

To successfully replicate, human adenovirus needs to carry out a rapid yet ordered transcriptional program that executes and drives viral replication. Early in infection, the viral E1A proteins are the key activators and regulators of viral transcription. Here we report, for the first time, that E1A works together with E4orf3 to perfect the viral transcriptional program and identify a novel mechanism by which the virus can adjust viral gene expression by modifying its genome’s nucleosomal organization via cooperation between E1A and E4orf3.
KEYWORDS: E1A, E4orf3, adenovirus, chromatin, histone
ABSTRACT
Human adenovirus expresses several early proteins that control various aspects of the viral replication program, including an orchestrated expression of viral genes. Two of the earliest viral transcriptional units activated after viral genome entry into the host cell nucleus are the E1 and E4 units, which each express a variety of proteins. Chief among these are the E1A proteins that function to reprogram the host cell and activate transcription of all other viral genes. The E4 gene encodes multiple proteins, including E4orf3, which functions to disrupt cellular antiviral defenses, including the DNA damage response pathway and activation of antiviral genes. Here we report that E1A directly interacts with E4orf3 via the conserved N terminus of E1A to regulate the expression of viral genes. We show that E4orf3 indiscriminately drives high nucleosomal density of viral genomes, which is restrictive to viral gene expression and which E1A overcomes via a direct interaction with E4orf3. We also show that during infection E1A colocalizes with E4orf3 to nuclear tracks that are associated with heterochromatin formation. The inability of E1A to interact with E4orf3 has a significant negative impact on overall viral replication, the ability of the virus to reprogram the host cell, and the levels of viral gene expression. Together these results show that E1A and E4orf3 work together to fine-tune the viral replication program during the course of infection and highlight a novel mechanism that regulates viral gene expression.
IMPORTANCE To successfully replicate, human adenovirus needs to carry out a rapid yet ordered transcriptional program that executes and drives viral replication. Early in infection, the viral E1A proteins are the key activators and regulators of viral transcription. Here we report, for the first time, that E1A works together with E4orf3 to perfect the viral transcriptional program and identify a novel mechanism by which the virus can adjust viral gene expression by modifying its genome’s nucleosomal organization via cooperation between E1A and E4orf3.
INTRODUCTION
During infection, human adenovirus (HAdV) carries out a highly coordinated and regulated transcriptional program to ensure a productive viral infection. Following entry of the viral genome into the host cell nucleus, the first protein-coding viral gene is expressed from the Early region 1A (E1A) (1). The E1A gene produces five different mRNAs that encode their cognate proteins. These include 13S and 12S mRNAs encoding E1A289R and E1A243R, the first E1A proteins expressed after infection, as well as mRNAs 11S, 10S, and 9S, which encode smaller variants expressed later in the infection, with the 10S mRNA and its 171-residue protein likely being the most abundant E1A in the late phase of viral infection (2). The E1A proteins have two primary functions: to drive expression from other viral genes and to reprogram quiescent cells to enter S phase so that the viral genomes can be replicated (3, 4).
Studies of E1A-mediated transcriptional regulation not only have elucidated how E1A regulates transcription of viral genes but also have shed important light on the mechanisms of eukaryotic transcription (1). Several important transcription factors or coregulators have been found thanks to studies of E1A transcriptional regulation, including several critical regulators such as p300 (5), C-terminal binding protein (CtBP) (6), and the retinoblastoma tumor suppressor (pRb) (7). Two regions within E1A have been found that play an important role in control of transcription, both cellular and viral. These are the N terminus (residues 1 to 25 in HAdV5) and conserved region 3 (CR3; residues 140 to 188) (3). Other regions of E1A play a role in regulating transcription as well, including CR1 (which together with the N terminus binds p300/CBP [8]), the area that binds pRb located within CR2 (9), and amino acids encoded within exon 2 of the gene, in particular CR4, which was found to bind to CtBP (6) and other transcriptional regulators such as FoxK1/K2 (10), DREF (11), and RuvBL1 (12).
Importantly, more recent studies have shown that there appears to be an interplay between the various E1A regions and isoforms in terms of their ability to transactivate viral and cellular promoters. For example, the long-standing mystery of why the N terminus of E1A243R repressed E1A289R-mediated transactivation was solved when it was discovered that the N terminus and CR3 regions of E1A compete for binding to p300/CBP (13). Essentially, E1A243R can sequester away p300/CBP from CR3, leading to reduced transactivation (13). A recent study has confirmed that two areas flanking the CR3 are capable of binding to p300/CBP, resulting in altered histone H3 acetylation at lysines (K) 18 and 27 (14). Studies have also shown that E1A is capable of tethering itself to cell-bound transcriptional repressors, which brings the powerful CR3 transactivation domain to repressed promoters, forcing expression of genes that they regulate (15, 16). Together, these findings paint a picture in which in E1A uses different isoforms to regulate gene expression in different ways in order to fine-tune the viral and cellular transcriptional program for the best possible outcome for the virus.
Here we report the identification of a novel interaction between HAdV5 E1A and E4orf3 proteins. E4orf3 is a small protein expressed early in infection from the E4 transcriptional unit located at the right end of the viral genome (1). The primary function of E4orf3 is to inactivate cellular stress response pathways, including interferon, p53, and the DNA damage response machinery (17–20). We describe the functional consequences of the interaction between E1A and E4orf3 and its effects on virus growth and viral gene expression.
RESULTS
The N terminus of E1A interacts with E4orf3.
We have previously studied the effects of E1A binding to the cellular kinase Nek9 (21, 22). Our studies showed that Nek9 is relocalized to nuclear, filamentous structures (termed tracks) in the nuclei of a proportion of the infected cells, which colocalize with E4orf3. We initially hypothesized that Nek9 may be localized to the tracks via an interaction with E4orf3; however, despite an extensive effort, we were unable to detect an interaction between these two proteins. Interestingly, while analyzing E4orf3 binding proteins, we identified E1A as an interaction partner (Fig. 1). Initial deletion mapping identified E1A residues 4 to 25 as essential for the interaction, these residues are deleted in E1A mutant dl1101 (23). E1A was able to bind to E4orf3 during the course of normal viral infection (Fig. 1B, D, and E) and when the two proteins were expressed together via cotransfection into uninfected cells (Fig. 1C). Finer deletion mapping identified residues 11 to 15 at the N terminus of E1A as being required for the interaction (Fig. 1C and D). We were also able to generate a viral mutant, with a deletion of residues 13 to 15 in E1A, that lost the ability to interact with E4orf3 in infected HT1080 cells (Fig. 1D) while retaining binding to p300 (Fig. 1D). Similarly, wild-type (wt) E1A was able to interact with E4orf3 in infected IMR-90 cells, while the mutant E1AΔ13-15 was deficient (Fig. 1E).
FIG 1.

