

The HSV-1 latency reactivation cycle is the cause of significant human pathology. The HSV-1 latency-associated transcript (LAT) functions by regulating latency and reactivation, in part by inhibiting apoptosis. However, the mechanism of this process is unknown. Here we show that LAT likely controls apoptosis via downregulation of several components in the JAK-STAT pathway. Furthermore, we provide evidence that immune exhaustion is not caused by the antiapoptotic activity of the LAT.
KEYWORDS: HSV-1, IFN, LAT, latency, ocular, TLR, reactivation
ABSTRACT
The herpes simplex virus (HSV-1) latency-associated transcript (LAT) has been shown to inhibit apoptosis via inhibiting activation of proapoptotic caspases. However, the mechanism of LAT control of apoptosis is unclear, because LAT is not known to encode a functional protein, and the LAT transcript is found largely in the nucleus. We hypothesized that LAT inhibits apoptosis by regulating expression of genes that control apoptosis. Consequently, we sought to establish the molecular mechanism of antiapoptosis functions of LAT at a transcriptional level during latent HSV-1 ocular infection in mice. Our results suggest the following. (i) LAT likely inhibits apoptosis via upregulation of several components of the type I interferon (IFN) pathway. (ii) LAT does not inhibit apoptosis via the caspase cascade at a transcriptional level or via downregulating Toll-like receptors (TLRs). (iii) The mechanism of LAT antiapoptotic effect is distinct from that of the baculovirus inhibitor of apoptosis (cpIAP) because replacement of LAT with the cpIAP gene resulted in a different gene expression pattern than in either LAT+ or LAT− viruses. (iv) Replacement of LAT with the cpIAP gene does not cause upregulation of CD8 or markers of T cell exhaustion despite their having similar levels of latency, further supporting that LAT and cpIAP function via distinct mechanisms.
IMPORTANCE The HSV-1 latency reactivation cycle is the cause of significant human pathology. The HSV-1 latency-associated transcript (LAT) functions by regulating latency and reactivation, in part by inhibiting apoptosis. However, the mechanism of this process is unknown. Here we show that LAT likely controls apoptosis via downregulation of several components in the JAK-STAT pathway. Furthermore, we provide evidence that immune exhaustion is not caused by the antiapoptotic activity of the LAT.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) infections are the cause of several diseases with mild to severe pathology, including oral and genital ulcers, virus-induced corneal scarring (CS) and blindness, and encephalitis (1–8). Initial infection occurs via the oral or genital mucosa or the eye, where the virus replicates. After initial replication, innate immune response begins, which helps in virus clearance from the eye. The virus then enters sensory neurons via microtubule-dependent retrograde transport and establishes lifelong latency in the trigeminal ganglia (TG), from where it can periodically reactivate, leading to immunopathology. CS is mainly caused by the immune response to reactivation of the latent virus and not the initial infection (9). It is well known that the latency-associated transcript (LAT), the only transcript that is detectable at high levels during HSV-1 latency, promotes latency reactivation (10–12).
LAT may promote reactivation, at least in part, via inhibiting apoptosis and hence promoting cell survival both in vitro and in vivo (13–19). Antiapoptotic activity of LAT is sufficient for reactivation, because replacement of LAT with any of three antiapoptotic genes can rescue reactivation to wild-type (WT) levels (20, 21). However, while restoring the antiapoptotic function of LAT by replacing LAT with antiapoptotic genes restores reactivation, these viruses still have reduced virulence, suggesting that LAT has other functions besides inhibiting apoptosis (22). The antiapoptotic function of LAT may be related to immune exhaustion seen during latency. Previously we found that latent HSV-1 infection leads to immune exhaustion in a LAT-dependent manner (9, 23–25). We showed that PD-1 and TIM-3, markers of immune exhaustion, were elevated in TG of latently infected mice, and this correlated with severity of CS (9). Later, we and others demonstrated that this was dependent on LAT, because mice latently infected with HSV-1 lacking LAT had significantly less immune exhaustion (23, 24).
The mechanism of LAT control of apoptosis in vivo is unclear. LAT has been shown to inhibit both the extrinsic and intrinsic pathways of apoptosis. For example, stable expression of LAT reduced activation of the intrinsic (i.e., mitochondrial) pathway of apoptosis by inhibiting AKT dephosphorylation (26). Furthermore, LAT reduced activation of apoptotic caspases 3, 8, and 9. In vitro, caspase 8 and caspase 9 cleavage was inhibited in Neuro 2A cells infected with WT virus, but not a LAT null mutant virus (14), suggesting that LAT functions via inhibiting activation of proapoptotic caspases. These data suggest that LAT controls activity of proapoptotic proteins. However, it is not known if LAT functions at the level of transcription, because it has not been shown that LAT encodes a functional protein (reviewed in references 22 and 27), and during latency, LAT is largely found in the nucleus (28). Therefore, it is possible that LAT inhibits apoptosis via a transcriptional mechanism, in addition to inhibiting cleavage of proapoptotic caspases.
In this study, we set out to establish the molecular mechanism of HSV-1 LAT inhibition of apoptosis and if antiapoptotic activity is necessary for immune exhaustion. We focused on components of the caspase cascade, Toll-like receptors (TLRs) and the type I interferon (IFN) pathway, because HSV-1 has been shown to activate these pathways (27). TLRs, which are an important part of the innate immune system, recognize a variety of pathogen-associated molecular patterns, and TLR2, TLR3, TLR7, and TLR9 were shown to be activated during an acute infection with HSV-1 (29–32). Another branch of the innate immune system, the type I IFN pathway (consisting mainly of IFN-α and -β), is activated by several HSV-1 glycoproteins (33). However, the HSV-1 LAT transcript was shown to delay expression of IFN-α and -β during an acute infection (34). Activation of the type I IFN pathway is transduced via Janus kinases (JAKs) and may result in transcription and translation of interferon-stimulated genes (ISGs), production of inflammatory cytokines, and growth arrest or apoptosis (35, 36), although the specific response may be dependent on the cell type (37, 38). ISGs exert antiviral activity via various mechanisms, including inhibiting viral entry, replication, egress, or budding (39). HSV-1, on the other hand, has developed mechanisms to inhibit several ISGs (40–43).
Our results suggest that LAT functions in the activation of the type I IFN pathway via downregulation of the Janus kinases during HSV-1 latency. We found that JAK-1 and JAK-2, as well as several downstream effectors of the JAK-STAT pathway, were downregulated in a LAT-dependent manner. We also found evidence of immune exhaustion in TG of mice infected with WT McKrae but not LAT-deficient virus. Together, these results suggest that the HSV-1 LAT likely affects apoptosis via downregulation of the JAK-STAT pathway components. Additionally, we show here that LAT does not function via the same mechanism as cpIAP, because replacement of LAT with the cpIAP gene resulted in a different gene expression pattern than in mice infected with either WT (LAT+) or LAT− virus. We also demonstrate that the antiapoptotic activity of LAT is not sufficient to cause immune exhaustion.
RESULTS
HSV-1 latent infection induces expression of proapoptotic genes in a LAT-dependent manner.
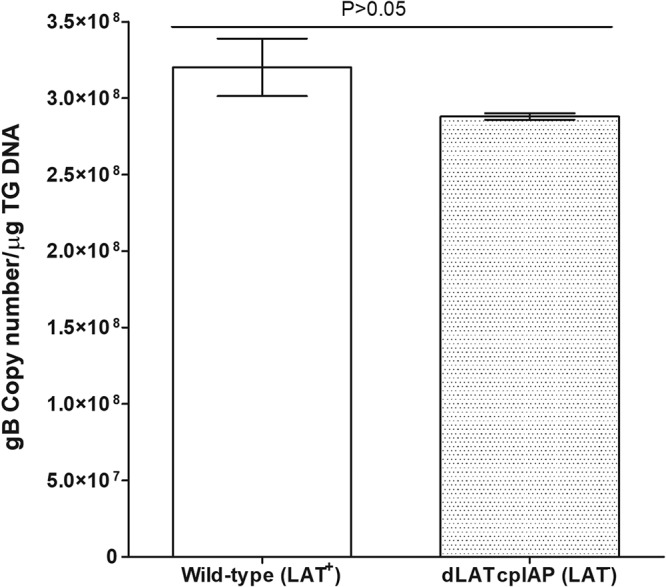
We and others have shown that LAT is necessary for efficient establishment of latency (10–12). To verify that latency is impaired under our experimental conditions, we collected trigeminal ganglia (TG) of C57BL6 mice 28 days after ocular infection with either the HSV-1 WT strain McKrae (LAT+) or the LAT null mutant dLAT2903 (LAT−). The amount of viral DNA in the TG of mice was determined by quantitative PCR (qPCR). Consistent with previous reports, mice infected with LAT+ virus contained approximately 6-fold more viral glycoprotein B (gB) DNA in TG than did mice infected with LAT− virus (P < 0.0001 [Fig. 1]). These results suggest that the level of latency is significantly reduced in mice infected with the LAT null mutant, as previously described (10–12).
