

Enteroviruses are important pathogens that can cause a variety of diseases in humans, including aseptic meningitis, myocarditis, hand-foot-and-mouth disease, conjunctivitis, and acute flaccid paralysis. Like many other viruses, enteroviruses must counteract antiviral cellular responses to establish an infection. It has been suggested that enterovirus proteases cleave cellular factors to perturb antiviral pathways, but the exact contribution of viral proteases 2Apro and 3Cpro remains elusive. Here, we show that 2Apro, but not 3Cpro, of all four human EV species (EV-A to EV-D) inhibits SG formation and IFN-β gene transcription. Our observations suggest that enterovirus 2Apro has a conserved function in counteracting antiviral host responses and thereby is the main enterovirus “security protein.” Understanding the molecular mechanisms of enterovirus immune evasion strategies may help to develop countermeasures to control infections with these viruses.
KEYWORDS: 2Apro, 3Cpro, CV-A21, CV-B3, EV-A71, EV-D68, SGs, enterovirus, stress granules, type I IFN
ABSTRACT
Most viruses have acquired mechanisms to suppress antiviral alpha/beta interferon (IFN-α/β) and stress responses. Enteroviruses (EVs) actively counteract the induction of IFN-α/β gene transcription and stress granule (SG) formation, which are increasingly implicated as a platform for antiviral signaling, but the underlying mechanisms remain poorly understood. Both viral proteases (2Apro and 3Cpro) have been implicated in the suppression of these responses, but these conclusions predominantly rely on ectopic overexpression of viral proteases or addition of purified viral proteases to cell lysates. Here, we present a detailed and comprehensive comparison of the effect of individual enterovirus proteases on the formation of SGs and the induction of IFN-α/β gene expression in infected cells for representative members of the enterovirus species EV-A to EV-D. First, we show that SG formation and IFN-β induction are suppressed in cells infected with EV-A71, coxsackie B3 virus (CV-B3), CV-A21, and EV-D68. By introducing genes encoding CV-B3 proteases in a recombinant encephalomyocarditis virus (EMCV) that was designed to efficiently activate antiviral responses, we show that CV-B3 2Apro, but not 3Cpro, is the major antagonist that counters SG formation and IFN-β gene transcription and that 2Apro’s proteolytic activity is essential for both functions. 2Apro efficiently suppressed SG formation despite protein kinase R (PKR) activation and α subunit of eukaryotic translation initiation factor 2 phosphorylation, suggesting that 2Apro antagonizes SG assembly or promotes its disassembly. Finally, we show that the ability to suppress SG formation and IFN-β gene transcription is conserved in the 2Apro of EV-A71, CV-A21, and EV-D68. Collectively, our results indicate that enterovirus 2Apro plays a key role in inhibiting innate antiviral cellular responses.
IMPORTANCE Enteroviruses are important pathogens that can cause a variety of diseases in humans, including aseptic meningitis, myocarditis, hand-foot-and-mouth disease, conjunctivitis, and acute flaccid paralysis. Like many other viruses, enteroviruses must counteract antiviral cellular responses to establish an infection. It has been suggested that enterovirus proteases cleave cellular factors to perturb antiviral pathways, but the exact contribution of viral proteases 2Apro and 3Cpro remains elusive. Here, we show that 2Apro, but not 3Cpro, of all four human EV species (EV-A to EV-D) inhibits SG formation and IFN-β gene transcription. Our observations suggest that enterovirus 2Apro has a conserved function in counteracting antiviral host responses and thereby is the main enterovirus “security protein.” Understanding the molecular mechanisms of enterovirus immune evasion strategies may help to develop countermeasures to control infections with these viruses.
INTRODUCTION
Picornaviruses are a large and diverse family of small (∼30-nm) nonenveloped viruses with a positive-sense RNA genome. Especially, the genus Enterovirus encompasses many human pathogens with implications for public health. The enteroviruses are categorized into 15 enterovirus (EV) species (EV-A to EV-L) and 3 rhinovirus (RV) species (RV-A to RV-C). Human pathogens are found among the EV-A to EV-D species and the three rhinovirus species. The best-known pathogenic enterovirus is poliovirus (PV), the causative agent of poliomyelitis. In addition, coxsackie A virus (CV-A), coxsackie B virus (CV-B), echoviruses, and several numbered enteroviruses (e.g., EV-A71 and EV-D68) cause a broad range of diseases, such as aseptic meningitis, myocarditis, hand-foot-and-mouth disease, conjunctivitis, and acute flaccid paralysis. Rhinoviruses are the leading cause of the common cold and are associated with exacerbations of asthma and chronic obstructive pulmonary disease (COPD) (reviewed in reference 1). The enterovirus genome is translated into one polyprotein that is autocatalytically processed into the structural proteins (VP1, VP2, VP3, VP4), the nonstructural proteins (2A, 2B, 2C, 3A, 3B, 3C, 3D), and several cleavage intermediates (e.g., 3CD) (reviewed in reference 1). While the structural proteins make up the viral capsid, the nonstructural proteins are involved in virus replication and/or interfere with antiviral responses. Nonstructural proteins 2A and 3C are the proteases involved in processing of the viral polyprotein. While 3Cpro is responsible for the majority of the cleavages needed for polyprotein processing, 2Apro cleaves at the junction between the structural proteins (P1 region) and its own N terminus (reviewed in reference 1). Besides their essential role in polyprotein processing, both proteases also cleave host cell factors to optimize virus replication. For example, 2Apro mediates host shutoff by cleaving eukaryotic translation initiation factor 4G (eIF4G) (2).
In response to virus infections, cells activate several antiviral responses. A well-known antiviral response is the induction of type I interferons (alpha/beta interferon [IFN-α/β]). Viral genome replication generates double-stranded RNA (dsRNA) replication intermediates, which can be recognized by cytoplasmic RIG-I-like receptors (RLRs). Enterovirus replication intermediates are predominantly recognized by MDA5 (3–5). However, recent evidence suggests that RIG-I may also contribute to the induction of IFN-α/β in certain cell types (6). Upon recognition of viral dsRNA, MDA5 interacts with MAVS, which subsequently activates TRAF3 and TBK1. TBK1 phosphorylates the transcription factors IRF3 and IRF7, resulting in their activation and dimerization. Simultaneously, TRAF3 activates the transcription factor NFκB. Upon activation, IRF3, IRF7, and NFκB localize to the nucleus, where they induce expression of IFN-α/β and other proinflammatory cytokines. Subsequent IFN-α/β signaling via the type I IFN receptor (IFNAR) and the JAK-STAT pathway induces the expression of hundreds of interferon-stimulated genes (ISGs). IFN-α/β signals in autocrine and paracrine ways to induce a tissue-wide antiviral state, thereby limiting viral spread (reviewed in references 7and 8).
One of the ISGs is dsRNA-dependent protein kinase R (PKR), a sensor protein of the integrated stress response (ISR). When PKR binds dsRNA ligands, it dimerizes, autophosphorylates, and subsequently phosphorylates the α subunit of eukaryotic translation initiation factor 2 (eIF2α). Phosphorylated eIF2α (p-eIF2α) represses translation and thereby impairs virus propagation. Stalled messenger ribonucleoproteins (mRNPs), consisting of mRNA, small ribosomal subunits, eukaryotic translation initiation factors (eIFs), and several auxiliary proteins, are stored in cytoplasmic aggregates known as stress granules (SG). SG formation is directed by SG scaffolding proteins, such as GTPase-activating protein-binding proteins 1 and 2 (G3BP1 and G3BP2) and T cell-restricted intracellular antigen 1 (TIA-1) (reviewed in reference 9). Rather than merely being an outcome of translation inhibition, SGs have also been suggested to serve as a platform for antiviral signaling. Multiple signaling molecules, including MDA5, RIG-I, PKR, and TRAF2, are recruited to SGs, and this localization has been suggested to regulate their activity (10–16). Further evidence for an antiviral function of SGs is the growing evidence that many viruses—apart from their effect on the upstream signaling cascade—have evolved mechanisms to counteract SG formation by targeting SG components (15, 17–20). The exact function of SGs, however, remains to be established.
