

Abstract
The bacterium Helicobacter pylori colonizes the gastric mucosa of half of the human population, resulting in chronic gastritis, ulcers, and cancer. We sequenced ten gene fragments from pairs of strains isolated sequentially at a mean interval of 1.8 years from 26 individuals. Several isolates had acquired small mosaic segments from other H. pylori or point mutations. The maximal mutation rate, the import size, and the frequency of recombination were calculated by using a Bayesian model. The calculations indicate that the last common ancestor of H. pylori existed at least 2,500–11,000 years ago. Imported mosaics have a median size of 417 bp, much smaller than for other bacteria, and recombination occurs frequently (60 imports spanning 25,000 bp per genome per year). Thus, the panmictic population structure of H. pylori results from very frequent recombination during mixed colonization by unrelated strains.
Keywords: Bayesian model‖horizontal genetic exchange‖genomic flux‖evolution
Helicobacter pylori colonizes the stomachs of more than one half of the world population. It is transmitted within families and occasionally from other sources (1). The sequence diversity of its housekeeping genes exceeds that of most other bacteria (2) and is associated with an exceptionally high frequency of distinct alleles. H. pylori sequences have a uniquely high homoplasy ratio (3), an indirect measure of genetic shuffling. These observations are thought to result from horizontal genetic exchange during mixed colonization by unrelated strains. Geographic structure exists within H. pylori and sequences are less related between isolates from different continents than between isolates from single countries (4).
Multiple recombinants have been isolated from one individual (5), but the frequency and other basic parameters of recombination have not yet been estimated. The age of H. pylori is unknown, and standard methods for estimating age based on sequence diversity cannot be used without an estimated molecular clock rate.
In this report we present data on the frequency of imports and mutations within paired sequential isolates from patients from two geographical areas. A model was developed that estimates recombination size, recombination rate, and mutation frequency from such data. This model can be used to set lower limits on the age of H. pylori and other bacteria with frequent recombination.
Materials and Methods
Bacterial Isolates.
Single colonies of H. pylori were isolated from sequential biopsies taken during two clinical trials. Louisiana (6): A clinical treatment trial in the 1980s in New Orleans involving patients at high risk of infection, most of whom were black. For those patients with multiple sequential isolates, the earliest and latest were used. Colombia (7): A trial addressing the effects of chemoprevention on the progression of precancerous lesions among Mestizos with multifocal atrophic gastritis in Narino in the Colombia Andes.
Nucleotide Sequencing and Sequence Analysis.
Core fragments of seven housekeeping genes (atpA, efp, mutY, ppa, trpC, ureI, yphC) and three virulence associated genes (flaA, flaB, vacA) (Fig. 1) were sequenced as described (3, 4). Regions flanking the core fragments were sequenced by using additional oligonucleotide primers (details available on request). All sequences have been deposited in the GenBank database (accession nos. AJ418065–AJ418366).
Figure 1.

Sequence comparisons of ten gene fragments from 26 pairs of isolates of H. pylori. The ten loci are indicated by tiny maps at the top of the figure (colors: core fragments, tan; flanking genes, gray; noncoding regions, white). The sources of the isolates are indicated by patient codes at the left (Colombia, three digits; Louisiana, four digits). The boxes indicate the lengths of the sequenced fragments, distinguished by color (white, identical sequences; yellow, SNPs; pink, clustered polymorphisms). Vertical lines within the boxes indicate the positions of sequence polymorphisms. The patients are separated into five groups (right) on the basis of all genetic changes between the paired isolates (1, no polymorphisms; 2, only SNPs; 3, clustered polymorphisms; 4, related isolates with numerous polymorphic fragments; 5, unrelated isolates). Mathematical analysis was only performed with data from patients in groups 1–3.
The Model.
We derived different formulas for the probabilities of three types of fragments. For each mosaic fragment, we use the lengths of three sequences: L, the polymorphic stretch, f1 and f2, the flanking sequences (Fig. 2A). The imported fragment must have spanned length L but could have been longer because of the import of nucleotides that were identical in donor and recipient. The model considers all possible imports extending to the left (d1) and right (d2) of the polymorphic stretch (Fig. 2A). The probability that the sequenced flanking stretches {min(d1, f1) [minimum of d1 and f1]; min(d2, f2)} are identical in donor, and recipient was estimated by the pident[n] function, which was derived from experimental data (see below). The summed probability of generating the observed distances for all possible import sizes is
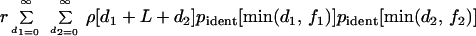 |
1 |
where r is the recombination rate and ρ[n] is a size distribution yielding the probability that a recombination event is of length n.
