 0.0001. (C) At 8 days p.i., increased GFAP (green) and reovirus σ3 (red) staining in virus-infected brains (Reo) compared to mock-infected controls can be seen in and around the hippocampus (hippo) and thalamus (thal), two areas targeted by reovirus.
TABLE 1.
Genes associated with astrocyte activation are upregulated in the brain following reovirus infection
| Function | Gene | Increase in expressiona
|
|
|---|---|---|---|
| Fold change | P | ||
| Astrocyte marker | GFAP | 5.44 | 9.93E–05 |
| Vim | 2.89 | 0.0003 | |
| Inflammation | TNFAIP9 | 22.4 | 0.005 |
| S1pr3 | 4.6 | 6.76E–05 | |
| Timp1 | 7.8 | 1.09E–05 | |
| Cxcl10 | 248.8 | 5.21E–07 | |
| Osmr | 4.9 | 0.0001 | |
| CCL5 | 195.9 | 0.0001 | |
| IL-6 | 31.4 | 0.0007 | |
| Innate immunity | Lcn2 | 386.2 | 7.61E–07 |
Increased expression of genes in reovirus-infected brains at 8 days p.i. compared to mock-infected controls was determined by microarray analysis (25).
Immunohistochemistry (IHC) was used to investigate the location of increased GFAP staining within infected brains. Brain sections prepared at 8 days p.i., when mice were showing clinical signs of CNS disease, were incubated with GFAP and reovirus antibodies (anti-sigma 3) or stained with hematoxylin and eosin (H&E). Increased GFAP staining was seen in the brain of reovirus-infected animals, compared to mock-infected controls, and was particularly noticeable in and around the hippocampus and thalamus, two regions of the brain that are specifically targeted by reovirus (Fig. 1C). The cortex, which is also targeted by reovirus, also showed increased GFAP staining following infection, but this was not as marked (not shown). Similar results were found using a second serotype 3 (T3Abney/T3A) reovirus strain (not shown).
Infection of ex vivo BSCs with reovirus also resulted in astrocyte activation, demonstrating that reovirus-induced astrocyte activation does not require the presence of immune cells that infiltrate from the periphery and instead can be brought about by factors intrinsic to the CNS (Fig. 2). For these experiments, BSCs were infected with reovirus (106 PFU/slice) and harvested after 8 days.
FIG 2.
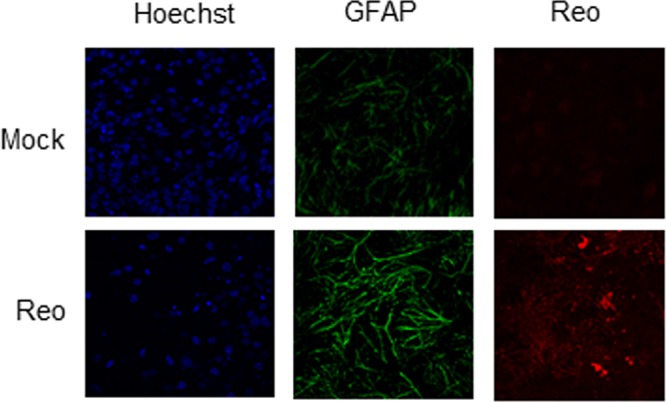
Expression of GFAP is increased in reovirus-infected ex vivo BSC. Ex vivo slice cultures were infected with reovirus (106 PFU/slice). At 8 days p.i., slices were prepared for IHC. Increased GFAP staining (green) is seen in virus-infected slices compared to mock-infected controls. The image shows infected cortex tissue.
Interferon activates primary astrocytes.
Although GFAP expression was increased in areas of the brain infected with reovirus, reovirus antigen did not colocalize with GFAP in individual cells, indicating that reovirus does not infect astrocytes (Fig. 3). This is consistent with our previous findings in reovirus-infected spinal cords (15), BSC (16) and primary glial cultures (17). Since reovirus does not appear to infect astrocytes, we wanted to see whether cytokines released following reovirus infection of neurons might be capable of activating astrocytes in a paracrine fashion. We have previously shown that reovirus infection of the CNS results in a robust IFN response (15, 18, 26, 27) that is protective (26) and which may be required for astrocyte activation (15). Treatment of primary astrocytes with IFN-β (100 U/ml) resulted in the upregulation of GFAP, as shown by Western blot analysis (Fig. 4A). Significant increases in protein levels were seen as early as 1 day following IFN-β treatment. In addition, reverse transcription-PCR (RT-PCR) was used to demonstrate that IFN-β treatment of primary astrocytes resulted in significant upregulation of cytokines that are associated with astrocyte activation (Fig. 4B). One day following IFN-β treatment of astrocytes, the expression of IL-6 and CCL5 increased ∼15-fold, and the expression of CXCL10 increased ∼2,000-fold. These data show that IFN-β, administered in a paracrine fashion, is capable of activating primary astrocytes in vitro. In contrast, there was no evidence of autocrine IFN signaling occurring in reovirus-exposed astrocytes since primary astrocytes did not support reovirus growth and did not produce IFN-β when exposed to reovirus (Fig. 4C and D),
FIG 3.
