

Abstract
Growing appreciation of the diversity of post-translational modifications (PTMs) in the mitochondria necessitates reevaluation of the roles these modifications play in both health and disease. Compared to the cytosol and nucleus, the mitochondrial proteome is highly acylated, and remodeling of the mitochondrial “acylome” is a key adaptive mechanism that regulates fundamental aspects of mitochondrial biology. It is clear that we need to understand the underlying chemistry that regulates mitochondrial acylation, as well as how chemical properties of the acyl chain impact biological functions. Here, we dissect the sources of PTMs in the mitochondria, review major mitochondrial pathways that control levels of PTMs, and highlight how sirtuin enzymes respond to the bioenergetic state of the cell via NAD+ availability to regulate mitochondrial biology. By providing a framework connecting the chemistry of these modifications, their biochemical consequences, and the pathways that regulate the levels of acyl PTMs, we will gain a deeper understanding of the physiological significance of mitochondrial acylation and its role in mitochondrial adaptation.
eTOC Blurb:
Lysine acyl modifications are highly enriched on mitochondrial proteins. In this review, Ringel et al. explore mechanisms that dynamically remodel the matrix “acylome” in response to nutrient cues and discuss central regulatory roles for mitochondrial sirtuins by coupling lysine deacylation to the metabolic state of the cell.
A brief history of lysine acylation
Histones were the first class of acetylated proteins to be identified and functionally characterized, when Allfrey et al. observed incorporation of radiolabeled acetate into calf thymus histone fractions in the mid 1960’s (Allfrey et al., 1964). Since then, lysine acetylation has become a hallmark of actively transcribed chromatin, where the acetylation of histones in gene promoters is associated with gene expression (Eberharter and Becker, 2002; Grunstein, 1997; Mizzen and Allis, 1998; Struhl, 1998). In the nucleus, lysine acetylation contributes in two major ways to transcriptional regulation. Overall, acetylation neutralizes the positive charge on lysine residues, breaking nucleosome-nucleosome contacts and opening up chromatin domains to transcriptional machinery (Bannister and Kouzarides, 2011; Shogren-Knaak et al., 2006). In addition, specific lysine acetylation marks are recognized by chromatin “reader” domains that recruit downstream transcriptional effectors (Musselman et al., 2012; Su and Denu, 2016; Yun et al., 2011). The development of increasingly sensitive mass spectrometry methods coupled with new separation and enrichment techniques has expanded the search for acetylated proteins beyond the nucleus, increasing the number of known acetylated proteins to ~1170 in mouse and ~3200 in human, spanning all major cellular compartments (Dittenhafer-Reed et al., 2015; Svinkina et al., 2015).
The first proteome-wide study of lysine acetylation was conducted in 2006, and proteome-scale surveys of other acyl modifications have closely followed their discovery in vivo (Kim et al., 2006). Within the next decade, reports of lysine propionylation (Chen et al., 2007; Cheng et al., 2009), butyrylation (Chen et al., 2007), crotonylation (Tan et al., 2011; Wei et al., 2017), succinylation (Weinert et al., 2013; Xie et al., 2012; Zhang et al., 2010), malonylation (Colak et al., 2015; Peng et al., 2011; Xie et al., 2012), glutarylation (Tan et al., 2014), 2-hydroxyisobutyrylation (Dai et al., 2014; Huang et al., 2018), and β-hydroxybutyrylation (Xie et al., 2016) significantly expanded the inventory of known acyl modifications (Ex. Peng et al. MCP 2011, Zhang et al. NCB 2011). These studies have uncovered the surprisingly broad range of lysine PTMs that comprise the lysine acylome, which differ with respect to steric properties (branching, acyl chain length) and charge (Figure 1). The discovery that a constellation of lysine modifications exists in cells set the stage for exploring lysine acylation as a regulatory modification and revealed that acetylation is just one example of a much larger class of lysine PTMs.
Figure 1.
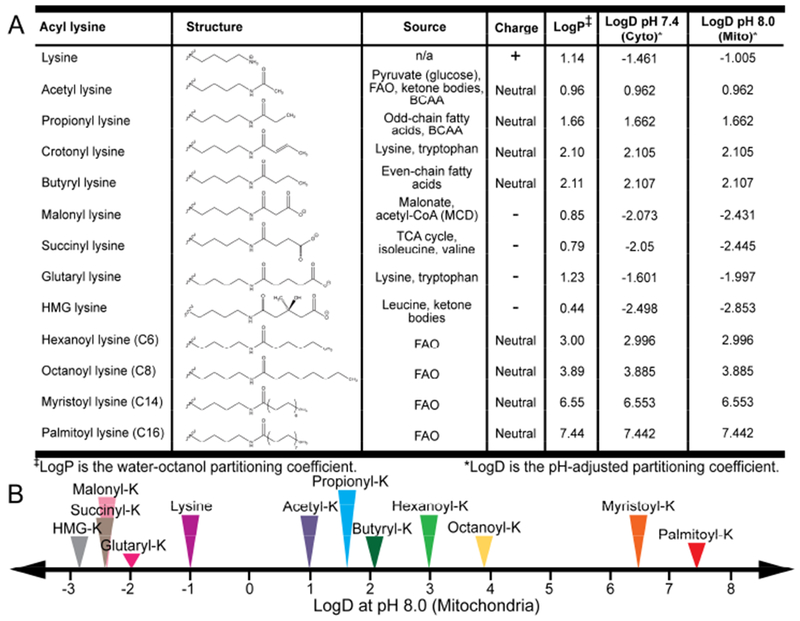
Lysine acyl modifications possess diverse chemical properties. (A) Table summarizing the chemical properties (structure, charge, hydrophobicility) and metabolic sources of lysine acyl PTMs. (B) The hydrophobicity of lysine acyl PTMs based on pH-adjusted water octanol partitioning coefficient varies widely. Abbreviations: FAO – fatty acid oxidation. BCAA – branched chain amino acid. MCD – malonyl-CoA decarboxylase.
Chemical diversity of lysine acyl modifications
The biological role of lysine acylation is impacted by the fundamental chemistry of the modification (Figure 1). Acylation changes the size, charge, and hydrophobicity of the lysine side chain, but the net effect of the modification depends on the chemical properties of the particular acyl modification. At physiological pH, protonation of the lysine epsilon amino group imparts a positive charge to the lysine side chain. Neutral acyl modifications contain saturated or unsaturated hydrocarbon chains connected through an amide bond linkage between the epsilon amino group of lysine and the acyl chain, neutralizing the charge on the epsilon amino group. Acetylation is a small acyl PTM that adds two carbon units to lysine. Longer acyl PTMs vary in length from three and four carbon units (propionyl and butyryl/crotonyl PTMs, respectively) to sixteen carbon units (palmitoylation), and thus are larger and more hydrophobic than acetyl modifications. The distinct physical properties of acylation can be quantified by calculated LogD values (pH-adjusted water-octanol partitioning coefficient) that increase proportionally with hydrocarbon chain length (Figure 1). Lysine malonylation, succinylation, and glutarylation belong to a subgroup of negatively charged modifications that contain a terminal carboxylate. So far, malonylation is the smallest of the negatively charged acyl PTMs to be identified, containing a three-carbon backbone. Succinylation and glutarylation are bulkier modifications, with four- and five-carbon backbones, respectively. Both negatively charged and branched modifications include 3-hydroxy-3-methylglutaryl (HMG) lysine, 3-methyl-glutaconyl (MGc) lysine, and 3-methylglutaryl (MG) lysine. As lysine acylation is a broad, chemically diverse family of PTMs, understanding the chemical properties of these modifications may reveal new insights into their biological consequences.
Acyl-CoA levels connect fuel availability with lysine acylation in the mitochondria
Since CoA metabolic intermediates provide activated acyl chains for lysine PTMs, the varieties of acyl-CoA metabolites present in the mitochondria dictates the chemical diversity of acyl lysine. Acyl-CoA pools fluctuate with nutrient levels and are compartmentalized in ways we still do not fully understand. Different arms of mitochondrial metabolism generate specific classes of acyl-CoA molecules, and cells depend on these metabolic pathways to varying extents in response to fuel availability (Figure 2A). For example, mitochondrial acetyl-CoA is derived from pyruvate, fatty acid oxidation (FAO), as well as ketone body catabolism in extra-hepatic tissues (Pietrocola et al., 2015). Fasting triggers a switch from glycolysis to FAO, which raises mitochondrial acetyl-CoA levels and, consequently, increases overall mitochondrial protein acetylation (Pougovkina et al., 2014). Thus, direct links exist among fuel switching from glucose oxidation to FAO, acetyl-CoA levels, and mitochondrial acetylation.
Figure 2.
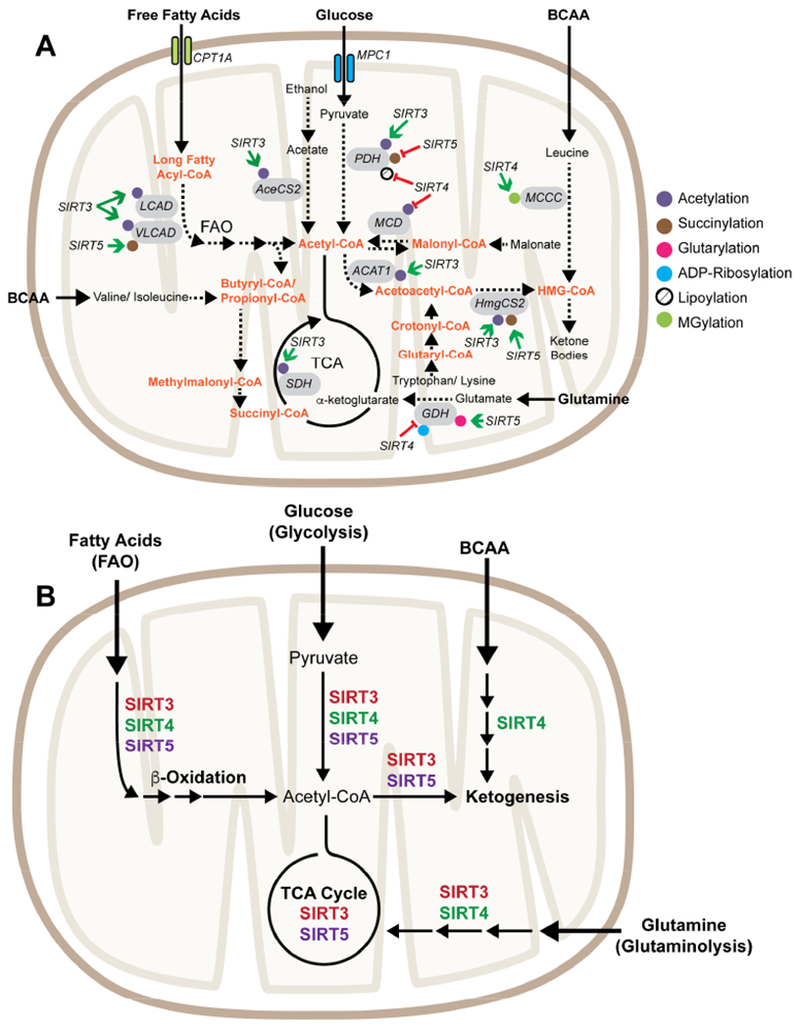
Overview of mitochondrial roles for sirtuins in metabolism. (A) Sirtuins remove lysine PTMs from enzymes that produce, consume, or interconvert mitochondrial acyl-CoA pools. Green arrows denote deacylation activities that enhance the catalytic activity of the substrate. Red bars highlight deacylation activities that inhibit substrate activity. Abbreviations: ACAT1 – Acetyl-CoA Acetyltransferase 1, AceCS2 – Acetyl-CoA Synthetase 2, BCAA – Branched chain amino acids, CPT1A – Carnitine Palmitoyltransferase 1A, GDH – Glutamate Dehydrogenase, HmgCS2 – 3-Hydroxy-3-Methylglutaryl-CoA Synthase 2, LCAD – Long Chain Acyl-CoA Dehydrogenase, MCCC – Methylcrotonyl-CoA Carboxylase, MCD – Malonyl-CoA decarboxylase, MPC1 – Mitochondrial Pyruvate Carrier 1, PDH – Pyruvate Dehydrogenase, SDH – Succinate Dehydrogenase, VLCAD – Very Long Chain Acyl-CoA Dehydrogenase. (B) Overview of metabolic pathways regulation by mitochondrial sirtuins.