Figure 2.

Sequence lengths used in the model as described in Materials and Methods.
Fragments with single polymorphisms can arise when L is 1 bp or by mutation. In this case, the total probability is Eq. 1 plus the mutation rate, μ.
The third formula was needed to calculate the probability of fragments that are identical between paired isolates. Under the assumption that multiple recombinational/mutational events per gene fragment are rare, the probability of identical fragments was estimated by subtracting from 1.0 the probability of observing mutational or recombinational events:
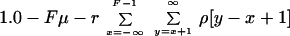 |
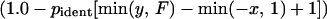 |
2 |
where F is the size of the sequenced fragment and x and y are the bounds of possible imported fragments (Fig. 2B).
pident[n] is the proportion of runs of identity between paired sequences that contain at least n nucleotides. Virtual sequences were constructed for each initial isolate consisting of all sequenced fragments joined end to end. pident[n] was estimated within and between geographic areas by pairwise comparisons from the first to the last polymorphic site within these virtual sequences. For mosaic fragments, runs of identity were tabulated within each polymorphic stretch between paired isolates.
The size distribution ρ model was implemented by using the exponential distribution.
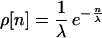 |
3 |
where λ is the mean recombination size. The exponential distribution is very similar to the geometric size distribution that has been used to model tract lengths for gene conversion in Drosophila melanogaster (8). The fit did not improve with a generalized gamma distribution, which contains an extra parameter.
Bayesian Parameter Estimation.
The total log likelihood for all data depends on the three parameters r, μ, and λ. The likelihood for individual combinations of these parameters was calculated by summing log likelihoods from the appropriate formula for each fragment. To facilitate numerical calculation, we assumed a maximum imported size of 20,000 bp. Median estimates and credibility regions were obtained from the likelihoods by using the Metropolis algorithm (9). The Metropolis algorithm wanders stochastically through parameter space, preferentially drifting toward combinations of parameters with higher probabilities. Probability calculations depend on priors that set a range of plausible parameter values. Because only 105 paired nucleotides had been sequenced, mutations would not have been detected at a frequency below 10−5. Discrete values of log10(μ) between −7 and −3 were used as a uniform prior such that half the weight is on detectable frequencies. Continuous values between −7 and −3 were used as a uniform prior on log10(r). A value of −7 would result in a 2% probability that one or more import ends had been observed while a value of −3 would result in over 100 ends, both of which are outside the range of the observed data. Continuous values of 1 to 4 were used as a uniform prior on log10(λ), which approximates the range of imported DNA in different organisms. The posterior was calculated as the amount of time spent by the Metropolis algorithm in each part of parameter space. Marginal likelihoods (posterior/prior) were calculated for each discrete value of μ. The marginal likelihood for each combination of r and λ was calculated as the sum of the likelihoods for each value of μ weighted by its posterior. Three repeated runs of 100,000 iterations each yielded values of r and λ that differed by less than 3%. Of these estimates, only that for μ is sensitive to the priors for the other parameters. Forced lower priors on r would have resulted in slightly higher estimates of μ. The program implementing this model is available on request.
Results
Sequence Differences in Sequential Isolates.
The genetic relationships of sequential isolates of H. pylori were investigated for 16 patients in New Orleans, LA (6) and 10 patients in Narino, Colombia (7). For each isolate, a total of 4,658 bp was sequenced from ten “core fragments” at unlinked chromosomal loci that encode seven housekeeping enzymes and three virulence-associated proteins. The same loci have also been sequenced for other isolates from diverse global sources (3, 4). For each core fragment that differed within a pair of isolates, ≈1 kb of flanking DNA was also sequenced to increase the reliability of the estimates of import lengths.
Of the pairs of isolates, 24 of 26 are closely related because large sequence stretches were identical in each pair (Fig. 1, parts 1–4). Indistinguishable whole-genome restriction enzyme patterns for 14 pairs from New Orleans (6) provide independent support for the relatedness within these pairs. The two remaining pairs from New Orleans (patients 1014 and 1040) must also contain closely related strains because of extensive sequence identities, even though they yielded distinct restriction enzyme patterns (6). In contrast, two pairs from Colombia contain genuinely unrelated isolates because their sequences differed at all 10 loci (Fig. 1, part 5); they were not investigated further.