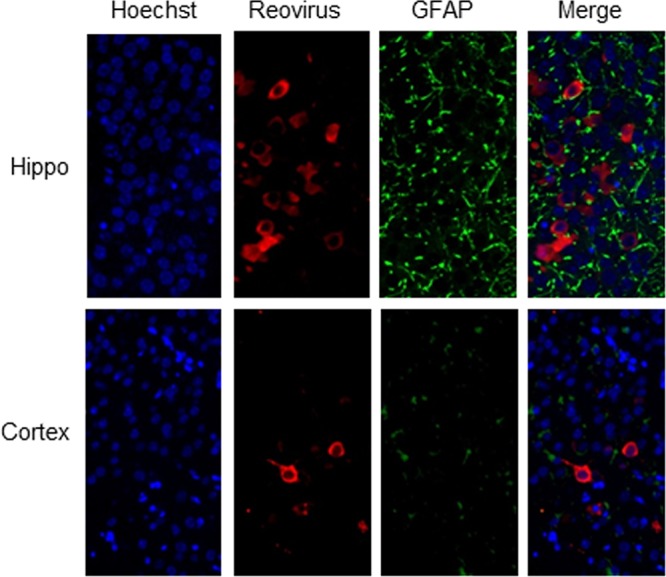
Viral antigen does not colocalize with GFAP in virus-infected brains. Two-day-old SW mice were infected with reovirus by i.c. inoculation of 100 PFU of reovirus (T3 Dearing/T3D). At 8 day p.i., mice were sacrificed, and brains were sectioned and prepared for IHC. Reovirus antigen (red) did not colocalize with GFAP (green) in sections from the hippocampus (Hippo) and cortex.
FIG 4.

Treatment with IFN-β activates primary astrocytes. Primary astrocytes were treated with IFN-β (100 U/ml). At 0, 1, 2, and 3 days posttreatment (DPT), the cells were harvested for Western blot analysis. (A) A representative blot shows that IFN-β treatment causes increased expression of GFAP in primary astrocytes. The graph shows combined data from three blots and demonstrates significant increases in relative intensity of GFAP staining in astrocytes treated with IFN-β. (B) Astrocytes were also collected for RT-PCR analysis. The graphs show the mean expression of chemokines/cytokines associated with astrocyte activation in IFN-β-treated and untreated astrocytes. (C) Astrocytes were infected with reovirus (MOI of 1). At 0, 1, and 3 days p.i., RNA was harvested from infected cells, and the viral titer was determined by quantitative PCR that was compared to a standard curve. The graph shows the mean viral titer in astrocytes immediately after infection (day 0) and after 1 and 3 days (black bars). Reovirus titers in L929 cells that were infected in parallel are shown for comparison (gray bars). (D) RNA from reovirus-exposed astrocytes was assayed for the presence of IFN-β RNA. The graph shows the mean expression compared to day 0 (black bars). Reovirus induced expression of IFN-β in L929 cells is shown for comparison (gray bars). All error bars represent the SEM. *, P < 0.05; **, P < 0.01; ****, P < 0.0001.
IFN antibodies block astrocyte activation following treatment of primary astrocytes with conditioned media from reovirus-infected BSCs.
To further demonstrate that factors released following reovirus infection of the CNS were capable of activating astrocytes, we performed supernatant transfer experiments, where conditioned media from reovirus-infected BSCs was used to activate primary astrocytes. Conditioned medium was collected from mock- or reovirus-infected BSC at 8 days p.i. and added to primary astrocytes (ratio of reovirus-conditioned medium [BSC supernatant] and fresh Dulbecco modified Eagle medium [DMEM] = 1/10). Three days after treatment, the astrocytes were stained with GFAP. Figure 5 demonstrates that reovirus-conditioned medium resulted in astrocyte activation, as determined by increased expression of GFAP in astrocytes exposed to conditioned medium compared to astrocytes exposed to medium from mock-infected BSCs. Astrocyte activation was decreased when the conditioned medium was first treated with anti-IFN-β antibody (10 µg/ml) but remained unchanged when the conditioned medium was treated with an isotype-matched antibody (10 µg/ml). Increased expression of GFAP in primary astrocytes treated with conditioned medum from reovirus-infected BSC compared to astrocytes treated with medium from mock-infected BSCs was also shown by Western blot analysis (Fig. 5C and D) and was again blocked in the presence of anti-IFN-β antibodies. Similar findings of increased expression of cytokines associated with astrocyte activation were also seen in primary astrocytes treated with conditioned media from reovirus-infected BSCs compared to astrocytes treated with media from mock-infected BSCs (Fig. 5E and F). These results show that IFN-β contributes to the activation of astrocytes by conditioned media from reovirus-infected BSCs.
FIG 5.

The ability of supernatants from reovirus-infected BSCs to activate primary astrocytes is blocked with anti-IFN-β antibody. Conditioned medium (CM) was collected from mock-infected and reovirus (Reo)-infected BSCs at 8 days p.i. and was added to primary astrocytes (ratio of reovirus conditioned medium [BSC supernatant] to fresh DMEM = 1/10) in the presence of anti-IFN-β antibodies or isotype-matched controls. Fresh BSC medium was also used as a control. (A) Three days following treatment, the astrocytes were stained with GFAP. Increased expression of GFAP (red) was seen in astrocytes exposed to reovirus-conditioned medium compared to astrocytes exposed to medum from mock-infected BSCs or medium alone. This activation was decreased when the conditioned medium was first treated with anti-IFN-β antibody (10 µg/ml) but remained unchanged when conditioned medium was treated with an isotype-matched antibody (10 µg/ml). (B) A graph shows the mean pixel intensity of images taken from the center of three wells of a chamber slide from astrocytes exposed to media from mock- or virus-infected primary astrocytes. (C to F) Increased expression of GFAP (C and D) and cytokines (E and F) in primary astrocytes treated with conditioned medium from reovirus-infected BSCs compared to astrocytes treated with medium from mock-infected BSCs, as shown by Western blotting (C and D) or RT-PCR analysis (E and F) and was blocked in the presence of anti-IFN-β antibodies. Graphs D to F show the mean expression of GFAP or chemokines/cytokines. Error bars represent the SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Activated astrocytes in highly infected/injured areas undergo apoptosis.