To establish a productive infection, enteroviruses actively suppress antiviral responses. Several studies have shown that enteroviruses suppress IRF3 dimerization and, thereby, IFN-α/β gene transcription (11, 21–25). Both viral proteases, 2Apro and 3Cpro, cleave various factors of the RLR signaling pathway, implicating them in the suppression of IFN-α/β gene transcription. Overexpression of 3Cpro of several enteroviruses has been shown to result in the cleavage of MAVS, IRF7, and IRF9 and, as a consequence, in the suppression of a coexpressed IFN reporter construct (24–28). However, in several of these studies, convincing evidence that the cleavage products observed upon 3Cpro overexpression are the same as those observed in enterovirus-infected cells is lacking. In fact, we and others have identified 2Apro to be the viral protease responsible for cleaving MAVS (21, 22). Addition of recombinant 2Apro, but not 3Cpro, to cell lysates resulted in the appearance of MAVS cleavage products of the same size as those observed in infected cells. These cleavage products were also observed when 2Apro, but not 3Cpro, was expressed by a recombinant encephalomyocarditis virus (EMCV), a picornavirus that by itself does not cleave components of the RLR pathway (21). Besides cleaving MAVS, 2Apro has also been suggested to cleave MDA5 (21), which contradicted earlier suggestions that MDA5 is degraded by the proteasome (29). Meanwhile, 3Cpro cleaves RIG-I (21, 30). Contradictory results on the mechanism of how enteroviruses interfere with IFN-α/β signaling have also been reported. Some studies reported that EV-A71 infection reduces the expression levels of the IFN receptor, blocks STAT1 and STAT2 phosphorylation, and suppresses expression of multiple ISGs through the action of 2Apro (31, 32). In another study, however, it was suggested that EV-A71 infection does not affect STAT1 and STAT2 phosphorylation but that 3Cpro prevents these molecules from entering the nucleus (33). Overall, these conflicting data indicate that the physiological role of the 2Apro- and/or 3Cpro-mediated cleavages of signaling molecules for the suppression of the RLR and JAK-STAT pathways requires further investigation.
Enteroviruses also actively suppress the ISR. Infection of cells with PV, CV-B3, and EV-A71 results in the formation of small SGs early in infection, but these disappear later in infection (34–37). Again, conflicting observations about the identity of the viral antagonist have been reported. Initially, the suppression of SGs was attributed to 3Cpro (34–36). 3Cpro was shown to cleave G3BP1 and suppress SG formation, and overexpression of a cleavage-resistant G3BP1 decreased virus replication, although this decrease was rather modest (34, 36, 38). Recently, however, evidence was presented that 2Apro from EV-A71, PV, or CV suppresses SG formation induced by infection, as well as that induced by sodium arsenite or heat shock (37).
In this study, we set out to directly compare the abilities of 2Apro and 3Cpro of multiple enteroviruses to suppress IFN-α/β induction and SG formation in infected cells. As described above, we previously introduced the genes encoding 2Apro and 3Cpro of several enteroviruses in EMCV to study 2Apro- and 3Cpro-mediated cleavages in infected cells (21). However, these recombinant EMCVs contained an intact antagonist of the antiviral responses, i.e., the leader (L) protein, and therefore, these viruses could not be used to study the effect of the enterovirus proteases on IFN-α/β mRNA levels and SG formation. To overcome this, we introduced the genes encoding the enterovirus proteases in a mutant EMCV containing mutations that inactivate the leader protein (EMCV-LZn) (3, 11, 39). Using these recombinant viruses, we provide a comprehensive and detailed analysis of the effect of the proteases of representative members of the enterovirus species EV-A to EV-D on the formation of SGs and the induction of IFN-α/β. We demonstrate that 2Apro, but not 3Cpro, of EV-A71, CV-B3, CV-A21, and EV-D68 strongly suppresses IFN-α/β induction and SG formation in virus-infected cells, thereby pinpointing 2Apro as a major antagonist of these antiviral responses.
RESULTS
Multiple enteroviruses suppress SG formation.
We first determined whether representative members of the enterovirus species EV-A to EV-D share the ability to suppress SGs. To this end, we infected HeLa cells with EV-A71, CV-B3, CV-A21, or EV-D68 and visualized the SGs. We also infected cells with EMCV-LZn, which induces SG formation (11) and which therefore served as a positive control. At 4 h and 6 h postinfection (hpi), cells were fixed and we performed immunofluorescence staining for dsRNA to visualize viral replication and the SG markers G3BP1 and eIF3. Infection with all the viruses led to an increase in the dsRNA signal, indicating that all viruses replicated efficiently. In the cells infected with the different enteroviruses, we observed small SGs at 4 hpi (Fig. 1). These SGs were no longer detected at 6 hpi in cells infected with CV-B3 or CV-A21, while the majority of the cells infected with EV-A71 and EV-D68 also lacked SGs at 6 hpi (Fig. 1). Suppression of SG formation was previously reported for PV (34), for CV-B3 (35), and—while this work was in progress—also for EV-A71 (36, 37). Here, we provide evidence that CV-A21, another member of the EV-C species, and EV-D68, an EV-D species member, also suppress the formation of SGs. Taken together, our data suggest that the ability to counteract SG formation is conserved among all human enterovirus species (i.e., EV-A to EV-D).
FIG 1.
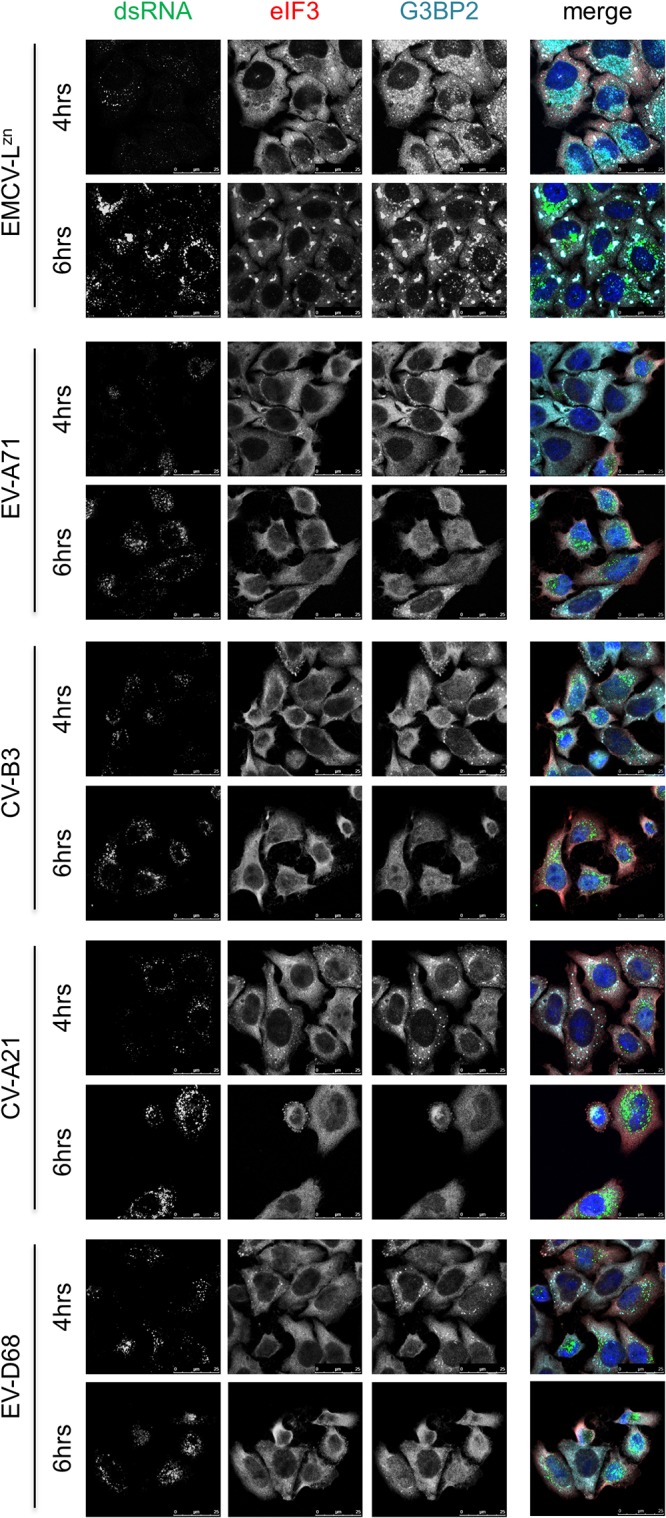
Multiple enteroviruses suppress SG formation. HeLa R19 cells were infected with EV-A71, CV-B3, CV-A21, EV-D68, or EMCV-LZn at an MOI of 10, and cells were fixed at 4 or 6 hpi. Subsequently, immunofluorescence staining was performed for the SG markers eIF3 and G3BP2, and viral replication was visualized by dsRNA staining.
Overexpression of enterovirus 2Apro suppresses sodium arsenite-induced SGs.
To better understand the effects of enterovirus proteases on SG formation, we overexpressed 2Apro and 3Cpro from EV-A71, CV-B3, or PV as enhanced green fluorescent protein (EGFP) fusion proteins and visualized SGs in the transfected cells. The cells were treated with sodium arsenite to induce SG formation. Afterwards we performed immunofluorescence staining against G3BP1 and TIA-1 (Fig. 2). In our hands, enterovirus 2Apro inhibited SG formation (Fig. 2A). In cells expressing EGFP-2Apro, we did not detect any G3BP1-positive SGs. However, we did observe a few TIA-1 granules. Meanwhile, expression of EGFP-3Cpro had no effect on SG formation (Fig. 2B). These findings suggest that enterovirus 2Apro is the major antagonist of SG formation.
FIG 2.

Overexpression of enterovirus 2Apro suppresses sodium arsenite-induced SGs. HeLa R19 cells were transfected with plasmids encoding 2Apro or 3Cpro of CV-B3, EV-A71, or PV N-terminally fused to EGFP. At 16 h posttransfection, cells were incubated with 500 μM sodium arsenite for 30 min. Subsequently, immunofluorescence staining was performed for the SG markers G3BP1 and TIA-1.
