

Abstract
In breast cancer cells, the linoleic acid (LA), an ω-6 essential polyunsaturated fatty acid, induces a variety of biological processes, including migration and invasion. Extracellular vesicles (EVs) are structures released by normal and malignant cells into extracellular space, and their function is dependent on their cargo and the cell type from which are secreted. Particularly, the EVs from MDA-MB-231 breast cancer cells treated with LA promote an epithelial-mesenchymal-transition (EMT)-like process in mammary non-tumorigenic epithelial cells MCF10A. Here, we found that EVs isolated from supernatants of MDA-MB-231 breast cancer cells stimulated with 90 μM LA induces activation of Akt2, FAK and ERK1/2 in MCF10A cells. In addition, EVs induces migration through a PI3K, Akt and ERK1/2-dependent pathway, whereas invasion is dependent on PI3K activity.
Electronic supplementary material
The online version of this article (10.1007/s12079-018-0490-2) contains supplementary material, which is available to authorized users.
Keywords: Breast cancer, Extracellular vesicles, Linoleic acid, PI3K/Akt, Migration, Invasion, MCF10A cells
Introduction
The free fatty acids (FFAs) are utilized as source of energy and synthesis of milk lipids in the epithelial cells of mammary gland. In breast cancer cells the FFAs, through activation of their receptors, are able to mediate a variety of cellular processes including proliferation, migration and invasion (Soto-Guzman et al. 2010; Yonezawa et al. 2004). The linoleic acid (LA) is an essential polyunsaturated fatty acid (PUFA), and it represents the main PUFA in the most occidental diets. Moreover, LA can be a precursor of eicosanoids, because it is able to be converted to arachidonic acid (AA), and then AA can be converted to a variety of eicosanoids. Therefore, LA is able to induce inappropriate inflammatory responses, which contribute to development of various chronic diseases (Calder 2001; Fritsche 2008; Simopoulos 2006). Particularly, LA induces proliferation, migration and invasion in breast cancer cells and an epithelial-mesenchymal-transition (EMT)-like process in MCF10A mammary non-tumorigenic epithelial cells (Byon et al. 2009; Espinosa-Neira et al. 2011; Serna-Marquez et al. 2013, 2017; Yonezawa et al. 2008).
Extracellular vesicles (EVs) are structures enclosed by a lipid bilayer that are released by normal and malignant cells into extracellular space. The EVs function is dependent on their cargo and the cell type from which are secreted (Muralidharan-Chari et al. 2010; Penfornis et al. 2016). The EVs are a broad and heterogeneous population of vesicles, which have been classically divided by size and origin in three groups: exosomes, microvesicles and apoptotic bodies (Ciardiello et al. 2016; Iraci et al. 2016; Minciacchi et al. 2015). Exosomes are the smaller vesicles (30–100 nm), homogeneous in size and released by endosomal compartments, whereas microvesicles are a heterogeneous population of larger vesicles (100–1000 nm), released from plasma membranes via membrane blebbing (Ciardiello et al. 2016; Kowal et al. 2014; Minciacchi et al. 2015). The EVs play important roles in the pathogenesis of various diseases, such as cancer (Ciardiello et al. 2016; Webber et al. 2015). Particularly, EVs from breast cancer patients and from MDA-MB-231 breast cancer cells stimulated with LA promote an EMT-like process in MCF10A cells (Galindo-Hernandez et al. 2014, 2015).
A variety of cellular processes in both normal and tumor cells are mediated by activation of the PI3K/Akt (phosphatidylinositol 3-kinase/protein kinase B) signaling pathway, such as proliferation, growth, survival and angiogenesis (Dillon et al. 2007; Yang et al. 2016). The PI3K family consists of three lipid kinases that originates phosphoinositol lipids, which act as second messengers in a number of intracellular signaling pathways, including PDK1 and Akt activation (Liu et al. 2009; Vanhaesebroeck et al. 2010). Akt family is the primary downstream mediator of PI3K, and it is constituted for three serine-threonine kinases, namely Akt1, Akt2 and Akt3 (Clark and Toker 2014; Matheny and Adamo 2009). Activation of Akt members is given by their phosphorylation at threonine (Thr)-308, Thr-309 and Thr-305, and their phosphorylation at serine (Ser)-473, Ser-474 and Ser-472 in Akt1, Akt2 and Akt3 respectively (Clark and Toker 2014; Dillon et al. 2007; Yang et al. 2016).
The present work demonstrate that EVs isolated from supernatants of MDA-MB-231 breast cancer cells treated with 90 μM LA induces activation of Akt2, FAK and ERK1/2 in mammary non-tumorigenic epithelial cells MCF10A. In addition, EVs induces migration via PI3K, Akt and ERK1/2 activity, whereas invasion requires PI3K activity.
Materials and methods
Materials
LA sodium salt (99% purity) and A6730 were from Sigma (St. Louis MO). Wortmannin and ERK inhibitor (3-(2-aminoethyl)-5-((4-ethoxyphenyl)methylene)-2,4 thiazolidinedione hydrochloride, HCl) were from Merck Millipore (Rockland, MA). FAK antibody (Ab) C-20 was from Calbiochem-Merck Millipore (Darmstadt, Germany). LY294002, Akt2 Ab F-7, Akt1 Ab 5C10, CD9 Ab C-4 and major histocompatibility complex class I (MHC-I) Ab BRA23/9 were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Flotillin-2 Ab and basement membrane matrix (BD Matrigel) were from BD Biosciences (Bedford, MA). Phosphospecific Ab to tyrosine (Tyr)-397 of FAK (anti-p-FAK Ab) was from Invitrogren (Waltham, MA). Phosphospecific Ab E10 to Thr-202 and Tyr-204 of ERK1/2 (anti-p-ERK1/2), phosphospecific Ab to Ser-473 of Akt (anti-p-Akt Ab) and ERK1/2 Ab were from Cell Signaling Technology (Beverly, MA). Actin Ab was kindly provided for Dr. Jose-Manuel Hernandez (Cinvestav-IPN).