In brain regions where reovirus infection was pronounced, as determined by high levels of virus and extensive injury (defined by significant neuronal loss), we observed an absence of GFAP staining (Fig. 6A and B). This is consistent with the possibility that astrocytes undergo activation-induced cell death (AICD) as a mechanism for curtailing inflammation. In contrast, GFAP staining was still present in uninfected areas. Prior studies have indicated that astrocyte apoptosis can be mediated by the proapoptotic Bcl-2 family protein Bak (28–34). In healthy cells, Bak is a monomer that resides in the outer mitochondrial membrane (OMM). During apoptosis, stress signals activate Bak causing a conformational change that exposes its N terminus and results in oligomerization and the formation of pores in the OMM (35, 36). Bak activation was investigated in the brains of reovirus-infected mice by IHC using an antibody specific to the N terminus of Bak (Bak-NT, 06-536; Millipore, Billerica, MA) that is only exposed following conformational change and was seen in activated astrocytes (Fig. 6C), suggesting that activated astrocytes undergo Bak-mediated apoptosis. As well as undergoing conformational activation, Bak mRNA levels are also upregulated (fold change, 1.4; P = 6.9E–05) following reovirus infection of the brain (18). Interestingly, active Bax was not seen in astrocytes in reovirus-infected brains and appeared to be confined to neurons (Fig. 6D), presumably as a consequence of reovirus infection (37). Apoptosis of astrocytes involving Bak, but not Bax, has previously been reported (33). Consistent with astrocytes undergoing activation-induced apoptosis, IHC was also used to show that cleaved (activated) caspase 3, the apoptotic executioner protein responsible for many of the morphological outcomes of apoptosis, also colocalized with GFAP in reovirus-infected brains (Fig. 6E). Activated Bak and cleaved caspase 3 were also found in cells that were not positive for GFAP and are likely reovirus-infected neurons, which are also known to undergo apoptosis during reovirus encephalitis (38).
FIG 6.

As infection progresses, astrocytes undergo apoptosis. Two-day-old SW mice were infected with reovirus (Reo) as described previously (Fig. 1). At 8 days p.i., mice were sacrificed, and brains were sectioned and prepared for IHC. (A) In areas with high levels of virus antigen (red staining) and injury (H&E staining), GFAP staining (green) is absent (arrows). (B) The red (reovirus antigen) and green (GFAP) pixel intensity for the CA2 region of the hippocampus of eight reovirus-infected mice was quantified using ImageJ. The graph shows high GFAP staining when reovirus antigen is present in relatively small amounts, but that GFAP expression drops off as levels of reovirus antigen increase. (C) Images from the thalamus demonstrating increased Bak-NT staining (red) in reovirus-infected sections compared to mock-infected controls. Bak-NT staining colocalized with GFAP staining (green) in some astrocytes (arrows). (D) Images from the hippocampus demonstrating that Bax-NT staining (green) increased in virus-infected sections compared to mock-infected controls but did not colocalize with GFAP (red). (E) Images from the hippocampus demonstrate that cleaved caspase 3 (CC3, red) colocalizes with GFAP (green) in some astrocytes (arrows). Cells staining for cleaved caspase 3 but not GFAP are likely reovirus-infected neurons.
DISCUSSION
Astrocytes are activated following infection of the CNS with a variety of different viruses, but their role in viral pathogenesis is not fully understood. Two important questions that remain unanswered include (i) how astrocytes are activated following infection of the CNS with viruses that predominantly infect neurons and (ii) how astrocytes influence virus-induced CNS disease. In this report we used reovirus encephalitis as a model system to investigate these issues.
We first show that GFAP expression is increased in reovirus-infected brains compared to mock-infected controls at both the mRNA and the protein level. GFAP is an intermediate filament protein expressed in astrocytes, and its upregulation is commonly used as a marker for astrocyte activation. Microarray analysis also demonstrated that increased expression of other genes associated with astrocyte activation was also seen in reovirus-infected brains, including Vim (another intermediate filament protein), along with numerous genes with functions related to inflammation (TNFAIP9, S1pr3, Timp1, Cxcl10, and Osmr) and innate immunity (Lcn2). These genes have been shown to be upregulated in astrocytes isolated from two different models of mouse CNS injury, ischemic stroke (where astrocytes are proposed to serve a protective function), and neuroinflammation (where astrocytes contribute to disease pathology) and represent a core set of genes that is upregulated in reactive astrocytes (19). Genes isolated from astrocytes from these models that were specific to stroke (cardiotrophin-like cytokine factor 1, IL-6, thrombospondin 1, and leukemia inhibitory factor) or neuroinflammation (complement genes) have been proposed to designate astrocytes with either “protective” or “destructive” functions. Genes from both groups were upregulated following i.c. reovirus injection (not shown), making it difficult to assign a protective or destructive role for astrocytes during reovirus encephalitis using gene expression analysis.