We are just beginning to realize the impact of metabolism on generating the cellular repertoire of acyl-CoA pools, and this is an exciting area of study. Oxidation of odd-chain fatty acids generates propionyl-CoA (in addition to acetyl-CoA); however, it is unknown how levels of mitochondrial propionyl-CoA fluctuate with fuel switching. Branched chain amino acid catabolism is another source of acyl-CoA metabolites in the mitochondria. Leucine is a ketogenic amino acid that is metabolized into HMG-CoA via two CoA intermediates (3-methylcrotonyl-CoA and 3-methylglutaconyl-CoA), and in extra-hepatic tissues further converted to acetyl-CoA (Figure 2A). Additionally, HMG-CoA is produced via ketogenesis in the liver, which is reversed by ketolysis in non-hepatic tissue to regenerate acetyl-CoA. Isoleucine and valine are metabolized first to propionyl-CoA and then to succinyl-CoA, which enters into the TCA cycle or is used for porphyrin synthesis. Lysine and tryptophan catabolism feed glutaryl-CoA pools, which are converted to crotonyl-CoA and then to acetyl-CoA. Other pathways that produce acyl-CoA in the mitochondria include ATP-dependent malonate detoxification via the mitochondrial enzyme ACSF3 to form malonyl-CoA (Bowman et al., 2017; Witkowski et al., 2011) and acetate condensation by AceCS2 (Fujino et al., 2001). In sum, acyl-CoA molecules are produced by many different arms of mitochondrial metabolism, and the relative composition of these acyl-CoA pools is a read-out of the metabolic state of the cell.
It is interesting to note that specific types of acyl modifications tend to be enriched within pathways that produce the corresponding acyl-CoA, which may experience elevated local concentrations of the reactive metabolite. For example, succinylation is enriched on enzymes invoved in succinyl-CoA producing pathways, such as the Val-Leu-Ile degradation pathway, TCA cycle, and propionate metabolic pathway (Park et al., 2013). A more recent study dissected the contribution of two enzymes involved in HMG-CoA versus glutaryl-CoA production to the enrichment of either lysine HMGylation or glutarylation on proteins involved in the respective pathways (Wagner et al., 2017). In hydroxymethylglutaryl-CoA lyase (HMGCL) knockout (KO) mice, which accumulate HMG-CoA, all the enzymes involved in the ketogenic pathway are enriched with HMG-lysine. By contrast, mice lacking glutaryl-CoA dehydrogenase (GCDH) accumulate glutaryl-CoA, and the corresponding acyl modification is enriched on proteins involved in lysine catabolism, which is the pathway that feeds glutaryl-CoA pools. Together, these observations hint that local pools of acyl-CoA molecules are a major source of lysine acyl PTMs.
Mitochondria provide a unique metabolic niche for acylation
The subcellular environment of the mitochondrial matrix provides a unique hotspot for lysine acylation. Thus far, specific enzymes to catalyze mitochondrial acyl transfer have not been identified, and current evidence suggests that mitochondrial acylation is nonenzymatic and depends on the abundance of acyl-CoA metabolites (Ghanta et al., 2013; Wagner and Hirschey, 2014). Conditions in the mitochondrial matrix favor nonenzymatic modification of lysine residues by reactive acyl-CoA molecules for several reasons. First, many acyl-CoA metabolites are present at high concentrations in the matrix; mitochondrial acetyl-CoA achieves millimolar concentrations and steady-state succinyl-CoA concentrations range between 0.1 to 0.6 mM (Garland et al., 1965; Hansford and Johnson, 1975). Second, proton pumping by complexes I, III, and IV of the electron transport chain (ETC) makes the matrix pH basic by moving H+ ions from the matrix to the intermembrane space. Since the pKa of free lysine is ~9, increasing pH promotes deprotonation of the lysine epsilon amino group, which increases the nucleophilicity and overall reactivity of lysine to acyl-CoA thioesters. Reproducing these conditions in vitro by incubating BSA, lysine-containing peptides, or mitochondrial extracts with acetyl-CoA at basic pH initiates robust dose- and time-dependent nonenzymatic acetylation (James et al., 2017; Simic et al., 2015; Wagner and Payne, 2013; Wagner et al., 2017). Finally, chemical properties that increase the intrinsic reactivity of acyl-CoA molecules also increase rates of nonenzymatic acylation. For example, classes of acyl-CoA molecules that can form reactive intermediates via intramolecular catalysis, such as succinyl-CoA, glutaryl-CoA, and HMG-CoA, readily modify proteins in vitro via nonenzymatic mechanisms, and do so at rates up to 150-fold faster than acetyl-CoA (Simic et al., 2015; Wagner et al., 2017). A recent study demonstrated that lysine acetylation can be potentiated by nearby S-acetylation of a cysteine residue, followed by intramolecular and irreversible SN-transfer to lysine (James et al., 2017). Thus, fundamental biochemical properties of the matrix produced by normal mitochondrial physiology support nonenzymatic lysine acylation.
In vivo studies mirror the concentration dependence of lysine PTMs on acyl-CoA pools, providing evidence that nonenzymatic mitochondrial acylation is physiologically relevant. For example, dietary interventions that increase FAO in the liver, such as calorie restriction (CR) and fasting, dial up both acetyl-CoA levels and mitochondrial lysine acetylation (Pougovkina et al., 2014; Schwer et al., 2009; Still et al., 2013). Mice lacking the key FAO enzymes adipose triglyceride lipase (ATGL) or long chain acyl-CoA dehydrogenase (LCAD) do not increase levels of mitochondrial acetylation upon fasting (Pougovkina et al., 2014; Weinert et al., 2015), suggesting that acetyl-CoA derived from FAO is the carbon source for lysine acetyl modifications. In addition, stress conditions, such as long-term high fat diet (HFD) and chronic alcohol consumption, also increase mitochondrial protein acetylation (Fritz et al., 2012; Hirschey et al., 2011; Kendrick et al., 2011). It is important to note that some metabolic enzymes that can bind acyl-CoAs for other purposes may moonlight as acyltransferases. A recent study revealed CPT1A, an enzyme better known as a carnitine acyltransferase, can also succinylate proteins (Kurmi et al., 2018). In addition, a-ketoglutarate dehydrogenase complex (KGDHC) activity has been reported to mediate lysine succinylation in yeast and mouse neurons, although whether this effect is direct or indirect via succinyl-CoA concentrations remains to be determined (Gibson et al., 2015; Weinert et al., 2013). Future studies may uncover additional categories of mitochondrial enzymes that are capable of modifying lysine residues with particular kinds of acyl chains, but do not belong to traditional classes of lysine acyltransferases.
Emerging roles for sirtuins removing chemically diverse acyl modifications
Yeast Sir2 is the founding member of the widely conserved sirtuin family of enzymes. Mechanistic insight into sirtuin biochemistry has continually evolved since the initial discovery of yeast sir2 from genetic screens for mating viability in the late 1970s (Haber and George, 1979; Klar et al., 1979; Rine and Herskowitz, 1987; Rine et al., 1979). The first clue that sirtuins might possess enzymatic activity came in 1998 with the realization that sirtuins share sequence homology with Salmonella typhimurium CobB, an enzyme that binds nicotinic acid mononucleotide (NaMN) (Trzebiatowski and Escalante-Semerena, 1997; Tsang and Escalante-Semerena, 1998), thus raising the possibility that sirtuins might bind metabolites related to NaMN such as NAD+. Two subsequent studies revealed weak ADP-ribosyltransferase activity (Frye, 1999; Tanny et al., 1999), followed soon after by the seminal discovery that acetylated histone tails could be deacetylated by sirtuins, and that this reaction was coupled to the consumption of NAD+ (Imai et al., 2000; Smith et al., 2000). These studies built upon one another to show that sirtuins hydrolyze NAD+ to catalyze transfer of the acetyl group from acetyl lysine to ADP-ribose, forming 2’/3’-O-acetyl-ADP-ribose, free nicotinamide (NAM), and free lysine (Landry et al., 2000; Tanner et al., 2000; Tanny and Moazed, 2001). The discovery of widespread lysine acylation on histone and non-histone proteins alike prompted a comprehensive reevaluation of sirtuin acyl chain specificity, which revealed that mammalian sirtuins remove a diverse array of acyl modifications from lysine (reviewed in (Bheda et al., 2016)). These studies have reclassified sirtuins as a family of lysine deacylases, each with unique acyl chain specificity, which collectively target a chemically diverse array of acyl PTMs for removal.
Mitochondrial sirtuins
In mammals, there are three sirtuin enzymes within the mitochondrial matrix, SIRT3-5, which catalyze the removal of lysine acyl modifications using NAD+ as an essential cofactor. The three mitochondrial sirtuins remove distinct types of acyl PTMs. SIRT3 is the best studied mitochondrial sirtuin and early studies of this enzyme demonstrated robust deacetylase activity that modulates mitochondrial acetylation levels (Lombard et al., 2007; Onyango et al., 2002). SIRT3 deletion causes an increase in lysine acetylation in mouse liver, and acetylation changes associated with SIRT3 loss are larger than those associated with dietary interventions like caloric restriction (CR), indicating that SIRT3 exerts a dominant effect suppressing mitochondrial acetylation (Dittenhafer-Reed et al., 2015; Hebert et al., 2013; Rardin et al., 2013a; Weinert et al., 2015). Proteomic surveys of mitochondrial acetylation revealed that SIRT3 deacetylates a subset of mitochondrial proteins involved in mitochondrial metabolism and stress response (Dittenhafer-Reed et al., 2015; Hebert et al., 2013; Rardin et al., 2013a). Furthermore, the SIRT3 interactome demonstrates that SIRT3 binding partners are enriched for acetylation sites (Yang et al., 2016). In total, biochemical studies reveal that SIRT3 influences the acetylation status of a coordinated set of metabolic pathways, and that SIRT3 deacetylates many targets simultaneously in the mitochondrial matrix to reestablish homeostasis in response to stress conditions.
By contrast, SIRT4 and SIRT5 possess weak to undetectable deacetylase activity in vitro. In 2011, the first evidence that mammalian sirtuins might possess alternate deacylating acitivity came to light. Structural and kinetic studies from Lin and coworkers revealed that SIRT5 preferentially removed succinylation and malonylation from lysine, with 29- to 1000-fold greater catalytic efficiency than that observed for acetylated substrates (Du et al., 2011). The structure of SIRT5 in complex with a succinylated peptide revealed that two residues, Tyr102 and Arg105, confer specificity for negatively charged acyl chains by interacting with the terminal succinyl carboxylate, and mutation of these residues significantly increased the SIRT5 KM for succinyl peptide (Du et al., 2011). Consistent with its specificity for negatively charged acyl modifications, SIRT5 also deglutarylates lysine in vivo and in vitro (Tan et al., 2014). Mitochondrial lysine succinylation, malonylation, and glutarylation increase in SIRT5 KO tissue and cells, mirroring the acyl chain specificity of SIRT5 (Nishida et al., 2015; Park et al., 2013; Rardin et al., 2013b; Tan et al., 2014).
The lack of a robust in vitro enzymatic activity for SIRT4 has made defining in vivo substrates challenging. Individual substrates targeted by SIRT4 have been identified corresponding to a variety of SIRT4 enzymatic activities, such as ADP-ribosylation of glutamate dehydrogenase (GDH) (Haigis et al., 2006), deacetylation of malonyl-CoA decarboxylase (MCD) (Laurent et al., 2013a), demethylglutarylation of methylcrotonyl-CoA carboxylase (MCCA) (Anderson et al., 2017), and delipoylation of pyruvate dehydrogenase (PDH) (Mathias et al., 2014). So far, all SIRT4 substrates validated in vivo produce, consume, or interconvert acyl-CoA pools, supporting a central role in metabolic regulation (Figure 2A). A recent study determined the structure of SIRT4, revealing that SIRT4 can remove hydroxymethylglutaryl chains from lysine (Pannek et al., 2017). Furthermore, the structure of SIRT4 revealed a hydrophobic channel near the active site. The observation that free lipoic acid inhibits SIRT4 deacetylation and de-HMGylation activity in vitro suggested that SIRT4 activity may be sensitive to endogenous metabolites. Interestingly, the nuclear sirtuin, SIRT6, also contains a hydrophobic channel, and its deacetylase activity is potently stimulated by free fatty acids in vitro (Feldman et al., 2013). It is possible that better acyl substrates for SIRT4 exist, particularly in the presence of activating metabolites.