Cell culture
The human MDA-MB-231 breast cancer cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 3.7 g/l sodium bicarbonate, 5% fetal bovine serum (FBS) and antibiotics. The human MCF10A mammary non-tumorigenic epithelial cells were cultured in DMEM/F12 medium (3:1) supplemented with 10% FBS, 0.5 μg/ml hydrocortisone, 20 ng/ml recombinant epidermal growth factor (EGF), 10 μg/ml insulin and antibiotics. Cells were cultured in a humidified atmosphere containing 5% CO2 and 95% air at 37 °C.
The MDA-MB-231 cells were starved in DMEM without FBS for 24 h; whereas MCF10A cells were starved in DMEM/F12 without FBS, hydrocortisone, EGF and insulin for 18 h, before treatment with LA or EVs respectively.
Stimulation of MDA-MB-231 cells with LA
Cell stimulation was performed as described previously (Galindo-Hernandez et al. 2014). After starvation, 8 × 106 MDA-MB-231 cells (confluent cultures) were stimulated without or with 90 μM LA for 48 h, and then conditioned medium was collected.
Isolation of EVs from conditioned medium
Isolation of EVs from conditioned medium of MDA-MB-231 cells stimulated with LA was performed as described previously (Abache et al. 2007; Escola et al. 1998). Conditioned medium was centrifuged twice for 10 min at 600 g and supernatants were obtained. Next, supernatants were carefully aspirated and then sequentially centrifuged at 2000 g twice for 15 min, once at 10,000 g for 30 min and once at 100,000 g for 60 min (EVs fraction). The EVs fraction obtained was enriched in exosomes and microvesicles.
The absolute number of EVs was determined by using TruCOUNT tubes as described previously (Galindo-Hernandez et al. 2014).
Stimulation of MCF10A cells with EV fractions
Cultures of MCF10A cells were washed twice with PBS and stimulated with EV fractions from 8 × 106 MDA-MB-231 cells unstimulated or stimulated with 90 μM LA for 48 h (~52,450 EVs / EV fraction / Experimental condition). After stimulation, medium was collected and cells were solubilized in 0.5 ml of ice-cold RIPA buffer (50 mM HEPES pH 7.4, 150 mM NaCl, 1 mM EGT4, 1 mM sodium orthovanadate, 100 mM NaF, 10 mM sodium pyrophosphate, 10% glycerol, 1% Triton X-100, 1% sodium deoxycholate, 1.5 mM MgCl2, 0.1% SDS and 1 mM PMSF) (Fig. 1S). The protein level of samples was determined by the micro-Bradford protein assay.
Transmission electron microscopy
Transmission electron microscopy (TEM) was performed as described previously (Baran et al. 2010). EV fractions were adsorbed for 5 min on carbon coated copper grids with mesh formvar (0.3%). The grids were exposed for 30 s on a drop of 2% uranyl acetate for negative staining, and excess of fluid was removed using filter paper. The grids were air dried and analyzed using a JEM-1400 transmission electron microscope (Jeol, Japan) operated at 80 kV and supplied with a digital camera Veleta (Olympus SIS, Germany).
Immunoprecipitation
Lysates were clarified by centrifugation at 12000 rpm for 10 min. Supernatants were transferred to fresh tubes and equal amounts of protein were immunoprecipitated overnight at 4 °C with protein A-agarose linked to anti-Akt1 Ab or anti-Akt2 Ab. Immunoprecipitates were washed three times with RIPA buffer and extracted in SDS-PAGE sample buffer by boiling 5 min and resolved by SDS-PAGE.
Western blotting
Equal amounts of protein were separated by SDS-PAGE using 10% separating gels followed by transfer to nitrocellulose membranes. After transfer, membranes were blocked using 5% non-fat dried milk in PBS pH 7.2/0.1% Tween 20 (wash buffer), and incubated overnight at 4 °C with primary Ab. Membranes were washed three times with wash buffer and incubated with secondary Ab (horseradish peroxidase-conjugated Abs) (1:5000) for 2 h at 22 °C. After washing three times with wash buffer, the immunoreactive bands were visualized using ECL detection reagent. Autoradiograms were scanned and the labeled bands were quantified using the ImageJ software (NIH, USA).
Scratch-wound assay
Confluent cultures of MCF10A cells were treated for 2 h with 12 μM mitomycin C to inhibit proliferation during the experiment. Cell cultures were scratch-wounded using a sterile 200 μl pipette tip, washed twice with PBS and re-fed with DMEM/F12 containing EV fractions from 8 × 106 MDA-MB-231 cells unstimulated or stimulated with LA for 48 h (~52,450 EVs / EV fraction / Experimental condition). The progress of cell migration into the wound was photographed at 48 h using an inverted microscope coupled to a camera. Migration was quantified using the ImageJ software (NIH, USA).
Chemotactic migration assay (Boyden chamber method)
Chemotactic migration assays were performed in 24-well plates containing 12 cell culture inserts with 8 μm pore size (Costar, Corning, Inc). MCF10A cells were treated for 2 h with 12 μM mitomycin C, and cells were re-suspended in DMEM/F12 and seeded into the upper chamber at 1 × 105 cells/well. EV fractions from 8 × 106 MDA-MB-231 cells unstimulated or stimulated with LA for 48 h (~52,450 EVs / EV fraction / Experimental condition) were added to the lower chamber. After 48 h of incubation at 37 °C, nonmigrated cells were removed from the upper side of the membrane with cotton swabs, and the cells on the lower surface of the membrane were fixed in cold methanol for 5 min. The membrane was stained with 0.1% crystal violet in PBS. The dye was eluted with 200 μl of 10% acetic acid, and the absorbance at 600 nm was measured. Background value was obtained from wells without cells.