CCL5 and IL-6, genes that have previously been shown to be upregulated in cultured astrocytes in response to viral infection and may be important for viral CNS pathogenesis (20–25), were also upregulated in reovirus-infected brains compared to mock-infected controls. These results suggest that although astrocytes are activated following reovirus infection and upregulate genes that encourage the influx of peripheral immune cells that function to clear the virus, this activation is not sufficient to prevent reovirus-induced CNS disease and associated mortality. Reovirus infection produces rapid death in baby mice, which typically die around 8 days p.i. It is possible that activated astrocytes might be more productive in fighting the infection if disease progressed more slowly, giving the immune response more time to react.
The fact that astrocytes are not infected by reovirus suggests that something produced by infected neurons might cause astrocyte activation. Viral infection of the brain generally results in a strong IFN response. Following infection, viral double-stranded viral RNA, produced as an intermediate during viral replication or, in the case of reovirus, its genetic material, is sensed as a danger signal in the infected cell by pattern recognition receptors, leading to the upregulation of type I IFNs (39, 40). These cytokines mediate antiviral effects following binding to the IFN-α receptor (IFNAR) that involves the upregulation of hundreds IFN-stimulated genes that can inhibit almost every step of the virus life cycle (41, 42). In mice, the type I IFN response is essential for protection against many viruses, including reovirus, West Nile virus, Japanese encephalitis virus, and Zika virus (43–47).
The CNS has been considered an immune-privileged tissue; however, recent studies have implicated the importance of intrinsic, innate antiviral responses within the CNS (48, 49), including following reovirus infection, where the lack of IFN signaling resulted in increased apoptosis in BSCs (26). Our prior studies have shown that reovirus infection of CNS tissue results in increased expression of primary IFNs, as well as IFN-stimulated genes (15, 18, 26). The IFN response is protective, and reovirus-induced CNS disease and survival are impaired in mice that are deficient in IFN signaling (26). The results presented here indicate that treatment of primary astrocytes with IFN-β results in astrocyte activation, as demonstrated by an increase in GFAP expression. The increases in GFAP levels are more pronounced in virus-infected brains compared to IFN-β-treated primary astrocytes (densitometric analysis revealed an ∼10-fold increase in GFAP expression in virus-infected, compared to mock-infected, brains at 7 days p.i., whereas there was a 2-fold increase in GFAP expression in IFB-β-treated astrocytes after 3 days: see Fig. 1 and 4, respectively). There are a couple of possible explanations for this. First, it is well established that astrocytes behave differently in culture than in the CNS (50). Second, it is possible that additional factors in addition to type I IFNs contribute to astrocyte activation. In addition to increased levels of GFAP in IFN-β-treated astrocytes, IL-6, CCL5, and CXCL10 expression also increased reflecting astrocyte activation. The expression of these genes peaked at 1 day posttreatment. The expression of CCL5 and CXCL10 then decreased (despite sustained GFAP levels) but was still significantly higher than the levels seen in untreated astrocytes. It is possible that decreased expression of CCL5 and CXCL10 occurs as part of a mechanism to control inflammation resulting from astrocyte activation. Further, although conditioned media from reovirus-infected BSCs activated primary astrocytes in vitro, this activation was reduced in the presence of anti-IFN antibodies. Our prior studies show that astrocytes are not activated in mice that are deficient in the expression of the IFN-α/β receptor (15), again suggesting that IFN contributes to astrocyte infection following reovirus infection of the CNS.
Areas of the brain that had pronounced infection, as determined by the widespread presence of viral antigen and severe tissue damage, had no GFAP staining. Apoptosis of activated astrocytes has been shown to occur both in vivo, in the CNS of patients with diseases such as Alzheimer’s disease (28), and following HIV infection (29), and there is a growing consensus that the apoptosis of activated astrocytes may serve as a self-regulatory mechanism (activation-induced cell death [AICD]) to control the severity of inflammation in the brain by limiting damage produced by their release of mediators with toxic effects on neurons. We now show that active Bak and cleaved (activated) caspase 3 colocalize in astrocytes during reovirus encephalitis suggesting that activated astrocytes in reovirus-infected brains undergo Bak-mediated apoptosis. Interestingly, we show that Bax is not activated in astrocytes during reovirus encephalitis but can be seen in neurons (37). These results are consistent with an earlier study demonstrating Bak-mediated, but not Bax-mediated, apoptosis in astrocytes (33). Activation-induced cell death has been proposed to be mediated by astrocyte-produced nitric oxide (31). These results suggest that possible detrimental actions of activated astrocytes due to the effects of the cytokines/chemokines and neurotoxic molecules they produce appear to be appropriately kept in check.
Interestingly, we did not see the death of primary astrocytes after IFN treatment. It is possible that astrocytes need a second signal to undergo apoptosis. The upregulation of mitochondrial proteins has previously been shown to sensitize cells to apoptosis (51). In this case Bak (but not Bax) was one of the proteins induced by IFN-γ and thought to contribute to the sensitization of cells to apoptosis induced by TNF-α and anti-Fas antibody.