E1A interacts with E4orf3. (A) U2OS-HA-E4orf3 cells were induced to express HA-E4orf3 with 2 μg/ml of doxycycline for 16 h and then infected with HAdV5 dl309 at an MOI of 30 for 24 h or mock infected for the same amount of time. Cells were then harvested and lysed, and immunoprecipitations were carried out for E1A using the M73 monoclonal antibody. Immunoprecipitations were resolved on a 4% to 12% Novex Bolt gel and Western blotted for HA or E1A as indicated. (B) U2OS-HA-E4orf3 cells were induced to express HA-E4orf3 with 2 μg/ml of doxycycline for 16 h, infected as for panel A with the indicated viruses (23), and immunoprecipitated for E4orf3 using the HA-tagged with the 3F10 rat anti-HA monoclonal antibody. Immunoprecipitations were resolved on a 4% to 12% Novex Bolt gel and Western blotted for HA or E1A as indicated. (C) HT1080 cells were transfected with expression vectors for the indicated E1A mutants (pCAN-E1A) and HA-E4orf3 (pCAN-HA-E4orf3) for 24 h. Immunoprecipitations were then carried out for HA using the rat 3F10 monoclonal antibody. Immunoprecipitations were resolved on a 4% to 12% Novex Bolt gel and Western blotted for HA or E1A as indicated. (D) HT1080 cells were infected with the indicated viruses at an MOI of 30 for 24 h. Cells were then harvested and lysed, and immunoprecipitations were then carried out for E4orf3 using the rat anti-E4orf3 monoclonal antibody 6A11. Immunoprecipitations were resolved on a 4% to 12% Novex Bolt gel and Western blotted for E4orf3 or E1A as indicated. (E) IMR-90 cells were infected with the indicated viruses at an MOI of 30 for 24 h. Cells were then harvested, lysed, and immunoprecipitated for E4orf3 using the rat monoclonal anti-E4orf3 antibody 6A11. Immunoprecipitations were resolved on a 4% to 12% Novex Bolt gel and blotted for E1A and E4orf3 as indicated. Input for E4orf3 shows the total E4orf3 immunoprecipitated in the assay.
To determine whether the interaction between E4orf3 and E1A was direct, we performed glutathione S-transferase (GST) pulldown assay with purified proteins from bacterial cells. Whereas a robust interaction was detected between GST-E4orf3 and His-tagged E1A289R, no such interaction was observed between GST and His-tagged E1A289R (Fig. 2). This observation demonstrates that the interaction between E1A and E4orf3 is likely direct.
FIG 2.
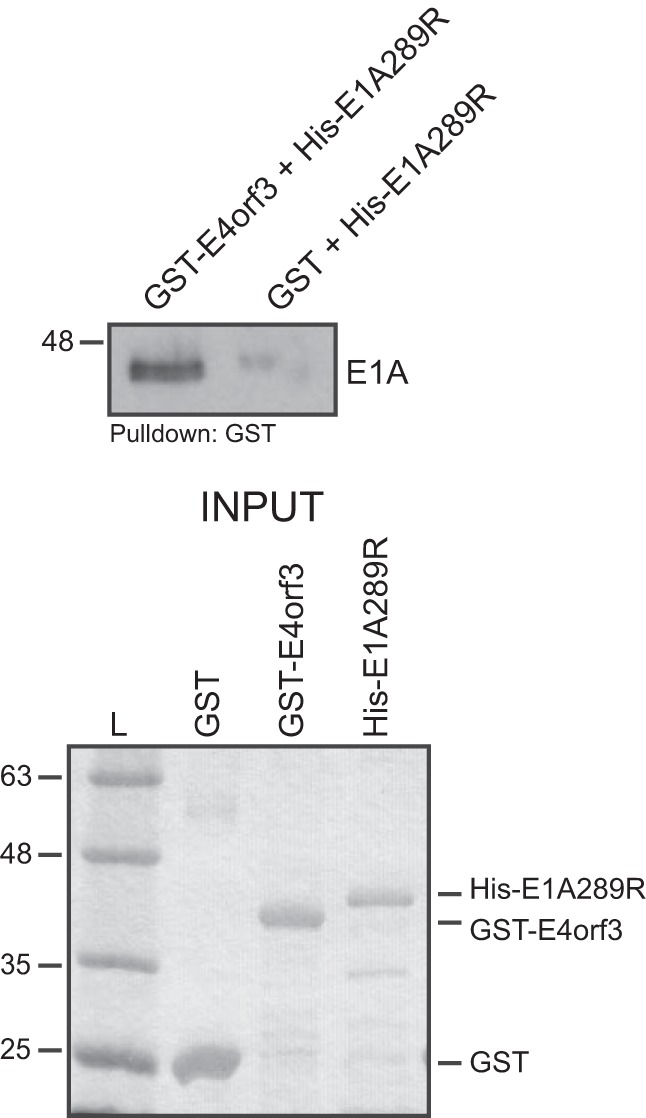
E1A binds directly to E4orf3. E. coli-expressed and -purified 6×His-E1A289R was mixed with either GST-E4orf3 or GST (also bacterially expressed and purified) and glutathione-agarose beads, incubated, washed, and resolved by SDS-PAGE. Pulldowns were blotted for E1A using M73 monoclonal antibody. Inputs for GST, GST-E4orf3, and 6×His-E1A289R (1 µg of each protein) were resolved by SDS-PAGE and stained with Coomassie. L, molecular weight ladder.
E1A and E4orf3 colocalize to nuclear tracks during infection.
During HAdV infection, E4orf3 forms nuclear tracks and disturbs the organization of promyelocytic nuclear bodies (PML-NB) (24, 25). E1A was previously shown to localize to PML bodies during viral infection (24) and indirectly shown to colocalize with E4orf3 nuclear tracks (24). The diffuse and strong nuclear staining observed when E1A is visualized in infected cells masks any subtle differences in its subnuclear distribution. To overcome this limitation, we employed a buffer as previously reported (26), which removes most proteins not physically attached to nuclear structures. Using this treatment, we observed three distinct phenotypes for E1A localization as shown in Fig. 3 and labeled as type I, II, or III. In most cells, E1A was completely removed from the nucleus by treatment with this buffer (type III), suggesting that it was not stably tethered to the nuclear structures. However, we also observed two other phenotypes in which E1A localized to nuclear structures distinct from E4orf3 tracks (type II) or localized with E4orf3 tracks (type I). Interestingly, deletion of the E4orf3-binding region of E1A significantly reduced its nuclear retention (Fig. 3B), as the E1AΔ13-15 mutant colocalized with E4orf3 only 10% to 20% of the time, versus 30% to 40% for wt E1A. Nevertheless, our results present direct evidence of E1A colocalization with E4orf3.
FIG 3.