Invasion assays
Invasion assays were performed by the modified Boyden chamber method in 24-well plates containing 12 cell culture inserts with 8 μm pore size (Costar, Corning, Inc). Briefly, 50 μl BD Matrigel was added into culture inserts and kept overnight at 37 °C. MCF10A cells were plated at 1 × 105 cells per insert in serum-free DMEM/F12 on the top chamber. The lower chamber contained 600 μl DMEM/F12 with EVs from 8 × 106 MDA-MB-231 cells untreated or treated with 90 μM LA (~52,450 EVs / EV fraction / Experimental condition). Boyden chamber were incubated for 72 h at 37 °C in a 5% CO2, atmosphere and then cells and matrigel on the upper surface of membrane were removed with cotton swabs, and the cells on the lower surface of the membrane were washed and fixed in methanol for 5 min. Number of invaded cells was estimated by staining with 0.1% crystal violet in PBS. The dye was eluted with 500 μl of 10% acetic acid, and the absorbance at 600 nm was measured. Background value was obtained from wells without cells.
EVs uptake assay
EVs were labeled using the CellMasK Orange plasma membrane stain kit, according to the manufacturer’s instructions. Briefly, EV fractions (~52,450 EVs / Experimental condition) were incubated in DMEM with CellMask Orange (2.5 μg/ml) for 30 min, and they were washed twice with DMEM by centrifugation at 100,000 g for 60 min. The MCF10A cells were treated with the labeled EVs for 0.5, 1 and 4 h in the absence or presence of inhibitors, and then they were rinsed twice with PBS and fixed with 4% paraformaldehyde. Cells were trypsinized and washed twice with PBS, re-suspended in PBS-1% BSA and analyzed with a cytometer Fortessa (Dako Cytomation). Data analysis was performed with the Summit 4.3 software.
Interference RNA
Akt2 expression was silenced in MCF10A cells by using the silencer siRNAs (0.3 μM Akt2 siRNAs) kit from Santa Cruz Biotechnology, according to the manufacture’s guidelines. One control of scramble siRNAs (0.3 μM) was included.
Statistical analysis
Results are expressed as mean ± S.D. of at least three independent experiments. Data were statistically analyzed using one-way ANOVA and Knewman-Keuls’s multiple comparison test. Statistical probability of P < 0.05 was considered significant.
Results
EVs from MDA-MB-231 cells stimulated with LA induce Akt2 activation in MCF10A cells
First, we isolated EVs from conditioned medium of MDA-MB-231 cells untreated or treated with 90 μM LA for 48 h by using differential centrifugation. To substantiate that those fractions were enriched in EVs, they were analyzed by TEM and Western blotting with anti-CD9 Ab, anti-Flotillin-2 Ab and anti-MHC-I Ab. As shown in Fig. 1a, EV fractions showed a homogeneous population of spherical vesicles with sizes ranging from 30 to 300 nm, and they express CD9, Flotilin-2 and MHC-I proteins. In contrast, these proteins were undetectable in depleted medium of EVs.
Fig. 1.
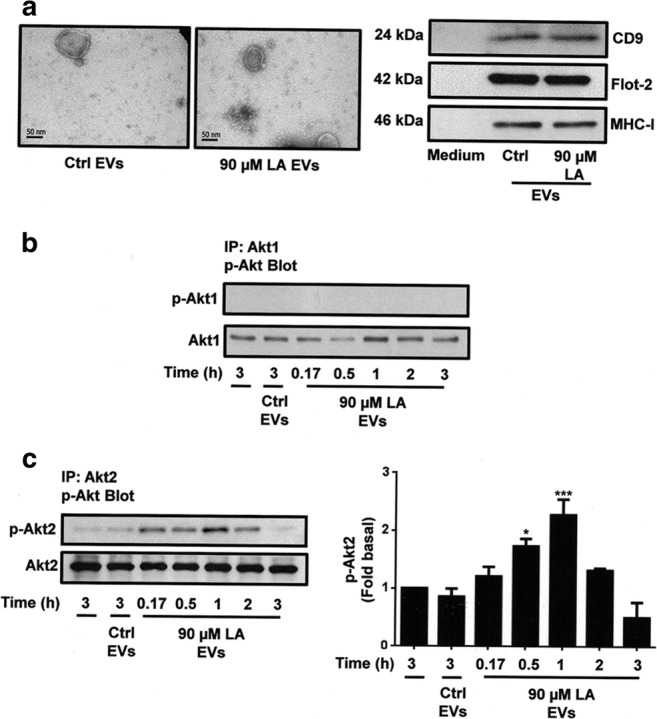
EVs from MDA-MB-231 cells treated with LA induces activation of Akt2 in MCF10A cells. Panel a. Analysis by transmission electron microscopy and Western blotting with anti-CD9 Ab, anti-Flotillin-2 (Flot-2) Ab and anti-MHC-I Ab of EV fractions from MDA-MB-231 cells unstimulated (Ctrl EVs) or stimulated with LA (90 μM LA EVs). Panel b and c. Lysates from MCF10A cells stimulated with EV fractions from MDA-MB-231 cells untreated or treated with LA for various times were analyzed by IP with anti-Akt1 Ab or anti-Akt2 Ab, and immunoprecipitates were analyzed by Western blotting with anti-p-Akt Ab. Membranes were reproved with anti-Akt1 Ab o anti-Akt2 Ab. One control of untreated MCF10A cells was included. Graph is the mean ± S.D. and is expressed as fold of p-Akt2 above Ctrl EVs value. Asterisks indicate comparisons made to Ctrl EVs. *P < 0.05, ***P < 0.001
We studied whether EVs from MDA-MB-231 cells stimulated with LA induced Akt1 and Akt2 activation, given by their phosphorylation at Ser-473 and Ser-474 respectively. Cultures of MCF10A cells were treated for various times with EV fractions from MDA-MB-231 cells unstimulated or stimulated with LA, and lysed. Cell lysates were immunoprecipitated (IP) with anti-Akt1 Ab or anti-Akt2 Ab and the immunocomplexes were analyzed by SDS-PAGE followed by Western blotting with anti-p-Akt Ab, which recognized phosphorylation at Ser-473 and Ser-474 of Akt1 and Akt2 respectively. Our findings demonstrated that treatment of MCF10A cells with EVs from MDA-MB-231 cells treated with LA did not induce an increase on Akt1 phosphorylation at Ser-473 (p-Akt1), whereas it induced an increase on Akt2 phosphorylation at Ser-474 (p-Akt2) (Fig. 1b and c, upper panel). Western blotting of same membranes with anti-Akt1 Ab and anti-Akt2 Ab were used as loading controls (Fig. 1b and c, lower panel).