Enterovirus 2Apro suppresses SG formation when introduced into recombinant EMCV containing an inactive stress antagonist.
Overexpression of enteroviral proteases, followed by sodium arsenite treatment to induce SGs, may not accurately represent the role of the proteases in an enterovirus-infected cell. Given the essential role of the viral proteases in polyprotein processing and RNA replication, it is difficult to study their role in suppressing SG formation by introducing proteolytically inactivating mutations in the viral genome. We previously generated recombinant EMCVs harboring 2Apro and 3Cpro of different enteroviruses upstream of the leader protein, to study the proteolytic activity of 2Apro and 3Cpro (21). Unfortunately, we could not use these viruses to study the effects of the enterovirus proteases on SG formation, as EMCV actively suppresses SG formation. It has been suggested that EMCV 3Cpro cleaves G3BP1 and that this prevents SG assembly, although this cleavage is observed only at very late stages of infection (10 to 12 hpi) (40). In contrast, we and others did not observe G3BP1 cleavage in cells infected with either EMCV or Theiler’s murine encephalomyelitis virus, and instead provided extensive evidence that the leader protein has an essential role in suppressing SG formation in cardiovirus-infected cells (11, 41, 42). As part of these studies, we generated EMCV-LZn, a mutant virus in which the leader protein is inactivated by mutations that disrupt its zinc finger domain and that, as a consequence, can no longer suppress SG formation. To investigate the role of enterovirus proteases in suppressing SG formation, we therefore inserted in EMCV-LZn the gene encoding CV-B3 2Apro (EMCV-2Apro) or CV-B3 3Cpro (EMCV-3Cpro) (Fig. 3A). In parallel, we generated EMCV-LZn encoding catalytically inactive mutants of CV-B3 2Apro (EMCV-2Am, mutation C109A) and 3Cpro (EMCV-3Cm, mutation C147A), allowing us to determine whether the effects of 2Apro and 3Cpro depend on their proteolytic activity. To ensure that CV-B3 2Apro and 3Cpro are active in the context of EMCV-LZn infection, we infected HeLa cells with the different chimeric EMCVs and performed Western blot analysis of eIF4G, a well-known 2Apro target, or G3BP1, a well-known 3Cpro target (Fig. 3B). Indeed, infection with EMCV-2Apro, but not EMCV-2Am, resulted in cleavage of eIF4G, while infection with EMCV-3Cpro resulted in G3BP1 cleavage. No G3BP1 cleavage was observed in cells infected with either EMCV-LZn, which is consistent with our previous observations (11, 41, 42), or EMCV-3Cm.
FIG 3.

Enterovirus 2Apro suppresses SG formation in EMCV-infected cells. (A) Schematic representation of the EMCV-LZn genome encoding 2Apro or 3Cpro. The endogenous stress response antagonist (leader) was inactivated by the insertion of point mutations in its Zn finger domain (C19A/C22A), and subsequently, the genes encoding 2Apro and 3Cpro were introduced at the 5′ end of the EMCV open reading frame. UTR, untranslated region. (B) HeLa R19 cells were infected with EMCV-LZn, EMCV-2Apro, EMCV-2Am, EMCV-3Cpro, or EMCV-3Cm at an MOI of 10, and cells were lysed at 8 hpi. Lysates were subjected to Western blot analysis for eIF4G and G3BP1, and tubulin served as a loading control. (C) HeLa R19 cells were infected with EMCV-LZn, EMCV-2Apro, EMCV-2Am, EMCV-3Cpro, or EMCV-3Cm at an MOI of 10, and the cells were fixed at 4 or 8 hpi. Subsequently, immunofluorescence staining was performed for the SG markers eIF3 and G3BP2. Viral replication was visualized by dsRNA staining. (D) HeLa R19 cells were infected with EMCV-LZn encoding the 2Apro or 3Cpro of EV-A71, CV-B3, CV-A21, or EV-D68 at an MOI of 10 and fixed at 8 hpi. Subsequently, immunofluorescence staining was performed for G3BP1 and the number of SGs per cell was analyzed for at least 50 cells per condition. (E) HeLa R19 cells wt for or deficient in PKR expression were infected with EMCV, EMCV-LZn, EMCV-2Apro, or EMCV-3Cpro at an MOI of 0.01. At 24 hpi, samples were frozen and subjected to 3 freeze-thaw cycles. Subsequently, the viral yield was determined by endpoint dilution on BHK21 cells. TCID50, 50% tissue culture infective dose.
Next, we determined the ability of EMCV-2Apro and EMCV-3Cpro to suppress SG formation. We infected HeLa cells with the different chimeric EMCVs, fixed the cells at 4 or 8 hpi, and performed immunofluorescence staining for the SG markers eIF3 and G3BP2 and dsRNA to identify the infected cells (Fig. 3C). Infection with EMCV-2Apro resulted in the formation of some small SGs at 4 hpi. Importantly, we did not observe these small SGs upon infection with the other chimeric EMCVs. This is consistent with previous reports that overexpression of 2Apro can induce the formation of some small SGs (31, 37, 38), most likely through its activity to cleave eIF4G and cause a host mRNA translation shutoff. Intriguingly, as infection with EMCV-2Apro progressed, these small SGs disappeared and we did not observe any SGs at 8 hpi. A similar pattern of SGs that first appear and subsequently disappear during the course of infection has frequently been reported for enteroviruses (34–37). Infection with EMCV-2Am did not induce small SGs at 4 hpi, nor did it suppress the formation of larger SGs at 8 hpi. This indicates that the catalytic activity of 2Apro is needed for both the induction of small SGs at 4 hpi and the subsequent suppression of the formation of larger SGs. In contrast to 2Apro expression (EMCV-2Apro), heterologous expression of 3Cpro (EMCV-3Cpro) had a small effect on SG formation. These data suggest that CV-B3 2Apro, rather than CV-B3 3Cpro, is the main antagonist of SG formation.
To test whether 2Apro is also the main stress antagonist of enteroviruses belonging to other species, we constructed EMCV-LZn viruses expressing either 2Apro or 3Cpro from EV-A71, CV-A21, and EV-D68 and determined whether these chimeric viruses were able to suppress SG formation. HeLa cells were infected with these recombinant EMCVs, and we visualized SGs by immunofluorescence staining. Subsequently, we quantified the number of SGs per cell (Fig. 3D). The different 2Apro-expressing viruses all efficiently reduced the number of SGs (∼90%), whereas infection with the different 3Cpro-expressing viruses had a less pronounced effect on the number of SGs (∼25 to 40% reduction). Overall, our data demonstrate that both enterovirus proteases have an effect on SG formation in HeLa cells but that 2Apro is the main antagonist of SG formation and that this function is conserved among enteroviruses belonging to the species EV-A to EV-D.
Expression of 2Apro restores the impaired replication of EMCV-LZn.
EMCV-LZn replication in HeLa cells is predominantly restricted by the ISR (43). We next asked whether heterologous expression of enterovirus 2Apro, which efficiently suppressed SG formation, restores the replication of EMCV-LZn. To investigate this, we compared the replication kinetics of EMCV-LZn, EMCV-2Apro, and EMCV-3Cpro (encoding the CV-B3 proteases) in a multicycle infection. We infected HeLa cells wild type (wt) for or deficient in PKR (PKR knockout [k.o.]) at a low multiplicity of infection (MOI) and determined the virus yields at 24 hpi (Fig. 3E). In agreement with previous results (39, 43), we observed a strong defect in the replication of EMCV-LZn in wt cells but not in PKR k.o. cells, confirming that replication in HeLa cells is predominantly restricted by the ISR. EMCV-2Apro replicated with a similar efficiency in both cell lines, illustrating that heterologous expression of 2Apro rescued EMCV-LZn replication by countering the antiviral function of PKR-activated ISR. In contrast, expression of 3Cpro failed to rescue EMCV-LZn replication. Collectively, these data point to an important antagonistic function of 2Apro in counteracting PKR-induced SG formation in virus-infected cells.
Enterovirus 2Apro does not affect PKR signaling.
2Apro depends on its catalytic activity to suppress SGs, which suggests that 2Apro cleaves a cellular factor to suppress SGs. 2Apro does not cleave known structural SG components like G3BP1, G3BP2, or TIA-1 (35, 37, 38). To investigate whether 2Apro affects PKR signaling, we infected HeLa cells with EMCV-LZn, EMCV-2Apro, or EMCV-2Am and determined the levels of phosphorylated PKR (p-PKR) and p-eIF2α at 8 hpi by flow cytometry (Fig. 4). Infection with EMCV-LZn activated the PKR signaling pathway, resulting in an increased level of p-PKR and p-eIF2α relative to that in mock-infected cells, consistent with previous results (43). Infection with EMCV-2Apro and EMCV-2Am resulted in similar increases in p-PKR and p-eIF2α levels, demonstrating that 2Apro does not affect the PKR signaling pathway. Thus, it is unlikely that 2Apro suppresses SG formation by cleaving a factor involved in PKR signaling. How 2Apro suppresses SG formation remains to be established.