FIG 1.

Latency in ocularly infected mice. Mice were infected with 2 × 105 PFU per eye of LAT+ or LAT− virus. Twenty-eight days p.i., TG from infected mice were isolated and quantitative PCR was performed on each individual mouse TG. In each experiment, an estimated relative copy number of the HSV-1 gB for viral DNA was calculated using standard curves generated from pGem-gB1. Briefly, the DNA template was serially diluted 10-fold such that 5 μl contained from 103 to 1011 copies of gB and then subjected to TaqMan PCR with the same set of primers. By comparing the normalized threshold cycle of each sample to the threshold cycle of the standard, the copy number for each reaction was determined. GAPDH expression was used to normalize the relative expression of viral (gB) DNA in the TG. Each bar is based on 20 TG from 2 separate experiments.
To determine if LAT controls apoptosis via inhibiting transcription of proapoptotic genes, we measured expression levels of several apoptotic genes. We found that caspases 3, 8, and 9 were mildly elevated in TG of LAT+ mice compared to those of uninfected mice (Fig. 2). This upregulation was dependent on LAT, because mice infected with LAT− virus had significantly lower expression levels of caspase 3, 8, and 9 than did LAT+ virus-infected mice (P < 0.01 [Fig. 2]). Proapoptotic markers BID, FasL, and Card10 were also upregulated in TG of mice infected with LAT+ HSV-1 (Fig. 2). However, this upregulation was independent of LAT, because mice infected with LAT− virus had levels of expression of BID, Card10, and FasL similar to those of mice infected with LAT+ HSV-1 (P > 0.05 [Fig. 2]). The antiapoptotic BCL2 gene was upregulated in TG of LAT+ virus-infected mice, while it was significantly decreased in LAT− virus-infected mice (P < 0.01 [Fig. 2]). The expression level of the Fas-associated death domain (FADD) gene was lower in both LAT+ and LAT− virus-infected mice than in uninfected mice, and the differences between the LAT+ and LAT− virus-infected TG were not statistically significant (P > 0.05 [Fig. 2]). These results suggest that LAT is unlikely to regulate apoptosis via downregulation of caspase pathway components.
FIG 2.

HSV-1 latent infection induces expression of proapoptotic genes. Mice were infected and TG collected as for Fig. 1. On day 28 p.i., mice were euthanized and TG from individual mice collected and combined. qRT-PCR was performed using total RNA. Expression of BCL2, BID, caspase 3, caspase 8, caspase 9, Card, FADD, and FasL in naive mice was used to estimate the relative expression of each transcript in TG, and GAPDH was used to normalize expression of each transcript in infected TG. Each point represents the mean ± SEM from 10 mice.
Upregulation of NF-κB and Toll-like receptors during HSV-1 latent infection occurs independently of LAT.
Previous studies have established that NF-κB is upregulated during HSV-1 acute infection (44–46). However, it is not known if this transcription factor is also upregulated during latency and, if so, whether this is dependent on LAT. We measured expression of NF-κB in latently infected mouse TG and found that NF-κB is upregulated similarly in TG of LAT+ and LAT− virus-infected mice (P > 0.05 [Fig. 3]). Thus, similar to the case with acute infection (44–46), NF-κB expression during latency is upregulated, and this upregulation is independent of LAT.
FIG 3.
NF-κB expression is upregulated during latent HSV-1 infection. Mice were infected and TG collected as for Fig. 1. On day 28 p.i., mice were euthanized and TG from individual mice collected and combined. qRT-PCR was performed using total RNA. NF-κB transcript levels in naive mice were used to estimate the relative expression of NF-κB in TG, and GAPDH was used to normalize expression of each transcript. Each point represents the mean ± SEM from 10 mice.
To test if HSV-1 escapes immune detection and apoptosis via downregulation of TLRs during latency, we measured expression levels of several TLRs. We found that TLR2, -3, -4, -7, and -9, as well as the TLR adaptor protein MyD88, were upregulated in TG of both LAT+ and LAT− virus-infected mice (Fig. 4). However, levels of TLR2, -3, -4, -7, and -9 and MyD88 were similar between mice infected with LAT+ and LAT− viruses (P > 0.05 [Fig. 4]). These results demonstrate that the TLR pathway is activated during HSV-1 latency. However, LAT does not mediate its antiapoptotic response via downregulating TLRs during latency.
FIG 4.

Effect of LAT on TLR expression during latent infection with HSV-1. Mice were infected and TG collected as for Fig. 1. On day 28 p.i., mice were euthanized and TG from individual mice collected and combined. qRT-PCR was performed using total RNA. Expression of TLR2, TLR3, TLR4, TLR7, TLR9, and MyD88 in naive mice was used to estimate the relative expression of each transcript in TG, and GAPDH was used to normalize expression of each transcript. Each point represents the mean ± SEM from 10 mice.
LAT controls apoptosis via downregulating the type I IFN pathway.
The type I interferon (IFN) signaling pathway is activated in response to viral infections. Activation of this pathway results in initiation of the inflammatory response and transcription of antiviral genes such as IFN-stimulated genes (ISGs). Depending on cell type and timing of IFN exposure, activation of the type I IFN pathway may result in stimulation or inhibition of apoptosis (reviewed in reference 47). HSV-1 has several methods to inhibit type I IFN signaling. For example, the HSV-1 immediate early (IE) protein 27 (ICP27) was shown to inhibit IFN signaling via blocking activation of the Janus kinase (JAK)-signal transducer and activator of transcription (STAT) in Vero cells (48). To test if LAT can inhibit apoptosis during latency via the JAK-STAT pathway, we measured expression levels of JAK-1 and JAK-2, the tyrosine kinases directly upstream of STAT. Indeed, we found that both JAK-1 and JAK-2 expression levels were downregulated in mouse TG latently infected with LAT+ virus but not in LAT− HSV-1-infected mice and that these differences were highly significant (P < 0.0001 [Fig. 5]).
FIG 5.
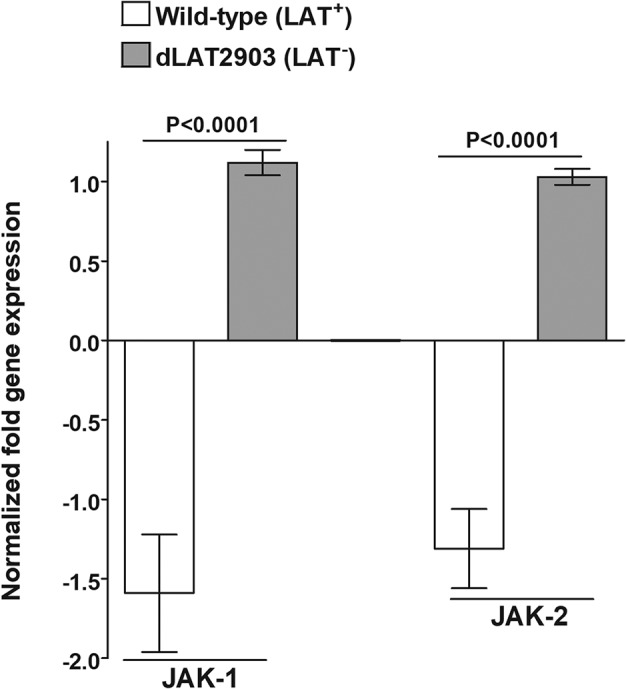
LAT regulates expression of JAK-1 and JAK-2. Mice were infected and TG collected as for Fig. 1. On day 28 p.i., mice were euthanized and TG from individual mice collected and combined. qRT-PCR was performed using total RNA. Expression of JAK-1 and JAK-2 in naive mice was used to estimate the relative expression of the same transcript in TG of infected mice, and GAPDH was used to normalize expression of each transcript. Each point represents the mean ± SEM from 10 mice.