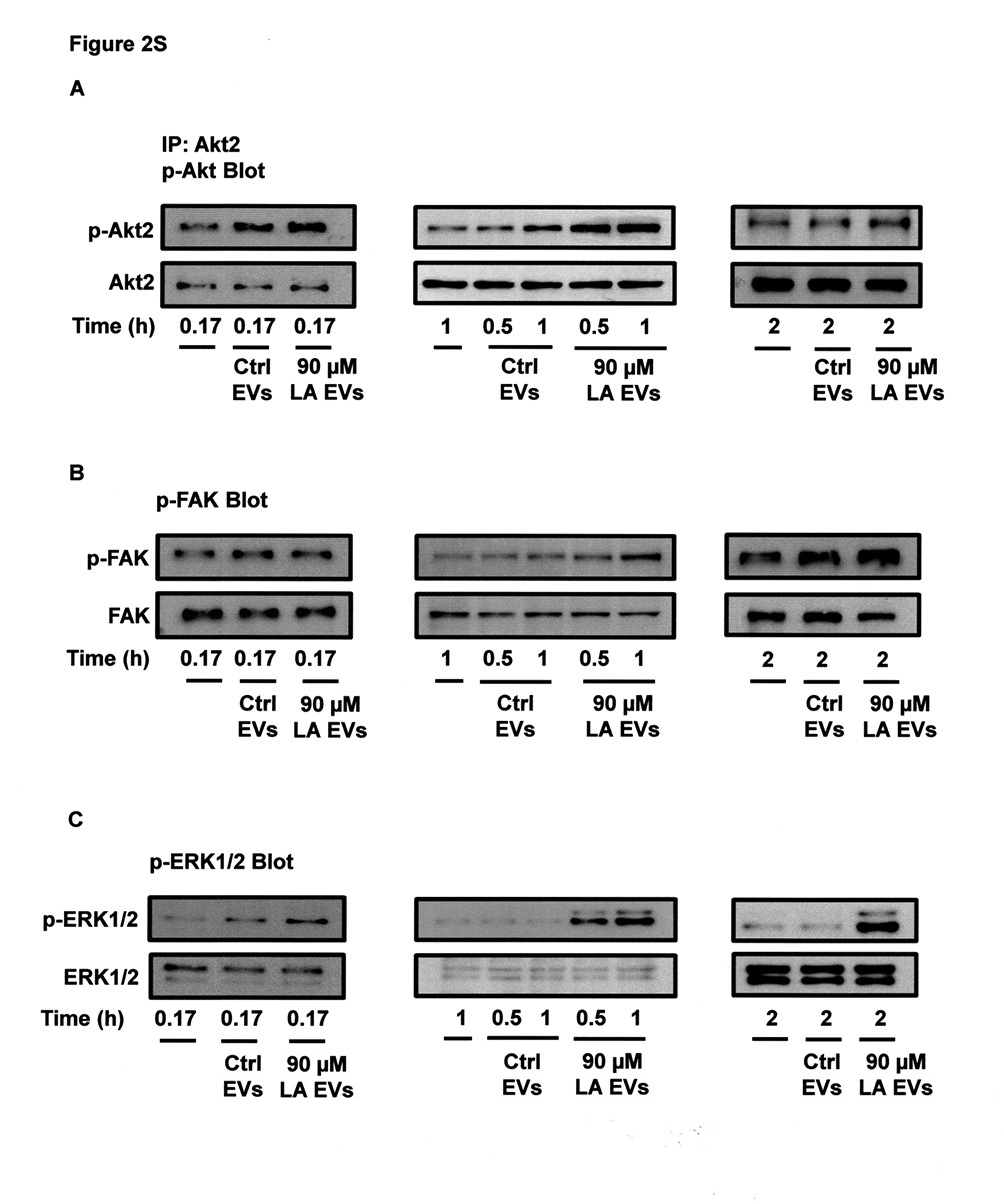
Since, EVs from MDA-MB-231 cells treated with LA induced Akt2 activation at 0.5 and 1 h of treatment; we determined whether EVs from untreated cells induced Akt2 activation at different times of stimulation. Our findings demonstrated that EVs from untreated cells induced a weak activation of Akt2 at 0.17, 0.5 and 1 h of treatment (Supplemental figure, 2S-A).
PI3K is required for Akt2 activation induced by EVs from MDA-MB-231 cells treated with LA in MCF10A cells
We determined whether Akt2 activation required PI3K activity, because Akt is a downstream mediator of PI3K. MCF10A cells were treated for 2 h with 60 nM wortmannin or 10 μM LY294002, which are specific inhibitors of PI3K activity, and then stimulated for 1 h with EVs from MDA-MB-231 cells treated with LA and lysed. Lysates were immunoprecipitated with anti-Akt2 Ab, followed by Western blotting with anti-p-Akt Ab. Our findings showed that EVs induced p-Akt2 and it required the activity of PI3K in MCF10A cells (Fig. 2a).
Fig. 2.