Sirtuins act as key sensors that integrate multiple external cues to regulate mitochondrial pathways (Figure 2B). Studies show that SIRT3 coordinates the physiological response to stress and nutrient deprivation. For example, SIRT3 coordinates the reduction in oxidative stress and damage associated with CR by deacetylating and activating the mitochondrial isoform of superoxide dismutase, SOD2 (Qiu et al., 2010). Likewise, SIRT3 protects hematopoietic stem cells (HSCs) from oxidative damage associated with stress or age (Brown et al., 2013). Increased reactive oxygen species (ROS) due to SIRT3 loss are pro-tumorigenic by stabilizing hypoxia-inducible factor-1α (HIF1α) (Finley et al., 2011b). SIRT3 localization within the mitochondria provides another level of regulation over SIRT3 activity. In polarized mitochondria, SIRT3 forms a complex with ATP synthase at the inner mitochondrial membrane, sequestering SIRT3 away from potential matrix substrates. Uncoupling disrupts this interaction via a pH-dependent mechanism. In this way, changes in membrane potential alter SIRT3 localization and facilitate the deacetylation of SIRT3 substrates (Yang et al., 2016). With respect to nutrient deprivation, CR and fasting are associated with both an increase in overall mitochondrial acetylation as well as upregulation of SIRT3 expression (Hallows et al., 2011; Hirschey et al., 2010; Schwer et al., 2009). As a result, while overall levels of mitochondrial acetylation rise with fasting, acetylation at SIRT3-targeted sites decreases or does not change (Weinert et al., 2015). SIRT3 KO mice gain more weight and are more insulin resistant on a HFD (Hirschey et al., 2011), which is in line with biochemical identification of TCA cycle, oxidative phosphorylation, and FAO proteins as SIRT3 targets (Dittenhafer-Reed et al., 2015; Hirschey et al., 2010; Sol et al., 2012). SIRT3 regulates basal energy homeostasis as well, since in the absence of SIRT3, levels of resting ATP are reduced in key metabolic organs such as the heart, liver, and kidney (Ahn et al., 2008). Thus, SIRT3 coordinates the metabolic response to fasting and stress by targeting multiple arms of metabolism.
In contrast to SIRT3, the activity of SIRT4 appears to be most physiologically important during times of nutrient abundance. Fasting reduces SIRT4 expression (Laurent et al., 2013b), and SIRT4 KO mice gain less weight on a HFD (Laurent et al., 2013a). Energy-sensing signaling pathways are also involved in regulating SIRT4 levels, as mTORC1 represses the transcription of SIRT4 (Csibi et al., 2013). The future identification of additional SIRT4 substrates and validation of robust SIRT4 deacylase activities will foster a deeper understanding of SIRT4 function in the mitochondria.
The physiological role of SIRT5 is not well understood either, although the discovery of SIRT5 desuccinylase activity has provided fresh insight and raised new questions regarding its biological roles. Fasting does not affect hepatic expression of SIRT5 (Nakagawa et al., 2009), even though succinylation levels increase under the same conditions (Park et al., 2013). SIRT5 redirects carbon metabolism in the mitochondria by desuccinylating and inhibiting PDH, although the relationship with nutrient deprivation has yet to be determined (Park et al., 2013). However, mitochondrial nitrogen metabolism has been connected to SIRT5 funtion on both molecular and organismal levels. SIRT5 activates GDH and CPS1 by deglutarylation and desuccinylation, respectively (Park et al., 2013; Wang et al., 2018; Weinert et al., 2013). GDH activation by SIRT5 increases the flux of glutamine carbon into the TCA cycle, which liberates free nitrogen that can be recaptured by CPS1 for detoxification. Mice lacking SIRT5 exhibit hyperammonemia with fasting, consistent with a role for SIRT5 regulating nitrogen metabolism (Nakagawa et al., 2009).
In cases where NAD+ concentrations are limiting for sirtuin activity, NAD+ levels may provide an additional layer of regulation. Mitochondrial NAD+ rises more than two-fold in the liver after a 48 hour fast (Nakagawa et al., 2009; Yang et al., 2007) spanning a concentration range that can stimulate sirtuin activity based on reported KM values for SIRT3-5 (Fischer et al., 2012; Hirschey et al., 2011; Laurent et al., 2013a). Mitochondrial NAD+ availability would be predicted to change much more quickly than transcription, providing a rapid regulatory switch for sirtuin activity. Indeed, mitochondrial lysine deacetylation has been observed only two hours after fasting/refeeding and well before any changes in SIRT3 protein expression occur (Still et al., 2013). Thus, sirtuins integrate information from multiple environmental cues (co-substrate availability and acylation burden) to act as sensors that can rapidly alter mitochondrial biology in response to fuel stress.
Sirtuins may participate in feedback mechanisms by regulating enzymes that control acyl-CoA levels. A growing number of studies have implicated deacylation of specific metabolic enzymes by SIRT3-5 in the regulation of mitochondrial pathways that control acyl-CoA pools. Deacetylation by SIRT3 regulates the activity of enzymes that change acetyl-CoA levels such as LCAD (Bharathi et al., 2013; Hirschey et al., 2010), VLCAD (Zhang et al., 2015), ACAT1 (Dittenhafer-Reed et al., 2015; Still et al., 2013), AceCS2 (Hallows et al., 2006; Schwer et al., 2006), and PDHA (Jing et al., 2013), as well as HMGCS2 (Shimazu et al., 2010) that makes HMG-CoA (Figure 2A). SIRT4 de-methylglutarylates and activates a subunit of the MCCC complex, which participates in the ketogenic catabolism of leucine to acetyl-CoA and directly consumes β-methylcrotonyl-CoA to produce β-methylglutaconyl-CoA (Anderson et al., 2017). SIRT4 also deacetylates and represses the activity of MCD, which converts malonyl-CoA to acetyl-CoA (Laurent et al., 2013a). Desuccinylation by SIRT5 regulates the activity of two enzymes involved in FAO, VLCAD and ECHA, which contribute to acetyl-CoA pools when cells burn fatty acids for energy (Boylston et al., 2015; Zhang et al., 2015). SIRT5 also regulates the activity of HMGCS2 by desuccinylating residues two lysine residues, one of which overlaps with the SIRT3-targeted sites (Rardin et al., 2013b). Furthermore, SIRT5 desuccinylates multiple subunits of the PDH complex to inhibit PDH activity, which reduces the number of carbon units from glycolysis that enter the TCA cycle (Park et al., 2013). SIRT5 also deglutarylates GDH, which increases flux through glutaminolysis (Wang et al., 2018). Since mitochondrial enzymes that regulate acyl-CoA pool sizes are also substrates for SIRT3-5, sirtuins are poised to balance nutrient abundance cues with the activation or inhibition of other metabolic pathways.
Given the unique metabolic demands and fuel preferences associated with specific tissues, it remains to be seen whether each mitochondrial sirtuin targets the same repertoire of acylated substrates and removes the same acyl PTMs among different tissues. The impact of SIRT3 loss on the acetyl proteome has been compared across five tissue types (brain, heart, kidney, liver, and skeletal muscle) to identify common regulated substrates as well as tissue-specific activities for SIRT3 (Dittenhafer-Reed et al., 2015). This study revealed that tissue-specific pathways regulated by SIRT3 tend to reflect the core metabolic processes that comprise distinguishing features of the tissue (i.e. ketone body utilization in the brain versus ketolysis in the liver) (Dittenhafer-Reed et al., 2015). Thus, tissue context changes SIRT3 activity and specificity. Whether tissue-specific functions are a common feature among all three mitochondrial sirtuins remains to be seen. Another area that is lacking in mechanistic understanding are the metabolic and/or cellular features that may regulate tissue-specific sirtuin activities. For example, acyl-CoA pool size differs among tissue types. Studies comparing the metabolite composition of mouse tissues have revealed that acyl-CoA levels not only differ among tissue types, but that these differences are mirrored in the mitochondrial lysine acylome (Sadhukhan et al., 2016). Pools of succinyl-CoA as well as levels of protein succinylation are much larger in the heart than in the liver, kidney, brain, or muscle(Sadhukhan et al., 2016). Accordingly, the increase in succinylation associated with SIRT5 loss is larger in cardiac tissue than in other tissue types (Sadhukhan et al., 2016). Even though SIRT5 can remove multiple kinds of negatively charged acyl chains from lysine, SIRT5 predominately acts as a desuccinylase in the heart, as malonylation and glutarylation levels are not altered to the same extent with SIRT5 loss (Hershberger et al., 2017; Sadhukhan et al., 2016). These data raise the exciting possibility that tissue physiology may tune sirtuin substrate preference as well as acyl chain specificity. Since all three mitochondrial sirtuins possess multiple deacylating activities (Table 1), future studies unraveling unique versus overlapping acyl chain and lysine site specificities for mitochondrial sirtuins will significantly advance this field.
Table 1:
Summary of the acyl PTMs targeted by mitochondrial sirtuins
| Sirtuin: | Acyl PTM targeted: | References: |
|---|---|---|
| SIRT3 | 2-Hydroxyisobutyryl Acetyl Crotonyl |
(Bao et al., 2014; Bharathi et al., 2013; Dai et al.,2014; Dittenhafer-Reed et al., 2015; Hallows et al.,2006; Hirschey et al., 2010; Jing et al., 2013; Schwer et al., 2006; Shimazu et al., 2010; Still et al., 2013; Zhang et al., 2015) |
| SIRT4 | Acetyl Hydroxymethylglutaryl Methylglutaryl |
(Anderson et al., 2017; Laurent et al., 2013a; Pannek et al., 2017) |
| SIRT5 | Acetyl Glutaryl Malonyl Succinyl |
(Nakagawa et al., 2009; Nishida et al., 2015; Park et al., 2013; Rardin et al., 2013b; Tan et al., 2014; Wang et al., 2018) |
Functional consequences of lysine acylation and deacylation by sirtuins in the mitochondria
It is interesting to speculate how acylation might change the activity of proteins at the molecular level, as well as mechanisms connecting these changes to biologically impactful outcomes. PTMs may alter biology downstream via numerous mechanisms, including by: altering protein multimerization, activity, or subcellular localization (Figure 3). For example, the chemical properties, charge, and hydrophobicity may initiate or inhibit protein complex assembly and stability. Bulky PTMs that occur near a protein active site or on a regulatory loop may directly alter enzyme activity in cis by altering substrate binding or changing the chemical properties of the active site. Alternatively, PTMs may drive differential subcellular localization, depending on the hydrophobicity of the modification. These mechanisms may have dramatic effects on protein function and significant regulatory consequences for mitochondrial biology.
Figure 3.

Mechanisms connecting the physical properties of lysine acyl PTMs to changes in macromolecular function.
The stoichiometry of mitochondrial acetylation tends to be lower overall than for PTMs that are present in other cellular compartments (Weinert et al., 2015). It is likely that many other acyl modifications are also present at low stoichiometries, although rigorous measurements still have to be performed. According to one study, fewer than 50 SIRT3-targeted acetylation sites attain stoichiometries >1% in SIRT3 KO tissue (Weinert et al., 2015). By contrast, PTMs that are catalyzed achieve much higher stoichiometries. For example, phosphorylation in mammalian cells can attain stoichiometries >30% (Tsai et al., 2015), and the acetylation stoichiometry at histone H3K14 and H4K16 in HeLa cells is 27.8% and 35.7%, respectively (Zheng et al., 2013). It is interesting to ponder how such low stoichiometry events may regulate substrate function and impact biological pathways. This is an area of sirtuin research that is lagging in mechanistic understanding. Whether PTM changes cause dominant or cumulative effects on biological functions is an important aspect of pathway regulation and may involve several mechanisms, including: (i) multivalent acylation collectively dials the activity of a protein up or down, (ii) small effects on multiple enzymes in the same pathway exert an aggregate effect on total flux through a single pathway, and (iii) modification sites exist in multi-subunit complexes that affect either cooperative complex assembly or allosterically change the activity of other subunits (Baeza et al., 2016; Yang et al., 2016). For example, modifications that promote the assembly of fully functional multi-protein complexes could exert dominant regulatory roles. In addition, removing a low stoichiometry modification that enhances activity can switch the pathway from on to off. While the majority of modifications that mitochondrial sirtuins remove appear to be inhibitory, sirtuins do remove a handful of activating modifications. For example, SIRT3 deacetylates and inactivates aconitase to repress flux through the TCA cycle (Fernandes et al., 2015). For a subset of sirtuin modifications, these mechanisms can explain the impact of low stoichiometry events on cellular physiology.