Thirteen pairs of isolates (Fig. 1, part 1) contained no sequence differences. Three pairs of isolates contained only single nucleotide polymorphisms (SNPs) that might reflect mutations or short imports (Fig. 1, part 2). Six pairs of isolates differed by multiple nucleotide exchanges at one or two of the gene fragments and are likely to reflect import of sequences from other bacteria during mixed colonization (Fig. 1, part 3). This set of 22 pairs of sequences comprises the data set that was used for calculations of recombination rate, import size, and mutation rate.
Two other pairs (Fig. 1, part 4) contain related isolates, but so many loci were polymorphic that some polymorphic stretches may reflect more than one import. This possibility is supported by unusually long stretches of sequence identity within their polymorphic stretches (data not shown). The data from these two pairs could lead to biased estimates of recombination size and they were excluded from mathematical analysis.
Length of Identical Sequences Between Unrelated Isolates.
A model was devised that calculates the likelihood of the data based on the assumption that each mosaic fragment results from a single event. The likelihood depends on various parameters, including the probability that imported mosaics include flanking nucleotide stretches with identical sequences. This probability, pident, reflects the diversity of the gene pool of imported sequences. Within-population estimates of pident based on pairwise comparisons between patients in Colombia and Louisiana were almost indistinguishable up to 200 bp (Fig. 3, Colombia and Louisiana). Between-population estimates were consistently lower (Fig. 3, Columbia vs. Louisiana) than the within-population estimates, indicating that the gene pools differ between these two areas.
Figure 3.

Probability of identity (pident) between sequences versus length. The black curves are pairwise comparisons between the initial isolates within Colombia or Louisiana. The weighted average of these curves is shown in blue. The green curve is from pairwise comparisons between Louisiana and Colombia. Data from within polymorphic stretches from paired isolates (Fig. 1, part 3) are in red.
pident from the imported mosaics (Fig. 3, Mosaics) resembled the between population estimate up to lengths of 100 bp and the within population estimates between 100 and 300 bp. This estimate of pident is not suitable for calculations because it does not extend beyond 300 bp due to a limited sample size; however, it does confirm that the mosaic sequences were indeed imported from H. pylori that are similar to the populations sampled here, and justifies the use of population-based pident estimates. The parameter estimates based on within- and between-population functions of pident differed by less than 5%. The following parameter estimates were obtained by using an average within population estimate, weighted by the number of pairwise comparisons from each area (Fig. 3, weighted average).
Parameter Estimates.
The model calculates the likelihood of the sequence changes and identities in each fragment by summing the probabilities of all possible events that could result in these combinations. The total likelihood for all data depends on the mean recombination size λ, the recombination rate r, and the mutation rate μ. The most probable estimates of r and λ fell into a reasonably narrow range (Fig. 4A), despite the presence of only eight mosaics in the data set (Fig. 1, part 3). Analysis of simulated data (see the supporting information, which is published on the PNAS web site, www.pnas.org) showed that the model can accurately estimate mean recombination size when several of the sequences contain at least one end of the imported DNA fragment. According to the pident distribution for H. pylori, the end of an import has probably been reached when it is flanked by identical sequences of over 200 bp. This situation applies to all but one of the sixteen ends in Fig. 1, part 3.
Figure 4.

Parameters estimated by the model for the data in Fig. 1, parts 1–3. (A) Contour plot of marginal likelihoods (posterior/prior) of recombination parameters. (B) Marginal likelihood of mutation rates. The arrow indicates a maximum below which different mutation rates were not distinguished at the 5% level.
The estimated value of mean recombination fragment size, λ, is 417 bp (95% credibility region of 259–732 bp). This number is considerably lower than estimates of recombination size after transformation or transduction in other bacteria and is comparable to estimates for gene conversion in D. melanogaster (Table 1).
Table 1.