E1A colocalizes with E4orf3 nuclear tracks during infection. (A) IMR-90 cells were infected with the indicated viruses for 24 h, treated with nuclear extraction buffer, fixed with 4% formaldehyde, and stained for E1A using the M73 mouse monoclonal antibody or for E4orf3 using the rat anti-E4orf3 monoclonal antibody. Alexa Fluor conjugates were used as secondary antibodies to visualize the staining. DAPI was used as a nuclear counterstain. Images were acquired on a Zeiss LSM700 laser confocal microscope using the 63× oil immersion lens. (B) Quantification of the observed E1A and E4orf3 colocalizations seen in panel A. Five random fields of view were acquired using a Molecular Devices ImageXpress Micro 4 high-content imager, representing no less than 100 cells. Images were analyzed using MetaXpress software and plotted as percentages of the three types of phenotypes observed. Statistically significant differences (P value ≤ 0.0005) between the two viruses are indicated with an asterisk. Error bars represent SDs between the five fields of view acquired.
E4orf3 binding to E1A is essential for efficient virus growth.
To investigate how binding of E4orf3 to E1A affects viral fitness, we generated a mutant virus expressing E1A with a deletion of residues 13 to 15 (Ad5.Δ13-15), which results in loss of binding to E4orf3 (Fig. 1D). Growth of this virus was determined in arrested IMR-90 cells at 48, 72, and 96 h after infection at a multiplicity of infection (MOI) of 50 and was compared to that of dl309 at the same MOI. Ad5.Δ13-15 growth was considerably reduced compared to that of dl309 (Fig. 4) and showed approximately 5- and 10-fold growth deficits at 72 h and 96 h, respectively, after infection compared to dl309 expressing wild-type E1A (Fig. 4A). Interestingly, the virus expressing mutant E1A plateaued in growth at 72 h after infection, which was not observed with dl309, which continued to grow exponentially past 72 h. The mutant virus also showed delayed appearance of cytopathic effect (CPE) (Fig. 4B). Appearance of CPE was delayed by approximately 24 h and progressed slowly. Whereas dl309 showed nearly complete CPE by 96 h after infection, Ad5.Δ13-15 had minimal changes in CPE observed between 72 and 96 h, which also corresponded with the observed plateauing of virus growth.
FIG 4.

Interaction of E1A with E4orf3 is necessary for efficient viral growth. (A) IMR-90 cells were grown until confluence, at which point medium was replaced and the cells were allowed to grow for another 72 h. At that point cells were infected with the indicated viruses at an MOI of 50 for 1 h, after which the saved medium originally removed from the cells was reapplied. Cells were then incubated for the indicated time, after which virus titer was determined by plaque assay on 293 cells. Statistically significant differences (P value ≤ 0.008) between the two viruses are indicated with an asterisk. Error bars represent SDs from three biological replicates. (B) Images of cells from panel A were acquired prior to harvest with the Bio-Rad ZOE cell imager using bright field and 20× lens objective. Zoom images represent a 1,000- by 1,000-pixel view of the top right corner of the original images.
Binding of E4orf3 to E1A enhances viral gene expression and reduces viral genome chromatinization.
Reduced viral growth suggested that the Ad5.Δ13-15 virus is impaired in its ability to replicate in arrested normal cells. To further elucidate the reasons behind the observed deficiencies, we analyzed the expression of viral genes during infection of arrested IMR-90 cells. In these cells, the mutant virus showed moderate reduction of expression of early viral genes, particularly E1B, E2, and E3, compared to the expression by dl309 (Fig. 5A). Interestingly, expression of E4orf3 (and E4orf6/7 [data not shown]) was either unaltered or slightly enhanced compared to the expression by dl309. Furthermore, expression of late genes was slightly higher, up to 2-fold, early in infection and similar to that for dl309 later in infection. These observations suggested that E4orf3 binding by E1A is important in regulating a subset of viral early genes, particularly E1, E2, and E3.
FIG 5.

E1A interaction with E4orf3 affect expression of viral early genes and influences viral chromatin structure. (A) IMR-90 cells were grown until confluence, at which point medium was replaced and the cells were allowed to grow for another 72 h. At that point cells were infected with either dl309 or Ad5.Δ13-15 at an MOI of 30 for 1 h, after which the saved medium originally removed from the cells was reapplied. Cells were then incubated for the indicated time, after which RNA was extracted using the TRIzol reagent, converted to cDNA using SuperScript VILO, and analyzed for expression using real-time quantitative PCR (qPCR) with the Bio-Rad CFX96 instrument and ABI SuperMix for CFX reagent. Analysis of expression was performed using the Pfaffl method, comparing the expression in Ad5.Δ13-15-infected cells to that in dl309-infected cells. Expression of GAPDH was used to normalize the results. Statistically significant differences (P value < 0.05) between the two viruses are indicated with an asterisk. Error bars represent SDs from three biological replicates. (B) IMR-90 cells treated in the same manner as for panel A were fixed with paraformaldehyde 24 h after infection, and chromatin was immunoprecipitated using an anti-E1A antibody cocktail consisting of an equal mix of M58 and M73 hybridoma supernatants or a rabbit anti-rat IgG control antibody. Promoter occupancy of E1A was assayed using real-time qPCR using ABI SuperMix for CFX with a Bio-Rad CFX96 real-time instrument. The inset shows total E1A immunoprecipitated from the samples. Results are represented as percentage of input. Statistically significant differences (P value < 0.0001) between the two viruses are indicated with an asterisk. Error bars represent SDs from two biological replicates. (C) The same cells as used for panel B for E1A immunoprecipitation were used to immunoprecipitate histone H3 or K9-acetylated histone H3. Data are represented as percentage of input, and error bars represent SDs from two biological replicates. Statistically significant differences (P value ≤ 0.0472) between the two viruses are indicated with an asterisk. (D) Data presented in panel C were used to calculate the ratio of K9-acetylated H3 (AcH3) to total H3, which is represented on the bar chart. Statistically significant differences (P value < 0.0001) between the two viruses are indicated with an asterisk. Error bars represent SDs from two biological replicates.