FIG 4.

Enterovirus 2Apro does not affect PKR signaling. HeLa R19 cells were infected with EMCV-LZn, EMCV-2Apro, or EMCV-2Am at an MOI of 10 and fixed at 8 hpi. Subsequently, the cells were stained with antibodies against dsRNA and p-PKR or dsRNA and p-eIF2α for flow cytometric analysis. Graphs represent the level of p-PKR or p-eIF2α in dsRNA-positive (infected) cells.
Enterovirus 2Apro suppresses the induction of type I IFN gene transcription.
In addition to the ISR, enteroviruses are known to suppress the induction of type I IFNs (11, 21–25). Indeed, infection of HeLa cells with EV-A71, CV-B3, CV-A21, or PV did not significantly induce the expression of IFN-β mRNA at 8 h p.i. (Fig. 5A). Various studies have shown that both 2Apro and 3Cpro can cleave proteins in the RLR signaling pathway and, thus, may be involved in suppressing IFN-β gene transcription in infected cells (21–27, 30, 44, 45). To evaluate the effect of the CV-B3 proteases on IFN-β mRNA induction in infected cells, we infected HeLa cells with EMCV-2Apro or EMCV-3Cpro and determined the induction of IFN-β mRNA at 8 hpi by reverse transcription (RT)-quantitative PCR (qPCR) analysis (Fig. 5B). As described previously (3, 11, 39), EMCV-LZn failed to suppress the RLR signaling pathway and resulted in high IFN-β mRNA levels. In contrast, infection with EMCV-2Apro hardly induced IFN-β mRNA levels, while infection with EMCV-2Am resulted in high IFN-β mRNA levels, indicating that 2Apro suppresses IFN-β gene transcription via its catalytic activity. Infection with EMCV-3Cpro resulted in high levels of IFN-β mRNA, indicating that 3Cpro is unable to suppress RLR-mediated signaling in HeLa cells. Collectively, these data suggest that enterovirus 2Apro is the viral protease that is responsible for suppressing IFN-β gene transcription in infected HeLa cells.
FIG 5.

Enterovirus 2Apro suppresses the induction of type I IFN gene expression. (A) HeLa R19 cells were infected with the indicated viruses at an MOI of 10, and the cells were lysed at 8 hpi, total cellular RNA was isolated, and RT-qPCR analysis was performed for IFN-β and actin mRNA. (B, C, D) HeLa R19 cells were infected with CV-B3, EMCV, EMCV-LZn, EMCV-2Apro, EMCV-2Am, EMCV-3Cpro, or EMCV-3Cm at an MOI of 10, and the cells were lysed at 8 hpi, total cellular RNA was isolated, and RT-qPCR analysis was performed for IFN-β, ISG56, MDA5, and actin mRNA. (E) HeLa R19 cells were infected with EMCV-LZn encoding the 2Apro or 3Cpro of EV-A71, CV-B3, CV-A21, or EV-D68 at an MOI of 10, and the cells were lysed at 8 hpi, total cellular RNA was isolated, and RT-qPCR analysis was performed for IFN-β and actin mRNA, as well as EMCV RNA. All graphs depict the fold induction of target gene mRNA levels compared to the levels in mock-infected cells, after correction for actin mRNA levels, with the exception of EMCV RNA, which is shown as the copy number per cell calculated from a plasmid standard. One-way ANOVA with the Dunnett post hoc test was used to determine statistical significance compared to the results for EMCV-infected (A to D) or EMCV-LZn-infected (E) cells (**, P < 0.01; ****, P < 0.0001). vRNA, viral RNA.
We also tested the effect of 2Apro and 3Cpro on JAK-STAT signaling by investigating the expression levels of ISGs. We determined the induction of ISG56 and MDA5 gene transcription via RT-qPCR analysis (Fig. 5C and D). Consistent with the ability of 2Apro to suppress IFN-β gene transcription, we did not observe an increase in the expression of these ISGs. 3Cpro had no effect on the induction of IFN-β gene transcription (Fig. 5B), yet we observed an ∼10-fold reduction in the expression of both ISG56 and MDA5, suggesting that 3Cpro affects JAK-STAT signaling rather than RLR signaling.
To determine whether the 2Apro of other enteroviruses also suppress IFN-β gene transcription, we infected cells with EMCVs expressing 2Apro or 3Cpro from EV-A71, CV-A21, and EV-D68 and determined IFN-β mRNA levels via RT-qPCR analysis (Fig. 5E). Indeed, 2Apro of these different enteroviruses suppressed IFN-β gene expression in the context of EMCV, while 3Cpro of these viruses did not. This demonstrates that the ability of 2Apro to suppress the induction of IFN-β gene expression is conserved among EV-A to EV-D species members.
Inhibition of SG formation by 2Apro unlikely accounts for the inhibition of type I IFN gene transcription.
It has been suggested that SG formation is critical for the induction of type I IFN (10, 40). This raised the possibility that 2Apro suppresses the induction of IFN-β mRNA via its ability to inhibit SG formation. To investigate the role of SGs in the induction of type I IFN, we used PKR k.o. cells, which cannot form SGs. Two types of PKR k.o. mouse embryo fibroblasts (MEFs) have been described (46–48); one lacks the RNA binding domain (RBD) (PKR RBD k.o. MEFs), while the other lacks the kinase domain (KD) (PKR KD k.o. MEFs) (Fig. 6A). Interestingly, while it was shown that the PKR RBD k.o. cells fail to induce IFN expression (10), we previously observed only a 5- to 10-fold reduction in IFN-β mRNA levels in PKR KD k.o. MEFs (11). To better understand these different results, we compared both types of PKR k.o. MEFs side by side under identical experimental conditions. We confirmed that both cell lines expressed truncated PKR, determined using Western blot analysis (Fig. 6B), and that both lost the ability to form SGs (Fig. 6C). Upon infection with EMCV-LZn, the PKR RBD k.o. cells showed no IFN-β mRNA induction, while the PKR KD k.o. cells showed only a relatively small reduction (5- to 10-fold) in IFN-β mRNA levels. Why the two cell lines yielded such different results remains to be elucidated. In addition to its role in the ISR, PKR has been suggested to play a role in the MDA5 signaling pathway (49). Possibly, the RBD is of greater importance for this function.
FIG 6.

Two types of PKR k.o. MEFs are both deficient in SG formation but differ in IFN-β induction. (A) Schematic representation of the PKR gene and the two different types of PKR k.o. MEFs used in this study. In the PKR KD k.o. cells, a premature stop codon was introduced before the kinase domain. In the PKR RBD k.o. cells, the initial start codon was removed, resulting in a PKR that lacks the RNA binding domain. (B) PKR Western blot analysis of lysates of PKR k.o. MEFs and the corresponding wt MEFs. Numbers to the left of the gel are molecular masses (in kilodaltons). (C) PKR k.o. and wt MEFs were infected with EMCV-LZn at an MOI of 10, fixed at 8 hpi, and stained for the SG marker G3BP1. (D) PKR k.o. and wt MEFs were infected with EMCV-LZn at an MOI of 10 and lysed at 6 and/or 10 hpi. Total RNA was isolated and subjected to RT-qPCR analysis for IFN-β, actin mRNA, and EMCV RNA. IFN-β mRNA is depicted as the fold induction of IFN-β mRNA levels compared to the levels in mock-infected cells, after correction for actin mRNA levels. EMCV RNA is shown as the copy number per cell, calculated from a plasmid standard. Two-way ANOVA with the Bonferroni post hoc test was used to calculate statistical significance (***, P < 0.001; ****, P < 0.0001).
The results described above did not provide a clear answer on the role of SGs in the induction of type I IFN. Therefore, we studied the role of SGs in IFN-β induction via an alternative, PKR-independent approach. To interfere with SG formation, we generated HeLa cells lacking expression of SG scaffolding protein G3BP1 and/or G3BP2 using the CRISPR/Cas9 technology (Fig. 7A). To characterize our newly generated cell lines, we determined whether they form SGs upon infection with EMCV-LZn or addition of sodium arsenite (Fig. 7B). While deletion of G3BP1 had a small effect on SG formation, G3BP1 and G3BP2 needed to be simultaneously depleted (G3BP1/2 k.o.) to prevent SG formation, consistent with the findings of a previous small interfering RNA-based study (50). To determine whether the PKR signaling pathway was affected by knocking out G3BP1 and/or G3BP2, we infected the different k.o. cell lines and performed Western blot analysis for PKR and p-PKR (Fig. 7C). Infection with EMCV-LZn resulted in the phosphorylation of PKR in all cell lines, indicating that the signaling pathway was not affected. Next, we determined the induction of IFN-β gene transcription upon infection with EMCV-LZn via RT-qPCR analysis (Fig. 7D). Only in the G3BP1/2 k.o. cells, which cannot form SGs, did we observe 5- to 10-fold lower IFN-β mRNA levels at 6 and 8 hpi. This reduction was similar to what we previously observed in the PKR KD k.o. MEFs. Collectively our data suggest that while SGs contribute to the induction of IFN-β mRNA, they do not play a critical role. This implies that it is unlikely that the nearly complete inhibition of type I IFN gene transcription by 2Apro is due to its ability to suppress SG formation.