Average sizes (bp) of recombined fragments in different organisms
| Species | Mean | Median | Source of data | Citation |
|---|---|---|---|---|
| Drosophila subobscura | 122 | Population-based, gene conversion | 10 | |
| D. melanogaster | 352 | Laboratory, gene conversion | 8 | |
| H. pylori | 417 | 290 | Sequential isolates, transformation | This paper |
| Streptococcus pneumoniae | 2,000 | Laboratory, transformation | 11 | |
| N. meningitidis | 5,100 | Population-based, transformation | 12 | |
| S. pneumoniae | 6,000 | Population-based, transformation | 13 | |
| Bacillus subtilis | 10,000 | Laboratory, transformation | 14 | |
| E. coli | 14,000 | Laboratory, transduction | 15 |
The recombination rate, r, is the rate with which recombination events start at any particular nucleotide. The estimated value of r (6.9 × 10−5; 95% credibility region 3.5 × 10−5 to 1.2 × 10−4) indicates that each pair of sequential isolates differs on average by 114 (58) recombination events. Based on the estimates of λ and r, 2.9% (1.5–4.9%) of the genome or a total of 48,000 nucleotides differ between each sequential pair of isolates because of imported DNA.
The mutation rate, μ, was estimated as being at most 4.1 × 10−5 (Fig. 4B), resulting in an r/μ ratio of at least 1. This is only a maximal estimate of μ (and a minimal estimate of r/μ) because the three SNPs that were found might have resulted from recombination rather than mutation according to the model parameters. All three SNPs led to amino acid changes (nonsynonymous exchanges).
r/μ (0.02 [r = 7 × 10−12; μ = 3 × 10−10]) for neutral genes from Escherichia coli (16), which possesses strong clonal population structure, is at least 50-fold lower than our estimate. r/μ estimates that are only slightly lower than our minimal ratio have been calculated for a porin gene under immune selection in panmictic Neisseria gonorrhoeae (ref. 17; range 0.1 to 1.4 in different populations) and for humans (refs. 18 and 19; ≈0.5 [1.3 × 10−8/2.5 × 10−8]).
Minimal Age of H. pylori.
Over long time periods, nonsynonymous mutations are removed by selection and synonymous mutations, which are more neutral, contribute most to divergence between strains. In H. pylori, synonymous sequence polymorphisms in housekeeping genes are four times as frequent between random pairs of isolates as are nonsynonymous differences (data not shown).
A maximal synonymous molecular clock rate (max μS) can be calculated for data from paired isolates according to
 |
4 |
where ni is the number of potential synonymous sites in each fragment, ti is the time between isolation, and m is the number of observed synonymous mutations. Excluding fragments with polymorphic sequence stretches, the data set in Fig. 1 (parts 1–4) contains 22,950 identical synonymous sites between paired isolates taken on average 1.8 years apart, which corresponds to 42,608 synonymous bp years. As described elsewhere (20, 21), the maximal mutation rate can be estimated from zero observed mutations according to the Poisson distribution of e−m by substituting m with 2.996 (95% confidence limit) or 0.693 (50% confidence limit). These substitutions yield corresponding maximal synonymous clock rates of 7 × 10−5 and 1.6 × 10−5, respectively.
max μS can be used to estimate the minimal age of H. pylori. To this end, we calculated mean DS (average pairwise difference at synonymous sites) from sequences of the core fragments from a globally representative collection [220–235 isolates except for flaA (72) and flaB (103); data not shown]. After Jukes–Cantor correction, the mean DS for H. pylori is 0.182. After division by the mutation rate, this yields a minimal estimate of 11,000 years (50% confidence limit) or 2,500 years (95% limit) since the existence of the last common ancestor of H. pylori.
The synonymous clock rate per nucleotide differs between bacterial species (22). The maximal clock rate estimated here is roughly four orders of magnitude faster than that of E. coli or Buchnera (22) (or mammals). If the true clock rate were as low as the E. coli rate, the diversity in H. pylori would indicate that the last common ancestor of all H. pylori existed 40 million years ago. This seems unlikely because H. pylori should then be isolated from numerous different species of mammals, which is not the case.
Discussion
Family studies are an invaluable tool for discerning patterns of genetic linkage among humans and other eukaryotes. The analysis of sequential bacterial isolates is conceptually similar to family studies and provides an exciting approach for determining basic evolutionary parameters. Bacterial adaptation to the human host can result in amino acid changes in exposed outer membrane proteins (23) and genomic rearrangements (24), but such studies had not yet been performed with selectively neutral genes. This study presents extensive sequence data on numerous pairs of sequential isolates and demonstrates that such data can be used to determine mutation rate, recombination size, and recombination rate.