To investigate the mechanism behind the reduced expression of viral genes, we investigated promoter occupancy by E1A at viral promoters 24 h after infection (Fig. 5B). E1A was found to occupy all viral promoters analyzed, with the lowest occupancy observed at the E4 promoter. Interestingly, E1A occupancy was substantially reduced in cells infected with Ad5.Δ13-15, with levels being only slightly higher than background levels observed with the IgG negative-control antibody, despite comparable levels of E1A immunoprecipitated in the assay (Fig. 5B, inset). E1A itself does not possess any intrinsic DNA binding activity but rather gains access to viral and cellular promoters via interactions with promoter-bound factors. Access of these promoter-bound factors is regulated by the state of chromatin at these sites, with heterochromatin being restrictive to promoter binding. Due to the role of E4orf3 in driving heterochromatin formation in order to silence gene expression (25), we hypothesized that in the absence of E1A binding to E4orf3, excess heterochromatin is formed, blocking transcription factor access to promoters and therefore reducing E1A access to these promoters. To test this hypothesis, we investigated the total level of chromatin at viral promoters and the level of relaxed chromatin by investigating H3 occupancy on viral promoters and degree of H3 acetylation on lysine 9 (K9) (Fig. 5C). We also compared the ratio of K9-acetylated histone H3 over total histone H3, which is a mark of active transcription and relaxed chromatin (27). In cells expressing E1A unable to bind to E4orf3, we observed increased H3 loading and low to undetectable levels of H3 K9 acetylation compared to those in cells expressing wt E1A (Fig. 5C). In dl309-infected IMR-90 cells, the ratio of AcK9 H3 over total H3 was ∼0.20 at the E2A promoter (Fig. 5D), but for cells infected with Ad5.Δ13-15, the ratio was less than 0.01, indicative of greater histone H3 association with the viral genome and reduced histone acetylation. This was also observed at the E3A and the major late promoter (MLP) but not the E4 promoter, which was expressed at similar levels in dl309- and Ad5.Δ13-15-infected cells (Fig. 5A). Importantly, the overall level of histone H3 at these promoters was higher in Ad5.Δ13-15-infected cells while, at the same time, the cells had a lower level of histone H3 K9 acetylation (Fig. 5C).
E4orf3 reduces E1A289R-mediated transactivation from viral promoters and inhibits E1A289R access to viral promoters.
To investigate how regulation of viral gene expression by E1A is affected by the presence of E4orf3, we used a cell line based on A549 cells that provides E1A289R in trans via stable retroviral transduction. This also ensures that E1A levels between all samples are identical and are not directly affected by mutations within the virus. To investigate how E4orf3 affects expression of viral genes, we infected A549-E1A289R cells with Ad-CMV, which expresses no E1A and carries a mutation in the E4orf3 open reading frame yielding no E4orf3 protein, or with Ad-CMV-E4orf3, which expresses hemagglutinin (HA)-tagged E4orf3 in place of E1A. We then analyzed gene expression at 16 h after infection prior to viral genome replication to ensure that there were no replication-related effects (Fig. 6A). We observed significantly reduced expression from all viral promoters in the presence of E4orf3. Expression of late genes was, overall, lower than that of the early genes, as would be expected at this early time point.
FIG 6.

E4orf3 inhibits viral gene expression and affects viral chromatin organization. (A) A549-E1A289R cells that stably express HAdV5 E1A289R were infected with either Ad-CMV, which expresses no E1A or E4orf3, or Ad-CMV-E4orf3, which expresses an HA-tagged E4orf3 protein. Sixteen hours after infection, total RNA was extracted using the TRIzol reagent, converted to cDNA using SuperScript VILO, and analyzed for expression using real-time qPCR with the Bio-Rad CFX96 instrument and ABI SuperMix for CFX reagent. Analysis of expression was determined as percentage of the GAPDH mRNA level. Since E4orf3 was provided by the virus and Ad-CMV has no extra copy of the E4orf3 gene, we used E4orf6/7 to analyze the expression from the E4 transcriptional unit. Statistically significant differences (P value ≤ 0.0123) between the two viruses are indicated with an asterisk. Error bars represent SDs from three biological replicates. (B) A549-E1A289R cells were infected at an MOI of 10 for 24 h. Cells were subsequently fixed with paraformaldehyde 24 h after infection, and chromatin was immunoprecipitated using an anti-E1A antibody cocktail consisting of an equal mix of M58 and M73 hybridoma supernatants or a rabbit anti-rat IgG control antibody. Promoter occupancy of E1A was assayed using real-time qPCR using ABI SuperMix for CFX with a Bio-Rad CFX96 real-time instrument. The inset shows total E1A immunoprecipitated from the samples. Results are represented as percentage of input. Statistically significant differences (P value < 0.0001) between the two viruses are indicated with an asterisk. Error bars represent SDs from two biological replicates. (C) The same cells as used for panel B for E1A immunoprecipitation were used to immunoprecipitate histone H3 or K9-acetylated histone H3. Data are represented as percentage of input, and error bars represent SDs from two biological replicates. Statistically significant differences (P value ≤ 0.0422) between the two viruses are indicated with an asterisk. (D) Data presented in panel C were used to calculate the ratio of AcK9 H3 to total H3, which is represented on the bar chart. Statistically significant differences (P value < 0.05) between the two viruses are indicated with an asterisk. Error bars represent SDs from two biological replicates.
To investigate whether E1A was impaired for binding to the viral genome in the presence of E4orf3, we performed chromatin immunoprecipitation (ChIP) for viral promoters after infection of A549-E1A289R cells with either Ad-CMV, which, as noted above, expresses no E1A and no E4orf3, or Ad-CMV-E4orf3, which expresses wt E4orf3 (Fig. 6B). Consistently and on all viral promoters, E1A levels were reduced in the presence of E4orf3. Furthermore, this correlated with higher histone H3 loading on viral promoters (Fig. 6C) and reduced histone H3 acetylation. Importantly, the ratio of acetylated H3 over total H3 was lower when E4orf3 was present (Fig. 6D), similar to what was observed with E1AΔ13-15-expressing virus (Fig. 5D).
DISCUSSION
Here we report, for the first time, the discovery of a direct interaction between the HAdV5 E1A and E4orf3 proteins. E1A was found to bind to E4orf3 via the N terminus, with residues 11 to 15 of E1A essential for the interaction. Deletion of the E4orf3 binding region in E1A lowered viral fitness, with reduced viral growth, delayed appearance of CPE, and lower viral gene expression. E1A was also found to colocalize with E4orf3 to E4orf3 tracks in infected cells. Importantly, E1A binding to E4orf3 enhanced viral gene expression and reduced chromatinization of the viral genome during infection. The inability of E1A to interact with E4orf3 was associated with higher nucleosomal density as judged by histone H3 loading onto viral genomes and reduced histone H3 acetylation on K9, which is a mark of active gene transcription (27). Together, these results suggest that E1A and E4orf3 work together during viral infection to fine-tune the viral transcriptional program and that E1A counteracts E4orf3-driven heterochromatin formation in order to efficiently transactivate viral promoters and potentially drive expression from cellular promoters. A model supported by our results is illustrated in Fig. 7.
FIG 7.

Proposed model of how the interaction between E1A and E4orf3 affects viral chromatin organization during infection. Brown discs represent histones, with the black line representing the viral DNA coiling around the core histones. mE1A, mutant E1A unable to bind to E4orf3; HDAC, histone deacetylase; HMT, histone methyl transferase; DNMT, DNA methyl transferase.