FIG 7.

SG formation-deficient HeLa G3BP1/2 k.o. cells show only a small impairment in IFN-β induction. (A) Lysates of HeLa G3BP1 k.o., G3BP2 k.o., and G3BP1/2 k.o. cells were subjected to Western blot analysis for G3BP1, G3BP2, and tubulin. (B) HeLa wt, G3BP1 k.o., G3BP2 k.o., and G3BP1/2 k.o. cells were infected with EMCV-LZn at an MOI of 10 or incubated with 500 μM sodium arsenite (NaArs). Cells were fixed at 8 hpi or 30 min after addition of sodium arsenite, and SGs were visualized by immunofluorescence staining for G3BP1, eIF3, and G3BP2. (C) HeLa wt, G3BP1 k.o., G3BP2 k.o., and G3BP1/2 k.o. cells were infected with EMCV-LZn at an MOI of 10 and lysed at 4, 6, and 8 hpi. Lysates were subjected to Western blot analysis for PKR, phosphorylated PKR, and tubulin. (D) HeLa wt, G3BP1 k.o., G3BP2 k.o. and G3BP1/2 k.o. cells were infected with EMCV-LZn at an MOI of 10 and lysed at 4, 6, and 8 hpi. Subsequently, RNA was isolated and RT-qPCR analysis was performed for IFN-β, actin mRNA, and EMCV RNA. IFN-β mRNA is depicted as the fold induction of IFN-β mRNA levels compared to the levels in mock-infected cells, after correction for actin mRNA levels. EMCV RNA is shown as the copy number per cell, calculated from a plasmid standard. Two-way ANOVA with the Bonferroni post hoc test was used to calculate statistical significance (****, P < 0.0001). In panels A and C, numbers to the left of the gels are molecular masses (in kilodaltons).
DISCUSSION
To establish an infection, viruses must navigate an intricate network of antiviral host responses. Enteroviruses suppress both SG formation and the induction of IFN-α/β. Since both enterovirus 2Apro and enterovirus 3Cpro are essential for viral polyprotein processing and virus replication, it is challenging to study the ability of these proteins to suppress SGs during enterovirus infection by impairing their function. Therefore, most previous work relies on overexpression data for 2Apro and 3Cpro. However, overexpression of a viral protease may not accurately mimic its function during infection (21), and this may explain the conflicting results on the role of these proteases in antagonizing the ISR and the IFN-α/β pathway that have been reported. In this study, we demonstrated that EV-A71, CV-B3, CV-A21, and EV-D68 suppress SG formation and IFN-β gene transcription in HeLa cells. To study the role of 2Apro and 3Cpro in suppressing SG formation and IFN-β gene transcription, we introduced the proteases of CV-B3 in a recombinant EMCV isolate that was designed to efficiently activate antiviral responses (EMCV-LZn). We demonstrated that 2Apro strongly inhibited SG formation and IFN-β gene transcription in infected cells, suggesting that the cleavages mediated by 2Apro are more important for suppressing antiviral host responses than those mediated by 3Cpro. Using the same system, we showed that 2Apro of EV-A71, CV-A21, and EV-D68 also suppressed SG formation and IFN-β gene transcription, indicating that the identity of the antagonist is conserved among enteroviruses belonging to the species EV-A to EV-D.
The ability of enteroviruses to suppress SG formation was initially attributed to the cleavage of G3BP1 by 3Cpro (34–36). In these studies, expression of a cleavage-resistant G3BP1 (G3BP1Q326E) led to an increase in the number of SGs (34, 35) and a small reduction in viral titers (34). We also observed that enterovirus 3Cpro had an effect on SG formation, but enterovirus 2Apro had a much stronger inhibitory effect on the number of SGs. 2Apro strongly inhibited the number of SGs during overexpression of the protease and in the context of EMCV infection. While the manuscript for this article was in preparation, another study also reported the essential role of 2Apro in the suppression of SG formation (37). Importantly, in that study it was shown that an EV-A71 mutant containing an internal ribosome entry site (IRES) between the P1 and P2 regions and a catalytically inactive 2Apro was no longer able to suppress SG formation (37). These data, combined with our data, strongly suggest that 2Apro, rather than 3Cpro, is the major enterovirus antagonist of the ISR.
How 2Apro suppresses the formation of SGs is unknown. Thus far, no cleavages of known SG components (e.g., G3BP1, G3BP2, and TIA-1) by 2Apro have been detected (35, 37, 38). An alternative mechanism for viruses to prevent SG formation is to interfere with the upstream PKR-dependent signaling (43, 51–55). However, this does not seem to be the case for enteroviruses. It has been reported that PKR and eIF2α are phosphorylated during infection with several enteroviruses (37, 56–58), indicating that the viruses do not block activation of the ISR. Consistently, we did not observe an effect of 2Apro on the phosphorylation of PKR and eIF2α. Furthermore, 2Apro can inhibit arsenite-induced SG formation (Fig. 2) (37), which is triggered via activation of another eIF2α kinase. This suggests that 2Apro targets SG assembly. While this article was in the submission process, it was reported that SG formation relies on a newly identified interaction between eIF4GI and G3BP1 and that this interaction is targeted by 2Apro to block SG formation (59). Whether 2Apro also targets other protein-protein interactions underlying SG formation remains to be established. Apart from this, it should be considered that the direct cleavage of eIF4G by 2Apro contributes to the suppression of SG formation. Cleavage of eIF4G impairs the recruitment of 40S ribosomes to mRNAs, thereby altering the composition of mRNPs, which, in turn, may affect their recruitment to SGs. The observation that 2Apro triggers SG formation by cleavage of eIF4G (31, 37, 38) seems counterintuitive, but these 2Apro-induced SGs are smaller in size and differ in composition from traditional SGs. Whether these small SGs fail to grow into traditional SGs because of the cleavage of eIF4G by 2Apro or the ability of 2Apro to disrupt the protein-protein interactions underlying SG formation, or both, remains unknown.
During enterovirus infection, several signaling molecules in the RLR pathway have been suggested to be cleaved by 2Apro (MDA5 and MAVS) and 3Cpro (RIG-I, MAVS, IRF7, and IRF9) (21, 23, 26, 27, 29, 44, 45). There are conflicting observations, and the question remained of which, if any, of the identified cleavages is important for inhibiting IFN-α/β gene transcription. For instance, several studies have shown that 3Cpro, when overexpressed, cleaves MAVS and suppresses IFN-α/β induction (24, 25), but we showed that 3Cpro, when expressed by a recombinant EMCV, failed to cleave MAVS (21). Here, by infecting cells with EMCV-LZn encoding 2Apro or 3Cpro, we were able to study the effect of these proteases on IFN-α/β mRNA levels in picornavirus-infected cells. The major advantages of this approach are that MDA-5 is activated by a natural ligand (i.e. EMCV dsRNA) and that the enterovirus protease (2Apro or 3Cpro) is dynamically expressed during infection, similar to enterovirus-infected cells. Using these recombinant viruses, we demonstrate that enterovirus 2Apro, but not 3Cpro, suppresses the induction of IFN-α/β gene transcription in HeLa cells. The possibility that 3Cpro, by cleaving RIG-I, has some role in suppressing IFN-α/β gene transcription in other cell types cannot be excluded. The exact mechanism(s) used by 2Apro to inhibit IFN-α/β gene transcription remains to be identified. We previously demonstrated that 2Apro cleaves MDA5 and MAVS (21). These cleavages are likely involved in the suppression of IFN-α/β induction, but we cannot formally exclude the possibility of contributions of other 2Apro-dependent mechanisms. For instance, 2Apro is known to affect nuclear-cytoplasmic transport via the cleavage of nuclear pore proteins (NUPs) (60–63), which could interfere with the translocation of transcription factors (such as IRF3, IRF7, and NF-κB) to the nucleus. The 2Apro-mediated inhibition of SG formation may also contribute to suppress IFN-α/β induction. In this study, we showed that cells that cannot form SGs (i.e., PKR KD k.o. MEFs and G3BP1/2 k.o. HeLa cells) showed a 5- to 10-fold reduction in IFN-β gene transcription. The observation that heterologous expression of 2Apro during EMCV infection (EMCV-2Apro) nearly completely blocked IFN-β gene transcription (∼500-fold reduction) indicates that it is unlikely that suppression of SG formation by 2Apro is the major contributing factor in the viral suppression of IFN-β gene transcription.
Besides 2A proteins as antagonists of antiviral responses, some picornaviruses (like EMCV) also encode an L protein. Many 2A and L proteins have been shown to interfere with antiviral host responses and have therefore collectively been referred to as “security proteins” (64). Our work demonstrates that for enteroviruses, which lack an L protein, 2Apro is essential for suppressing both SG formation and the induction of IFN-β gene transcription, and we demonstrate that these two antagonizing functions are conserved in 2Apro of different enteroviruses belonging to the species EV-A to EV-D. These data support the idea that 2Apro is a major enteroviral security protein.