The data were obtained with pairs of bacteria isolated from adults (mean age of 49 years). The time interval between each pair of isolates and their common ancestor is at least the interval between isolation dates (Fig. 1; average of 1.8 years). This time would be greater if the adults had been colonized with both strains over a longer time period, such as since childhood, or if the strains had already coexisted in the source of infection. Thus, our recombination and mutation clock rates are both maximal estimates.
Recombination.
On average, pairs of bacteria differed by ≈100 DNA imports, corresponding to three percent of the genome or 50 kb. By further extrapolation from the average time of 1.8 years between isolation, half of the genome would have been replaced by import within 41 years (1.8 × 22.5; calculated by solving 0.97x = proportion of genome that is unrecombined). Even if the pairs of isolates were derived from a common ancestor that last existed in early childhood, half of the genome would have been replaced within 2,200 years, a surprisingly short time interval. By comparison, 10–100 million years were needed to replace 60% of the E. coli genome (25).
Recombination in other bacteria is less frequent. For example, excluding the import of sequences flanking the tbpB gene due to selection by the immune system, only one case of import was detected among three gene fragments from 200 isolates of Neisseria meningitidis during several years of epidemic and endemic disease (12). Similarly, only three recombination events over 100 kb distinguish five isolates of E. coli that are thought to have diverged within the last 2,400 years (26). No recombinations (or point mutations) were detected in six housekeeping gene fragments among 36 isolates of Yersinia pestis that have diverged in the last 1,500 years (21). Thus, the recombination frequency within H. pylori is extraordinarily high!
Import Size.
The mean size of imported fragments in H. pylori is unusually small for bacteria and is comparable to the size of gene conversion in Drosophila (Table 1). The unusually small size of imported fragments might reflect digestion of naked DNA in the gastric mucosal environment populated by H. pylori, the presence of multiple restriction endonucleases in the H. pylori genome (27), or still other factors. Combined with the high frequency of import, small imported fragments suggest that H. pylori may be lacking mechanisms that restrict DNA import from unrelated organisms. Specific sequences, such as DNA uptake sequences in Hemophilus influenzae and the neisseriae (28) or chi-sequences in diverse bacteria (29), ensure that import of DNA is more efficient from related than from unrelated organisms. Such sequences are probably lacking in H. pylori (30) and would also not be expected to occur routinely within the short coding fragments that were imported. In enteric bacteria, mismatch repair provides a barrier against import from unrelated organisms (31), but a complete mismatch repair system has also not been identified in H. pylori (32).
Possibly H. pylori does not need to defend against import of DNA from unrelated organisms. It lives under a protective mucus layer (33) in an isolated, sterile environment without microbial competitors. It has been suggested that DNA import is important for adaptation to the individual host (34) and indeed one import detected here resulted in an inactive vacA gene, due to an imported stop codon (data not shown). However, extensive sequence based evidence for the importance of import to host adaptation is still lacking (35). Furthermore, imports were not concentrated on any particular gene, unlike the situation with N. meningitidis (12). Thus, the biological significance of frequent import of short fragments remains uncertain and may largely represent neutral events.
Age of H. pylori.
No synonymous mutations were detected during a total of 42,608 synonymous bp years, leading to a maximal synonymous clock rate of 2–7 × 10−5 and a corresponding minimal age since the last common ancestor of 2,500–11,000 years. Refined estimates could be obtained through sequencing additional fragments from the same 22 pairs of isolates and/or by comparison of paired isolates from families that have been separated for longer times.
H. pylori has been isolated from humans across the globe and sequence differences between different continents indicate that these bacterial populations have been separated for millenia (4). The length of time with which H. pylori has been associated with humans is interesting in the context of important human milestones, such as the ≈13,000 years of agriculture since the end of the last ice age and the ≈50,000 years since global colonization by anatomically modern man (36). The current minimal age estimate of 2,500–11,000 years needs to be refined by one to two orders of magnitude for comparison with these milestones.
Summary.
The results presented here provide a paradigm for estimating basic evolutionary parameters of bacteria based on the use of sequential isolates. A method is described that can reliably estimate recombination rates and mean imported fragment size based on limited numbers of events, as long as the ends of the imports are included within the sequenced fragments. The method also has the potential to accurately determine mutation clock rates with larger data sets than were used in this analysis.