The results presented here demonstrate that reovirus infection of the mouse brain results in astrogliosis, which appears to be triggered in part by IFN released from infected neurons, although it is also possible that the production of IFN by astrocytes, perhaps following the ingestion of infected neurons or debris, amplifies astrocyte activation in an autocrine manner. We also present evidence of activation-induced cell death of astrocytes in reovirus-infected brains. An increased understanding of astrocyte activation following virus infection of the CNS may lead to novel therapies for virus-induced CNS disease.
MATERIALS AND METHODS
Viral stocks.
Reoviruses serotype 3 strain Dearing (T3D) and serotype 3 strain Abney (T3A) are laboratory stocks derived via plaque purification and double passage in L929 (ATCC CCL1) cells. Viruses were further purified via high-speed cesium chloride density gradient centrifugation to remove medium components and diluted in phosphate-buffered saline (PBS). Mock infections were carried out using PBS.
In vivo studies.
SW outbred mice were obtained from Harlan Laboratories (Indianapolis, IN). Two-day-old mice were i.c. inoculated with 100 (or as otherwise indicated) PFU of T3D diluted in 10 μl of PBS. Mock-infected mice were i.c. injected with PBS only at equal volumes. All animal experiments were approved by the Institutional Animal Care and Use Committee.
Western blot analysis.
Immediately upon harvesting, whole brains were triturated in lysis buffer in the presence of phosphatase inhibitor cocktail (Thermo Scientific, Rockford, IL). Lysates were cleared, electrophoresed through 10% polyacrylamide gels at a constant 120 V, and transferred to polyvinylidene difluoride membranes (Amersham Biosciences, Pittsburgh, PA). Immunoblotting was performed with anti-GFAP (catalog no. AB7260; Abcam, Cambridge, MA), anti-tubulin (catalog no. 2144; Cell Signaling, Boston, MA) and anti-actin (Calbiochem, Sunnyvale, CA) antibodies. After washes and 1 h of incubation with appropriate horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA), images were obtained on a FluorochemQ MultiImage III imaging workstation (Cell Biosciences, Santa Clara, CA), and densitometry quantification was performed using Alphaview v3.0 software.
Histological studies and immunocytochemistry staining.
Freshly harvested brain tissue was immersed in 10% buffered formalin for no less than 12 h, embedded in paraffin, serially sectioned into 6-μm-thick sections, and mounted onto slides. Sections were desiccated at 50°C for 15 min and then rehydrated in PBS for 30 min. Tissue was subjected to antigen retrieval (antigen unmasking solution; Vector Laboratories, Burlingame, CA) and permeabilization/blocking (5% fetal bovine serum and 5% normal goat serum in 0.3% Triton/PBS) prior to incubation with primary antibodies: monoclonal reovirus sigma 3 antibody (4F2; 1:100), rabbit polyclonal reovirus antibody (1:200), rabbit anti-GFAP (1:200, catalog no. AB7260; Abcam, Cambridge, MA), mouse anti-GFAP (1:100, catalog no. G3893; Millipore/Sigma, Burlington, MA), rabbit anti-Bak-NT (1:100, catalog no. 06-536;Millipore/Sigma), and rabbit anti-Bax-NT (1:100, catalog no. 06-499; Upstate, Lake Placid, NY). Sections were then incubated with secondary antibodies: Alexa Fluor 488-conjugated IgG and Alexa Fluor 568-conjugated IgG. Nuclei were Hoechst stained (10 µg/ml; Immunochemistry Technology, Bloomington, MN) for immunofluorescence imaging. Sections were mounted with VectorShield mounting medium (Vector Laboratories), and slides were imaged either on a Marianas fluorescence imaging workstation (Intelligent Imaging Innovation, Inc., Denver, CO) based on a Zeiss 300M inverted microscope or on Olympus FV1000 confocal microscope with Olympus FluoViews software (Olympus, Center Valley, PA).
Organotypic brain slice culture studies.
BSCs were prepared from 2- to 3-day-old mice and infected with 106 PFU of T3A reovirus as previously described (23). T3A reovirus was used for BSC infections since it is more virulent than T3D in vitro. Briefly, four 400-µm coronal sections of cerebrum were made from a single animal using a vibrating-blade microtome (VT1000S; Leica, Bannockburn, IL). Slices were maintained in a humidified incubator at 5% carbon dioxide (CO2) and 36.5°C on a semiporous membrane insert (PICMORG50; Millipore) positioned in a 35-mm tissue culture well with 1.1 ml of medium containing 10% fetal bovine serum (FBS). Immediately after plating, slices were infected by the dropwise addition of 106 PFU of T3A (diluted in 20 µl of PBS) or of PBS only to each slice. The medium was refreshed with 5% FBS-containing medium approximately 12 h after slicing, and subsequent medium changes were made with serum-free medium every 3 days thereafter. The conditioned medium from reovirus-infected or mock-infected BSCs was collected at 8 days p.i. for supernatant transfer experiments with primary astrocytes.
Primary astrocyte culture.