As we observed LAT-mediated downregulation of JAK-1 and JAK-2, we predicted that downstream effector gene expression is also affected. To test this hypothesis, we measured the expression levels of several known IFN-stimulated genes that were shown to be regulated by HSV-1 during an acute infection (40–43). We found that several interferon-stimulated genes (ISGs), such as ISG56 (or IFIT1) and the genes for interferon regulatory factor 1 (IRF-1), myxoma resistance protein 1 (Mx-1), and class II major histocompatibility complex transactivator (CIITA), were downregulated in mice infected with LAT+ virus but upregulated in mice infected with LAT− virus (Fig. 6). These differences between LAT+ and LAT− virus-infected TG were statistically significant (P < 0.0001 [Fig. 6]). In contrast, the level of oligoadenylate synthetase 3 (OAS-3) was elevated in LAT+ virus-infected mice, while it was lower in TG of LAT− virus-infected mice (P < 0.0001 [Fig. 6]). The expression levels of ISG15, ISG54, and complement component 3 (C3) were lower in infected than in uninfected TG, but similar in both LAT+ and LAT− infected TG (P < 0.05 for ISG15 and ISG54 and P > 0.05 for C3 [Fig. 6]). Finally, interferon-inducible cytokine 10 (IP-10), IRF-3, and guanylate binding protein 1 (GBP-1) expression levels increased in TG of infected mice independent of the presence or absence of LAT, and the difference between LAT+ and LAT− virus-infected mice was not statistically significant (P > 0.05 [Fig. 6]). Overall, these results indicate that LAT may control apoptosis via downregulating JAKs and several, but not all, IFN-regulated genes.
FIG 6.

LAT downregulates several type I IFN pathway components. Mice were infected and TG collected as for Fig. 1. On day 28 p.i., mice were euthanized and TG from individual mice collected and combined. qRT-PCR was performed using total RNA. Expression of ISG15, ISG54, ISG56, IP-10, OAS-3, IRF-1, IRF-3, Mx-1, CIITA, C3, and GBP-1 in naive mice was used to estimate the relative expression of each transcript in TG, and GAPDH was used to normalize expression of each transcript. Each point represents the mean ± SEM from 10 mice.
LAT and cpIAP function via distinct mechanisms.
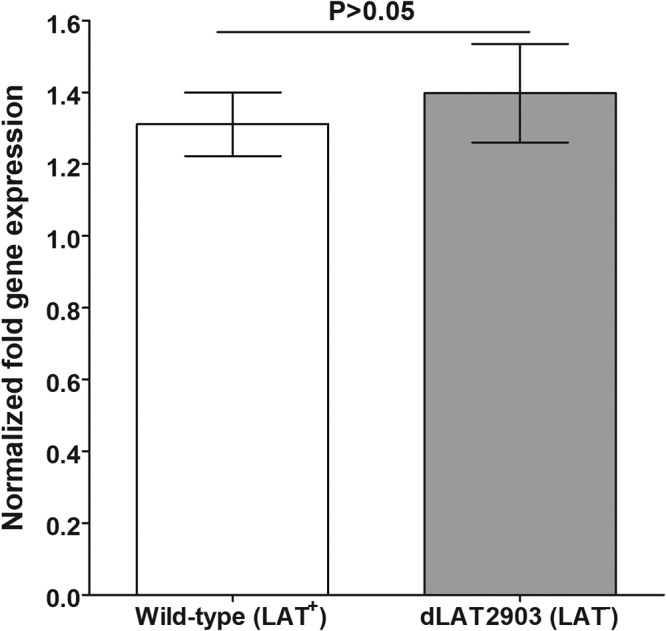
LAT antiapoptotic activity enhances HSV-1 reactivation because replacement of LAT with either the baculovirus inhibitor of apoptosis (cpIAP), cellular FLIP, or bovine herpesvirus 1 (BHV-1) latency-related (LR) gene rescued the reactivation defect seen in LAT− virus-infected mouse TG to WT levels (15, 20, 49, 50). To verify that antiapoptotic activity is sufficient to rescue latency in the absence of LAT, we ocularly infected mice with either WT HSV-1 strain McKrae or a mutant virus in which LAT was replaced with the baculovirus cpIAP gene (dLATcpIAP). At 28 days after ocular infection, we collected mouse TG and measured gB levels as an indicator of viral load using qPCR. There was no significant difference in gB copy numbers of mice infected with WT and dLATcpIAP viruses, suggesting similar levels of latency (P > 0.05 [Fig. 7]). This lack of difference could reflect an increased number of neurons surviving due to the antiapoptotic activity of cpIAP.
FIG 7.
Replacement of LAT with the cpIAP gene rescues latency in LAT cells. Mice were infected with LAT+ or dLATcpIAP (LAT−) virus as for Fig. 1. Twenty-eight days p.i., TG from infected mice were isolated and quantitative PCR was performed on each individual mouse TG. In each experiment, an estimated relative copy number of the HSV-1 gB for viral DNA was calculated using standard curves generated from pGem-gB1 as for Fig. 1. By comparing the normalized threshold cycle of each sample to the threshold cycle of the standard, the copy number for each reaction was determined. GAPDH expression was used to normalize the relative expression of viral (gB) DNA in the TG. Each bar is based on 20 TG.
To examine if cpIAP and LAT control apoptosis using a similar mechanism, we measured expression levels of caspase 8, BCL2, BID, and FADD in TG of mice latently infected with WT HSV-1 and dLATcpIAP virus. Mice infected with virus containing cpIAP had significantly lower expression levels of caspase 8, BCL2, BID, and FADD genes than mice infected with WT virus (P < 0.0001 [Fig. 8]). These results suggest that while both LAT and cpIAP can regulate expression of apoptotic genes, they function via distinct mechanisms.
FIG 8.

LAT and cpIAP function via distinct mechanisms. Mice were infected with LAT+ or dLATcpIAP (LAT−) virus as for Fig. 1. On day 28 p.i., mice were euthanized and TG from individual mice collected and combined. qRT-PCR was performed using total RNA. Expression of caspase 8, BCL2, BID, and FADD in naive mice was used to estimate the relative expression of each transcript in TG, and GAPDH was used to normalize expression of each transcript. Each point represents the mean ± SEM from 10 mice.
Previously we have shown that HSV-1 latent infection is correlated with CD8+ T cell exhaustion in a LAT-dependent manner (9, 23). We hypothesized that the antiapoptotic activity of LAT is the mechanism that leads to CD8+ T cell exhaustion. If this is the case, we predicted that replacement of LAT with a different antiapoptotic gene will rescue the loss of CD8+ T cell exhaustion associated with LAT deficiency. To test this hypothesis, we infected mice ocularly with WT McKrae, LAT−, or dLATcpIAP (LAT+) virus. We found that expression levels of CD8, PD-1, and TIM-3 mRNAs were significantly lower in TG of mice infected with either the LAT− virus or a recombinant virus in which LAT was replaced with the cpIAP gene (dLATcpIAP) than in LAT+ virus-infected mice (P < 0.0001 [Fig. 9]). Because replacement of LAT with cpIAP does not restore expression levels of CD8, PD-1, and TIM-3 to those of LAT+ virus, we conclude that LAT and cpIAP function via distinct mechanisms and that exhaustion seen in infection with WT HSV-1 is not due to the antiapoptotic function of LAT.
FIG 9.

Markers of immune exhaustion are upregulated in LAT+ virus-infected mice. Mice were infected with LAT+, LAT−, or dLATcpIAP (LAT−) virus as for Fig. 1. On day 28 p.i., mice were euthanized and TG from individual mice collected and combined. qRT-PCR was performed using total RNA. Expression of CD8, PD-1, and TIM-3 in naive mice was used to estimate the relative expression of each transcript in TG, and GAPDH was used to normalize expression of each transcript. Each point represents the mean ± SEM from 10 mice.
DISCUSSION
Despite some early skepticism, the ability of the HSV-1 LAT to inhibit apoptosis has been well documented in various infection models and is now widely accepted (13–19). Furthermore, antiapoptotic activity of LAT likely plays a role in controlling the latency reactivation cycle. However, the mechanism by which LAT controls apoptosis is unclear. Since LAT is not known to encode a functional protein, we hypothesized that it likely functions at the level of transcription. In this study, we took a systematic approach to test several pathways that have been shown to be activated in response to HSV-1 infections. We demonstrated that various components of type I IFN pathway are downregulated in a LAT-dependent manner.
Viral infections trigger the innate immune response. Viral antigens such as DNA or RNA bind to a pattern recognition receptor (PRR), such as Toll-like receptor 3 (TLR3), the cytoplasmic retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), or the nucleotide oligomerization domain (NOD)-like receptors (NLRs) (reviewed in reference 51). This results in expression and secretion of IFN-α and -β. In the canonical type I IFN pathway, binding of IFN-α or -β to the interferon α receptor (IFNAR) results in activation of Janus kinase proteins 1 and 2, which then phosphorylate their effector, signal transducer and activator of transcription 1 (STAT1), resulting in transcription and translation of interferon-stimulated genes (ISGs) (39). On a cellular level, activation of the type I IFN pathway results in growth arrest and/or apoptosis (35, 36), although this may be cell type specific (37, 38).