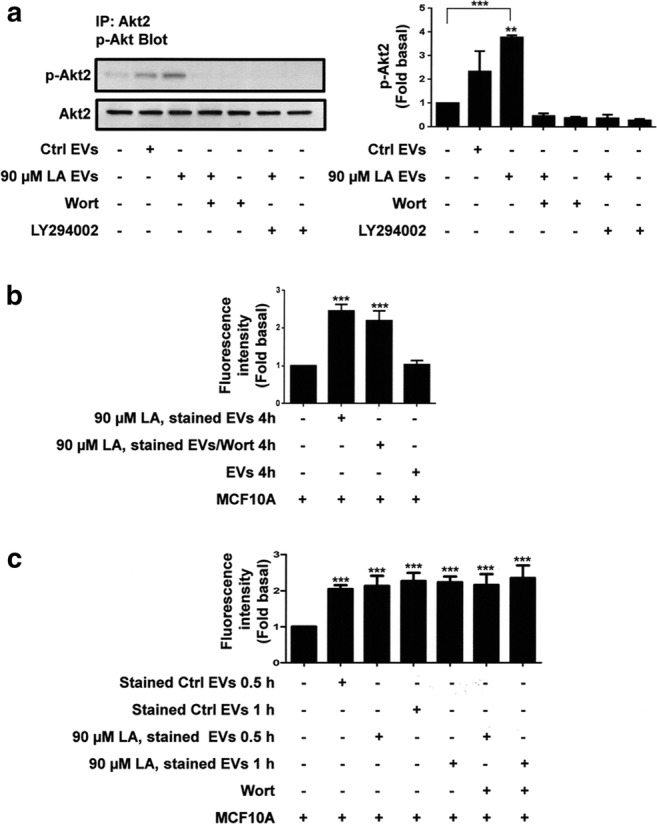
Role of PI3K in Akt2 activation induced by EVs from MDA-MB-231 cells stimulated with LA in MCF10A cells. Panel a. Lysates from MCF10A cells treated with Wortmannin (Wort) or LY294002 and stimulated with EVs from MDA-MB-231 cells untreated or treated with LA were analyzed by IP with anti-Akt2 Ab, and immunoprecipitates were analyzed by Western blotting with anti-p-Akt Ab. Membranes were reproved with anti-Akt2 Ab. One control of untreated MCF10A cells was included. Panel b. Flow cytometric analysis of MCF10A cells pretreated with Wort and treated with unstained (EVs) or stained EVs from MDA-MB-231 cells stimulated with LA for 4 h. Panel c. Flow cytometric analysis of MCF10A cells pretreated with Wort and treated with stained EVs from MDA-MB-231 cells stimulated with LA for 0.5 and 1 h. Graphs are the mean ± S.D. and are expressed as fold of p-Akt2 or mean fluorescence intensities above Ctrl EVs or unstained EVs. Asterisks indicate comparisons made to Ctrl EVs and control (unstimulated cells). **P < 0.01, ***P < 0.001
Next, we studied whether EVs from MDA-MB-231 cells treated with LA were taken up by MCF10A cells and the role of PI3K. We labeled EVs from MDA-MB-231 cells treated with LA with CellMasK Orange dye. Next, cultures of MCF10A cells were untreated or treated with 60 nM wortmannin for 2 h and then they were treated with stained EVs for 4 h. Cells were analyzed for their fluorescent intensity by flow cytometry. As illustrated in Fig. 2b, EVs from MDA-MB-231 cells treated with LA were taken up by MCF10A cells, whereas treatment with wortmannin did not inhibit the uptake of EVs (Fig. 2b).
To further substantiate that EVs were taken up by MCF10A cells at different times, we performed taken up assays using MCF10A cells untreated or treated with wortmannin and with stained EVs from MDA-MB-231 cells untreated and treated with LA for 0.5 and 1 h. Our findings demonstrated that EVs from MDA-MB-231 cells untreated and treated with LA were taken up by MCF10A cells at 0.5 and 1 of treatment, and incubation with wortmannin did not inhibit the uptake of EVs (Fig. 2c).
Role of PI3K/Akt2 in migration induced by EVs from MDA-MB-231 cells stimulated with LA in MCF10A cells
EVs from MDA-MB-231 cells treated with LA induce migration of MCF10A cells (Galindo-Hernandez et al. 2014). We studied the role of PI3K and Akt on migration. Cultures of MCF10A cells were treated with two specific PI3K inhibitors (60 nM wortmannin and 10 μM LY294002) and one specific inhibitor of Akt1/2 (2 μM A6730) for 1 h, and then they were scratch-wounded and stimulated with EVs from MDA-MB-231 cells treated with LA. Our findings showed that EVs from MDA-MB-231 cells treated with LA induced migration through a PI3K- and Akt1/2-dependent pathway in MCF10A cells (Fig. 3a and b).
Fig. 3.