Primary astrocyte cultures were prepared from neocortical tissues of new born mice within 24 h. Cortex tissue was isolated after carefully removing the hippocampus, basal ganglia, and meninges. Cortex tissue was then minced with a scalpel and further triturated by pipetting. The minced tissue was dissociated with trypsin for 15 min at 37°C. Dissociated cells were centrifuged at 1,200 rpm for 5 min and resuspended in DMEM containing 10% FBS, 1× GlutaMAX, and 1% penicillin-streptomycin. Cells were seeded onto flasks and grown at 37°C in 5% CO2 with a change of medium once a week. At confluence (around 2 weeks), the primary cells were split and expanded for treatment. The resultant primary astrocytes were treated either with IFN-β (100 U/ml, catalog no. 12400; PBL Assay Science, Piscataway, NJ) or conditioned medium from reovirus-infected BSCs (10% volume of fresh culture media). Anti-IFN-β antibody (10 µg/ml; catalog no. 12400; PBL Assay Science) or an isotype control antibody (10 µg/ml, catalog no. 37415; Abcam) was added as described.
Statistical analysis.
All bar graphs are presented as means ± the standard deviations. Numbers of independent experiments are indicated by the n value. All statistical analyses were performed using Prism software (GraphPad Software, Inc., San Diego, CA). Statistical comparisons of mean measurement between two groups (e.g., mock versus infected) were made using a two-tailed, unpaired t test.
ACKNOWLEDGMENTS
This study was supported by the Department of Veteran’s Affairs and the National Institutes of Health (R01: NS076512; R33: AI101064). K.L.T. is the Louise Baum endowed Professor and Chair.
REFERENCES
- 1.Belanger M, Magistretti PJ. 2009. The role of astroglia in neuroprotection. Dialogues Clin Neurosci 11:281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Buffo A, Rolando C, Ceruti S. 2010. Astrocytes in the damaged brain: molecular and cellular insights into their reactive response and healing potential. Biochem Pharmacol 79:77–89. doi: 10.1016/j.bcp.2009.09.014. [DOI] [PubMed] [Google Scholar]
- 3.Burda JE, Sofroniew MV. 2014. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 81:229–248. doi: 10.1016/j.neuron.2013.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kigerl KA, de Rivero Vaccari JP, Dietrich WD, Popovich PG, Keane RW. 2014. Pattern recognition receptors and central nervous system repair. Exp Neurol 258:5–16. doi: 10.1016/j.expneurol.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Farina C, Aloisi F, Meinl E. 2007. Astrocytes are active players in cerebral innate immunity. Trends Immunol 28:138–145. doi: 10.1016/j.it.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 6.Brambilla R, Bracchi-Ricard V, Hu WH, Frydel B, Bramwell A, Karmally S, Green EJ, Bethea JR. 2005. Inhibition of astroglial nuclear factor κB reduces inflammation and improves functional recovery after spinal cord injury. J Exp Med 202:145–156. doi: 10.1084/jem.20041918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gelpi E, Preusser M, Garzuly F, Holzmann H, Heinz FX, Budka H. 2005. Visualization of Central European tick-borne encephalitis infection in fatal human cases. J Neuropathol Exp Neurol 64:506–512. doi: 10.1093/jnen/64.6.506. [DOI] [PubMed] [Google Scholar]
- 8.Kornyey S. 1978. Contribution to the histology of tick-borne encephalitis. Acta Neuropathol 43:179–183. doi: 10.1007/BF00685013. [DOI] [PubMed] [Google Scholar]
- 9.German AC, Myint KS, Mai NT, Pomeroy I, Phu NH, Tzartos J, Winter P, Collett J, Farrar J, Barrett A, Kipar A, Esiri MM, Solomon T. 2006. A preliminary neuropathological study of Japanese encephalitis in humans and a mouse model. Trans R Soc Trop Med Hyg 100:1135–1145. doi: 10.1016/j.trstmh.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 10.van Marle G, Antony J, Ostermann H, Dunham C, Hunt T, Halliday W, Maingat F, Urbanowski MD, Hobman T, Peeling J, Power C. 2007. West Nile virus-induced neuroinflammation: glial infection and capsid protein-mediated neurovirulence. J Virol 81:10933–10949. doi: 10.1128/JVI.02422-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Detje CN, Lienenklaus S, Chhatbar C, Spanier J, Prajeeth CK, Soldner C, Tovey MG, Schlüter D, Weiss S, Stangel M, Kalinke U. 2015. Upon intranasal vesicular stomatitis virus infection, astrocytes in the olfactory bulb are important interferon beta producers that protect from lethal encephalitis. J Virol 89:2731–2738. doi: 10.1128/JVI.02044-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reinert LS, Harder L, Holm CK, Iversen MB, Horan KA, Dagnæs-Hansen F, Ulhøi BP, Holm TH, Mogensen TH, Owens T, Nyengaard JR, Thomsen AR, Paludan SR. 2012. TLR3 deficiency renders astrocytes permissive to herpes simplex virus infection and facilitates establishment of CNS infection in mice. J Clin Invest 122:1368–1376. doi: 10.1172/JCI60893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kallfass C, Ackerman A, Lienenklaus S, Weiss S, Heimrich B, Staeheli P. 2012. Visualizing production of beta