HSV-1 inhibits the type I IFN pathway during the lytic cycle (43). A study from the Jones laboratory showed that LAT delays several IFN-α subtypes, including IFN-α1, IFN-α4, IFN-α5, IFN-α6, and IFN-α14, as well as IFN-β, during an acute infection in vitro, and IFN-α4 and IFN-β in vivo (34). Additionally, the JAK-STAT pathway was shown to have a protective role during acute HSV-1 infection in vivo (52). A recent study also reported a correlation between high expression levels of JAK-STAT pathway components and a lack of HSV-1 symptoms in humans (53). We have shown previously that JAK-STAT pathway is involved in blocking HSV-1 replication in vitro during an acute infection in macrophages and dendritic cells (54). Finally, in vitro studies by the Smiley group showed that several ISGs, including ISG15, ISG56, IP-10 (CXCL10), OAS-3, and Mx-1, were stimulated upon infection with HSV-1 lacking functional ICP0 and VP16 but not with WT HSV-1 (41). These results, and studies by others, suggested that while HSV-1 infection triggers activation of type I IFN pathway, HSV-1 encodes several proteins, including ICP0 and ICP27, that inhibit the type I IFN pathway components during the acute phase of infection (40–43). We hypothesized that LAT could utilize a similar mechanism to inhibit apoptosis during latency in mice. Indeed, we found JAK-1 and JAK-2 to be downregulated in mice latently infected with WT but not LAT-deficient HSV-1. Although we did not directly test if protein levels of JAK-1 and JAK-2 were also affected, we observed a concomitant downregulation of several downstream ISGs (ISG15, ISG54, ISG56, IRF-1, Mx-1, CIITA, and C3) in mice infected with WT McKrae, suggesting that the activity of this pathway was reduced. Further experiments, such as phosphorylation status and nuclear localization of STATs, would help support these findings.
Our results are in agreement with previous reports that showed an immediate early (IE) gene expression-dependent downregulation of these factors during an acute infection (40–43). ISG56, IRF-1, Mx-1, and CIITA were downregulated in a LAT-dependent manner, suggesting that similarly to IE genes during an acute infection, LAT can regulate some, but not all, of the type I IFN pathway during latency. Interestingly, we also found a LAT-dependent upregulation of OAS-3. As OAS-3 functions in limiting virus replication (55, 56), it is tempting to speculate that LAT’s mechanism to maintain latency could involve upregulation of OAS-3. Considering that only a small fraction of the cells in TG are latently infected, the fact that we see significant differences in gene expression suggests that gene expression changes within those infected cells must be substantial. This could be tested by examining expression levels of these genes in individual cells, for example, by using RNA fluorescent in situ hybridization (FISH).
What property of the LAT inhibits the type I IFN pathway?
LAT partially overlaps and lies antisense to the ICP0 gene (57). ICP0, on the other hand, can prevent IFN-mediated repression of HSV-1 during acute infection (52). Because ICP0 is still intact in the LAT null virus dLAT2903 used in this study, the effects seen in this study are not due to lack of ICP0 expression. However, LAT may repress expression of ICP0 during latency (58), although conflicting evidence has also been reported (59). Therefore, it is possible that continued expression of ICP0 in the absence of the LAT could contribute to downregulation of JAK-1 and JAK-2. It would be interesting to test if this is the case by using a LAT-deficient virus in which ICP0 is also disrupted. Unfortunately, ICP0 mutant viruses have lowered virulence, which could be a confounding factor.
An alternative hypothesis could be made that LAT control of type I IFN genes is an indirect effect of lower production of viral particles and, thus, lowered immunogenic response. Previous studies have shown that LAT inhibits expression of lytic genes, possibly via an epigenetic mechanism during latency (60–63). While this is an attractive hypothesis, one would further predict that loss of LAT would phenocopy the ISG expression pattern seen during lytic cycle. However, based on the data presented here, we cannot completely discount this possibility, and further in vitro experiments could help in making such a distinction.
LAT is not known to encode a functional protein to regulate latency reactivation cycle or apoptosis. Therefore, it is possible that LAT exerts its effect as an RNA molecule. The primary LAT transcript of 8.3 kb is spliced to a stable 2-kb LAT and an unstable 6.3-kb LAT. Several microRNAs are encoded within the primary LAT, at least two of which reside in the LAT promoter region (reviewed in reference 8). Additionally, two small noncoding RNAs (sncRNAs) within the 2-kb LAT which are not present in the LAT− virus have been described (64). These sncRNAs have been implicated in protection from apoptosis (19) and affect upregulation of HVEM (herpesvirus entry mediator) and, thus, increase in reactivation from latency (65). Whether these two sncRNAs mediate this LAT-dependent inhibition of JAK-1 and JAK-2 is under investigation.
Previous work from various groups has demonstrated that LAT controls apoptosis via inhibiting activation of key caspases (14, 17, 18, 26, 66). Interestingly, we found that caspase 3, 8, and 9 transcript levels were reduced in mice infected with LAT null mutant compared to those in mice infected with WT HSV-1. These results are in contrast to several published works reporting a LAT-dependent inhibition of caspase 3, 8, and 9 activation (17, 66, 67). However, as we did not test if these caspases are activated, it is likely that the LAT mediates activation of the caspase pathway only at the level of protein activation, not gene expression. In principle, this could occur via BCL2, because we observed a slight, LAT-dependent upregulation of the antiapoptotic gene Bcl2 (Fig. 2). BCL2 inhibits apoptosis via regulating the activity of the proapoptotic BCL2 family members, such as BAD, BID, and BIM (68). Additionally, BCL2 inhibits activation of caspase 3 and caspase 9 indirectly via inhibiting cytochrome c release from the mitochondria (68).
NF-κB is upregulated early after infection with HSV-1 (44, 45). Upon viral entry, HSV-1 was shown to redirect NF-κB to ICP0 gene to promote viral replication and to block NF-κB recruitment to the promoter of Iκ-Bα in vitro (44). Additionally, expression of gD or treatment of cells with soluble gD protected cells from Fas-mediated apoptosis via upregulation of NF-κB (69). Later studies from the Mastino lab established that gD binding to the HVEM mediates upregulation of NF-κB (46). Although the mechanism of NF-κB activation during early infection with HSV-1 has been established, it is not known if NF-κB is also affected during latency and, if so, whether this is LAT dependent. Here we show that while NF-κB transcription is upregulated in latently infected mice, this is not dependent on LAT expression.
TLR2 and TLR3 have been found to mediate apoptotic signaling in response to bacterial or viral infections (70, 71). During acute HSV-1 infection viral glycoproteins are recognized by TLR2 (72). In addition, TLR3 has been shown to play a protective role during HSV-1 acute infection. TLR3 deficiency was identified in two patients with HSV-1 encephalitis (HSE) (73–75), and Lafaille et al. showed that induced pluripotent stem cells (iPSCs) from patients lacking TLR3 were more susceptible to HSV-1 infection (76). TLR3 is necessary for CD8 T cell immunity to HSV-1 (77). Based on these findings, we hypothesized that LAT could inhibit apoptosis via downregulating the TLR pathway during latency. However, we did not observe a downregulation in any of the TLRs tested, suggesting that, at least during latency, LAT does not function via this pathway to inhibit apoptosis.
LAT mechanism of control of apoptosis is distinct from baculovirus antiapoptotic gene cpIAP.
LAT antiapoptotic function is necessary for the HSV-1 latency reactivation cycle, and replacement of LAT with the unrelated, baculovirus antiapoptotic gene cpIAP restores latency and reactivation to WT HSV-1 levels (21). However, it is not known if LAT inhibits apoptosis in a manner similar to that of other viral antiapoptotic genes. The LAT sequence necessary to promote cell survival is contained within a stable 1.5-kb intron, which also functions to promote spontaneous reactivation, whereas the baculovirus cpIAP gene encodes a functional protein (78). Therefore, we hypothesized that the LAT mechanism of action may be different from that of cpIAP. Here we show evidence that replacement of LAT with the baculovirus cpIAP causes a drastic downregulation of caspase 8, BCL2, BID, and FADD transcripts, while WT HSV-1 causes a significantly lesser downregulation of FADD and upregulation of caspase 8, BCL2, and BID (Fig. 2 and 8). These results suggest that LAT and cpIAP inhibit apoptosis using different mechanisms.
Antiapoptotic activity is not sufficient to cause immune exhaustion. We have reported that HSV-1 latency is associated with immune exhaustion and that this is dependent on expression of LAT and subclinical reactivation of virus (23). This finding suggests that instead of true latency, HSV-1 may establish a chronic, low-level infection or cause continuous abortive reactivations. Several of LAT’s functions could contribute toward this activity: inhibition of apoptosis, immune evasion, and immune exhaustion. Here we show that apoptosis does not contribute toward establishing a state of immune exhaustion, because replacement of LAT with an unrelated antiapoptotic gene cpIAP did not result in immune exhaustion.