MATERIALS AND METHODS
Cells and viruses.
HeLa R19 and BHK21 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% (vol/vol) fetal calf serum. PKR KD and RBD k.o. MEFs have been described previously (46–48). HeLa R19 PKR k.o. cells have been described elsewhere (43). HeLa R19 G3BP1, G3BP2, and G3BP1/2 k.o. cells were made using the CRISPR/Cas9 methodology as described previously (43, 65). G3BP1 was targeted with guide RNA (gRNA) sequences 5′-TAGTCCCCTGCTGGTCGGGC-3′ and 5′-TATTACACACTGCTGAACC-3′, and G3BP2 was targeted with gRNA sequences 5′-CGCCCTACAAGCAGCGG-3′ and 5′-AAGCTCCGGAATATTTACAC-3′. EV-D68, EV-A71 (BrCr), and CV-A21 (Coe) were obtained from the National Institute for Public Health and Environment (RIVM, The Netherlands). The poliovirus 1 Sabin reference strain was obtained from J. Martin (NIBSC, United Kingdom). All enteroviruses were passaged on HeLa R19 cells and subsequently concentrated by ultracentrifugation through a 30% sucrose cushion at 140,000 × g for 16 h in an SW32Ti rotor and stored at −80°C. Recombinant EMCVs were generated by cloning the genes of interest into the XhoI/NotI restriction sites from the pM16.1-VFETQG-Zn infectious clone, which was described previously (43). The Strep2 tag was omitted in the viruses used in this study. Viruses were recovered by transfection of runoff RNA transcripts into BHK-21 cells. Subsequently, viruses were concentrated by ultracentrifugation and stored at −80°C.
Expression plasmids.
The 2Apro and 3Cpro genes from different enteroviruses were obtained by PCR of enterovirus viral RNA. The oligonucleotides used for these PCRs encode flanking XhoI and NotI restriction sites that were used to ligate the PCR products into the desired plasmids. The 3Cpro genes were ligated into the pcDNA-GFP vector, while the 2Apro genes were ligated into the pIRES-EGFP-MCS plasmid, both of which plasmids were described previously (42).
Antibodies.
The following antibodies were used for immunofluorescence analysis (IFA) staining procedures: anti-dsRNA (English & Scientific Consulting), anti-G3BP1 (Aviva Systems Biology), anti-G3BP2 (Bethyl Laboratories), anti-eIF3 (Santa Cruz), and anti-TIA-1 (Santa Cruz). Alexa Fluor 488-, Alexa Fluor 594-, and Alexa Fluor 647-conjugated secondary antibodies (Invitrogen) were used for detection. For flow cytometry staining, we used anti-dsRNA (English & Scientific Consulting), anti-p-PKR (Abcam), and anti-p-eIF2α (Abcam) antibodies and Alexa Fluor 488- or Alexa Fluor 647-conjugated (Invitrogen) secondary antibodies. For Western blotting, we used the antibodies anti-G3BP1 (Aviva Systems Biology), anti-eIF4G (Bethyl Laboratories), anti-PKR (Santa Cruz), anti-p-PKR (Abcam) and anti-tubulin (Sigma). Respective IRdye680- or IRdye800-conjugated secondary antibodies (LiCOR) were used for detection.
Immunofluorescence analysis.
HeLa R19 cells were grown on 12-mm glass coverslips and on the next day infected with the viruses indicated above (MOI, 10) or transfected with 1 μg of the plasmids indicated above using the FuGENE 6 reagent (Promega) according to the manufacturer’s instructions. At the time points indicated above, the cells were washed in phosphate-buffered saline (PBS) before being fixed in 4% paraformaldehyde for 30 min. Residual paraformaldehyde was washed away using PBS plus 10 mM glycine. Cells were permeabilized in PBS plus 0.1% Triton X-100 and subsequently incubated in blocking buffer (PBS plus 0.1% Tween 20 and 3% bovine serum albumin [BSA]) for 2 h. All subsequent steps were performed in blocking buffer. Samples were incubated with primary antibody for 1 h and incubated with secondary antibodies and DAPI (4′,6-diamidino-2-phenylindole) for 30 min. After antibody incubations, samples were washed three times with PBS plus 0.1% Tween 20. Before mounting coverslips on microscopy slides with FluorSafe reagent (Calbiochem), they were washed once more in Milli-Q water. Cells were examined by confocal microscopy (Leica SPE-II) and Leica Application Suite Advanced Fluorescence (LAS-AF) software.
Quantification of SGs.
The number of SGs and their surface area in ∼100 cells per condition were analyzed by the use of ImageJ software, using a combined total of 10 to 20 images. For each image, the background signal was removed by creating a blurred duplicate and subtracting it from the original image. Subsequently, the remaining diffuse (cytoplasmic) SG marker signal was removed via weak blurring, adjustment of the contrast settings, and application of a black-and-white threshold. In the resulting image, the number and average surface area of the SGs (shown in black on a white background) were quantified. The macro used is available upon request. Subsequent statistical analyses were performed using GraphPad Prism software. Error bars represent standard deviations, and P values were calculated using one-way analysis of variance (ANOVA) analysis with the Bonferroni post hoc test (infections with recombinant EMCVs).
Flow cytometry analysis.
HeLa R19 cells were seeded in 12-well plates and on the next day were infected with the indicated viruses (MOI, 10). A 6 hpi, cells were trypsinized and resuspended in fluorescent-activated cell sorting (FACS) buffer (PBS plus 1% BSA). The cells were fixed for 30 min in 2% paraformaldehyde in FACS buffer and subsequently fixed in ice-cold methanol for 10 min. All subsequent steps were performed in FACS buffer. Cells were stained with primary antibodies for 1 h. Subsequently, the cells were washed three times and incubated in secondary antibodies in the dark for 30 min. The cells were washed three times and kept in 1% paraformaldehyde until analysis on a FACSCanto II flow cytometer (BD Biosciences) using BD FACSDiva software. Data analysis was performed using FlowJo software (TreeStar).
Western blot analysis.
HeLa R19 cells were seeded in 10-cm dishes and on the next day were infected with the viruses indicated above (MOI, 10). At 8 hpi, cells were released using trypsin, washed once in PBS, and lysed in 100 μl lysis buffer (100 mM Tris, pH 8.0, 1 mM EDTA, 50 mM NaCl, 1% NP-40, protease inhibitor mix [Roche]). Postnuclear lysate was obtained by centrifugation at 15,000 × g at 4°C for 15 min. The amount of total protein in the lysates was determined using a bicinchoninic acid assay (Thermo Fisher). Fifty micrograms of protein from the cleared cell lysates was resolved using reducing sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and the proteins were transferred to 0.2-μm-pore-size nitrocellulose membranes by wet electrophoretic transfer. The membranes were incubated for 1 h in blocking buffer (PBS plus 0.1% Tween 20 and 2% BSA), successively incubated overnight with primary antibodies diluted in blocking buffer, and then incubated for 30 min with the respective secondary antibodies diluted in blocking buffer. After the antibody incubations, the membranes were washed three times with PBS plus 0.1% Tween 20. Finally, the membranes were washed once with PBS and scanned using an Odyssey imager (LiCOR).
RT-PCR analysis.
HeLa R19 cells were seeded in 24-well plates and on the next day were infected with the indicated viruses at an MOI of 10. At 8 hpi, the cells were lysed and cellular RNA was isolated using a total RNA isolation kit (Macherey-Nagel) according to the manufacturer’s instructions. Reverse transcription was set up using TaqMan reverse transcription reagents (Applied Biosystems) before performing qPCR analysis with SYBR green (Roche) as described previously (43).
ACKNOWLEDGMENTS
The work was supported by a Vici grant (NWO-918.12.628) from The Netherlands Organization for Scientific Research. Martijn A. Langereis was supported by a Veni grant (NWO-863.13.008) and Linda J. Visser is supported by the NWO graduate program Infection and Immunity (NWO-022.004.018), both from the Netherlands Organization for Scientific Research.