It has also been suggested that mitochondrial sirtuins may clean up “carbon stress”, when nonenzymatic modification of lysine residues produces spurious acylation that inhibits host protein function (Wagner and Hirschey, 2014). Thus, sirtuin remodeling of the mitochondrial acylome may impact a hierarchy of biological functions, ranging from site-specific deacylation events that promote or inhibit the activity of individual enzymes to guarding the mitochondrial proteome against carbon lesions that impair mitochondrial homeostasis.
The intersection between mitochondrial NAD+ metabolism, NAD+ availability and sirtuin activity
Sirtuins are the only class of enzymes catalyze the removal of lysine acyl PTMs in the mitochondria. Thus, the combined activities of SIRT3, SIRT4, and SIRT5 are the only known cellular factors that are capable of remodeling the mitochondrial lysine acylome. The sirtuin reaction mechanism explicitly depends on the availability of NAD+. In contrast to other NAD+-dependent enzymes, which are predominantly involved in redox transactions, sirtuins couple NAD+ consumption to lysine deacylation by cleaving off nicotinamide and covalently linking the acyl chain to ADP-ribose (ADPr). As a result, mitochondrial NAD+ levels simultaneously regulate and are regulated by sirtuin activity. Thus, the availability of mitochondrial NAD+ and the status of the mitochondrial acylome are inextricably linked through sirtuins.
Sirtuins require NAD+ for catalysis, which connects the regulation of the mitochondrial acylome to all other cellular pathways that use NAD+ as an essential cofactor. NAD+ is utilized by diverse metabolic pathways ranging from bioenergetics to transcriptional regulation (Canto et al., 2012; Cerutti et al., 2014; Mouchiroud et al., 2013). Although NAD+ is traditionally appreciated as a redox cofactor that acts as a hydride acceptor, NAD+ can also be funneled into other kinds of cellular pathways. For example, approximately 10% of cellular NAD+ is utilized as a precursor for NADP+/NADPH, which is used for reductive biosynthetic pathways and ROS detoxification (Ying, 2008). In addition to sirtuins, NAD+ can also be consumed by several classes of enzymes that include poly ADP-ribose polymerases (PARPs) and cyclic ADP-ribose synthases CD38/CD157 (Aksoy et al., 2006; Chambon et al., 1963; De Flora et al., 2004; Haigis and Sinclair, 2010). Collectively, the balance between NAD+ synthesis, salvage, redox state, and consumption regulates compartmentalized NAD+ levels.
Regulation and maintenance of mitochondrial NAD+
In mitochondria the maintenance of NAD+ levels as well as the NAD+/NADH ratio is crucial for homeostatic mitochondrial function and ATP production. The mitochondrial NAD+ pool is maintained separately from the nuclear and cytoplasmic NAD+ pools, and the matrix houses distinct biosynthetic and regulatory pathways (Pittelli et al., 2010; Yang et al., 2007). Cytoplasmic and nuclear NAD+/NADH ratios are typically maintained between 60 and 700 in eukaryotes depending on cell type, while the mitochondrial ratio is much lower, around 7-8 (Veech et al., 1972; Williamson et al., 1967; Zhang et al., 2002). Additionally, mitochondria contain a large proportion of cellular NAD+, with estimates ranging from 40-70% of the total cellular NAD+ pool (Alano et al., 2007; Di Lisa et al., 2001; Tischler et al., 1977). Both the low mitochondrial NAD+/NADH ratio and high concentration of NAD+ relative to other cellular compartments are requirements for optimal oxidative phosphorylation and ATP production. Due to the critical energy-producing and redox roles that mitochondria perform, cells guard the pool of mitochondrial NAD+ from stressors that disrupt total cellular NAD+ levels. For example, although PARP activation downstream of genotoxic stress depletes cytosolic and nuclear NAD+, mitochondrial pools of NAD+ are preserved to support the pro-survival functions of SIRT3 and SIRT4 (Yang et al., 2007).
Cellular NAD+ pools are fed by two major biosynthetic pathways: de novo synthesis and regeneration (Figure 4). De novo synthesis from tryptophan occurs in the cytosol and contributes to a small fraction of the overall NAD+ pool (Krehl et al., 1945; Liu et al., 2018; Powanda and Wannemacher, 1970; Schutz and Feigelson, 1972). Three pathways regenerate NAD+ from different precursor molecules: NAM via the salvage pathway, nicotinic acid (NA) or niacin through the Preiss-Handler pathway, or synthesized from nicotinamide riboside (NR) (Bieganowski and Brenner, 2004; Magni et al., 1999; Priess and Handler, 1958b, 1958a). Sirtuins generate NAM as a reaction byproduct, which can be used by the salvage pathway to regenerate NAD+ through conversion to nicotinamide mononucleotide (NMN) by nicotinamide phosphoribosyltransferase (NAMPT) and subsequent conversion of NMN to NAD+ by NMN adenylyltransferases 1-3 (NMNAT1-3) (Berger et al., 2005). Unlike NAD+, in mammalian cells NMN can cross the inner mitochondrial membrane and has been shown to undergo conversion to NAD+ in isolated mitochondria (Barile et al., 1996; Nikiforov et al., 2011). While yeast transport NAD+ across the inner mitochondrial membrane via two nucleoside deoxyribosyltransferases, Ndt1 and Ndt2, a mammalian mitochondrial NAD+ transporter has yet to be identified (Todisco et al., 2006). Therefore, the current model for mitochondrial NAD+ biosynthesis is that NAD+ precursors and intermediates in the cytoplasm undergo conversion to NMN, which is transported into mitochondria for NAD+ biosynthesis by mitochondria-specific NMNAT3. However, since exogenous NAD+ increases mitochondrial NAD+ levels more than cytoplasmic levels, it is likely that undiscovered mitochondrial transport mechanisms exist for NAD+ precursors, intermediates, or even NAD+ itself (Billington et al., 2008; Pittelli et al., 2010, 2011; Yang et al., 2007). Indeed, stable isotope tracing studies have revealed that NAD+ can be directly imported into mammalian mitochondria, and that the majority of mitochondrial NAD+ originates from the cytoplasmic pool rather than from imported NMN (Davila et al., 2018). To better understand how mammalian cells maintain mitochondrial NAD+ levels, further studies are needed to identify the specific transporters that move NAD+, NMN and other NAD+ precursors into the mitochondria.
Figure 4.
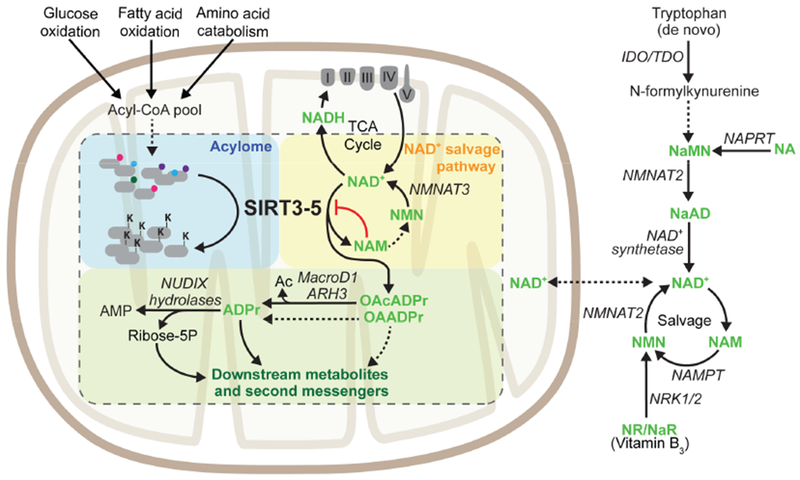
The activity of mitochondrial sirtuins connects fluctuations in NAD+ metabolism to changes in protein acylation. Abbreviations: Ac – Acetate, ADPr – ADPribose, AMP – adenosine monophosphate, NAD+ – Nicotinamide Adenine Dinucleotide, NADH – Nicotinamide Adenine Dinucleotide Hydride, NA – Nicotinic Acid, NaAD – Nicotinic Acid Adenine Dinucleotide, NAM – Nicotinamide, NaMN – Nicotinic Acid Mononucleotide, NMN – Nicotinamide Mononucleotide, NR – Nicotinamide Riboside, OAcADPr – O-Acetyl-ADPribose, OAADPr – O-Acyl-ADPribose.
In addition to total NAD+ levels, the NAD+/NADH ratio is both highly regulated and dynamic. Mitochondrial health is tightly linked to the NAD+/NADH ratio, which plays a role in regulating ROS, mitochondrial permeabilization, and membrane potential (Green and Reed, 1998; Song et al., 2017). Mitchondria are also the site of the TCA cycle and oxidative phosphorylation by the ETC, processes which both rely on the mitochondrial balance of NAD+ versus NADH. To maintain the appropriate NAD+/NADH ratio, reducing equivalents are shuttled across the inner mitochondrial membrane through the activity of two NAD+/NADH redox shuttles. NADH generated by glycolysis can be transferred into the mitochondrial matrix through the malate/aspartate and glycerol-3-phosphate shuttles, which move reducing equivalents into the mitochondria in the form of NADH or FADH2, respectively (LaNoue and Tischler, 1974; Ying, 2008). Cells employ multiple layers of regulation to manage NAD+ pools, which are sensitive to metabolic flux via the NAD+/NADH ratio as well as the availability of precursor metabolites.
Diet and aging: key modulators of NAD+
A variety of nutritional and environmental cues, as well as circadian oscillation, modulates NAD+ biosynthesis as well as the ratio of NAD+ to NADH, highlighting that NAD+ levels are dynamic and tightly regulated. NAD+ levels are sensitive to dietary cues. In the liver and skeletal muscle, several studies have shown that mitochondrial NAD+ levels rise in response to CR and fasting (Canto et al., 2010; Fulco et al., 2008; Nakagawa et al., 2009; Yang et al., 2007). In contrast, HFD reduces overall NAD+ levels in the liver and white adipose tissue (WAT) (Yoshino et al., 2011). Alterations in NAMPT expression parallel the changes in NAD+ observed with fasting, CR, or HFD. Whereas fasting, CR, and excercise induce NAMPT expression two- to threefold, HFD reduces NAMPT protein levels in the liver and WAT (Yoshino et al., 2011). Likewise, the mitochondrial sirtuins are transcriptionally regulated by diet and stress, as discussed above. NAD+ levels are also under the control of circadian transcription factors, which regulate the activity of sirtuins (Ramsey et al., 2009). During the daytime, NAD+ levels increase as a result of NAMPT expression (Ramsey et al., 2009). Rising NAD+ levels periodically activate SIRT3, resulting in circadian patterns of mitochondrial protein acetylation that promote oxidative phosphorylation (Mauvoisin et al., 2017; Peek et al., 2013; Sato et al., 2017). Finally, aging dramatically reduces NAD+ levels by decreasing NAMPT expression (Braidy et al., 2011; Gomes et al., 2013; Massudi et al., 2012; Mouchiroud et al., 2013; Someya et al., 2010; Zhang et al., 2016). Sirtuin activity fluctuates with NAD+ availability, which connects NAD+ metabolism to biological adaptations mediated by sirtuins.
Biological functions for metabolites downstream of sirtuin biochemistry
All sirtuins, including SIRT3-5, use NAD+ as an acyl chain acceptor, resulting in the conversion of NAD+ to NAM and the production of 2’/3’-O-acyl-ADPribose (OAADPr). Most measurements of mitochondrial NAD+ report concentrations of at least 250 μM, which can rise to millimolar levels (Nakagawa et al., 2009; Yang et al., 2007). Dependence on NAD+ as a cosubstrate raises the possibility that sirtuins act as NAD+ sensors. Reported NAD+ KM values for SIRT3 (880 μM) and SIRT5 (980 μM) fall within the range of physiological changes in NAD+ (Fischer et al., 2012; Hirschey et al., 2011). On the other hand, NAD+ availability is unlikely to regulate SIRT4 activity, since the SIRT4 KM for NAD+ is an order of magnitude lower than the other mitochondrial sirtuins, and thus may be below physiological relevant concentrations (Laurent et al., 2013a; Pannek et al., 2017).