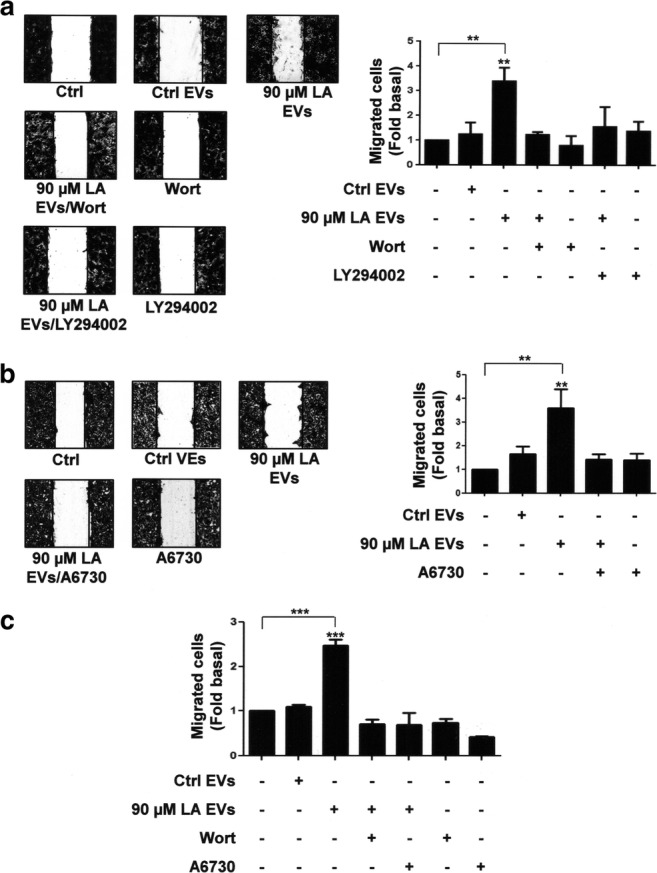
Role of PI3K and Akt in migration induced by EVs from MDA-MB-231 cells stimulated with LA in MCF10A cells. Panel a and b. Cultures of MCF10A cells were scratch-wounded and pretreated with Wort, LY294002 or A6730 and then treated with EVs from MDA-MB-231 cells untreated or treated with LA. Panel c. Migration assays were performed by using the Boyden chamber method and MCF10A cells treated with Wort and LY294002 and then stimulated with EVs from MDA-MB-231 cells untreated or treated with LA. One control of untreated MCF10A cells was included. Graphs are the mean ± S.D. and are expressed as fold of migrated cells above Ctrl EVs. Asterisks indicate comparisons made to Ctrl EVs and control. **P < 0.01, ***P < 0.001
To further substantiate our findings, cell migration assays were performed by using the Boyden chamber method and MCF10A cells treated with wortmannin and A6730, followed of stimulation with EVs from MDA-MB-231 cells treated with LA. As illustrated in Fig. 3c, migration induced by EVs requires the activity of PI3K and Akt1/2 in MCF10A cells. Next we studied the role of Akt2 in migration mediated by EVs. Akt2 expression was knocked down by using siRNA against Akt2 in MCF10A cells (Fig. 4a). Next, these MCF10A cells were scratch-wounded and stimulated with EVs from MDA-MB-231 cells untreated or treated with LA. As illustrated in Fig. 4b, migration induced by EVs was partly dependent in Akt2 expression in MCF10A cells.
Fig. 4.
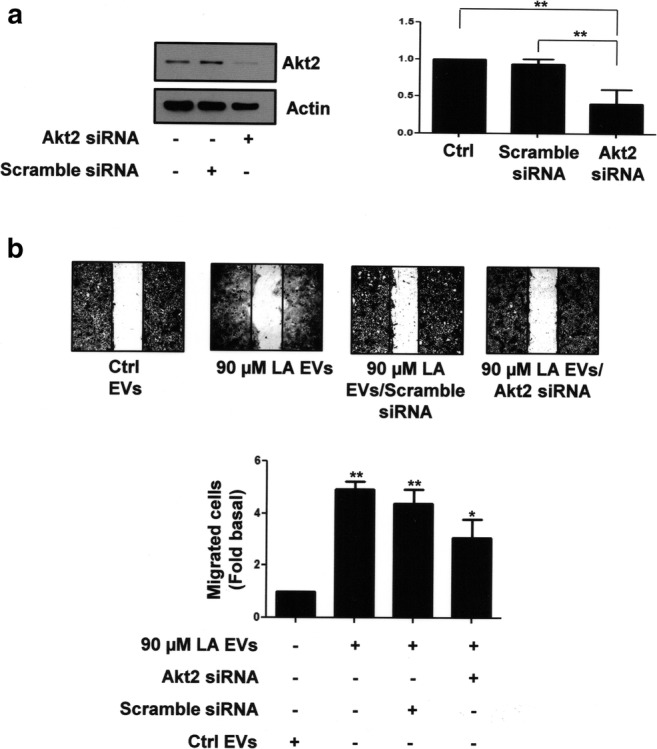
Role of Akt2 in migration induced by EVs from MDA-MB-231 cells treated with LA in MCF10A cells. Panel a. Akt2 expression of MCF10A cells transfected with Akt2-specific or scramble siRNAs was analyzed by Western blotting with anti-Akt2 Ab. One control of actin was included. Graph is the mean ± S.D. and is expressed as fold of Akt2 under Ctrl. Panel b. Migration assays were performed with MCF10A cells transfected with Akt2 siRNAs or scramble siRNAs and treated with EVs from MDA-MB-231 cells untreated or treated with LA. Graph is the mean ± S.D. and is expressed as fold of migrated cells above Ctrl EVs. Asterisks indicate comparisons made to Ctrl, scramble siRNA and Ctrl EVs. *P < 0.05, **P < 0.01
EVs from MDA-MB-231 cells stimulated with LA induce invasion in MCF10A cells
Since EVs from MDA-MB-231 cells stimulated with LA induced invasion in MCF10A cells (Galindo-Hernandez et al. 2014), we determined the role of PI3K in the invasion process. By using the Boyden chamber method, invasion assays were performed with MCF10A cells treated with 60 nM wortmannin for 2 h and stimulated with EVs from MDA-MB-231 cells treated with LA. Our findings showed that treatment with wortmannin inhibited the invasion induced by EVs in MCF10A cells.
FAK and ERK1/2 play a pivotal role in migration and invasion (Di et al. 2015; Zhao and Guan 2011). We determined whether EVs from MDA-MB-231 cells treated with LA were able to induce activation of FAK and ERK1/2 in MCF10A cells. Cultures of MCF10A cells were treated for various times with EVs from MDA-MB-231 cells stimulated with LA and lysed. Lysates were analyzed by Western blotting with anti-p-FAK Ab and anti-p-ERK1/2 Ab. As illustrated in Fig. 5b and c (upper panel), EVs from MDA-MB-231 cells treated with LA induced activation of FAK and ERK1/2, given by their Tyr phosphorylation, in MCF10A cells. Membranes were reproved with anti-FAK Ab and anti-ERK1/2 Ab as loading controls (Fig. 5b and c, lower panel).
Fig. 5.