E4orf3 is not the first HAdV protein to be reported to interact with E1A, with a recent study showing that E4orf6 binds to E1A, and a complex, which E4orf6 is a part of, enhances E1A-mediated transactivation of E2F-regulated cellular promoters (28). Nevertheless, our study is only the second such study which shows that E1A is capable of interacting with other viral proteins during infection. The region of E1A that was found to bind to E4orf3 is located within the N terminus of E1A, an area that binds to a wide array of cellular factors, many of which regulate transcription (1, 3). Our initial analysis of E1A mutants identified residues 4 to 25, lost in a dl1101 E1A mutant, as essential to the binding. However, this mutant also loses the ability to bind to many cellular factors, including p300/CBP (29, 30), the E2F/DP-1 complex (16), Nek9 (21, 22), pCAF (30, 31), GCN5 (32), and many others. This plethora of binding partners makes it difficult to decipher what contribution each makes, as deletions in E1A that abolish binding to one may affect another. Therefore, we generated smaller deletions that allowed mutants to retain binding to at least some of these factors while losing the ability to bind E4orf3. We should note here that some of the observed effects on viral growth seen with E1AΔ13-15-expressing HAdV may be due to loss of binding by this E1A mutant to other factors, which we did not test beyond p300. We wanted to retain E4orf3 expression in order to investigate the effects of the interaction between E1A and E4orf3 rather than the consequences of the loss of E4orf3, which has previously been well characterized and loss of which has significant effects on virus growth (19, 33, 34). Despite the deletion of three amino acids, Ad5.Δ13-15 grew considerably worse than dl309, with approximately 10-fold reduction in overall titers observed 96 h after infection and with plateauing observed starting at 72 h after infection. This is considerably lower than what was previously observed for a virus with a deletion of E4orf3 in HeLa cells (33) and likely reflects the transformed nature of HeLa cells partially complementing the loss of E4orf3. The plateauing of growth of Ad5.Δ13-15 may be caused by persistent infection (35) due to potential defects in the ability of the virus to modulate interferon signaling. Indeed, E4orf3 was previously shown to affect transcription of interferon-stimulated genes (18). Interestingly, although we observed colocalization of E1A to E4orf3 nuclear tracks, this was not the predominant phenotype (Fig. 3). It is possible that this occurs at specific times during infection. It is also possible that most of the E1A interacting with E4orf3 and localizing to tracks was washed out during our treatment to visualize the colocalization, and a resistant interaction is less frequent. Lastly, the observed differences in colocalization may be caused by random MOI differences in the infected cells causing protein levels to be variable, and having one virus, specifically mutant Ad5.Δ13-15, proceed through the infection at a lower rate. Nevertheless, not only were we able to observe a biochemically stable interaction between E1A and E4orf3, but also we also confirmed that this is a direct interaction using a GST pulldown assay.
Analysis of viral fitness showed significant reduction in the expression of viral genes during infection with mutant Ad5.Δ13-15, particularly E1, E2, and E3 transcriptional units (Fig. 5A). This was caused, at least in part, by considerably reduced E1A occupancy at viral promoters (Fig. 5B). Interestingly, although E1A occupancy was reduced at all viral promoters tested, only some of them were affected in terms of gene expression. The E4 transcriptional unit, for example, was not affected by the deletion of E4orf3 binding region in E1A, while structural genes showed slightly higher levels of expression than in a virus expressing wild-type E1A. Unaltered expression of E4 may be explained by lack of changes in viral chromatin organization measured by histone H3 loading and acetylation. Unlike other transcriptional units, which showed dramatic reduction in histone H3 acetylation while at the same time showing enhanced histone H3 loading in Ad5.Δ13-15-infected cells, E4 was almost unchanged (Fig. 5D). This may be due to the overall spatial position of this promoter at the end of the genome, or it may be due to a difference in how E1A affects transcription from this promoter. Similarly, although we observed alteration in chromatin organization at the viral major late promoter (Fig. 5D), this had little effect on the expression of late genes. As with E4, this may reflect a difference in how this promoter is regulated distinctly from the early promoters (36). Alternatively, it is possible that other histone modifications drive expression from the MLP or E4 promoter; for example, p300 drives E1A CR3-mediated acetylation of K18 and K27 in histone H3 (14), which we did not investigate in this study. The deletion of residues 13 to 15 within E1A is in close proximity to a region that binds pCAF (37); a recent study (38) has shown that E1A fragment consisting of residues 1 to 33 binds to the pCAF catalytic site and inhibits its acetyltransferase activity. It is possible that deletion of amino acids 13 to 15 within E1A enhances pCAF binding and may drive some of the observed reduction in acetylated H3, as shown in Fig. 5.
Heterochromatin formation is one of the major functions of E4orf3, as a mechanism to silence stress-responsive genes and aid viral replication (18). It is unclear how E4orf3 specifically traps only certain promoters/genes while leaving others active. However, our data suggest that this may be a nonspecific mechanism of gene silencing that unwittingly traps some viral genomes into a heterochromatin-like state that is restrictive to transcription. Indeed, loss of E4orf3 is associated with higher overall E1A promoter occupancy (Fig. 6B) and viral gene expression (Fig. 6A), despite overall loss of virus growth. This may also represent another means by which the virus controls its transcriptional program for optimal growth. By having factors that drive gene expression, and those that silence it, the virus can tune transcription of its own genes as well as the host’s in order to orchestrate a perfectly conducted program for optimal virus growth. We have observed this mode of action before, where expression of different E1A isoforms, some of which are activators while others are repressors, can control and tune gene expression (13, 15, 16). Current models (25) of how the E4orf3 polymer drives silencing of cellular genes do not explain the selectivity for one gene over another, only suggesting that this may be governed by specific interactions with chromatin-proximal factors, such as coregulators. Our data suggest that other viral proteins, such as E1A, may work together with E4orf3 in order to drive expression of those cellular genes that the virus needs and which may otherwise be silenced by E4orf3-induced heterochromatin. However, further investigation is needed to fully elucidate this possibility.
In conclusion, our study has identified a novel interaction between E1A and E4orf3 of HAdV5. The functional consequences of this interaction regulate viral gene expression, enhancing E1A promoter occupancy and increasing viral gene expression during infection. The interaction between E1A and E4orf3 appears to play a role in perfecting the viral transcriptional program in order to maximize viral replication. An intriguing possibility is that these two proteins work in similar manners on cellular genes and that E1A may be one of the means by which E4orf3-induced heterochromatin is relaxed at cellular promoters that need to be active for a productive infection to happen, a hypothesis that merits future investigation.
MATERIALS AND METHODS
Antibodies.
Mouse monoclonal anti-E1A M73 and M58 antibodies were previously described (39) and were grown in-house and used as the hybridoma supernatant. Rat monoclonal anti-E4orf3 antibody was previously described (40) and was a generous gift from Thomas Dobner. Anti-histone H3 and anti-acetyl K9 H3 antibodies were purchased from Abcam (catalg no. ab180727 and ab10812, respectively) and were used according to the manufacturer’s specifications. Rat anti-HA antibody (Roche), clone 3F10, was used at a dilution of 1:5,000 for Western blotting. Secondary antibodies were purchased from Jackson ImmunoResearch.