The data presented here provide direct evidence that the panmictic population structure of H. pylori is caused by very frequent recombination during mixed colonization by unrelated strains. H. pylori is characterized by the highly unusual combination of high import frequency and low import size. Recombination is so frequent that appreciable fractions of the entire genome are exchanged during the colonization of a single human, resulting in a highly flexible genome content and frequent shuffling of sequence polymorphisms throughout the local gene pool.
Supplementary Material
Acknowledgments
We appreciate helpful comments on the manuscript by Christine Josenhans, Giovanna Morelli, and Helga Gressmann, and expert technical assistance by Susanne Friedrich, Anne Wirsing, and Elena Katzowitsch. This work was supported by grants SU 133/3-2 and SFB 479/A5 from the Deutsche Forschungsgemeinschaft, and by the EBP network (EU contract QLK2-CT-2000-01536).
Abbreviation
- SNP
single nucleotide polymorphism
Footnotes
References
- 1.Feldman R A. In: Helicobacter pylori: Molecular and Cellular Biology. Achtman M, Suerbaum S, editors. Norfolk, England: Horizon Scientific Press; 2001. pp. 29–51. [Google Scholar]
- 2.Achtman M. In: Helicobacter pylori: Molecular and Cellular Biology. Achtman M, Suerbaum S, editors. Wymondham, U.K.: Horizon Scientific Press; 2001. pp. 311–321. [Google Scholar]
- 3.Suerbaum S, Maynard Smith J, Bapumia K, Morelli G, Smith N H, Kunstmann E, Dyrek I, Achtman M. Proc Natl Acad Sci USA. 1998;95:12619–12624. doi: 10.1073/pnas.95.21.12619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Achtman M, Azuma T, Berg D E, Ito Y, Morelli G, Pan Z-J, Suerbaum S, Thompson S, van der Ende A, van Doorn L J. Mol Microbiol. 1999;32:459–470. doi: 10.1046/j.1365-2958.1999.01382.x. [DOI] [PubMed] [Google Scholar]
- 5.Kersulyte D, Chalkauskas H, Berg D E. Mol Microbiol. 1999;31:31–43. doi: 10.1046/j.1365-2958.1999.01140.x. [DOI] [PubMed] [Google Scholar]
- 6.Taylor N S, Fox J G, Akopyants N S, Berg D E, Thompson N, Shames B, Yan L, Fontham E, Janney F, Hunter F M, et al. J Clin Microbiol. 1995;33:918–923. doi: 10.1128/jcm.33.4.918-923.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Correa P, Fontham E T, Bravo J C, Bravo L E, Ruiz B, Zarama G, Realpe J L, Malcom G T, Li D, Johnson W D, et al. J Natl Cancer Inst. 2000;92:1881–1888. doi: 10.1093/jnci/92.23.1881. [DOI] [PubMed] [Google Scholar]
- 8.Hilliker A J, Harauz G, Reaume A G, Gray M, Clark S H, Chovnick A. Genetics. 1994;137:1019–1026. doi: 10.1093/genetics/137.4.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Metropolis N, Rosenbluth A W, Rosenbluth M N, Teller A H, Teller E. J Chem Phys. 1953;21:1087–1092. [Google Scholar]
- 10.Betran E, Rozas J, Navarro A, Barbadilla A. Genetics. 1997;146:89–99. doi: 10.1093/genetics/146.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guild W R, Cato A, Jr, Lacks S. Cold Spring Harbor Symp Quant Biol. 1968;33:643–645. doi: 10.1101/sqb.1968.033.01.072. [DOI] [PubMed] [Google Scholar]
- 12.Linz B, Schenker M, Zhu P, Achtman M. Mol Microbiol. 2000;36:1049–1058. doi: 10.1046/j.1365-2958.2000.01932.x. [DOI] [PubMed] [Google Scholar]
- 13.Enright M C, Spratt B G. Mol Biol Evol. 1999;16:1687–1695. doi: 10.1093/oxfordjournals.molbev.a026082. [DOI] [PubMed] [Google Scholar]
- 14.Fornili S L, Fox M S. J Mol Biol. 1977;113:181–191. doi: 10.1016/0022-2836(77)90048-1. [DOI] [PubMed] [Google Scholar]