MATERIALS AND METHODS
Virus and mice.
Three HSV-1 strains were used in this study: wild-type (WT) HSV-1 McKrae (LAT+), dLAT2903 (LAT−), in which both LAT promoters in each viral long repeat and the first 1,667 nucleotides (nt) of the LAT are deleted (11), and dLATcpIAP (LAT−), which is similar to dLAT2903 except that it contains the complete baculovirus inhibitor of apoptosis protein gene (cpIAP) open reading frame (ORF) in place of the LAT (50). The WT and mutant viruses are depicted in Fig. S1 in the supplemental material. Plaque-purified viruses were grown in rabbit skin (RS) monolayer cells and maintained in 5% fetal bovine serum (FBS) in minimal essential medium (MEM) as described previously (11). WT C57BL/6 female mice (Jackson Laboratories) were infected with 2 × 105 PFU suspended in 2 μl of MEM per eye of each virus without corneal scarification. All animal procedures adhered to the Association for Research in Vision and Ophthalmology (ARVO) statement for the Use of Animals in Ophthalmic and Vision Research and institutional animal care and use guidelines.
DNA extraction and PCR analysis for HSV-1 genomic DNA.
DNA was isolated from homogenized individual TG of infected mice on day 28 postinfection (p.i.) using the commercially available DNeasy blood and tissue kit (catalog number 69506; Qiagen, Stanford, CA) according to the manufacturer’s instructions. PCR analyses were done using gB-specific primers (forward, 5′-AACGCGACGCACATCAAG-3′; reverse, 5′-CTGGTACGCGATCAGAAAGC-3′; and probe, 5-FAM-CAGCCGCAGTACTACC-3′, where FAM is 6-carboxyfluorescein). The amplicon length for this primer set is 72 bp. Relative copy numbers for the gB DNA were calculated using standard curves generated from plasmid pAc-gB1 (79).
RNA extraction, cDNA synthesis, and TaqMan RT-PCR.
Infected and mock-infected mice were euthanized 28 days p.i., and TG were harvested and suspended in RNAlater (Qiagen) and stored at −80°C until processed. Tissue processing and RNA extraction were done using QIAzol RNA reagent (Qiagen) and 1-bromo-2 chloropropane (BCP) as described previously (80–82). One microgram of RNA was reverse transcribed using random-hexamer primers and murine leukemia virus (MuLV) reverse transcriptase from a high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA), using the manufacturer’s recommendations. Expression levels of various RNAs were determined using TaqMan gene expression assays (Applied Biosystems). Primer-probe sets consisted of two unlabeled PCR primers and the FAM dye-labeled TaqMan MGB probe in a single mixture. Additionally, all amplicons included an intron-exon junction to eliminate signal from genomic DNA contamination.
The mRNA expression levels of the genes of interest were evaluated using TaqMan gene expression assays (Applied Biosystems) and are listed in Table 1.
TABLE 1.
TaqMan gene expression assaysa
| Gene | Gene expression assay no. | Amplicon size (bp) |
|---|---|---|
| CD8α | Mm01182108_m1 | 67 |
| PD-1 | Mm00435532_m1 | 65 |
| TIM-3 | Mm00454540_m1 | 98 |
| BCL2 | Mm00477631_m1 | 85 |
| BID | Mm00432073_m1 | 108 |
| Caspase 3 | Mm01195085_m1 | 70 |
| Caspase 8 | Mm00802247_m1 | 96 |
| Caspase 9 | Mm00516563_m1 | 68 |
| FADD | Mm00438861_m1 | 98 |
| FasL | Mm00438864_m1 | 84 |
| NF-κB | Mm00476361_m1 | 70 |
| TLR2 | Mm00442346_m1 | 69 |
| TLR3 | Mm01207404_m1 | 121 |
| TLR4 | Mm00445273_m1 | 87 |
| TLR7 | Mm00446590_m1 | 125 |
| TLR9 | Mm00446193_m1 | 60 |
| Myd88 | Mm00440338_m1 | 90 |
| JAK-1 | Mm00600614_m1 | 72 |
| JAK-2 | Mm00434561_m1 | 74 |
| ISG15 | Mm01705338_s1 | 107 |
| IP-10 | Mm99999072_m1 | 62 |
| CIITA | Mm00482919_m1 | 93 |
| IRF-1 | Mm01288579_m1 | 95 |
| IRF-3 | Mm01203177_m1 | 55 |
| Card10 | Mm00459941_m1 | 77 |
| ISG56 | Mm00515153_m1 | 80 |
| ISG54 | Mm00492606_m1 | 66 |
| Mx-1 | Mm01218004_m1 | 106 |
| OAS-3 | Mm00460944_m1 | 87 |
| C3 | Mm01232779_m1 | 88 |
| GBP-1 | Mm00657086_m1 | 110 |
| GAPDH | Mm99999915_g1 | 107 |
Primers used for RT-PCR assay of each gene transcript in TG of latently infected mice.
Quantitative reverse transcription-PCR (qRT-PCR) was performed using QuantStudio 5 (Applied Biosystems) in 384-well plates, as we described previously (83, 84). Real-time PCR was performed in triplicate for each tissue sample. The threshold cycle (CT) values, which represent the PCR cycles at which there is a noticeable increase in the reporter fluorescence above baseline, were determined using SDS, version 2.2 software. In all experiments, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used for normalization of transcripts. Expression in naive mice was used as a baseline control to estimate the relative expression of each transcript in TG of latently infected mice.
Statistical analysis.
Student’s t test and analysis of variance (ANOVA) were performed using Prism (GraphPad, San Diego, CA). Results were considered significant when the P value was <0.05.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by Public Health Service NIH grants 1R01EY013615, 1RO1EY026944, and 1RO1EY029160.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.00103-19.
REFERENCES
- 1.Barron BA, Gee L, Hauck WW, Kurinij N, Dawson CR, Jones DB, Wilhelmus KR, Kaufman HE, Sugar J, Hyndiuk RA. 1994. Herpetic eye disease study. A controlled trial of oral acyclovir for herpes simplex stromal keratitis. Ophthalmology 101:1871–1882. doi: 10.1016/S0161-6420(13)31155-5. [DOI] [PubMed] [Google Scholar]
- 2.Wilhelmus KR, Dawson CR, Barron BA, Bacchetti P, Gee L, Jones DB, Kaufman HE, Sugar J, Hyndiuk RA, Laibson PR, Stulting RD, Asbell PA. 1996. Risk factors for herpes simplex virus epithelial keratitis recurring during treatment of stromal keratitis or iridocyclitis. Herpetic Eye Disease Study Group. Br J Ophthalmol 80:969–972. doi: 10.1136/bjo.80.11.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liesegang TJ. 2001. Herpes simplex virus epidemiology and ocular importance. Cornea 20:1–13. doi: 10.1097/00003226-200101000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Hill TJ. 1987. Ocular pathogenicity of herpes simplex virus. Curr Eye Res 6:1–7. doi: 10.3109/02713688709020060. [DOI] [PubMed] [Google Scholar]
- 5.Roberts CM, Pfister JR, Spear SJ. 2003. Increasing proportion of herpes simplex virus type 1 as a cause of genital herpes infection in college students. Sex Transm Dis 30:797–800. doi: 10.1097/01.OLQ.0000092387.58746.C7. [DOI] [PubMed] [Google Scholar]
- 6.Auslander BA, Biro FM, Rosenthal SL. 2005. Genital herpes in adolescents. Semin Pediatr Infect Dis 16:24–30. doi: 10.1053/j.spid.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 7.Singh AE, Romanowski B, Wong T, Gourishankar S, Myziuk L, Fenton J, Preiksaitis JK. 2005. Herpes simplex virus seroprevalence and risk factors in 2 Canadian sexually transmitted disease clinics. Sex Transm Dis 32:95–100. doi: 10.1097/01.olq.0000151415.78210.85. [DOI] [PubMed] [Google Scholar]