Cell and virus culture.
IMR-90 (ATCC CCL-186), A549-E1A289R, U2OS-HA-E4orf3 (41), and HT1080 (ATCC CCL-121) cells were grown in Dulbecco’s modified Eagle’s medium (DMEM; HyClone) supplemented with 10% fetal bovine serum (Seradigm) and streptomycin and penicillin (HyClone). All virus infections were carried out in serum-free media for 1 h, after which saved complete medium was added without removal of the infection media. Induction of HA-E4orf3 expression in U2OS-HA-E4orf3 cells was carried out by addition of 2 μg/ml of doxycycline to the growth media for at least 16 h or as indicated in the figure legends. U2OS-HA-E4orf3 cells, when induced, express levels of E4orf3 similar to those of HAdV5-infected cells at 24 h after infection (41). A549-E1A289R cells were generated by transduction with a retrovirus expressing HAdV5 E1A289R and selection with G418. For retrovirus generation, E1A289R-encoding cDNA was amplified by PCR and cloned into the LNSX retrovirus backbone.
Chromatin immunoprecipitation.
Chromatin immunoprecipitation (ChIP) was carried out essentially as previously described (13). IMR-90 cells were infected with the desired adenoviruses at an MOI of 30 and harvested 24 h after infection for ChIP analysis.
PCRs were carried out for HAdV5 early and major late promoters using EvaGreen master mix for digital droplet PCR (ddPCR) (Bio-Rad) according to the manufacturer’s directions using 3% total ChIP DNA as the template and a Bio-Rad QX200 digital droplet PCR instrument. The annealing temperature used was between 55°C and 65°C, depending on the primer set, and 40 cycles were run. Primers for viral promoters were previously described (11).
Immunofluorescence.
IMR-90 cells were plated at low density (∼40,000 cells per chamber) on chamber slides (Nalgene Nunc) and subsequently infected as described above. Twenty-four hours after infection, cells were treated with nuclear extraction (cytoskeleton) buffer (26) [10 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES; pH 6.8), 100 mM NaCl, 3 mM MgCl2, 1 mM EGTA, 0.5% Triton X-100] on ice for 5 min and washed twice in 1× phosphate-buffered saline (PBS). Cells were subsequently fixed in 4% paraformaldehyde, blocked in blocking buffer (1% normal goat serum, 1% bovine serum albumin [BSA], 0.2% Tween 20 in PBS), and stained with specific primary antibodies. M73 was used neat (hybridoma supernatant), E4orf3 antibody was used at a dilution of 1:800, and Alexa Fluor 488 and 594 secondary antibodies (Jackson ImmunoResearch) were used at a dilution of 1:600. After staining and extensive washing, slides were mounted using Prolong gold with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen) and imaged using a Zeiss LSM700 confocal laser scanning microscope. Images were analyzed using the Zeiss ZEN software package.
Immunoprecipitation.
Transfected HT1080 cells were lysed in NP-40 lysis buffer (0.5% NP-40, 50 mM Tris [pH 7.8], 150 mM NaCl) supplemented with a protease inhibitor cocktail (Sigma). Cell lysate containing 1 mg of total protein was used for IP with the monoclonal M73 anti-E1A antibody, rat 3F10 anti-HA antibody, or rat anti-E4orf3 antibody, depending on the specific experiment.
Statistical analysis.
Statistical analysis for Fig. 3 and 4 was performed using a two-tailed Student t test. Statistical analysis for Fig. 5 and 6 was conducted using two-way analysis of variance (ANOVA) followed by post hoc comparison using the Tukey test. P values of <0.05 were considered statistically significant.
PCR primers.
Plasmids.
The expression plasmid pcDNA3.1-E1A was previously described (22), and it expresses all E1A isoforms. The expression plasmid pGEX-6P-1-E4orf3 was generated by cloning E4orf3 in frame with the GST protein. pCAN-HA is a neomycin resistance expressing vector, expressing an N-terminal HA tag, generated by cloning HA into pCAN-Myc. The pCAN-HA-E4orf3 vector was generated by cloning E4orf3 int, pCAN-HA. Plasmids expressing mutant E1A proteins (pCAN-E1A Δ11-13, pCAN-E1A Δ17-19, pCAN-E1A Δ20-22, pCAN-E1A Δ23-25, and pCAN-E1A Δ26-28) were generated by inverse PCR, oligonucleotide primers that contained the E1A deletion were used to PCR amplify E1A, and the PCR products were then inserted into pCAN-Myc (Myc tag was removed during insertion).
Protein purification and GST pulldown assay.
Glutathione S-transferase fusion of E4orf3 was made by subcloning the cDNA into pGEX-6P1 (GE Healthcare Life Sciences) in frame with the N-terminal GST tag. His-tagged E1A289R was made by subcloning the entire E1A289R cDNA into the pET42 vector (Novagen) in frame with a C-terminal 6×His tag. Proteins were expressed in Escherichia coli strain BL21(DE3) and purified on their respective resins according to the manufacturer’s specifications. GST pulldown assay was carried out as previously described (13, 37).
Real-time gene expression analysis.
IMR-90 cells, or others as indicated, were infected with dl309 (45) at an MOI of 30. Total RNA was extracted using the TRIzol reagent (Sigma) at the desired time points according to manufacturer’s instructions. A total of 1.25 μg of total RNA was used in a reverse transcriptase reaction using SuperScript VILO reverse transcriptase (Invitrogen) according to the manufacturer’s guidelines using random hexanucleotides for priming. The cDNA was subsequently used for real-time expression analysis using the Bio-Rad CFX96 real-time thermocycler. Analysis of expression data was carried out using the Pfaffl method (46) and was normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA levels. Total E1A was detected as previously described (2).
Transfections.
Cells were plated in 10-cm plates at a density of 2.0 × 106/plate 24 h prior to transfection. Transfections were prepared by mixing 1 ml of serum-free DMEM, 10 μg of total plasmid DNA, and 20 μl of linear 1-mg/ml solution of polyethylenimine (25 kDa) reagent from Polysciences (catalog no. 23966-2). This was vortexed for 10 s and incubated at room temperature for 20 min. The complexes were then added to the cells and incubated for 24 to 48 h.
Viruses.