A number of metabolites with structural similarity to NAD+ are found in the mitochondria, which provides additional layers of regulation for sirtuin activity that intersect with the metabolic state of the cell. For example, the sirtuin reaction byproduct, NAM, is a non-competitive inhibitor for all three mitochondrial sirtuins at physiological concentrations, although the potency in vivo remains to be determined (Feldman et al., 2015; Pannek et al., 2017). Reduction of NAD+ by the TCA cycle and other redox pathways produces NADH, which can compete with NAD+ for the sirtuin NAD+ binding pocket. Physiological NADH levels range from 50 to 100 μM in skeletal muscle, which are far below the millimolar concentrations of NADH required to inhibit SIRT3 and SIRT5 in vitro (Madsen et al., 2016; Pirinen et al., 2014). SIRT4 is better tuned to respond to NADH levels, since the IC50 of SIRT4 for NADH is 142 ± 54 uM, a concentration where NADH regulation of SIRT4 activity becomes plausible (Pannek et al., 2017). However, the significance of NADH-mediated inhibition of SIRT4 in vivo has yet to be addressed. Thus, environmental cues that change NAD+ metabolism in different ways can precipitate distinct functional outcomes for each of the three mitochondrial sirtuins. It is also important to consider the regulatory consequences of the ratios of NAD+-like metabolites, which are produced and consumed by parallel metabolic pathways within the mitochondria. For example, changes in the NAD+/NAM ratio affect the activity of all three mitochondrial sirtuins, and is a read-out of sirtuin activity (via NAM production) versus the rate of NAD+ salvage (Feldman et al., 2015). Further studies elucidating mitochondrial mechanisms of NAD+ salvage from NAM are necessary to better understand the fate of mitochondrial NAM and its biological relevance as a mitochondrial sirtuin inhibitor. The unique biochemical properties of the mitochondrial sirtuins could provide insight into the distinct roles SIRT3-5 play in adaptation to metabolic changes.
Biological roles for another byproduct of the sirtuin-catalyzed reaction, OAADPr, are not well understood, although studies in yeast and HEK293 cells have shown that 2’/3’-O-acetyl-ADP-ribose (OAcADPr) affects gene silencing and gating of the TRPM2 cation channel, respectively (Grubisha et al., 2006; Liou et al., 2005; Martino et al., 2009; Perraud et al., 2005). The discovery that sirtuins remove a number of chemically diverse acyl chains, producing acylated ADPr and not just acetylated ADPr, has significantly expanded the possible metabolic functions for these molecules. Several mitochondrial enzymes are reported to metabolize OAcADPr, such as macrodomain proteins, glycohydrolases and NUDIX hydrolases (Figure 4). Among macrodomain proteins, MacroD1 is the only known example that localizes to the mitochondria (Agnew et al., 2018). Likewise, ARH3 is the only member of the glycohydrolase family found in the mitochondria to date (Ono et al., 2006). MacroD1 and ARH3 hydrolyze acetyl groups from OAcADPr to generate ADPr and free acetate (Chen et al., 2011; Ono et al., 2006). NUDIX hydrolases cleave ADPR into 2’/3’-O-acetyl ribose-5-phosphate and AMP (Rafty et al., 2002). Whether enzymes exist that break down other acylated forms of OAADPr remains to be seen and the ultimate fates of metabolites generated from these hydrolysis reactions remain unknown, especially whether and where the acyl chain carbon reenters metabolism. Taken together, these findings are highly suggestive of still undiscovered fates and new cellular roles for the rapidly expanding class of OAADPr species as secondary messengers.
Coupling protein deacylation with NAD+ levels facilitates cellular adaptation to stress
Importantly, a body of work supports the hypothesis that NAD+ biosynthesis and changing mitochondrial NAD+ levels regulate biological processes by changing the activity of mitochondrial sirtuins. For example, Nampt upregulation and rising mitochondrial NAD+ levels protect against cell death caused by genotoxic stress, a phenotype that requires intact SIRT3 and SIRT4 (Yang et al., 2007). Genotoxic agents induce the expression of SIRT4 as well, which coordinates a metabolic checkpoint to DNA damage (Jeong et al., 2013). In addition, CR protects mice from aging-dependent deafness, but this effect is lost upon SIRT3 deletion, thus suggesting the wildtype aging-dependent deafness on normal diet was a result of decreased mitochondrial NAD+ levels (Someya et al., 2010). SIRT3 overexpression or administration of NR also protects mice from trauma-induced hearing loss, indicating that NAD+ levels are limiting for SIRT3-mediated neuroprotective activity (Brown et al., 2014). Since SIRT3-5 are implicated in numerous metabolic and aging-related pathologies such as diabetes, cancer, and neurodegenerative diseases, further studies on the intersection between NAD+ homeostasis and mitochondrial sirtuin activity are of central importance.
Conclusions and future perspectives
The unique chemical properties of the mitrochondria facilitate lysine acylation on the mitochondrial proteome, and changing patterns of acyl modifications regulate nearly every aspect of mitochondrial biology. The mitochondrial lysine acylome is emerging as a new factor that may be altered in disease, due either to changes in sirtuin activity that catalyze acyl PTM removal or to differences in acyl-CoA pool sizes that nonenzymatically modify lysine side chains. Since SIRT3 and SIRT4 expression levels are often reduced in neoplasia, mitochondrial sirtuins have been primarily studied as tumor suppressors. For example, SIRT3 loss, which is widely observed across multiple human cancers, promotes tumorgenicity by increasing ROS levels that: (i) ultimately stabilize HIF1α (Bell et al., 2011; Finley et al., 2011a) and (ii) enhance cell migration via the activation of Src/focal adhesion kinase signaling (Lee et al., 2018). SIRT3 loss also increases cellular glutamine consumption and de novo nucleotide biosynthesis by promoting mechanistic target of rapamycin complex 1 (mTORC1) signaling (Gonzalez Herrera et al., 2018). Decreased SIRT4 expression has been shown to promote the growth of tumor cells by enhancing glutaminolysis via the regulation of GDH activity (Jeong et al., 2013, 2014). A number of reviews exploring roles for sirtuins in cancer have been published, and the topic will not be explored in greater depth here (Chalkiadaki and Guarente, 2015; German and Haigis, 2015; Kumar and Lombard, 2015). Overall, these pro-tumorigenic effects observed downstream of sirtuin loss were historically linked to changes in specific pathways or individual substrates regulated by sirtuins. However, the impact of genetic and environmental factors that drive tumorigenesis on global remodeling of the mitochondrial acylome is a relatively new and exciting field of study. For example, mitochondrial proteins from IDH1 mutant cancers that produce high levels of the oncometabolite R-2HG are hypersuccinylated, since R-2HG inhibits SDH and increases local pools of succinyl-CoA. These hypersuccinylated cells are more resistant to apoptotic stimuli, which can be reversed by SIRT5 overexpresson (Li et al., 2015). The tumor microenvironment can also provide external stimuli that impact the mitochondrial acylome. Mitochondrial acetylation levels increase with tumor microenvironment acidosis, due to metabolic reprogramming that causes cancer cells to rely on FAO (Corbet et al., 2016). These studies highlight how the mitochondrial acylome can be remodeled in response to both cell-intrinsic and environmental cues, and that this remodeling can be pathogenic (as in cancer) or adaptive (as in response to fuel switching).
Biochemical and in vivo studies have revealed that the mitochondrial lysine acylation landscape is shaped by the activity of sirtuin enzymes, and defined sites of lysine acylation that sirtuins regulate. Sirtuins coordinate competing arms of mitochondrial metabolism by remodeling the lysine acylome in response to fluctuations in nutrient availability and stress. By integrating bioenergetic information from multiple sources, mitochondrial sirtuins execute pivotal regulatory roles in the matrix. The revelation that sirtuins are deacylases rather than deacetylases has raised a number of critical questions going forward. First, defining roles for particular acyl PTMs or classes of acyl PTMs that are linked to chemical properties of the acyl modification will shed light on new regulatory mechanisms that operate in the mitrochondrial matrix. Second, it is not known whether NAD+-like metabolites regulate sirtuin deacylation activities to the same degree, and whether the sensitivity to inhibition for a specific sirtuin differs among acyl chain substrates. Third, sirtuin crystal structures have revealed active site features that may bind metabolites that modulate sirtuin activity. Determining whether sirtuins can sense concentrations of small molecules linked to metabolic pathways is an open area of research. Fourth, dissecting mechanisms by which cells recycle the acyl carbon on OAADPr will reveal new connections between fuel sourcing, mitochondrial carbon stress, and metabolism. Finally, understanding whether and to what extent the activity of each sirtuin contributes to lysine deacylation under different conditions will prove highly relevant in studies of pathophysiologies that alter matrix acyl PTMs.
Acknowledgments
We thank all members of the Haigis lab for their comments and discussion on the manuscript. This review is not meant to be a comprehensive summary of all research on regulatory mechanisms for mitochondrial acyl modifications, and we apologize for all the primary literature and work we could not cite due to space limitations. A.E.R. is supported by a postdoctoral fellowship from the American Cancer Society (130373-PF-17-132-01-CCG). S.A.T. is supported by a predoctoral T32 fellowship through the Joslin Diabetes Center (DK07260-042). M.C.H. is supported by the Ludwig Center at Harvard, the Glenn Foundation for Medical Research, and the National Institute of Diabetes and Digestive and Kidney Diseases (1R01DK103295-01A1).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agnew T, Munnur D, Crawford K, Palazzo L, Mikoc A, and Ahel I (2018). MacroD1 Is a Promiscuous ADP-Ribosyl Hydrolase Localized to Mitochondria. Front. Microbiol 9, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn B-H, Kim H-S, Song S, Lee IH, Liu J, Vassilopoulos A, Deng C-X, and Finkel T (2008). A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. U. S. A 105, 14447–14452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aksoy P, White TA, Thompson M, and Chini EN (2006). Regulation of intracellular levels of NAD: a novel role for CD38. Biochem. Biophys. Res. Commun 345, 1386–1392. [DOI] [PubMed] [Google Scholar]
- Alano CC, Tran A, Tao R, Ying W, Karliner JS, and Swanson RA (2007). Differences among cell types in NAD(+) compartmentalization: a comparison of neurons, astrocytes, and cardiac myocytes. J. Neurosci. Res 85, 3378–3385. [DOI] [PubMed] [Google Scholar]
- Allfrey VG, Faulkner R, and Mirsky AE (1964). Acetylation and Methylation of Histones and Their Possible Role in the Regulation of RNA Synthesis. Proc. Natl. Acad. Sci. U. S. A 51, 786–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KA, Huynh FK, Fisher-Wellman K, Stuart JD, Peterson BS, Douros JD, Wagner GR, Thompson JW, Madsen AS, Green MF, et al. (2017). SIRT4 Is a Lysine Deacylase that Controls Leucine Metabolism and Insulin Secretion. Cell Metab. 25, 838–855.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeza J, Smallegan MJ, and Denu JM (2016). Mechanisms and Dynamics of Protein Acetylation in Mitochondria. Trends Biochem. Sci. 41, 231–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister AJ, and Kouzarides T (2011). Regulation of chromatin by histone modifications. Cell Res. 21, 381–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao X, Wang Y, Li X, Li X-M, Liu Z, Yang T, Wong CF, Zhang J, Hao Q, and Li XD (2014). Identification of “erasers” for lysine crotonylated histone marks using a chemical proteomics approach. Elife 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barile M, Passarella S, Danese G, and Quagliariello E (1996). Rat liver mitochondria can synthesize nicotinamide adenine dinucleotide from nicotinamide mononucleotide and ATP via a putative matrix nicotinamide mononucleotide adenylyltransferase. Biochem. Mol. Biol. Int 38, 297–306. [PubMed] [Google Scholar]