EVs from MDA-MB-231 cells stimulated with LA induces invasion of MCF10A cells through a PI3K-dependent pathway. Panel a. Invasion assays were performed with MCF10A cells pretreated with Wort and stimulated with EVs from MDA-MB-231 cells untreated or treated with LA. Representative pictures of Boyden chamber membranes are included. Panel b and c. Lysates from MCF10A cells stimulated with EVs from MDA-MB-231 cells untreated or treated with LA for various times were analyzed by Western blotting with anti-p-FAK Ab or anti-p-ERK1/2 Ab. Membranes were further analyzed with anti-FAK Ab and anti-ERK1/2 Ab. A control of untreated MFC10A cells was included. Panel d. Migration assays were performed by using the Boyden chamber method and MCF10A cells pretreated with ERK inhibitor and stimulated with EVs from MDA-MB-231 cells untreated or treated with LA. Graphs are the mean ± S.D. and are expressed as the fold of invasion, p-FAK, p-ERK1/2 or migrated cells above Ctrl EVs. Asterisks indicate comparisons made to Ctrl EVs and control. *P < 0.05, **P < 0.01, ***P < 0.001
Since, EVs from MDA-MB-231 cells treated with LA induced FAK activation at 1 h of treatment and ERK1/2 activation at 0.5, 1 and 2 h of treatment; we determined whether EVs from untreated cells also induced activation of FAK and ERK1/2 at different times of stimulation. Our findings demonstrated that EVs from untreated cells did not induce activation of FAK and ERK1/2 at 0.17, 0.5, 1 and 2 h of treatment (Supplemental figure, 2S-B,C).
Next, we studied the role of ERK1/2 in migration. By using the Boyden chamber method, migration assays were performed with MCF10A cells treated for 1 h with 100 μM ERK inhibitor, and then treated with EVs from MDA-MB-231 cells stimulated with LA. As shown in Fig. 5d, EVs from MDA-MB-231 cells treated with LA induced migration through an ERK1/2-dependent pathway in MCF10A cells.
Discussion
An increased risk of breast cancer has been related with a diet rich in fatty acids including saturated fatty acids, monounsaturated fatty acids and ω-6 PUFA (Kim et al. 2006; Schulz et al. 2008). The major PUFA in occidental diets is the LA, which has been estimated to be consumed at 15–20 g per day per person, with a plasma concentration of ~275 μM (Anderson et al. 2009; Ferrucci et al. 2006). In MDA-MB-231 breast cancer cells, stimulation with 90 μM LA induces migration and invasion (Serna-Marquez et al. 2013, 2017).
Cancer cells share proteins and genetic information through secretion and uptake of EVs, which are able to transfer bioactive molecules including nucleic acids, chemokine receptors, growth factor receptors, functional transcription factors, and other transmembrane proteins (Ciardiello et al. 2016; Minciacchi et al. 2015). We previously reported that EVs from MDA-MB-231 cells stimulated with 90 μM LA promotes an EMT-like process in MCF10A cells (Galindo-Hernandez et al. 2014). However, the signal transduction pathways that mediate migration and invasion induced by EVs from MDA-MB-231 cells treated with LA in MCF10A cells remain to be studied.
The PI3K/Akt pathway mediates several biological processes including survival and cancer development (Toker 2012). Particularly, PI3Ks mediate a variety of cellular processes, such as migration and invasion (Cantley 2002; Vivanco and Sawyers 2002). We demonstrate here that EVs from MDA-MB-231 cells treated with LA mediates migration and invasion through a PI3K-dependent activity in MCF10A cells. Interestingly, PI3K activity does not mediate the uptake of EVs from MDA-MB-231 cell untreated and treated with LA in MCF10A cells. Since, treatment with EVs from untreated MDA-MB-231 cells does not induce migration and invasion, we propose that these EVs do not induce PI3K activation.
In addition, EVs from MDA-MB-231 cells untreated and treated with LA are uptaken for MCF10A cells at similar amounts. Therefore, we propose that treatment of MDA-MB-231 cells with LA induces the presence of cargos in the EVs, which are able to mediate migration and invasion through a PI3K-dependent pathway in MCF10A cells. Supporting our proposal, exosomes derived from the human gastric cancer cell line SGC7901 induce Akt activation and proliferation in SGC7901 and BGC823 human gastric cancer cells, whereas proliferation is dependent of PI3K activity (Qu et al. 2009).
Studies in genetically engineered mouse models and breast cancer cell lines demonstrate that Akt members mediate isoform specific cell functions. In a mouse model of mammary tumorigenesis, Akt1 promotes tumorigenesis and suppresses invasion; whereas Akt2 increases invasion, metastasis and promotes an EMT process in breast and ovarian cancer cells (Clark and Toker 2014; Irie et al. 2005). We demonstrate that just the EVs from MDA-MB-231 cells treated with LA induce Akt2 activation and it requires PI3K activity in MCF10A cells. In addition, EVs induce migration, and treatment with an Akt1/2 inhibitor completely inhibits migration. However, inhibition of Akt2 expression partly inhibits migration in MCF10A cells. We propose that EVs induce migration through an Akt2-dependent pathway, but inhibition of Akt2 expression promotes that Akt1 partly mediates the migration induced by EVs in MCF10A cells. Supporting our proposal, exosomes from hepatocellular carcinoma cell lines induces migration and Akt1/2 activation in an immortalized hepatocyte line (He et al. 2015).
During EMT process, epithelial cells gain the capacity to migrate and invade through extracellular matrix, which is mediated by activation of cell surface receptors and non-receptor kinases, including FAK and ERK1/2 (Avizienyte and Frame 2005). Particularly, FAK has been associated with tumorigenesis and metastasis, because it regulates survival, migration, invasion, angiogenesis and the EMT process (Zhao and Guan 2011). In addition, ERK1/2 are associated with migration, because they phosphorylate FAK at Ser or Thr residues (Huang et al. 2004). Our results demonstrate that EVs from MDA-MB-231 cells treated with LA induce FAK and ERK1/2 activation, and that migration is dependent on ERK1/2 activity in MCF10A cells. In agreement with our findings, embryonic stem cells secrete microvesicles, which promote migration and FAK and JNK activation in trophoblast during the implantation process (Desrochers et al. 2016). Moreover, exosomes derived from SW480 colorectal cancer cells induce ERK1/2 activation and migration in hepatocellular cancer cells HepG2, whereas EVs from colorectal cancer cells induce migration via ERK1/2 and JNK activation in endothelial cells (Chiba et al. 2016; Yoon et al. 2014).