HAdV5 mutant dl309 (45) expressing wt E1A but with a deletion of much of the E3 region was generously donated by Joe Mymryk. Ad5.Δ13-15 was generated by cloning E1A with a deletion of residues 13 to 15 into pXC1 and cotransfecting this plasmid together with pJM17 into low-passage-number 293 cells. After 3 weeks, total virus was collected, plaque purified by two rounds of plaque purification, and sequenced to verify the deletion. All viruses were amplified in low-passage-number 293 cells, and virus titers were determined on these 293 cells prior to performing assays. All infections were carried out in serum-free medium for 1 h at an MOI of 10 unless otherwise specified in the figure legends. Ad5-CMV and Ad5-CMV-E4orf3 carry a deletion of HAdV5 nucleotides 451 to 3330, deleting all of E1A and part of E1B, and consequently express no E1A nor E1B proteins. The endogenous viral E1A promoter was replaced with the cytomegalovirus (CMV) promoter/enhancer, and HA-tagged E4orf3 was cloned into the virus to generated Ad-CMV-E4orf4. Both viruses carry mutations in the endogenous E4orf3 gene and were previously described (19).
Virus growth assay.
Contact inhibited IMR-90 cells were infected with HAdV5 dl309 or Ad5.Δ13-15 at an MOI of 50 in serum-free media. Virus was adsorbed for 1 h at 37°C under 5% CO2. Virus titers were determined 48, 72, and 96 h after infection by plaque assays performed on 293 cells by serial dilution.
ACKNOWLEDGMENTS
This work was supported by a grant from the Natural Sciences and Engineering Research Council (grant number RGPIN/435375-2013) to P.P., a Cancer Research Society operating grant (grant number 23106) to P.P., and a Research Manitoba operating grant to P.P. A.M.S. was supported by the NSERC Canada Graduate Scholarship for master’s students. L.C. was supported by a University of Manitoba graduate fellowship.
We thank Joe Mymryk for countless reagents and Thomas Dobner for the E4orf3 rat monoclonal antibody. P.P. thanks Stanisława Pełka for invaluable support and assistance and Ryszard Pełka for assistance and curiosity.
REFERENCES
- 1.Berk AJ. 2013. Chapter 55. Adenoviridae In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed (electronic) Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Radko S, Jung R, Olanubi O, Pelka P. 2015. Effects of adenovirus type 5 E1A isoforms on viral replication in arrested human cells. PLoS One 10:e0140124. doi: 10.1371/journal.pone.0140124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pelka P, Ablack JN, Fonseca GJ, Yousef AF, Mymryk JS. 2008. Intrinsic structural disorder in adenovirus E1A: a viral molecular hub linking multiple diverse processes. J Virol 82:7252–7263. doi: 10.1128/JVI.00104-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.King CR, Zhang A, Tessier TM, Gameiro SF, Mymryk JS. 2018. Hacking the cell: network intrusion and exploitation by adenovirus E1A. mBio 9:e00390-18. doi: 10.1128/mBio.00390-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harlow E, Whyte P, Franza BR Jr, Schley C. 1986. Association of adenovirus early-region 1A proteins with cellular polypeptides. Mol Cell Biol 6:1579–1589. doi: 10.1128/MCB.6.5.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boyd JM, Subramanian T, Schaeper U, La Regina M, Bayley S, Chinnadurai G. 1993. A region in the C-terminus of adenovirus 2/5 E1a protein is required for association with a cellular phosphoprotein and important for the negative modulation of T24-ras mediated transformation, tumorigenesis and metastasis. EMBO J 12:469–478. doi: 10.1002/j.1460-2075.1993.tb05679.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Whyte P, Buchkovich KJ, Horowitz JM, Friend SH, Raybuck M, Weinberg RA, Harlow E. 1988. Association between an oncogene and an anti-oncogene: the adenovirus E1A proteins bind to the retinoblastoma gene product. Nature 334:124–129. doi: 10.1038/334124a0. [DOI] [PubMed] [Google Scholar]
- 8.Barbeau D, Charbonneau R, Whalen SG, Bayley ST, Branton PE. 1994. Functional interactions within adenovirus E1A protein complexes. Oncogene 9:359–373. [PubMed] [Google Scholar]
- 9.Whyte P, Williamson NM, Harlow E. 1989. Cellular targets for transformation by the adenovirus E1A proteins. Cell 56:67–75. doi: 10.1016/0092-8674(89)90984-7. [DOI] [PubMed] [Google Scholar]
- 10.Komorek J, Kuppuswamy M, Subramanian T, Vijayalingam S, Lomonosova E, Zhao LJ, Mymryk JS, Schmitt K, Chinnadurai G. 2010. Adenovirus type 5 E1A and E6 proteins of low-risk cutaneous beta-human papillomaviruses suppress cell transformation through interaction with FOXK1/K2 transcription factors. J Virol 84:2719–2731. doi: 10.1128/JVI.02119-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Radko S, Koleva M, James KM, Jung R, Mymryk JS, Pelka P. 2014. Adenovirus E1A targets the DREF nuclear factor to regulate virus gene expression, DNA replication, and growth. J Virol 88:13469–13481. doi: 10.1128/JVI.02538-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olanubi O, Frost JR, Radko S, Pelka P. 2017. Suppression of type I interferon signaling by E1A via RuvBL1/pontin. J Virol 91:e02484-16. doi: 10.1128/JVI.02484-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pelka P, Ablack JN, Torchia J, Turnell AS, Grand RJ, Mymryk JS. 2009. Transcriptional control by adenovirus E1A conserved region 3 via p300/CBP. Nucleic Acids Res 37:1095–1106. doi: 10.1093/nar/gkn1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hsu E, Pennella MA, Zemke NR, Eng C, Berk AJ. 2018. Adenovirus E1A activation domain regulates H3 acetylation affecting varied steps in transcription at different viral promoters. J Virol 92:e00805-18. doi: 10.1128/JVI.00805-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bruton RK, Pelka P, Mapp KL, Fonseca GJ, Torchia J, Turnell AS, Mymryk JS, Grand RJ. 2008. Identification of a second CtBP binding site in adenovirus type 5 E1A conserved region 3. J Virol 82:8476–8486. doi: 10.1128/JVI.00248-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pelka P, Miller MS, Cecchini M, Yousef AF, Bowdish DM, Dick F, Whyte P, Mymryk JS. 2011. Adenovirus E1A directly targets the E2F/DP-1 complex. J Virol 85:8841–8851. doi: 10.1128/JVI.00539-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ullman AJ, Reich NC, Hearing P. 2007. Adenovirus E4 ORF3 protein inhibits the interferon-mediated antiviral response. J Virol 81:4744–4752. doi: 10.1128/JVI.02385-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soria C, Estermann FE, Espantman KC, O’Shea CC. 2010. Heterochromatin silencing of p53 target genes by a small viral protein. Nature 466:1076–1081. doi: 10.1038/nature09307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Evans JD, Hearing P. 2003. Distinct roles of the adenovirus E4 ORF3 protein in viral DNA replication and inhibition of genome concatenation. J Virol 77:5295–5304. doi: 10.1128/JVI.77.9.5295-5304.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karen KA, Hoey PJ, Young CS, Hearing P. 2009. Temporal regulation of the Mre11-Rad50-Nbs1 complex during adenovirus infection. J Virol 83:4565–4573. doi: 10.1128/JVI.00042-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jung R, Radko S, Pelka P. 2016. The dual nature of Nek9 in adenovirus replication. J Virol 90:1931–1943. doi: 10.1128/JVI.02392-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pelka P, Scime A, Mandalfino C, Joch M, Abdulla P, Whyte P. 2007. Adenovirus E1A proteins direct subcellular redistribution of Nek9, a NimA-related kinase. J Cell Physiol 212:13–25. doi: 10.1002/jcp.20983. [DOI] [PubMed] [Google Scholar]