- 8.Nicoll MP, Proenca JT, Connor V, Efstathiou S. 2012. Influence of herpes simplex virus 1 latency-associated transcripts on the establishment and maintenance of latency in the ROSA26R reporter mouse model. J Virol 86:8848–8858. doi: 10.1128/JVI.00652-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mott KR, Bresee CJ, Allen SJ, BenMohamed L, Wechsler SL, Ghiasi H. 2009. Level of herpes simplex virus type 1 latency correlates with severity of corneal scarring and exhaustion of CD8+ T cells in trigeminal ganglia of latently infected mice. J Virol 83:2246–2254. doi: 10.1128/JVI.02234-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leib DA, Bogard CL, Kosz-Vnenchak M, Hicks KA, Coen DM, Knipe DM, Schaffer PA. 1989. A deletion mutant of the latency-associated transcript of herpes simplex virus type 1 reactivates from the latent state with reduced frequency. J Virol 63:2893–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perng GC, Dunkel EC, Geary PA, Slanina SM, Ghiasi H, Kaiwar R, Nesburn AB, Wechsler SL. 1994. The latency-associated transcript gene of herpes simplex virus type 1 (HSV-1) is required for efficient in vivo spontaneous reactivation of HSV-1 from latency. J Virol 68:8045–8055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steiner I, Spivack JG, Lirette RP, Brown SM, MacLean AR, Subak-Sharpe JH, Fraser NW. 1989. Herpes simplex virus type 1 latency-associated transcripts are evidently not essential for latent infection. EMBO J 8:505–511. doi: 10.1002/j.1460-2075.1989.tb03404.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perng GC, Jones C, Ciacci-Zanella J, Stone M, Henderson G, Yukht A, Slanina SM, Hofman FM, Ghiasi H, Nesburn AB, Wechsler SL. 2000. Virus-induced neuronal apoptosis blocked by the herpes simplex virus latency-associated transcript. Science 287:1500–1503. doi: 10.1126/science.287.5457.1500. [DOI] [PubMed] [Google Scholar]
- 14.Henderson G, Peng W, Jin L, Perng GC, Nesburn AB, Wechsler SL, Jones C. 2002. Regulation of caspase 8- and caspase 9-induced apoptosis by the herpes simplex virus type 1 latency-associated transcript. J Neurovirol 8:103–111. doi: 10.1080/13550280290101085. [DOI] [PubMed] [Google Scholar]
- 15.Perng GC, Maguen B, Jin L, Mott KR, Osorio N, Slanina SM, Yukht A, Ghiasi H, Nesburn AB, Inman M, Henderson G, Jones C, Wechsler SL. 2002. A gene capable of blocking apoptosis can substitute for the herpes simplex virus type 1 latency-associated transcript gene and restore wild-type reactivation levels. J Virol 76:1224–1235. doi: 10.1128/JVI.76.3.1224-1235.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Branco FJ, Fraser NW. 2005. Herpes simplex virus type 1 latency-associated transcript expression protects trigeminal ganglion neurons from apoptosis. J Virol 79:9019–9025. doi: 10.1128/JVI.79.14.9019-9025.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carpenter D, Hsiang C, Brown DJ, Jin L, Osorio N, BenMohamed L, Jones C, Wechsler SL. 2007. Stable cell lines expressing high levels of the herpes simplex virus type 1 LAT are refractory to caspase 3 activation and DNA laddering following cold shock induced apoptosis. Virology 369:12–18. doi: 10.1016/j.virol.2007.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang X, Chentoufi AA, Hsiang C, Carpenter D, Osorio N, BenMohamed L, Fraser NW, Jones C, Wechsler SL. 2011. The herpes simplex virus type 1 latency-associated transcript can protect neuron-derived C1300 and Neuro2A cells from granzyme B-induced apoptosis and CD8 T-cell killing. J Virol 85:2325–2332. doi: 10.1128/JVI.01791-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.da Silva LF, Jones C. 2013. Small non-coding RNAs encoded within the herpes simplex virus type 1 latency associated transcript (LAT) cooperate with the retinoic acid inducible gene I (RIG-I) to induce beta-interferon promoter activity and promote cell survival. Virus Res 175:101–109. doi: 10.1016/j.virusres.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jin L, Carpenter D, Moerdyk-Schauwecker M, Vanarsdall AL, Osorio N, Hsiang C, Jones C, Wechsler SL. 2008. Cellular FLIP can substitute for the herpes simplex virus type 1 latency-associated transcript gene to support a wild-type virus reactivation phenotype in mice. J Neurovirol 14:389–400. doi: 10.1080/13550280802216510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jin L, Perng GC, Carpenter D, Mott KR, Osorio N, Naito J, Brick DJ, Jones C, Wechsler SL. 2007. Reactivation phenotype in rabbits of a herpes simplex virus type 1 mutant containing an unrelated antiapoptosis gene in place of latency-associated transcript. J Neurovirol 13:78–84. doi: 10.1080/13550280601164333. [DOI] [PubMed] [Google Scholar]
- 22.Jones C. 2013. Bovine herpes virus 1 (BHV-1) and herpes simplex virus type 1 (HSV-1) promote survival of latently infected sensory neurons, in part by inhibiting apoptosis. J Cell Death 6:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Allen SJ, Hamrah P, Gate DM, Mott KR, Mantopoulos D, Zheng L, Town T, Jones C, von Andrian UH, Freeman GJ, Sharpe AH, Benmohamed L, Ahmed R, Wechsler SL, Ghiasi H. 2011. The role of LAT in increased CD8+ T cell exhaustion in trigeminal ganglia of mice latently infected with herpes simplex virus type 1. J Virol 85:4184–4197. doi: 10.1128/JVI.02290-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chentoufi AA, Kritzer E, Tran MV, Dasgupta G, Lim CH, Yu DC, Afifi RE, Jiang X, Carpenter D, Osorio N, Hsiang C, Nesburn AB, Wechsler SL, BenMohamed L. 2011. The herpes simplex virus 1 latency-associated transcript promotes functional exhaustion of virus-specific CD8+ T cells in latently infected trigeminal ganglia: a novel immune evasion mechanism. J Virol 85:9127–9138. doi: 10.1128/JVI.00587-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.St Leger AJ, Jeon S, Hendricks RL. 2013. Broadening the repertoire of functional herpes simplex virus type 1-specific CD8+ T cells reduces viral reactivation from latency in sensory ganglia. J Immunol 191:2258–2265. doi: 10.4049/jimmunol.1300585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carpenter D, Hsiang C, Jiang X, Osorio N, BenMohamed L, Jones C, Wechsler SL. 2015. The herpes simplex virus type 1 (HSV-1) latency-associated transcript (LAT) protects cells against cold-shock-induced apoptosis by maintaining phosphorylation of protein kinase B (AKT). J Neurovirol 21:568–575. doi: 10.1007/s13365-015-0361-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Phelan D, Barrozo ER, Bloom DC. 2017. HSV1 latent transcription and non-coding RNA: a critical retrospective. J Neuroimmunol 308:65–101. doi: 10.1016/j.jneuroim.2017.03.002. [DOI] [PubMed] [Google Scholar]
- 28.Inman M, Perng GC, Henderson G, Ghiasi H, Nesburn AB, Wechsler SL, Jones C. 2001. Region of herpes simplex virus type 1 latency-associated transcript sufficient for wild-type spontaneous reactivation promotes cell survival in tissue culture. J Virol 75:3636–3646. doi: 10.1128/JVI.75.8.3636-3646.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kurt-Jones EA, Chan M, Zhou S, Wang J, Reed G, Bronson R, Arnold MM, Knipe DM, Finberg RW. 2004. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc Natl Acad Sci U S A 101:1315–1320. doi: 10.1073/pnas.0308057100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sato A, Linehan MM, Iwasaki A. 2006. Dual recognition of herpes simplex viruses by TLR2 and TLR9 in dendritic cells. Proc Natl Acad Sci U S A 103:17343–17348. doi: 10.1073/pnas.0605102103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li H, Zhang J, Kumar A, Zheng M, Atherton SS, Yu FS. 2006. Herpes simplex virus 1 infection induces the expression of proinflammatory cytokines, interferons and TLR7 in human corneal epithelial cells. Immunology 117:167–176. doi: 10.1111/j.1365-2567.2005.02275.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lund J, Sato A, Akira S, Medzhitov R, Iwasaki A. 2003. Toll-like receptor 9-mediated recognition of herpes simplex virus-2 by plasmacytoid dendritic cells. J Exp Med 198:513–520. doi: 10.1084/jem.20030162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Randall RE, Goodbourn S. 2008. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J Gen Virol 89:1–47. doi: 10.1099/vir.0.83391-0. [DOI] [PubMed] [Google Scholar]