- 15.McKane M, Milkman R. Genetics. 1995;139:35–43. doi: 10.1093/genetics/139.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Milkman R, Bridges M M. Genetics. 1990;126:505–517. doi: 10.1093/genetics/126.3.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Posada D, Crandall K A, Nguyen M, Demma J C, Viscidi R P. Mol Biol Evol. 2000;17:423–436. doi: 10.1093/oxfordjournals.molbev.a026322. [DOI] [PubMed] [Google Scholar]
- 18.Yu A, Zhao C, Fan Y, Jang W, Mungall A J, Deloukas P, Olsen A, Doggett N A, Ghebranious N, Broman K W, et al. Nature (London) 2001;409:951–953. doi: 10.1038/35057185. [DOI] [PubMed] [Google Scholar]
- 19.Nachman M W, Crowell S L. Genetics. 2000;156:297–304. doi: 10.1093/genetics/156.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rich S M, Licht M C, Hudson R R, Ayala F J. Proc Natl Acad Sci USA. 1998;95:4425–4430. doi: 10.1073/pnas.95.8.4425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Achtman M, Zurth K, Morelli G, Torrea G, Guiyoule A, Carniel E. Proc Natl Acad Sci USA. 1999;96:14043–14048. doi: 10.1073/pnas.96.24.14043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ochman H, Elwyn S, Moran N A. Proc Natl Acad Sci USA. 1999;96:12638–12643. doi: 10.1073/pnas.96.22.12638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Groeneveld K, van Alphen L, Voorter C, Eijk P P, Jansen H M, Zanen H C. Infect Immun. 1989;57:3038–3044. doi: 10.1128/iai.57.10.3038-3044.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Römling U, Schmidt K D, Tümmler B. FEMS Microbiol Lett. 1997;150:149–156. doi: 10.1111/j.1574-6968.1997.tb10363.x. [DOI] [PubMed] [Google Scholar]
- 25.Lawrence J G, Ochman H. J Mol Evol. 1997;44:383–397. doi: 10.1007/pl00006158. [DOI] [PubMed] [Google Scholar]
- 26.Guttman D S, Dykhuizen D E. Science. 1994;266:1380–1383. doi: 10.1126/science.7973728. [DOI] [PubMed] [Google Scholar]
- 27.Tomb J F, White O, Kerlavage A R, Clayton R A, Sutton G G, Fleischmann R D, Ketchum K A, Klenk H P, Gill S, Dougherty B A, et al. Nature (London) 1997;388:539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- 28.Goodman S D, Scocca J J. J Bacteriol. 1991;173:5921–5923. doi: 10.1128/jb.173.18.5921-5923.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.El Karoui M, Biaudet V, Schbath S, Gruss A. Res Microbiol. 1999;150:579–587. doi: 10.1016/s0923-2508(99)00132-1. [DOI] [PubMed] [Google Scholar]
- 30.Saunders N J, Peden J F, Moxon E R. Microbiology. 1999;145:3523–3528. doi: 10.1099/00221287-145-12-3523. [DOI] [PubMed] [Google Scholar]
- 31.Denamur E, Lecointre G, Darlu P, Tenaillon O, Acquaviva C, Sayada C, Sunjevaric I, Rothstein R, Elion J, Taddei F, et al. Cell. 2000;103:711–721. doi: 10.1016/s0092-8674(00)00175-6. [DOI] [PubMed] [Google Scholar]
- 32.Wang G, Humayun M Z, Taylor D E. Trends Microbiol. 1999;7:488–493. doi: 10.1016/s0966-842x(99)01632-7. [DOI] [PubMed] [Google Scholar]
- 33.Schreiber S, Stuben M, Josenhans C, Scheid P, Suerbaum S. Infect Immun. 1999;67:5151–5156. doi: 10.1128/iai.67.10.5151-5156.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Montecucco C, Rappuoli R. Nat Rev Mol Cell Biol. 2001;2:457–466. doi: 10.1038/35073084. [DOI] [PubMed] [Google Scholar]
- 35.Kuipers E J, Israel D A, Kusters J G, Gerrits M M, Weel J, van Der E A, Der Hulst R W, Wirth H P, Hook-Nikanne J, Thompson S A, et al. J Infect Dis. 2000;181:273–282. doi: 10.1086/315173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Diamond J. Guns, Germs and Steel. London: Jonathan Cape; 1997. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.