- Bell EL, Emerling BM, Ricoult SJ, and Guarente L (2011). SirT3 suppresses hypoxia inducible factor 1alpha and tumor growth by inhibiting mitochondrial ROS production. Oncogene 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger F, Lau C, Dahlmann M, and Ziegler M (2005). Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J. Biol. Chem 280, 36334–36341. [DOI] [PubMed] [Google Scholar]
- Bharathi SS, Zhang Y, Mohsen A-W, Uppala R, Balasubramani M, Schreiber E, Uechi G, Beck ME, Rardin MJ, Vockley J, et al. (2013). Sirtuin 3 (SIRT3) protein regulates long-chain acyl-CoA dehydrogenase by deacetylating conserved lysines near the active site. J. Biol. Chem 288, 33837–33847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bheda P, Jing H, Wolberger C, and Lin H (2016). The Substrate Specificity of Sirtuins. Annu. Rev. Biochem 85, 405–429. [DOI] [PubMed] [Google Scholar]
- Bieganowski P, and Brenner C (2004). Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell 117, 495–502. [DOI] [PubMed] [Google Scholar]
- Billington RA, Travelli C, Ercolano E, Galli U, Roman CB, Grolla AA, Canonico PL, Condorelli F, and Genazzani AA (2008). Characterization of NAD uptake in mammalian cells. J. Biol. Chem 283, 6367–6374. [DOI] [PubMed] [Google Scholar]
- Bowman CE, Rodriguez S, Selen Alpergin ES, Acoba MG, Zhao L, Hartung T, Claypool SM, Watkins PA, and Wolfgang MJ (2017). The Mammalian Malonyl-CoA Synthetase ACSF3 Is Required for Mitochondrial Protein Malonylation and Metabolic Efficiency. Cell Chem. Biol 24, 673–684.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boylston JA, Sun J, Chen Y, Gucek M, Sack MN, and Murphy E (2015). Characterization of the cardiac succinylome and its role in ischemia-reperfusion injury. J. Mol. Cell. Cardiol 88, 73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braidy N, Guillemin GJ, Mansour H, Chan-Ling T, Poljak A, and Grant R (2011). Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS One 6, e19194–e19194. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Brown K, Xie S, Qiu X, Mohrin M, Shin J, Liu Y, Zhang D, Scadden DT, and Chen D (2013). SIRT3 Reverses Aging-associated Degeneration. Cell Rep. 3, 319–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KD, Maqsood S, Huang J-Y, Pan Y, Harkcom W, Li W, Sauve A, Verdin E, and Jaffrey SR (2014). Activation of SIRT3 by the NAD(+) precursor nicotinamide riboside protects from noise-induced hearing loss. Cell Metab. 20, 1059–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Jiang LQ, Deshmukh AS, Mataki C, Coste A, Lagouge M, Zierath JR, and Auwerx J (2010). Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab. 11, 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, Fernandez-Marcos PJ, Yamamoto H, Andreux PA, Cettour-Rose P, et al. (2012). The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 15, 838–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerutti R, Pirinen E, Lamperti C, Marchet S, Sauve AA, Li W, Leoni V, Schon EA, Dantzer F, Auwerx J, et al. (2014). NAD(+)-dependent activation of Sirt1 corrects the phenotype in a mouse model of mitochondrial disease. Cell Metab. 19, 1042–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalkiadaki A, and Guarente L (2015). The multifaceted functions of sirtuins in cancer. Nat. Rev. Cancer 15, 608. [DOI] [PubMed] [Google Scholar]
- Chambon P, Weill JD, and Mandel P (1963). Nicotinamide mononucleotide activation of new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem. Biophys. Res. Commun 11, 39–43. [DOI] [PubMed] [Google Scholar]
- Chen D, Vollmar M, Rossi MN, Phillips C, Kraehenbuehl R, Slade D, Mehrotra PV, von Delft F, Crosthwaite SK, Gileadi O, et al. (2011). Identification of macrodomain proteins as novel O-acetyl-ADP-ribose deacetylases. J. Biol. Chem 286, 13261–13271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Sprung R, Tang Y, Ball H, Sangras B, Kim SC, Falck JR, Peng J, Gu W, and Zhao Y (2007). Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol. Cell. Proteomics 6, 812–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z, Tang Y, Chen Y, Kim S, Liu H, Li SSC, Gu W, and Zhao Y (2009). Molecular characterization of propionyllysines in non-histone proteins. Mol. Cell. Proteomics 8, 45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colak G, Pougovkina O, Dai L, Tan M, Te Brinke H, Huang H, Cheng Z, Park J, Wan X, Liu X, et al. (2015). Proteomic and Biochemical Studies of Lysine Malonylation Suggest Its Malonic Aciduria-associated Regulatory Role in Mitochondrial Function and Fatty Acid Oxidation. Mol. Cell. Proteomics 14, 3056–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbet C, Pinto A, Martherus R, Santiago de Jesus JP, Polet F, and Feron O (2016). Acidosis Drives the Reprogramming of Fatty Acid Metabolism in Cancer Cells through Changes in Mitochondrial and Histone Acetylation. Cell Metab. 24, 311–323. [DOI] [PubMed] [Google Scholar]
- Csibi A, Fendt S-M, Li C, Poulogiannis G, Choo AY, Chapski DJ, Jeong SM, Dempsey J, Parkhitko A, Morrison T, et al. (2013). The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell 153, 840–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai L, Peng C, Montellier E, Lu Z, Chen Y, Ishii H, Debernardi A, Buchou T, Rousseaux S, Jin F, et al. (2014). Lysine 2-hydroxyisobutyrylation is a widely distributed active histone mark. Nat. Chem. Biol 10, 365–370. [DOI] [PubMed] [Google Scholar]
- Davila A, Liu L, Chellappa K, Redpath P, Nakamaru-Ogiso E, Paolella LM, Zhang Z, Migaud ME, Rabinowitz JD, and Baur JA (2018). Nicotinamide adenine dinucleotide is transported into mammalian mitochondria. Elife 7, e33246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittenhafer-Reed KE, Richards AL, Fan J, Smallegan MJ, Siahpirani AF, Kemmerer ZA, Prolla TA, Roy S, Coon JJ, and Denu JM (2015). SIRT3 mediates multi-tissue coupling for metabolic fuel switching. Cell Metab. 21, 637–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, Kim J, Woo J, Kim JH, Choi BH, et al. (2011). Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 334, 806–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberharter A, and Becker PB (2002). Histone acetylation: a switch between repressive and permissive chromatin: Second in review series on chromatin dynamics. EMBO Rep. 3, 224–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman JL, Baeza J, and Denu JM (2013). Activation of the protein deacetylase SIRT6 by long-chain fatty acids and widespread deacylation by mammalian sirtuins. J. Biol. Chem 288, 31350–31356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman JL, Dittenhafer-Reed KE, Kudo N, Thelen JN, Ito A, Yoshida M, and Denu JM (2015). Kinetic and Structural Basis for Acyl-Group Selectivity and NAD(+) Dependence in Sirtuin-Catalyzed Deacylation. Biochemistry 54, 3037–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes J, Weddle A, Kinter CS, Humphries KM, Mather T, Szweda LI, and Kinter M (2015). Lysine Acetylation Activates Mitochondrial Aconitase in the Heart. Biochemistry 54, 4008–4018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley LW, Carracedo A, Lee J, Souza A, Egia A, Zhang J, Teruya-Feldstein J, Moreira PI, Cardoso SM, Clish CB, et al. (2011a). SIRT3 opposes reprogramming of cancer cell metabolism through HIF1α destabilization. Can Cell 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley LWS, Carracedo A, Lee J, Souza A, Egia A, Zhang J, Teruya-Feldstein J, Moreira PI, Cardoso SM, Clish CB, et al. (2011b). SIRT3 opposes reprogramming of cancer cell metabolism through HIF1alpha destabilization. Cancer Cell 19, 416–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer F, Gertz M, Suenkel B, Lakshminarasimhan M, Schutkowski M, and Steegborn C (2012). Sirt5 deacylation activities show differential sensitivities to nicotinamide inhibition. PLoS One 7, e45098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Flora A, Zocchi E, Guida L, Franco L, and Bruzzone S (2004). Autocrine and paracrine calcium signaling by the CD38/NAD+/cyclic ADP-ribose system. Ann. N. Y. Acad. Sci 1028, 176–191. [DOI] [PubMed] [Google Scholar]
- Fritz KS, Galligan JJ, Hirschey MD, Verdin E, and Petersen DR (2012). Mitochondrial Acetylome Analysis in a Mouse Model of Alcohol-Induced Liver Injury Utilizing SIRT3 Knockout Mice. J. Proteome Res. 11, 1633–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frye RA (1999). Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem. Biophys. Res. Commun 260, 273–279. [DOI] [PubMed] [Google Scholar]
- Fujino T, Kondo J, Ishikawa M, Morikawa K, and Yamamoto TT (2001). Acetyl-CoA Synthetase 2, a Mitochondrial Matrix Enzyme Involved in the Oxidation of Acetate. J. Biol. Chem 276, 11420–11426. [DOI] [PubMed] [Google Scholar]
- Fulco M, Cen Y, Zhao P, Hoffman EP, McBurney MW, Sauve AA, and Sartorelli V (2008). Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev. Cell 14, 661–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garland PB, Shepherd D, and Yates DW (1965). Steady-state concentrations of coenzyme A, acetyl-coenzyme A and long-chain fatty acyl-coenzyme A in rat-liver mitochondria oxidizing palmitate. Biochem. J 97, 587 LP–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- German NJ, and Haigis MC (2015). Sirtuins and the metabolic hurdles in cancer. Curr. Biol 25, R569–R583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghanta S, Grossmann RE, and Brenner C (2013). Mitochondrial protein acetylation as a cell-intrinsic, evolutionary driver of fat storage: chemical and metabolic logic of acetyl-lysine modifications. Crit. Rev. Biochem. Mol. Biol 48, 561–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson GE, Xu H, Chen H-L, Chen W, Denton T, and Zhang S (2015). Alpha-ketoglutarate dehydrogenase complex-dependent succinylation of proteins in neurons and neuronal cell lines. J. Neurochem 134, 86–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes AP, Price NL, Ling AJY, Moslehi JJ, Montgomery MK, Rajman L, White JP, Teodoro JS, Wrann CD, Hubbard BP, et al. (2013). Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 155, 1624–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez Herrera KN, Zaganjor E, Ishikawa Y, Spinelli JB, Yoon H, Lin J-R, Satterstrom FK, Ringel A, Mulei S, Souza A, et al. (2018). Small-Molecule Screen Identifies De Novo Nucleotide Synthesis as a Vulnerability of Cells Lacking SIRT3. Cell Rep. 22, 1945–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR, and Reed JC (1998). Mitochondria and apoptosis. Science 281, 1309–1312. [DOI] [PubMed] [Google Scholar]
- Grubisha O, Rafty LA, Takanishi CL, Xu X, Tong L, Perraud A-L, Scharenberg AM, and Denu JM (2006). Metabolite of SIR2 reaction modulates TRPM2 ion channel. J. Biol. Chem 281, 14057–14065. [DOI] [PubMed] [Google Scholar]
- Grunstein M (1997). Histone acetylation in chromatin structure and transcription. Nature 389, 349–352. [DOI] [PubMed] [Google Scholar]
- Haber JE, and George JP (1979). A mutation that permits the expression of normally silent copies of mating-type information in Saccharomyces cerevisiae. Genetics 93, 13–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigis MC, and Sinclair DA (2010). Mammalian sirtuins: biological insights and disease relevance. Annu. Rev. Pathol 5, 253–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigis MC, Mostoslavsky R, Haigis KM, Fahie K, Christodoulou DC, Murphy AJ, Valenzuela DM, Yancopoulos GD, Karow M, Blander G, et al. (2006). SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 126, 941–954. [DOI] [PubMed] [Google Scholar]
- Hallows WC, Lee S, and Denu JM (2006). Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc. Natl. Acad. Sci. U. S. A 103, 10230–10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallows WC, Yu W, Smith BC, Devries MK, Ellinger JJ, Someya S, Shortreed MR, Prolla T, Markley JL, Smith LM, et al. (2011). Sirt3 promotes the urea cycle and fatty acid oxidation during dietary restriction. Mol. Cell 41, 139–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansford RG, and Johnson RN (1975). The steady state concentrations of coenzyme A-SH and coenzyme A thioester, citrate, and isocitrate during tricarboxylate cycle oxidations in rabbit heart mitochondria. J. Biol. Chem 250, 8361–8375. [PubMed] [Google Scholar]