- 23.Jelsma TN, Howe JA, Evelegh CM, Cunniff NF, Skiadopoulos MH, Floroff MR, Denman JE, Bayley ST. 1988. Use of deletion and point mutants spanning the coding region of the adenovirus 5 E1A gene to define a domain that is essential for transcriptional activation. Virology 163:494–502. doi: 10.1016/0042-6822(88)90290-5. [DOI] [PubMed] [Google Scholar]
- 24.Carvalho T, Seeler JS, Ohman K, Jordan P, Pettersson U, Akusjarvi G, Carmo-Fonseca M, Dejean A. 1995. Targeting of adenovirus E1A and E4-ORF3 proteins to nuclear matrix-associated PML bodies. J Cell Biol 131:45–56. doi: 10.1083/jcb.131.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ou HD, Kwiatkowski W, Deerinck TJ, Noske A, Blain KY, Land HS, Soria C, Powers CJ, May AP, Shu X, Tsien RY, Fitzpatrick JA, Long JA, Ellisman MH, Choe S, O'Shea CC. 2012. A structural basis for the assembly and functions of a viral polymer that inactivates multiple tumor suppressors. Cell 151:304–319. doi: 10.1016/j.cell.2012.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mirzoeva OK, Petrini JH. 2001. DNA damage-dependent nuclear dynamics of the Mre11 complex. Mol Cell Biol 21:281–288. doi: 10.1128/MCB.21.1.281-288.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rothbart SB, Strahl BD. 2014. Interpreting the language of histone and DNA modifications. Biochim Biophys Acta 1839:627–643. doi: 10.1016/j.bbagrm.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dallaire F, Schreiner S, Blair GE, Dobner T, Branton PE, Blanchette P. 2016. The human adenovirus type 5 E4orf6/E1B55K E3 ubiquitin ligase complex enhances E1A functional activity. mSphere 1:e00015-15. doi: 10.1128/mSphere.00015-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eckner R, Ewen ME, Newsome D, Gerdes M, DeCaprio JA, Lawrence JB, Livingston DM. 1994. Molecular cloning and functional analysis of the adenovirus E1A-associated 300-kD protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes Dev 8:869–884. doi: 10.1101/gad.8.8.869. [DOI] [PubMed] [Google Scholar]
- 30.Pelka P, Ablack JN, Shuen M, Yousef AF, Rasti M, Grand RJ, Turnell AS, Mymryk JS. 2009. Identification of a second independent binding site for the pCAF acetyltransferase in adenovirus E1A. Virology 391:90–98. doi: 10.1016/j.virol.2009.05.024. [DOI] [PubMed] [Google Scholar]
- 31.Reid JL, Bannister AJ, Zegerman P, Martínez-Balbás MA, Kouzarides T. 1998. E1A directly binds and regulates the P/CAF acetyltransferase. EMBO J 17:4469–4477. doi: 10.1093/emboj/17.15.4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lang SE, Hearing P. 2003. The adenovirus E1A oncoprotein recruits the cellular TRRAP/GCN5 histone acetyltransferase complex. Oncogene 22:2836–2841. doi: 10.1038/sj.onc.1206376. [DOI] [PubMed] [Google Scholar]
- 33.Huang MM, Hearing P. 1989. Adenovirus early region 4 encodes two gene products with redundant effects in lytic infection. J Virol 63:2605–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bridge E, Ketner G. 1989. Redundant control of adenovirus late gene expression by early region 4. J Virol 63:631–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng Y, Stamminger T, Hearing P. 2016. E2F/Rb family proteins mediate interferon induced repression of adenovirus immediate early transcription to promote persistent viral infection. PLoS Pathog 12:e1005415. doi: 10.1371/journal.ppat.1005415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dery CV, Herrmann CH, Mathews MB. 1987. Response of individual adenovirus promoters to the products of the E1A gene. Oncogene 2:15–23. [PubMed] [Google Scholar]
- 37.Shuen M, Avvakumov N, Walfish PG, Brandl CJ, Mymryk JS. 2002. The adenovirus E1A protein targets the SAGA but not the ADA transcriptional regulatory complex through multiple independent domains. J Biol Chem 277:30844–30851. doi: 10.1074/jbc.M201877200. [DOI] [PubMed] [Google Scholar]
- 38.Shi S, Liu K, Chen Y, Zhang S, Lin J, Gong C, Jin Q, Yang XJ, Chen R, Ji Z, Han A. 2016. Competitive inhibition of lysine acetyltransferase 2B by a small motif of the adenoviral oncoprotein E1A. J Biol Chem 291:14363–14372. doi: 10.1074/jbc.M115.697300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harlow E, Franza BR Jr, Schley C. 1985. Monoclonal antibodies specific for adenovirus early region 1A proteins: extensive heterogeneity in early region 1A products. J Virol 55:533–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nevels M, Tauber B, Kremmer E, Spruss T, Wolf H, Dobner T. 1999. Transforming potential of the adenovirus type 5 E4orf3 protein. J Virol 73:1591–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vink EI, Zheng Y, Yeasmin R, Stamminger T, Krug LT, Hearing P. 2015. Impact of adenovirus E4-ORF3 oligomerization and protein localization on cellular gene expression. Viruses 7:2428–2449. doi: 10.3390/v7052428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frost JR, Olanubi O, Cheng SK, Soriano A, Crisostomo L, Lopez A, Pelka P. 2017. The interaction of adenovirus E1A with the mammalian protein Ku70/XRCC6. Virology 500:11–21. doi: 10.1016/j.virol.2016.10.004. [DOI] [PubMed] [Google Scholar]
- 43.Crisostomo L, Soriano AM, Frost JR, Olanubi O, Mendez M, Pelka P. 2017. The influence of E1A C-terminus on adenovirus replicative cycle. Viruses 9:387. doi: 10.3390/v9120387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Crisostomo L, Soriano AM, Mendez M, Graves D, Pelka P. 2019. Temporal dynamics of adenovirus 5 gene expression in normal human cells. PLoS One 14:e0211192. doi: 10.1371/journal.pone.0211192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jones N, Shenk T. 1979. Isolation of adenovirus type 5 host range deletion mutants defective for transformation of rat embryo cells. Cell 17:683–689. doi: 10.1016/0092-8674(79)90275-7. [DOI] [PubMed] [Google Scholar]
- 46.Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]