interferon by astrocytes and microglia in brain of La Crosse virus-infected mice. J Virol 86:11223–11230. doi: 10.1128/JVI.01093-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lindqvist R, Mundt F, Gilthorpe JD, Wölfel S, Gekara NO, Kröger A, Överby AK. 2016. Fast type I interferon response protects astrocytes from flavivirus infection and virus-induced cytopathic effects. J Neuroinflammation 13:277. doi: 10.1186/s12974-016-0748-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schittone SA, Dionne KR, Tyler KL, Clarke P. 2012. Activation of innate immune responses in the central nervous system during reovirus myelitis. J Virol 86:8107–8118. doi: 10.1128/JVI.00171-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dionne KR, Leser JS, Lorenzen KA, Beckham JD, Tyler KL. 2011. A brain slice culture model of viral encephalitis reveals an innate CNS cytokine response profile and the therapeutic potential of caspase inhibition. Exp Neurol 228:222–231. doi: 10.1016/j.expneurol.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goody RJ, Hoyt CC, Tyler KL. 2005. Reovirus infection of the CNS enhances iNOS expression in areas of virus-induced injury. Exp Neurol 195:379–390. doi: 10.1016/j.expneurol.2005.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tyler KL, Leser JS, Phang TL, Clarke P. 2010. Gene expression in the brain during reovirus encephalitis. J Neurovirol 16:56–71. doi: 10.3109/13550280903586394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, Barres BA. 2012. Genomic analysis of reactive astrogliosis. J Neurosci 32:6391–6410. doi: 10.1523/JNEUROSCI.6221-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Verma S, Kumar M, Nerurkar VR. 2011. Cyclooxygenase-2 inhibitor blocks the production of West Nile virus-induced neuroinflammatory markers in astrocytes. J Gen Virol 92:507–515. doi: 10.1099/vir.0.026716-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hussmann KL, Fredericksen BL. 2014. Differential induction of CCL5 by pathogenic and nonpathogenic strains of West Nile virus in brain endothelial cells and astrocytes. J Gen Virol 95:862–867. doi: 10.1099/vir.0.060558-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheeran MC, Hu S, Sheng WS, Rashid A, Peterson PK, Lokensgard JR. 2005. Differential responses of human brain cells to West Nile virus infection. J Neurovirol 11:512–524. doi: 10.1080/13550280500384982. [DOI] [PubMed] [Google Scholar]
- 23.Ng YP, Lee SM, Cheung TK, Nicholls JM, Peiris JS, Ip NY. 2010. Avian influenza H5N1 virus induces cytopathy and proinflammatory cytokine responses in human astrocytic and neuronal cell lines. Neuroscience 168:613–623. doi: 10.1016/j.neuroscience.2010.04.013. [DOI] [PubMed] [Google Scholar]
- 24.Quintana A, Molinero A, Borup R, Nielsen FC, Campbell IL, Penkowa M, Hidalgo J. 2008. Effect of astrocyte-targeted production of IL-6 on traumatic brain injury and its impact on the cortical transcriptome. Dev Neurobiol 68:195–208. doi: 10.1002/dneu.20584. [DOI] [PubMed] [Google Scholar]
- 25.Chen CJ, Ou YC, Chang CY, Pan HC, Liao SL, Raung SL, Chen SY. 2011. TNF-α and IL-1β mediate Japanese encephalitis virus-induced RANTES gene expression in astrocytes. Neurochem Int 58:234–242. doi: 10.1016/j.neuint.2010.12.009. [DOI] [PubMed] [Google Scholar]
- 26.Dionne KR, Galvin JM, Schittone SA, Clarke P, Tyler KL. 2011. Type I interferon signaling limits reoviral tropism within the brain and prevents lethal systemic infection. J Neurovirol 17:314–326. doi: 10.1007/s13365-011-0038-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clarke P, Leser JS, Bowen RA, Tyler KL. 2014. Virus-induced transcriptional changes in the brain include the differential expression of genes associated with interferon, apoptosis, interleukin 17 receptor A, and glutamate signaling as well as flavivirus-specific upregulation of tRNA synthetases. mBio 5:e00902-14. doi: 10.1128/mBio.00902-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smale G, Nichols NR, Brady DR, Finch CE, Horton WJ. 1995. Evidence for apoptotic cell death in Alzheimer’s disease. Exp Neurol 133:225–230. doi: 10.1006/exnr.1995.1025. [DOI] [PubMed] [Google Scholar]
- 29.Petito CK, Roberts B. 1995. Evidence of apoptotic cell death in HIV encephalitis. Am J Pathol 146:1121–1130. [PMC free article] [PubMed] [Google Scholar]
- 30.Saas P, Boucraut J, Quiquerez AL, Schnuriger V, Perrin G, Desplat-Jego S, Bernard D, Walker PR, Dietrich PY. 1999. CD95 (Fas/Apo-1) as a receptor governing astrocyte apoptotic or inflammatory responses: a key role in brain inflammation? J Immunol 162:2326–2333. [PubMed] [Google Scholar]
- 31.Suk K, Lee J, Hur J, Kim YS, Lee M, Cha S, Yeou Kim S, Kim H. 2001. Activation-induced cell death of rat astrocytes. Brain Res 900:342–347. doi: 10.1016/S0006-8993(01)02326-5. [DOI] [PubMed] [Google Scholar]
- 32.Rossi D. 2015. Astrocyte physiopathology. At the crossroads of intercellular networking, inflammation and cell death. Prog Neurobiol 30:86–120. doi: 10.1016/j.pneurobio.2015.04.003. [DOI] [PubMed] [Google Scholar]