- Hebert AS, Dittenhafer-Reed KE, Yu W, Bailey DJ, Selen ES, Boersma MD, Carson JJ, Tonelli M, Balloon AJ, Higbee AJ, et al. (2013). Calorie restriction and SIRT3 trigger global reprogramming of the mitochondrial protein acetylome. Mol. Cell 49, 186–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberger KA, Abraham DM, Martin AS, Mao L, Liu J, Gu H, Locasale JW, and Hirschey MD (2017). Sirtuin 5 is required for mouse survival in response to cardiac pressure overload. J. Biol. Chem 292, 19767–19781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, et al. (2010). SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464, 121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschey MD, Shimazu T, Jing E, Grueter CA, Collins AM, Aouizerat B, Stancakova A, Goetzman E, Lam MM, Schwer B, et al. (2011). SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol. Cell 44, 177–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Luo Z, Qi S, Huang J, Xu P, Wang X, Gao L, Li F, Wang J, Zhao W, et al. (2018). Landscape of the regulatory elements for lysine 2-hydroxyisobutyrylation pathway. Cell Res. 28, 111–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Armstrong CM, Kaeberlein M, and Guarente L (2000). Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403, 795–800. [DOI] [PubMed] [Google Scholar]
- James AM, Hoogewijs K, Logan A, Hall AR, Ding S, Fearnley IM, and Murphy MP (2017). Non-enzymatic N-acetylation of Lysine Residues by AcetylCoA Often Occurs via a Proximal S-acetylated Thiol Intermediate Sensitive to Glyoxalase II. Cell Rep. 18, 2105–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong SM, Xiao C, Finley LWS, Lahusen T, Souza AL, Pierce K, Li Y-H, Wang X, Laurent G, German NJ, et al. (2013). SIRT4 has tumor-suppressive activity and regulates the cellular metabolic response to DNA damage by inhibiting mitochondrial glutamine metabolism. Cancer Cell 23, 450–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong SM, Lee A, Lee J, and Haigis MC (2014). SIRT4 Protein Suppresses Tumor Formation in Genetic Models of Myc-induced B Cell Lymphoma. J. Biol. Chem 289, 4135–4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing E, O’Neill BT, Rardin MJ, Kleinridders A, Ilkeyeva OR, Ussar S, Bain JR, Lee KY, Verdin EM, Newgard CB, et al. (2013). Sirt3 regulates metabolic flexibility of skeletal muscle through reversible enzymatic deacetylation. Diabetes 62, 3404–3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendrick AA, Choudhury M, Rahman SM, McCURDY CE, Friederich M, Van Hove JLK, Watson PA, Birdsey N, Bao J, Gius D, et al. (2011). Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation. Biochem. J 433, 505–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao H, Xiao L, et al. (2006). Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell 23, 607–618. [DOI] [PubMed] [Google Scholar]
- Klar AJ, Fogel S, and Macleod K (1979). MAR1-a Regulator of the HMa and HMalpha Loci in SACCHAROMYCES CEREVISIAE. Genetics 93, 37–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krehl WA, Teply LJ, Sarma PS, and Elvehjem CA (1945). Growth-Retarding Effect of Corn in Nicotinic Acid-Low Rations and its Counteraction by Tryptophane. Science 101, 489–490. [DOI] [PubMed] [Google Scholar]
- Kumar S, and Lombard DB (2015). Mitochondrial Sirtuins and Their Relationships with Metabolic Disease and Cancer. Antioxid. Redox Signal. 22, 1060–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurmi K, Hitosugi S, Wiese EK, Boakye-Agyeman F, Gonsalves WI, Lou Z, Karnitz LM, Goetz MP, and Hitosugi T (2018). Carnitine Palmitoyltransferase 1A Has a Lysine Succinyltransferase Activity. Cell Rep. 22, 1365–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry J, Sutton A, Tafrov ST, Heller RC, Stebbins J, Pillus L, and Sternglanz R (2000). The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc. Natl. Acad. Sci. U. S. A 97, 5807–5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaNoue KF, and Tischler ME (1974). Electrogenic characteristics of the mitochondrial glutamate-aspartate antiporter. J. Biol. Chem 249, 7522–7528. [PubMed] [Google Scholar]
- Laurent G, German NJ, Saha AK, de Boer VCJ, Davies M, Koves TR, Dephoure N, Fischer F, Boanca G, Vaitheesvaran B, et al. (2013a). SIRT4 coordinates the balance between lipid synthesis and catabolism by repressing malonyl CoA decarboxylase. Mol. Cell 50, 686–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent G, de Boer VCJ, Finley LWS, Sweeney M, Lu H, Schug TT, Cen Y, Jeong SM, Li X, Sauve AA, et al. (2013b). SIRT4 represses peroxisome proliferator-activated receptor alpha activity to suppress hepatic fat oxidation. Mol. Cell. Biol 33, 4552–4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JJ, van de Ven RAH, Zaganjor E, Ng MR, Barakat A, Demmers JJPG, Finley LWS, Gonzalez Herrera KN, Hung YP, Harris IS, et al. (2018). Inhibition of epithelial cell migration and Src/FAK signaling by SIRT3. Proc. Natl. Acad. Sci [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, He X, Ye D, Lin Y, Yu H, Yao C, Huang L, Zhang J, Wang F, Xu S, et al. (2015). NADP+-IDH Mutations Promote Hypersuccinylation that Impairs Mitochondria Respiration and Induces Apoptosis Resistance. Mol. Cell 60, 661–675. [DOI] [PubMed] [Google Scholar]
- Liou G-G, Tanny JC, Kruger RG, Walz T, and Moazed D (2005). Assembly of the SIR complex and its regulation by O-acetyl-ADP-ribose, a product of NAD-dependent histone deacetylation. Cell 121, 515–527. [DOI] [PubMed] [Google Scholar]
- Di Lisa F, Menabo R, Canton M, Barile M, and Bernardi P (2001). Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J. Biol. Chem 276, 2571–2575. [DOI] [PubMed] [Google Scholar]
- Liu L, Su X, Quinn WJ 3rd, Hui S, Krukenberg K, Frederick DW, Redpath P, Zhan L, Chellappa K, White E, et al. (2018). Quantitative Analysis of NAD Synthesis-Breakdown Fluxes. Cell Metab. 27, 1067–1080.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard DB, Alt FW, Cheng H-L, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A, et al. (2007). Mammalian Sir2 Homolog SIRT3 Regulates Global Mitochondrial Lysine Acetylation . Mol. Cell. Biol 27, 8807–8814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen AS, Andersen C, Daoud M, Anderson KA, Laursen JS, Chakladar S, Huynh FK, Colaco AR, Backos DS, Fristrup P, et al. (2016). Investigating the Sensitivity of NAD+-dependent Sirtuin Deacylation Activities to NADH. J. Biol. Chem 291, 7128–7141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magni G, Amici A, Emanuelli M, Raffaelli N, and Ruggieri S (1999). Enzymology of NAD+ synthesis. Adv. Enzymol. Relat. Areas Mol. Biol 73, 135–82, xi. [DOI] [PubMed] [Google Scholar]
- Martino F, Kueng S, Robinson P, Tsai-Pflugfelder M, van Leeuwen F, Ziegler M, Cubizolles F, Cockell MM, Rhodes D, and Gasser SM (2009). Reconstitution of yeast silent chromatin: multiple contact sites and O-AADPR binding load SIR complexes onto nucleosomes in vitro. Mol. Cell 33, 323–334. [DOI] [PubMed] [Google Scholar]
- Massudi H, Grant R, Braidy N, Guest J, Farnsworth B, and Guillemin GJ (2012). Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS One 7, e42357–e42357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathias RA, Greco TM, Oberstein A, Budayeva HG, Chakrabarti R, Rowland EA, Kang Y, Shenk T, and Cristea IM (2014). Sirtuin 4 is a lipoamidase regulating pyruvate dehydrogenase complex activity. Cell 159, 1615–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauvoisin D, Atger F, Dayon L, Nunez Galindo A, Wang J, Martin E, Da Silva L, Montoliu I, Collino S, Martin F-P, et al. (2017). Circadian and Feeding Rhythms Orchestrate the Diurnal Liver Acetylome. Cell Rep. 20, 1729–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizzen CA, and Allis CD (1998). Linking histone acetylation to transcriptional regulation. Cell. Mol. Life Sci 54, 6–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Canto C, Mottis A, Jo Y-S, Viswanathan M, Schoonjans K, et al. (2013). The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell 154, 430–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musselman CA, Lalonde M-E, Cote J, and Kutateladze TG (2012). Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol 19, 1218–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Lomb DJ, Haigis MC, and Guarente L (2009). SIRT5 Deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle. Cell 137, 560–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikiforov A, Dolle C, Niere M, and Ziegler M (2011). Pathways and subcellular compartmentation of NAD biosynthesis in human cells: from entry of extracellular precursors to mitochondrial NAD generation. J. Biol. Chem 286, 21767–21778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida Y, Rardin MJ, Carrico C, He W, Sahu AK, Gut P, Najjar R, Fitch M, Hellerstein M, Gibson BW, et al. (2015). SIRT5 Regulates both Cytosolic and Mitochondrial Protein Malonylation with Glycolysis as a Major Target. Mol. Cell 59, 321–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono T, Kasamatsu A, Oka S, and Moss J (2006). The 39-kDa poly(ADP-ribose) glycohydrolase ARH3 hydrolyzes O-acetyl-ADP-ribose, a product of the Sir2 family of acetyl-histone deacetylases. Proc. Natl. Acad. Sci. U. S. A 103, 16687–16691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onyango P, Celic I, McCaffery JM, Boeke JD, and Feinberg AP (2002). SIRT3, a human SIR2 homologue, is an NAD-dependent deacetylase localized to mitochondria. Proc. Natl. Acad. Sci. U. S. A 99, 13653–13658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannek M, Simic Z, Fuszard M, Meleshin M, Rotili D, Mai A, Schutkowski M, and Steegborn C (2017). Crystal structures of the mitochondrial deacylase Sirtuin 4 reveal isoform-specific acyl recognition and regulation features. Nat. Commun 8, 1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Chen Y, Tishkoff DX, Peng C, Tan M, Dai L, Xie Z, Zhang Y, Zwaans BMM, Skinner ME, et al. (2013). SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol. Cell 50, 919–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peek CB, Affinati AH, Ramsey KM, Kuo H-Y, Yu W, Sena LA, Ilkayeva O, Marcheva B, Kobayashi Y, Omura C, et al. (2013). Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science 342, 1243417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng C, Lu Z, Xie Z, Cheng Z, Chen Y, Tan M, Luo H, Zhang Y, He W, Yang K, et al. (2011). The first identification of lysine malonylation substrates and its regulatory enzyme. Mol. Cell. Proteomics 10, M111.012658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perraud A-L, Takanishi CL, Shen B, Kang S, Smith MK, Schmitz C, Knowles HM, Ferraris D, Li W, Zhang J, et al. (2005). Accumulation of free ADP-ribose from mitochondria mediates oxidative stress-induced gating of TRPM2 cation channels. J. Biol. Chem 280, 6138–6148. [DOI] [PubMed] [Google Scholar]
- Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F, and Kroemer G (2015). Acetyl Coenzyme A: A Central Metabolite and Second Messenger. Cell Metab. 21, 805–821. [DOI] [PubMed] [Google Scholar]
- Pirinen E, Canto C, Jo YS, Morato L, Zhang H, Menzies KJ, Williams EG, Mouchiroud L, Moullan N, Hagberg C, et al. (2014). Pharmacological Inhibition of poly(ADP-ribose) polymerases improves fitness and mitochondrial function in skeletal muscle. Cell Metab. 19, 1034–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittelli M, Formentini L, Faraco G, Lapucci A, Rapizzi E, Cialdai F, Romano G, Moneti G, Moroni F, and Chiarugi A (2010). Inhibition of nicotinamide phosphoribosyltransferase: cellular bioenergetics reveals a mitochondrial insensitive NAD pool. J. Biol. Chem 285, 34106–34114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittelli M, Felici R, Pitozzi V, Giovannelli L, Bigagli E, Cialdai F, Romano G, Moroni F, and Chiarugi A (2011). Pharmacological effects of exogenous NAD on mitochondrial bioenergetics, DNA repair, and apoptosis. Mol. Pharmacol 80, 1136–1146. [DOI] [PubMed] [Google Scholar]
- Pougovkina O, te Brinke H, Ofman R, van Cruchten AG, Kulik W, Wanders RJA, Houten SM, and de Boer VCJ (2014). Mitochondrial protein acetylation is driven by acetyl-CoA from fatty acid oxidation. Hum. Mol. Genet 23, 3513–3522. [DOI] [PubMed] [Google Scholar]
- Powanda MC, and Wannemacher RWJ (1970). Evidence for a linear correlation between the level of dietary tryptophan and hepatic NAD concentration and for a systematic variation in tissue NAD concentration in the mouse and the rat. J. Nutr 100, 1471–1478. [DOI] [PubMed] [Google Scholar]
- Priess J, and Handler P (1958a). Biosynthesis of diphosphopyridine nucleotide. II. Enzymatic aspects. J. Biol. Chem 233, 493–500. [PubMed] [Google Scholar]
- Priess J, and Handler P (1958b). Biosynthesis of diphosphopyridine nucleotide. I. Identification of intermediates. J. Biol. Chem 233, 488–492. [PubMed] [Google Scholar]
- Qiu X, Brown K, Hirschey MD, Verdin E, and Chen D (2010). Calorie Restriction Reduces Oxidative Stress by SIRT3-Mediated SOD2 Activation. Cell Metab. 12, 662–667. [DOI] [PubMed] [Google Scholar]