REFERENCES
- 1.Tapparel C, Siegrist F, Petty TJ, Kaiser L. 2013. Picornavirus and enterovirus diversity with associated human diseases. Infect Genet Evol 14:282–293. doi: 10.1016/j.meegid.2012.10.016. [DOI] [PubMed] [Google Scholar]
- 2.Ventoso I, MacMillan SE, Hershey JWB, Carrasco L. 1998. Poliovirus 2A proteinase cleaves directly the eIF-4G subunit of eIF-4F complex. FEBS Lett 435:79–83. doi: 10.1016/S0014-5793(98)01027-8. [DOI] [PubMed] [Google Scholar]
- 3.Feng Q, Hato SVV, Langereis MAA, Zoll J, Virgen-Slane R, Peisley A, Hur S, Semler BLL, van Rij RP, van Kuppeveld F. 2012. MDA5 detects the double-stranded RNA replicative form in picornavirus-infected cells. Cell Rep 2:1187–1196. doi: 10.1016/j.celrep.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang JP, Cerny A, Asher DR, Kurt-Jones EA, Bronson RT, Finberg RW. 2010. MDA5 and MAVS mediate type I interferon responses to coxsackie B virus. J Virol 84:254–260. doi: 10.1128/JVI.00631-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuo R-L, Kao L-T, Lin S-J, Wang R-L, Shih S-R. 2013. MDA5 plays a crucial role in enterovirus 71 RNA-mediated IRF3 activation. PLoS One 8:e63431. doi: 10.1371/journal.pone.0063431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Francisco E, Suthar M, Gale M, Rosenfeld AB, Racaniello VR. 2019. Cell-type specificity and functional redundancy of RIG-I-like receptors in innate immune sensing of coxsackievirus B3 and encephalomyocarditis virus. Virology 528:7–18. doi: 10.1016/j.virol.2018.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ivashkiv LB, Donlin LT. 2014. Regulation of type I interferon responses. Nat Rev Immunol 14:36–49. doi: 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goubau D, Deddouche S, Reis e Sousa C. 2013. Cytosolic sensing of viruses. Immunity 38:855–869. doi: 10.1016/j.immuni.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pakos‐Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. 2016. The integrated stress response. EMBO Rep 17:1374–1395. doi: 10.15252/embr.201642195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Onomoto K, Jogi M, Yoo JS, Narita R, Morimoto S, Takemura A, Sambhara S, Kawaguchi A, Osari S, Nagata K, Matsumiya T, Namiki H, Yoneyama M, Fujita T. 2012. Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity. PLoS One 7:e43031. doi: 10.1371/journal.pone.0043031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Langereis MA, Feng Q, van Kuppeveld FJ. 2013. MDA5 localizes to stress granules, but this localization is not required for the induction of type I interferon. J Virol 87:6314–6325. doi: 10.1128/JVI.03213-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim WJ, Back SH, Kim V, Ryu I, Jang SK. 2005. Sequestration of TRAF2 into stress granules interrupts tumor necrosis factor signaling under stress conditions. Mol Cell Biol 25:2450–2462. doi: 10.1128/MCB.25.6.2450-2462.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reineke LC, Lloyd RE. 2015. The stress granule protein G3BP1 recruits protein kinase R to promote multiple innate immune antiviral responses. J Virol 89:2575–2589. doi: 10.1128/JVI.02791-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reineke LC, Dougherty JD, Pierre P, Lloyd RE. 2012. Large G3BP-induced granules trigger eIF2 phosphorylation. Mol Biol Cell 23:3499–3510. doi: 10.1091/mbc.E12-05-0385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McCormick C, Khaperskyy DA. 2017. Translation inhibition and stress granules in the antiviral immune response. Nat Rev Immunol 17:647–660. doi: 10.1038/nri.2017.63. [DOI] [PubMed] [Google Scholar]
- 16.Kedersha N, Ivanov P, Anderson P. 2013. Stress granules and cell signaling: more than just a passing phase? Trends Biochem Sci 38:494–506. doi: 10.1016/j.tibs.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Humoud MN, Doyle N, Royall E, Willcocks MM, Sorgeloos F, van Kuppeveld F, Roberts LO, Goodfellow IG, Langereis MA, Locker N. 2016. Feline calicivirus infection disrupts assembly of cytoplasmic stress granules and induces G3BP1 cleavage. J Virol 90:6489–6501. doi: 10.1128/JVI.00647-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Panas MD, Varjak M, Lulla A, Eng KE, Merits A, Karlsson Hedestam GB, McInerney GM. 2012. Sequestration of G3BP coupled with efficient translation inhibits stress granules in Semliki Forest virus infection. Mol Biol Cell 23:4701–4712. doi: 10.1091/mbc.E12-08-0619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scholte FEM, Tas A, Albulescu IC, Žusinaite E, Merits A, Snijder EJ, van Hemert MJ. 2015. Stress granule components G3BP1 and G3BP2 play a proviral role early in Chikungunya virus replication. J Virol 89:4457–4469. doi: 10.1128/JVI.03612-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fros JJ, Domeradzka NE, Baggen J, Geertsema C, Flipse J, Vlak JM, Pijlman GP. 2012. Chikungunya virus nsP3 blocks stress granule assembly by recruitment of G3BP into cytoplasmic foci. J Virol 86:10873–10879. doi: 10.1128/JVI.01506-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feng Q, Langereis MA, Lork M, Nguyen M, Hato SV, Lanke K, Emdad L, Bhoopathi P, Fisher PB, Lloyd RE, van Kuppeveld F. 2014. Enterovirus 2Apro targets MDA5 and MAVS in infected cells. J Virol 88:3369–3378. doi: 10.1128/JVI.02712-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang B, Xi X, Lei X, Zhang X, Cui S, Wang J, Jin Q, Zhao Z. 2013. Enterovirus 71 protease 2Apro targets MAVS to inhibit anti-viral type I interferon responses. PLoS Pathog 9:e1003231. doi: 10.1371/journal.ppat.1003231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Drahos J, Racaniello VR. 2009. Cleavage of IPS-1 in cells infected with human rhinovirus. J Virol 83:11581–11587. doi: 10.1128/JVI.01490-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mukherjee A, Morosky SA, Delorme-Axford E, Dybdahl-Sissoko N, Oberste MS, Wang T, Coyne CB. 2011. The coxsackievirus B 3Cpro protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoS Pathog 7:e1001311. doi: 10.1371/journal.ppat.1001311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pang L, Yuan X, Shao C, Li M, Wang Y, Wang H, Xie G, Xie Z, Yuan Y, Zhou D, Sun X, Zhang Q, Xin Y, Li D, Duan Z. 2017. The suppression of innate immune response by human rhinovirus C. Biochem Biophys Res Commun 490:22–28. doi: 10.1016/j.bbrc.2017.05.169. [DOI] [PubMed] [Google Scholar]
- 26.Lei X, Xiao X, Xue Q, Jin Q, He B, Wang J. 2013. Cleavage of interferon regulatory factor 7 by enterovirus 71 3C suppresses cellular responses. J Virol 87:1690–1698. doi: 10.1128/JVI.01855-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiang Z, Liu L, Lei X, Zhou Z, He B, Wang J. 2016. 3C protease of enterovirus D68 inhibits cellular defense mediated by interferon regulatory factor 7. J Virol 90:1613–1621. doi: 10.1128/JVI.02395-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hung HC, Wang HC, Shih SR, Teng IF, Tseng CP, Hsu J. 2011. Synergistic inhibition of enterovirus 71 replication by interferon and rupintrivir. J Infect Dis 203:1784–1790. doi: 10.1093/infdis/jir174. [DOI] [PubMed] [Google Scholar]
- 29.Barral PM, Morrison JM, Drahos J, Gupta P, Sarkar D, Fisher PB, Racaniello VR. 2007. MDA-5 is cleaved in poliovirus-infected cells. J Virol 81:3677–3684. doi: 10.1128/JVI.01360-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barral PM, Sarkar D, Fisher PB, Racaniello VR. 2009. RIG-I is cleaved during picornavirus infection. Virology 391:171–176. doi: 10.1016/j.virol.2009.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang W, Zhang L, Wu Z, Tien P. 2014. Differential interferon pathway gene expression patterns in rhabdomyosarcoma cells during enterovirus 71 or coxsackievirus A16 infection. Biochem Biophys Res Commun 447:550–555. doi: 10.1016/j.bbrc.2014.04.021. [DOI] [PubMed] [Google Scholar]
- 32.Lu J, Yi L, Zhao J, Yu J, Chen Y, Lin MC, Kung H-F, He M-L. 2012. Enterovirus 71 disrupts interferon signaling by reducing the level of interferon receptor 1. J Virol 86:3767–3776. doi: 10.1128/JVI.06687-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang C, Sun M, Yuan X, Ji L, Jin Y, Cardona CJ, Xing Z. 2017. Enterovirus 71 suppresses interferon responses by blocking Janus kinase (JAK)/signal transducer and activator of transcription (STAT) signaling through inducing karyopherin-α1 degradation. J Biol Chem 292:10262–10274. doi: 10.1074/jbc.M116.745729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.White JP, Cardenas AM, Marissen WE, Lloyd RE. 2007. Inhibition of cytoplasmic mRNA stress granule formation by a viral proteinase. Cell Host Microbe 2:295–305. doi: 10.1016/j.chom.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 35.Fung G, Ng CS, Zhang J, Shi J, Wong J, Piesik P, Han L, Chu F, Jagdeo J, Jan E, Fujita T, Luo H. 2013. Production of a dominant-negative fragment due to G3BP1 cleavage contributes to the disruption of mitochondria-associated protective stress granules during CVB3 infection. PLoS One 8:e79546. doi: 10.1371/journal.pone.0079546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Y, Yao L, Xu X, Han H, Li P, Zou D, Li X, Zheng L, Cheng L, Shen Y, Wang X, Wu X, Xu J, Song B, Xu S, Zhang H, Cao H. 2018. Enterovirus 71 inhibits cytoplasmic stress granule formation during the late stage of infection. Virus Res 255:55–67. doi: 10.1016/j.virusres.2018.07.006. [DOI] [PubMed] [Google Scholar]