- 33.Kitamura Y, Ota T, Matsuoka Y, Tooyama I, Kimura H, Shimohama S, Nomura Y, Gebicke-Haerter PJ, Taniguchi T. 1999. Hydrogen peroxide-induced apoptosis mediated by p53 protein in glial cells. Glia 25:154–164. doi:. [DOI] [PubMed] [Google Scholar]
- 34.Beretti F, Ardizzoni A, Cermelli C, Guida M, Maraldi T, Pietrosemoli P, Paulone S, De Pol A, Blasi E, Portolani M. 2017. Apoptosis and inflammatory response in human astrocytes are induced by a transmissible cytotoxic agent of neurological origin. New Microbiol 40:27–32. [PubMed] [Google Scholar]
- 35.Griffiths GJ, Dubrez L, Morgan CP, Jones NA, Whitehouse J, Corfe BM, Dive C, Hickman JA. 1999. Cell damage-induced conformational changes of the proapoptotic protein Bak in vivo precede the onset of apoptosis. J Cell Biol 144:903–914. doi: 10.1083/jcb.144.5.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Korsmeyer SJ, Wei MC, Saito M, Weiler S, Oh KJ, Schlesinger PH. 2000. Proapoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ 7:1166–1173. doi: 10.1038/sj.cdd.4400783. [DOI] [PubMed] [Google Scholar]
- 37.Berens HM, Tyler KL. 2011. The proapoptotic Bcl-2 protein Bax plays an important role in the pathogenesis of reovirus encephalitis. J Virol 85:3858–3871. doi: 10.1128/JVI.01958-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Richardson-Burns SM, Kominsky DJ, Tyler KL. 2002. Reovirus-induced neuronal apoptosis is mediated by caspase 3 and is associated with the activation of death receptors. J Neurovirol 8:365–380. doi: 10.1080/13550280260422677. [DOI] [PubMed] [Google Scholar]
- 39.Stetson DB, Medzhitov R. 2006. Type I interferons in host defense. Immunity 25:373–381. doi: 10.1016/j.immuni.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 40.Takeuchi O, Akira S. 2009. Innate immunity to virus infection. Immunol Rev 227:75–86. doi: 10.1111/j.1600-065X.2008.00737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gonzalez-Navajas JM, Lee J, David M, Raz E. 2012. Immunomodulatory functions of type I interferons. Nat Rev Immunol 12:125–135. doi: 10.1038/nri3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Platanias LC. 2005. Mechanisms of type-I- and type-II-interferon-mediated signaling. Nat Rev Immunol 5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 43.Weber E, Finsterbusch K, Lindquist R, Nair S, Lienenklaus S, Gekara NO, Janik D, Weiss S, Kalinke U, Overby AK, Kroger A. 2014. Type I interferon protects mice from fatal neurotropic infection with Langat virus by systemic and local antiviral responses. J Virol 88:12202–12212. doi: 10.1128/JVI.01215-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lazear HM, Pinto AK, Vogt MR, Gale M Jr, Diamond MS. 2011. Beta interferon controls West Nile virus infection and pathogenesis in mice. J Virol 85:7186–7194. doi: 10.1128/JVI.00396-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Samuel MA, Diamond MS. 2005. Alpha/beta interferon protects against lethal West Nile virus infection by restricting cellular tropism and enhancing neuronal survival. J Virol 79:13350–13361. doi: 10.1128/JVI.79.21.13350-13361.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aoki K, Shimada S, Simantini DS, Tun MM, Buerano CC, Morita K, Hayasaka D. 2014. Type-I interferon response affects an inoculation dose-independent mortality in mice following Japanese encephalitis virus infection. Virol J 11:105. doi: 10.1186/1743-422X-11-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lazear HM, Govero J, Smith AM, Platt DJ, Fernandez E, Miner JJ, Diamond MS. 2016. A mouse model of Zika virus pathogenesis. Cell Host Microbe 19:720–730. doi: 10.1016/j.chom.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harris MG, Hulseberg P, Ling C, Karman J, Clarkson BD, Harding JS, Zhang M, Sandor A, Christensen K, Nagy A, Sandor M, Fabry Z. 2014. Immune privilege of the CNS is not the consequence of limited antigen sampling. Sci Rep 4:4422. doi: 10.1038/srep04422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nair S, Michaelsen-Preusse K, Finsterbusch K, Stegemann-Koniszewski S, Bruder D, Grashoff M, Korte M, Köster M, Kalinke U, Hauser H, Kröger A. 2014. Interferon regulatory factor-1 protects from fatal neurotropic infection with vesicular stomatitis virus by specific inhibition of viral replication in neurons. PLoS Pathog 10:e1003999. doi: 10.1371/journal.ppat.1003999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lange SC, Bak LK, Waagepetersen HS, Schousboe A, Norenberg MD. 2012. Primary cultures of astrocytes: their value in understanding astrocytes in health and disease. Neurochem Res 37:2569–2588. doi: 10.1007/s11064-012-0868-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ossina NK, Cannas A, Powers VC, Fitzpatrick PA, Knight JD, Gilbert JR, Shekhtman EM, Tomei LD, Umansky SR, Kiefer MC. 1997. Interferon-gamma modulates a p53-independent apoptotic pathway and apoptosis-related gene expression. J Biol Chem 272:16351–16357. doi: 10.1074/jbc.272.26.16351. [DOI] [PubMed] [Google Scholar]