- Rafty LA, Schmidt MT, Perraud A-L, Scharenberg AM, and Denu JM (2002). Analysis of O-acetyl-ADP-ribose as a target for Nudix ADP-ribose hydrolases. J. Biol. Chem 277, 47114–47122. [DOI] [PubMed] [Google Scholar]
- Ramsey KM, Yoshino J, Brace CS, Abrassart D, Kobayashi Y, Marcheva B, Hong H-K, Chong JL, Buhr ED, Lee C, et al. (2009). Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science 324, 651–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rardin MJ, Newman JC, Held JM, Cusack MP, Sorensen DJ, Li B, Schilling B, Mooney SD, Kahn CR, Verdin E, et al. (2013a). Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proc. Natl. Acad. Sci 110, 6601 LP–6606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rardin MJ, He W, Nishida Y, Newman JC, Carrico C, Danielson SR, Guo A, Gut P, Sahu AK, Li B, et al. (2013b). SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. 18, 920–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rine J, and Herskowitz I (1987). Four genes responsible for a position effect on expression from HML and HMR in Saccharomyces cerevisiae. Genetics 116, 9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rine J, Strathern JN, Hicks JB, and Herskowitz I (1979). A suppressor of mating-type locus mutations in Saccharomyces cerevisiae: evidence for and identification of cryptic mating-type loci. Genetics 93, 877–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadhukhan S, Liu X, Ryu D, Nelson OD, Stupinski JA, Li Z, Chen W, Zhang S, Weiss RS, Locasale JW, et al. (2016). Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function. Proc. Natl. Acad. Sci 113, 4320 LP–4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S, Solanas G, Peixoto FO, Bee L, Symeonidi A, Schmidt MS, Brenner C, Masri S, Benitah SA, and Sassone-Corsi P (2017). Circadian Reprogramming in the Liver Identifies Metabolic Pathways of Aging. Cell 170, 664–677.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutz G, and Feigelson P (1972). Purification and properties of rat liver tryptophan oxygenase. J. Biol. Chem 247, 5327–5332. [PubMed] [Google Scholar]
- Schwer B, Bunkenborg J, Verdin RO, Andersen JS, and Verdin E (2006). Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc. Natl. Acad. Sci. U. S. A 103, 10224–10229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B, Eckersdorff M, Li Y, Silva JC, Fermin D, Kurtev MV, Giallourakis C, Comb MJ, Alt FW, and Lombard DB (2009). Calorie restriction alters mitochondrial protein acetylation. Aging Cell 8, 604–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimazu T, Hirschey MD, Hua L, Dittenhafer-Reed KE, Schwer B, Lombard DB, Li Y, Bunkenborg J, Alt FW, Denu JM, et al. (2010). SIRT3 deacetylates mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 and regulates ketone body production. Cell Metab. 12, 654–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shogren-Knaak M, Ishii H, Sun J-M, Pazin MJ, Davie JR, and Peterson CL (2006). Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 311, 844–847. [DOI] [PubMed] [Google Scholar]
- Simic Z, Weiwad M, Schierhorn A, Steegborn C, and Schutkowski M (2015). The varepsilon-Amino Group of Protein Lysine Residues Is Highly Susceptible to Nonenzymatic Acylation by Several Physiological Acyl-CoA Thioesters. Chembiochem 16, 2337–2347. [DOI] [PubMed] [Google Scholar]
- Smith JS, Brachmann CB, Celic I, Kenna MA, Muhammad S, Starai VJ, Avalos JL, Escalante-Semerena JC, Grubmeyer C, Wolberger C, et al. (2000). A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein family. Proc. Natl. Acad. Sci. U. S. A 97, 6658–6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sol EM, Wagner SA, Weinert BT, Kumar A, Kim H-S, Deng C-X, and Choudhary C (2012). Proteomic Investigations of Lysine Acetylation Identify Diverse Substrates of Mitochondrial Deacetylase Sirt3. PLoS One 7, e50545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, Tanokura M, Denu JM, and Prolla TA (2010). Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 143, 802–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song SB, Jang S-Y, Kang HT, Wei B, Jeoun U-W, Yoon GS, and Hwang ES (2017). Modulation of Mitochondrial Membrane Potential and ROS Generation by Nicotinamide in a Manner Independent of SIRT1 and Mitophagy. Mol. Cells 40, 503–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Still AJ, Floyd BJ, Hebert AS, Bingman CA, Carson JJ, Gunderson DR, Dolan BK, Grimsrud PA, Dittenhafer-Reed KE, Stapleton DS, et al. (2013). Quantification of mitochondrial acetylation dynamics highlights prominent sites of metabolic regulation. J. Biol. Chem . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl K (1998). Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 12, 599–606. [DOI] [PubMed] [Google Scholar]
- Su Z, and Denu JM (2016). Reading the Combinatorial Histone Language. ACS Chem. Biol. 11, 564–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svinkina T, Gu H, Silva JC, Mertins P, Qiao J, Fereshetian S, Jaffe JD, Kuhn E, Udeshi ND, and Carr SA (2015). Deep, Quantitative Coverage of the Lysine Acetylome Using Novel Anti-acetyl-lysine Antibodies and an Optimized Proteomic Workflow. Mol. Cell. Proteomics 14, 2429–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, Buchou T, Cheng Z, Rousseaux S, Rajagopal N, et al. (2011). Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 146, 1016–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M, Peng C, Anderson KA, Chhoy P, Xie Z, Dai L, Park J, Chen Y, Huang H, Zhang Y, et al. (2014). Lysine Glutarylation Is a Protein Posttranslational Modification Regulated by SIRT5. Cell Metab. 19, 605–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner KG, Landry J, Sternglanz R, and Denu JM (2000). Silent information regulator 2 family of NAD-dependent histone/protein deacetylases generates a unique product, 1-O-acetyl-ADP-ribose. Proc. Natl. Acad. Sci. U. S. A. 97, 14178–14182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanny JC, and Moazed D (2001). Coupling of histone deacetylation to NAD breakdown by the yeast silencing protein Sir2: Evidence for acetyl transfer from substrate to an NAD breakdown product. Proc. Natl. Acad. Sci. U. S. A 98, 415–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanny JC, Dowd GJ, Huang J, Hilz H, and Moazed D (1999). An enzymatic activity in the yeast Sir2 protein that is essential for gene silencing. Cell 99, 735–745. [DOI] [PubMed] [Google Scholar]
- Tischler ME, Friedrichs D, Coll K, and Williamson JR (1977). Pyridine nucleotide distributions and enzyme mass action ratios in hepatocytes from fed and starved rats. Arch. Biochem. Biophys 184, 222–236. [DOI] [PubMed] [Google Scholar]
- Todisco S, Agrimi G, Castegna A, and Palmieri F (2006). Identification of the mitochondrial NAD+ transporter in Saccharomyces cerevisiae. J. Biol. Chem 281, 1524–1531. [DOI] [PubMed] [Google Scholar]
- Trzebiatowski JR, and Escalante-Semerena JC (1997). Purification and characterization of CobT, the nicotinate-mononucleotide:5,6-dimethylbenzimidazole phosphoribosyltransferase enzyme from Salmonella typhimurium LT2. J. Biol. Chem 272, 17662–17667. [DOI] [PubMed] [Google Scholar]
- Tsai C-F, Wang Y-T, Yen H-Y, Tsou C-C, Ku W-C, Lin P-Y, Chen H-Y, Nesvizhskii AI, Ishihama Y, and Chen Y-J (2015). Large-scale determination of absolute phosphorylation stoichiometries in human cells by motif-targeting quantitative proteomics. Nat. Commun 6, 6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang AW, and Escalante-Semerena JC (1998). CobB, a new member of the SIR2 family of eucaryotic regulatory proteins, is required to compensate for the lack of nicotinate mononucleotide:5,6-dimethylbenzimidazole phosphoribosyltransferase activity in cobT mutants during cobalamin biosynthesis in Salm. J. Biol. Chem 273, 31788–31794. [DOI] [PubMed] [Google Scholar]
- Veech RL, Guynn R, and Veloso D (1972). The time-course of the effects of ethanol on the redox and phosphorylation states of rat liver. Biochem. J 127, 387–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner GR, and Hirschey MD (2014). Nonenzymatic protein acylation as a carbon stress regulated by sirtuin deacylases. Mol. Cell 54, 5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner GR, and Payne RM (2013). Widespread and enzyme-independent Ne-acetylation and Ne-succinylation in the chemical conditions of the mitochondrial matrix. J. Biol. Chem . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner GR, Bhatt DP, O’Connell TM, Thompson JW, Dubois LG, Backos DS, Yang H, Mitchell GA, Ilkayeva OR, Stevens RD, et al. (2017). A Class of Reactive Acyl-CoA Species Reveals the Non-enzymatic Origins of Protein Acylation. Cell Metab. 25, 823–837.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y-Q, Wang H-L, Xu J, Tan J, Fu L-N, Wang J-L, Zou T-H, Sun D-F, Gao Q-Y, Chen Y-X, et al. (2018). Sirtuin5 contributes to colorectal carcinogenesis by enhancing glutaminolysis in a deglutarylation-dependent manner. Nat. Commun 9, 545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Mao A, Tang B, Zeng Q, Gao S, Liu X, Lu L, Li W, Du JX, Li J, et al. (2017). Large-Scale Identification of Protein Crotonylation Reveals Its Role in Multiple Cellular Functions. J. Proteome Res. 16, 1743–1752. [DOI] [PubMed] [Google Scholar]
- Weinert BT, Scholz C, Wagner SA, Iesmantavicius V, Su D, Daniel JA, and Choudhary C (2013). Lysine Succinylation Is a Frequently Occurring Modification in Prokaryotes and Eukaryotes and Extensively Overlaps with Acetylation. Cell Rep. 4, 842–851. [DOI] [PubMed] [Google Scholar]
- Weinert BT, Moustafa T, Iesmantavicius V, Zechner R, and Choudhary C (2015). Analysis of acetylation stoichiometry suggests that SIRT3 repairs nonenzymatic acetylation lesions. EMBO J. 34, 2620–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson DH, Lund P, and Krebs HA (1967). The redox state of free nicotinamide-adenine dinucleotide in the cytoplasm and mitochondria of rat liver. Biochem. J 103, 514–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkowski A, Thweatt J, and Smith S (2011). Mammalian ACSF3 protein is a malonyl-CoA synthetase that supplies the chain extender units for mitochondrial fatty acid synthesis. J. Biol. Chem 286, 33729–33736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Dai J, Dai L, Tan M, Cheng Z, Wu Y, Boeke JD, and Zhao Y (2012). Lysine succinylation and lysine malonylation in histones. Mol. Cell. Proteomics 11, 100–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Zhang D, Chung D, Tang Z, Huang H, Dai L, Qi S, Li J, Colak G, Chen Y, et al. (2016). Metabolic Regulation of Gene Expression by Histone Lysine beta-Hydroxybutyrylation. Mol. Cell 62, 194–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, Lamming DW, Souza-Pinto NC, Bohr VA, Rosenzweig A, et al. (2007). Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell 130, 1095–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Nagasawa K, Munch C, Xu Y, Satterstrom K, Jeong S, Hayes SD, Jedrychowski MP, Vyas FS, Zaganjor E, et al. (2016). Mitochondrial Sirtuin Network Reveals Dynamic SIRT3-Dependent Deacetylation in Response to Membrane Depolarization. Cell 167, 985–1000.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying W (2008). NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid. Redox Signal. 10, 179–206. [DOI] [PubMed] [Google Scholar]
- Yoshino J, Mills KF, Yoon MJ, and Imai S (2011). Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 14, 528–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun M, Wu J, Workman JL, and Li B (2011). Readers of histone modifications. Cell Res. 21, 564–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Ryu D, Wu Y, Gariani K, Wang X, Luan P, D’Amico D, Ropelle ER, Lutolf MP, Aebersold R, et al. (2016). NAD(+) repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 352, 1436–1443. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Piston DW, and Goodman RH (2002). Regulation of corepressor function by nuclear NADH. Science 295, 1895–1897. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Bharathi SS, Rardin MJ, Uppala R, Verdin E, Gibson BW, and Goetzman ES (2015). SIRT3 and SIRT5 regulate the enzyme activity and cardiolipin binding of very long-chain acyl-CoA dehydrogenase. PLoS One 10, e0122297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Tan M, Xie Z, Dai L, Chen Y, and Zhao Y (2010). Identification of lysine succinylation as a new post-translational modification. Nat. Chem. Biol 7, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Thomas PM, and Kelleher NL (2013). Measurement of acetylation turnover at distinct lysines in human histones identifies long-lived acetylation sites. Nat. Commun 4, 2203. [DOI] [PMC free article] [PubMed] [Google Scholar]