- 34.Peng W, Henderson G, Inman M, BenMohamed L, Perng GC, Wechsler SL, Jones C. 2005. The locus encompassing the latency-associated transcript of herpes simplex virus type 1 interferes with and delays interferon expression in productively infected neuroblastoma cells and trigeminal ganglia of acutely infected mice. J Virol 79:6162–6171. doi: 10.1128/JVI.79.10.6162-6171.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Apelbaum A, Yarden G, Warszawski S, Harari D, Schreiber G. 2013. Type I interferons induce apoptosis by balancing cFLIP and caspase-8 independent of death ligands. Mol Cell Biol 33:800–814. doi: 10.1128/MCB.01430-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chin YE, Kitagawa M, Kuida K, Flavell RA, Fu XY. 1997. Activation of the STAT signaling pathway can cause expression of caspase 1 and apoptosis. Mol Cell Biol 17:5328–5337. doi: 10.1128/MCB.17.9.5328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yadav A, Kalita A, Dhillon S, Banerjee K. 2005. JAK/STAT3 pathway is involved in survival of neurons in response to insulin-like growth factor and negatively regulated by suppressor of cytokine signaling-3. J Biol Chem 280:31830–31840. doi: 10.1074/jbc.M501316200. [DOI] [PubMed] [Google Scholar]
- 38.Wang T, Yuan W, Liu Y, Zhang Y, Wang Z, Zhou X, Ning G, Zhang L, Yao L, Feng S, Kong X. 2015. The role of the JAK-STAT pathway in neural stem cells, neural progenitor cells and reactive astrocytes after spinal cord injury. Biomed Rep 3:141–146. doi: 10.3892/br.2014.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schoggins JW, Rice CM. 2011. Interferon-stimulated genes and their antiviral effector functions. Curr Opin Virol 1:519–525. doi: 10.1016/j.coviro.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nicholl MJ, Robinson LH, Preston CM. 2000. Activation of cellular interferon-responsive genes after infection of human cells with herpes simplex virus type 1. J Gen Virol 81:2215–2218. doi: 10.1099/0022-1317-81-9-2215. [DOI] [PubMed] [Google Scholar]
- 41.Mossman KL, Macgregor PF, Rozmus JJ, Goryachev AB, Edwards AM, Smiley JR. 2001. Herpes simplex virus triggers and then disarms a host antiviral response. J Virol 75:750–758. doi: 10.1128/JVI.75.2.750-758.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eidson KM, Hobbs WE, Manning BJ, Carlson P, DeLuca NA. 2002. Expression of herpes simplex virus ICP0 inhibits the induction of interferon-stimulated genes by viral infection. J Virol 76:2180–2191. doi: 10.1128/jvi.76.5.2180-2191.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Christensen MH, Jensen SB, Miettinen JJ, Luecke S, Prabakaran T, Reinert LS, Mettenleiter T, Chen ZJ, Knipe DM, Sandri-Goldin RM, Enquist LW, Hartmann R, Mogensen TH, Rice SA, Nyman TA, Matikainen S, Paludan SR. 2016. HSV-1 ICP27 targets the TBK1-activated STING signalsome to inhibit virus-induced type I IFN expression. EMBO J 35:1385–1399. doi: 10.15252/embj.201593458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Amici C, Rossi A, Costanzo A, Ciafre S, Marinari B, Balsamo M, Levrero M, Santoro MG. 2006. Herpes simplex virus disrupts NF-kappaB regulation by blocking its recruitment on the IkappaBalpha promoter and directing the factor on viral genes. J Biol Chem 281:7110–7117. doi: 10.1074/jbc.M512366200. [DOI] [PubMed] [Google Scholar]
- 45.Patel A, Hanson J, McLean TI, Olgiate J, Hilton M, Miller WE, Bachenheimer SL. 1998. Herpes simplex type 1 induction of persistent NF-kappa B nuclear translocation increases the efficiency of virus replication. Virology 247:212–222. doi: 10.1006/viro.1998.9243. [DOI] [PubMed] [Google Scholar]
- 46.Sciortino MT, Medici MA, Marino-Merlo F, Zaccaria D, Giuffre-Cuculletto M, Venuti A, Grelli S, Bramanti P, Mastino A. 2008. Involvement of gD/HVEM interaction in NF-kB-dependent inhibition of apoptosis by HSV-1 gD. Biochem Pharmacol 76:1522–1532. doi: 10.1016/j.bcp.2008.07.030. [DOI] [PubMed] [Google Scholar]
- 47.Welsh RM, Bahl K, Marshall HD, Urban SL. 2012. Type 1 interferons and antiviral CD8 T-cell responses. PLoS Pathog 8:e1002352. doi: 10.1371/journal.ppat.1002352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Johnson KE, Knipe DM. 2010. Herpes simplex virus-1 infection causes the secretion of a type I interferon-antagonizing protein and inhibits signaling at or before Jak-1 activation. Virology 396:21–29. doi: 10.1016/j.virol.2009.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mott KR, Osorio N, Jin L, Brick DJ, Naito J, Cooper J, Henderson G, Inman M, Jones C, Wechsler SL, Perng GC. 2003. The bovine herpesvirus-1 LR ORF2 is critical for this gene’s ability to restore the high wild-type reactivation phenotype to a herpes simplex virus-1 LAT null mutant. J Gen Virol 84:2975–2985. doi: 10.1099/vir.0.19421-0. [DOI] [PubMed] [Google Scholar]
- 50.Jin L, Perng GC, Mott KR, Osorio N, Naito J, Brick DJ, Carpenter D, Jones C, Wechsler SL. 2005. A herpes simplex virus type 1 mutant expressing a baculovirus inhibitor of apoptosis gene in place of latency-associated transcript has a wild-type reactivation phenotype in the mouse. J Virol 79:12286–12295. doi: 10.1128/JVI.79.19.12286-12295.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schulz KS, Mossman KL. 2016. Viral evasion strategies in type I IFN signaling—a summary of recent developments. Front Immunol 7:498. doi: 10.3389/fimmu.2016.00498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Halford WP, Weisend C, Grace J, Soboleski M, Carr DJ, Balliet JW, Imai Y, Margolis TP, Gebhardt BM. 2006. ICP0 antagonizes Stat 1-dependent repression of herpes simplex virus: implications for the regulation of viral latency. Virol J 3:44. doi: 10.1186/1743-422X-3-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vahed H, Agrawal A, Srivastava R, Prakash S, Coulon PA, Roy S, BenMohamed L. 2018. Unique type I interferon, expansion/survival cytokines and JAK/STAT gene signatures of multi-functional HSV-specific effector memory CD8(+) TEM cells are associated with asymptomatic ocular herpes in humans. J Virol doi: 10.1128/JVI.01882-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mott KR, Underhill D, Wechsler SL, Town T, Ghiasi H. 2009. A role for the JAK-STAT1 pathway in blocking replication of HSV-1 in dendritic cells and macrophages. Virol J 6:56. doi: 10.1186/1743-422X-6-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Priya R, Dhanwani R, Patro IK, Rao PV, Parida MM. 2013. Differential regulation of TLR mediated innate immune response of mouse neuronal cells following infection with novel ECSA genotype of Chikungunya virus with and without E1:A226V mutation. Infect Genet Evol 20:396–406. doi: 10.1016/j.meegid.2013.09.030. [DOI] [PubMed] [Google Scholar]
- 56.Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. 1998. How cells respond to interferons. Annu Rev Biochem 67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 57.Rock DL, Nesburn AB, Ghiasi H, Ong J, Lewis TL, Lokensgard JR, Wechsler SL. 1987. Detection of latency-related viral RNAs in trigeminal ganglia of rabbits latently infected with herpes simplex virus type 1. J Virol 61:3820–3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mador N, Goldenberg D, Cohen O, Panet A, Steiner I. 1998. Herpes simplex virus type 1 latency-associated transcripts suppress viral replication and reduce immediate-early gene mRNA levels in a neuronal cell line. J Virol 72:5067–5075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen SH, Lee LY, Garber DA, Schaffer PA, Knipe DM, Coen DM. 2002. Neither LAT nor open reading frame P mutations increase expression of spliced or intron-containing ICP0 transcripts in mouse ganglia latently infected with herpes simplex virus. J Virol 76:4764–4772. doi: 10.1128/JVI.76.10.4764-4772.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nicoll MP, Hann W, Shivkumar M, Harman LE, Connor V, Coleman HM, Proenca JT, Efstathiou S. 2016. The HSV-1 latency-associated transcript functions to repress latent phase lytic gene expression and suppress virus reactivation from latently infected neurons. PLoS Pathog 12:e1005539. doi: 10.1371/journal.ppat.1005539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bloom DC, Giordani NV, Kwiatkowski DL. 2010. Epigenetic regulation of latent HSV-1 gene expression. Biochim Biophys Acta 1799:246–256. doi: 10.1016/j.bbagrm.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cliffe AR, Garber DA, Knipe DM. 2009. Transcription of the herpes simplex virus latency-associated transcript promotes the formation of facultative heterochromatin on lytic promoters. J Virol 83:8182–8190. doi: 10.1128/JVI.00712-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang QY, Zhou C, Johnson KE, Colgrove RC, Coen DM, Knipe DM. 2005. Herpesviral latency-associated transcript gene promotes assembly of heterochromatin on viral lytic-gene promoters in latent infection. Proc Natl Acad Sci U S A 102:16055–16059. doi: 10.1073/pnas.0505850102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Peng W, Vitvitskaia O, Carpenter D, Wechsler SL, Jones C. 2008. Identification of two small RNAs within the first 1.5-kb of the herpes simplex virus type 1-encoded latency-associated transcript. J Neurovirol 14:41–52. doi: 10.1080/13550280701793957. [DOI] [PubMed] [Google Scholar]