- 37.Yang X, Hu Z, Fan S, Zhang Q, Zhong Y, Guo D, Qin Y, Chen M. 2018. Picornavirus 2A protease regulates stress granule formation to facilitate viral translation. PLoS Pathog 14:e1006901. doi: 10.1371/journal.ppat.1006901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dougherty JD, Tsai WC, Lloyd RE. 2015. Multiple poliovirus proteins repress cytoplasmic RNA granules. Viruses 7:6127–6140. doi: 10.3390/v7122922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hato SV, Ricour C, Schulte BM, Lanke KHW, de Bruijn M, Zoll J, Melchers WJG, Michiels T, van Kuppeveld F. 2007. The mengovirus leader protein blocks interferon-alpha/beta gene transcription and inhibits activation of interferon regulatory factor 3. Cell Microbiol 9:2921–2930. doi: 10.1111/j.1462-5822.2007.01006.x. [DOI] [PubMed] [Google Scholar]
- 40.Ng CS, Jogi M, Yoo J-S, Onomoto K, Koike S, Iwasaki T, Yoneyama M, Kato H, Fujita T. 2013. Encephalomyocarditis virus disrupts stress granules, the critical platform for triggering antiviral innate immune responses. J Virol 87:9511–9522. doi: 10.1128/JVI.03248-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Borghese F, Michiels T. 2011. The leader protein of cardioviruses inhibits stress granule assembly. J Virol 85:9614–9622. doi: 10.1128/JVI.00480-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Visser LJ, Medina GN, Rabouw HH, de Groot RJ, Langereis MA, de Los Santos T, van Kuppeveld F. 2019. Foot-and-mouth disease virus leader protease cleaves G3BP1 and G3BP2 and inhibits stress granule formation. J Virol 93:e00922-18. doi: 10.1128/JVI.00922-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rabouw HH, Langereis MA, Knaap RCM, Dalebout TJ, Canton J, Sola I, Enjuanes L, Bredenbeek PJ, Kikkert M, de Groot RJ, van Kuppeveld F. 2016. Middle East respiratory coronavirus accessory protein 4a inhibits PKR-mediated antiviral stress responses. PLoS Pathog 12:e1005982. doi: 10.1371/journal.ppat.1005982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xiang Z, Li L, Lei X, Zhou H, Zhou Z, He B, Wang J. 2014. Enterovirus 68 3C protease cleaves TRIF to attenuate antiviral responses mediated by Toll-like receptor 3. J Virol 88:6650–6659. doi: 10.1128/JVI.03138-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lei X, Sun Z, Liu X, Jin Q, He B, Wang J. 2011. Cleavage of the adaptor protein TRIF by enterovirus 71 3C inhibits antiviral responses mediated by Toll-like receptor 3. J Virol 85:8811–8818. doi: 10.1128/JVI.00447-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abraham N, Stojdl DF, Duncan PI, Méthot N, Ishii T, Dubé M, Vanderhyden BC, Atkins HL, Gray DA, McBurney MW, Koromilas AE, Brown EG, Sonenberg N, Bell JC. 1999. Characterization of transgenic mice with targeted disruption of the catalytic domain of the double-stranded RNA-dependent protein kinase, PKR. J Biol Chem 274:5953–5962. doi: 10.1074/jbc.274.9.5953. [DOI] [PubMed] [Google Scholar]
- 47.Yang YL, Reis LF, Pavlovic J, Aguzzi A, Schäfer R, Kumar A, Williams BR, Aguet M, Weissmann C. 1995. Deficient signaling in mice devoid of double-stranded RNA-dependent protein kinase. EMBO J 14:6095–6106. doi: 10.1002/j.1460-2075.1995.tb00300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baltzis D, Li S, Koromilas AE. 2002. Functional characterization of PKR gene products expressed in cells from mice with a targeted deletion of the N terminus or C terminus domain of PKR. J Biol Chem 277:38364–38372. doi: 10.1074/jbc.M203564200. [DOI] [PubMed] [Google Scholar]
- 49.Pham AM, Santa Maria FG, Lahiri T, Friedman E, Marié IJ, Levy DE. 2016. PKR transduces MDA5-dependent signals for type I IFN induction. PLoS Pathog 12:e1005489. doi: 10.1371/journal.ppat.1005489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kedersha N, Panas MD, Achorn CA, Lyons S, Tisdale S, Hickman T, Thomas M, Lieberman J, McInerney GM, Ivanov P, Anderson P. 2016. G3BP-Caprin1-USP10 complexes mediate stress granule condensation and associate with 40S subunits. J Cell Biol 212:845–860. doi: 10.1083/jcb.201508028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lu Y, Wambach M, Katze MG, Krug RM. 1995. Binding of the influenza virus NS1 protein to double-stranded RNA inhibits the activation of the protein kinase that phosphorylates the eIF-2 translation initiation factor. Virology 214:222–228. doi: 10.1006/viro.1995.9937. [DOI] [PubMed] [Google Scholar]
- 52.Chang HW, Watson JC, Jacobs BL. 1992. The E3L gene of vaccinia virus encodes an inhibitor of the interferon-induced, double-stranded RNA-dependent protein kinase. Proc Natl Acad Sci U S A 89:4825–4829. doi: 10.1073/pnas.89.11.4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Feng Z, Cerveny M, Yan Z, He B. 2007. The VP35 protein of Ebola virus inhibits the antiviral effect mediated by double-stranded RNA-dependent protein kinase PKR. J Virol 81:182–192. doi: 10.1128/JVI.01006-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cruz JLG, Sola I, Becares M, Alberca B, Plana J, Enjuanes L, Zuñiga S. 2011. Coronavirus gene 7 counteracts host defenses and modulates virus virulence. PLoS Pathog 7:e1002090. doi: 10.1371/journal.ppat.1002090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li Y, Zhang C, Chen X, Yu J, Wang Y, Yang Y, Du M, Jin H, Ma Y, He B, Cao Y. 2011. ICP34.5 protein of herpes simplex virus facilitates the initiation of protein translation by bridging eukaryotic initiation factor 2alpha (eIF2alpha) and protein phosphatase 1. J Biol Chem 286:24785–24792. doi: 10.1074/jbc.M111.232439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Du Y, Bi J, Liu J, Liu X, Wu X, Jiang P, Yoo D, Zhang Y, Wu J, Wan R, Zhao X, Guo L, Sun W, Cong X, Chen L, Wang J. 2014. 3Cpro of foot-and-mouth disease virus antagonizes the interferon signaling pathway by blocking STAT1/STAT2 nuclear translocation. J Virol 88:4908–4920. doi: 10.1128/JVI.03668-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Black TL, Safer B, Hovanessian A, Katze MG. 1989. The cellular 68,000-Mr protein kinase is highly autophosphorylated and activated yet significantly degraded during poliovirus infection: implications for translational regulation. J Virol 63:2244–2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chang Y-H, Lau KS, Kuo R-L, Horng J-T. 2017. dsRNA binding domain of PKR is proteolytically released by enterovirus A71 to facilitate viral replication. Front Cell Infect Microbiol 7:284. doi: 10.3389/fcimb.2017.00284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang X, Hu Z, Zhang Q, Fan S, Zhong Y, Guo D, Qin Y, Chen M. 2019. SG formation relies on eIF4GI-G3BP interaction which is targeted by picornavirus stress antagonists. Cell Discov 5:1. doi: 10.1038/s41421-018-0068-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Park N, Skern T, Gustin KE. 2010. Specific cleavage of the nuclear pore complex protein Nup62 by a viral protease. J Biol Chem 285:28796–28805. doi: 10.1074/jbc.M110.143404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Park N, Katikaneni P, Skern T, Gustin KE. 2008. Differential targeting of nuclear pore complex proteins in poliovirus-infected cells. J Virol 82:1647–1655. doi: 10.1128/JVI.01670-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gustin KE, Sarnow P. 2001. Effects of poliovirus infection on nucleo-cytoplasmic trafficking and nuclear pore complex composition. EMBO J 20:240–249. doi: 10.1093/emboj/20.1.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Watters K, Palmenberg AC. 2011. Differential processing of nuclear pore complex proteins by rhinovirus 2A proteases from different species and serotypes. J Virol 85:10874–10883. doi: 10.1128/JVI.00718-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Agol VI, Gmyl AP. 2010. Viral security proteins: counteracting host defences. Nat Rev Microbiol 8:867–878. doi: 10.1038/nrmicro2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Langereis MA, Rabouw HH, Holwerda M, Visser LJ, van Kuppeveld F. 2015. Knockout of cGAS and STING rescues virus infection of plasmid DNA-transfected cells. J Virol 89:11169–11173. doi: 10.1128/JVI.01781-15. [DOI] [PMC free article] [PubMed] [Google Scholar]