- 65.Allen SJ, Rhode-Kurnow A, Mott KR, Jiang X, Carpenter D, Rodriguez-Barbosa JI, Jones C, Wechsler SL, Ware CF, Ghiasi H. 2014. Regulatory interactions between herpesvirus entry mediator (TNFRSF14) and latency associated transcript (LAT) during HSV-1 latency. J Virol 88:1961–1971. doi: 10.1128/JVI.02467-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Peng W, Jin L, Henderson G, Perng GC, Brick DJ, Nesburn AB, Wechsler SL, Jones C. 2004. Mapping herpes simplex virus type 1 latency-associated transcript sequences that protect from apoptosis mediated by a plasmid expressing caspase-8. J Neurovirol 10:260–265. doi: 10.1080/13550280490468690. [DOI] [PubMed] [Google Scholar]
- 67.Henderson G, Perng GC, Nesburn AB, Wechsler SL, Jones C. 2004. The latency-related gene encoded by bovine herpesvirus 1 can suppress caspase 3 and caspase 9 cleavage during productive infection. J Neurovirol 10:64–70. doi: 10.1080/13550280490261716. [DOI] [PubMed] [Google Scholar]
- 68.Shamas-Din A, Kale J, Leber B, Andrews DW. 2013. Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb Perspect Biol 5:a008714. doi: 10.1101/cshperspect.a008714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Medici MA, Sciortino MT, Perri D, Amici C, Avitabile E, Ciotti M, Balestrieri E, De Smaele E, Franzoso G, Mastino A. 2003. Protection by herpes simplex virus glycoprotein D against Fas-mediated apoptosis: role of nuclear factor kappaB. J Biol Chem 278:36059–36067. doi: 10.1074/jbc.M306198200. [DOI] [PubMed] [Google Scholar]
- 70.Aliprantis AO, Yang RB, Weiss DS, Godowski P, Zychlinsky A. 2000. The apoptotic signaling pathway activated by Toll-like receptor-2. EMBO J 19:3325–3336. doi: 10.1093/emboj/19.13.3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sun R, Zhang Y, Lv Q, Liu B, Jin M, Zhang W, He Q, Deng M, Liu X, Li G, Li Y, Zhou G, Xie P, Xie X, Hu J, Duan Z. 2011. Toll-like receptor 3 (TLR3) induces apoptosis via death receptors and mitochondria by up-regulating the transactivating p63 isoform alpha (TAP63alpha). J Biol Chem 286:15918–15928. doi: 10.1074/jbc.M110.178798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ma Y, He B. 2014. Recognition of herpes simplex viruses: Toll-like receptors and beyond. J Mol Biol 426:1133–1147. doi: 10.1016/j.jmb.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Casrouge A, Zhang SY, Eidenschenk C, Jouanguy E, Puel A, Yang K, Alcais A, Picard C, Mahfoufi N, Nicolas N, Lorenzo L, Plancoulaine S, Senechal B, Geissmann F, Tabeta K, Hoebe K, Du X, Miller RL, Heron B, Mignot C, de Villemeur TB, Lebon P, Dulac O, Rozenberg F, Beutler B, Tardieu M, Abel L, Casanova JL. 2006. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science 314:308–312. doi: 10.1126/science.1128346. [DOI] [PubMed] [Google Scholar]
- 74.Guo Y, Audry M, Ciancanelli M, Alsina L, Azevedo J, Herman M, Anguiano E, Sancho-Shimizu V, Lorenzo L, Pauwels E, Philippe PB, Perez de Diego R, Cardon A, Vogt G, Picard C, Andrianirina ZZ, Rozenberg F, Lebon P, Plancoulaine S, Tardieu M, Valerie D, Jouanguy E, Chaussabel D, Geissmann F, Abel L, Casanova JL, Zhang SY. 2011. Herpes simplex virus encephalitis in a patient with complete TLR3 deficiency: TLR3 is otherwise redundant in protective immunity. J Exp Med 208:2083–2098. doi: 10.1084/jem.20101568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang SY, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, Segal D, Sancho-Shimizu V, Lorenzo L, Puel A, Picard C, Chapgier A, Plancoulaine S, Titeux M, Cognet C, von Bernuth H, Ku CL, Casrouge A, Zhang XX, Barreiro L, Leonard J, Hamilton C, Lebon P, Heron B, Vallee L, Quintana-Murci L, Hovnanian A, Rozenberg F, Vivier E, Geissmann F, Tardieu M, Abel L, Casanova JL. 2007. TLR3 deficiency in patients with herpes simplex encephalitis. Science 317:1522–1527. doi: 10.1126/science.1139522. [DOI] [PubMed] [Google Scholar]
- 76.Lafaille FG, Pessach IM, Zhang SY, Ciancanelli MJ, Herman M, Abhyankar A, Ying SW, Keros S, Goldstein PA, Mostoslavsky G, Ordovas-Montanes J, Jouanguy E, Plancoulaine S, Tu E, Elkabetz Y, Al-Muhsen S, Tardieu M, Schlaeger TM, Daley GQ, Abel L, Casanova JL, Studer L, Notarangelo LD. 2012. Impaired intrinsic immunity to HSV-1 in human iPSC-derived TLR3-deficient CNS cells. Nature 491:769–773. doi: 10.1038/nature11583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Davey GM, Wojtasiak M, Proietto AI, Carbone FR, Heath WR, Bedoui S. 2010. Cutting edge: priming of CD8 T cell immunity to herpes simplex virus type 1 requires cognate TLR3 expression in vivo. J Immunol 184:2243–2246. doi: 10.4049/jimmunol.0903013. [DOI] [PubMed] [Google Scholar]
- 78.Jin L, Peng W, Perng GC, Brick DJ, Nesburn AB, Jones C, Wechsler SL. 2003. Identification of herpes simplex virus type 1 latency-associated transcript sequences that both inhibit apoptosis and enhance the spontaneous reactivation phenotype. J Virol 77:6556–6561. doi: 10.1128/JVI.77.11.6556-6561.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ghiasi H, Kaiwar R, Nesburn AB, Wechsler SL. 1992. Expression of herpes simplex virus type 1 glycoprotein B in insect cells. Initial analysis of its biochemical and immunological properties. Virus Res 22:25–39. doi: 10.1016/0168-1702(92)90087-P. [DOI] [PubMed] [Google Scholar]
- 80.Mott KR, Osorio Y, Brown DJ, Morishige N, Wahlert A, Jester JV, Ghiasi H. 2007. The corneas of naive mice contain both CD4+ and CD8+ T cells. Mol Vis 13:1802–1812. [PubMed] [Google Scholar]
- 81.Mott KR, Osorio Y, Maguen E, Nesburn AB, Wittek AE, Cai S, Chattopadhyay S, Ghiasi H. 2007. Role of anti-glycoproteins D (anti-gD) and K (anti-gK) IgGs in pathology of herpes stromal keratitis in humans. Invest Ophthalmol Vis Sci 48:2185–2193. doi: 10.1167/iovs.06-1276. [DOI] [PubMed] [Google Scholar]
- 82.Mott KR, Perng GC, Osorio Y, Kousoulas KG, Ghiasi H. 2007. A recombinant herpes simplex virus type 1 expressing two additional copies of gK is more pathogenic than wild-type virus in two different strains of mice. J Virol 81:12962–12972. doi: 10.1128/JVI.01442-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Matundan HH, Mott KR, Allen SJ, Wang S, Bresee CJ, Ghiasi YN, Town T, Wechsler SL, Ghiasi H. 2016. Interrelationship of primary virus replication, level of latency, and time to reactivation in the trigeminal ganglia of latently infected mice. J Virol 90:9533–9542. doi: 10.1128/JVI.01373-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mott KR, Gate D, Matundan HH, Ghiasi YN, Town T, Ghiasi H. 2016. CD8+ T cells play a bystander role in mice latently infected with herpes simplex virus 1. J Virol 90:5059–5067. doi: 10.1128/JVI.00255-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.