We propose that treatment of MDA-MB-231 cells with LA promotes changes in molecular composition of EVs, which induces Akt2 activation and the migration/invasion process through a PI3K/Akt-dependent pathway in MCF10A cells. Supporting our proposal, modifications in culture conditions modify the composition of EVs in endothelial and tumor cells, whereas radiation alters the cargo of exosomes released from squamous head and neck cancer cells, which promote migration of target cells (Colombo et al. 2014; Mutschelknaus et al. 2017).
In summary, our findings demonstrate, for the first time, that EVs from MDA-MB-231 cells stimulated with LA induce migration through a PI3K/Akt- and ERK1/2-dependent pathway in MCF10A cells. Moreover, EVs also promote FAK and ERK1/2 activation and invasion through a PI3K-dependent pathway. In contrast, EVs from unstimulated MDA-MB-231 cells do not induce activation of PI3K, Akt, FAK and ERK in MCF10A cells. Our findings are depicted in Fig. 6.
Fig. 6.

Model of role of PI3K/Akt and/or ERK1/2 on migration and invasion induced by treatment with EVs from MDA-MB-231 cells treated with LA in MCF10A cells
Electronic supplementary material
{kind=link}
(PNG 421 kb)
{kind=link}
(PNG 203 kb)
Acknowledgements
This work was financed by grants from CONACYT-Mexico (255429) and CONACYT-FOSISS-Mexico (Salud-2015-1-261637). E. L-O and J. R-R were financed by Predoctoral training grants from CONACYT-Mexico.
Compliance with ethical standards
Declaration of interest
Authors declare that there is not conflict of interest.
References
- Abache T, Le Naour F, Planchon S, Harper F, Boucheix C, Rubinstein E. The transferrin receptor and the tetraspanin web molecules CD9, CD81, and CD9P-1 are differentially sorted into exosomes after TPA treatment of K562 cells. J Cell Biochem. 2007;102:650–664. doi: 10.1002/jcb.21318. [DOI] [PubMed] [Google Scholar]
- Anderson SG, Sanders TA, Cruickshank JK. Plasma fatty acid composition as a predictor of arterial stiffness and mortality. Hypertension. 2009;53:839–845. doi: 10.1161/HYPERTENSIONAHA.108.123885. [DOI] [PubMed] [Google Scholar]
- Avizienyte E, Frame MC. Src and FAK signalling controls adhesion fate and the epithelial-to-mesenchymal transition. Curr Opin Cell Biol. 2005;17:542–547. doi: 10.1016/j.ceb.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Baran J, Baj-Krzyworzeka M, Weglarczyk K, Szatanek R, Zembala M, Barbasz J, Czupryna A, Szczepanik A, Zembala M (2010) Circulating tumour-derived microvesicles in plasma of gastric cancer patients. Cancer Immunol Immunother 59:841–850 [DOI] [PMC free article] [PubMed]
- Byon CH, Hardy RW, Ren C, Ponnazhagan S, Welch DR, McDonald JM, Chen Y. Free fatty acids enhance breast cancer cell migration through plasminogen activator inhibitor-1 and SMAD4. Lab Investig. 2009;89:1221–1228. doi: 10.1038/labinvest.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calder PC. Polyunsaturated fatty acids, inflammation, and immunity. Lipids. 2001;36:1007–1024. doi: 10.1007/s11745-001-0812-7. [DOI] [PubMed] [Google Scholar]
- Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- Chiba M, Watanabe N, Watanabe M, Sakamoto M, Sato A, Fujisaki M, Kubota S, Monzen S, Maruyama A, Nanashima N, Kashiwakura I, Nakamura T. Exosomes derived from SW480 colorectal cancer cells promote cell migration in HepG2 hepatocellular cancer cells via the mitogen-activated protein kinase pathway. Int J Oncol. 2016;48:305–312. doi: 10.3892/ijo.2015.3255. [DOI] [PubMed] [Google Scholar]
- Ciardiello C, Cavallini L, Spinelli C, Yang J, Reis-Sobreiro M, de Candia P, Minciacchi VR, Di Vizio D (2016) Focus on extracellular vesicles: new frontiers of cell-to-cell communication in cancer. Int J Mol Sci 17:175 [DOI] [PMC free article] [PubMed]
- Clark AR, Toker A. Signalling specificity in the Akt pathway in breast cancer. Biochem Soc Trans. 2014;42:1349–1355. doi: 10.1042/BST20140160. [DOI] [PubMed] [Google Scholar]
- Colombo M, Raposo G, Thery C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. 2014;30:255–289. doi: 10.1146/annurev-cellbio-101512-122326. [DOI] [PubMed] [Google Scholar]
- Desrochers LM, Bordeleau F, Reinhart-King CA, Cerione RA, Antonyak MA. Microvesicles provide a mechanism for intercellular communication by embryonic stem cells during embryo implantation. Nat Commun. 2016;7:11958. doi: 10.1038/ncomms11958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di J, Huang H, Qu D, Tang J, Cao W, Lu Z, Cheng Q, Yang J, Bai J, Zhang Y, Zheng J. Rap2B promotes proliferation, migration, and invasion of human breast cancer through calcium-related ERK1/2 signaling pathway. Sci Rep. 2015;5:12363. doi: 10.1038/srep12363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon RL, White DE, Muller WJ. The phosphatidyl inositol 3-kinase signaling network: implications for human breast cancer. Oncogene. 2007;26:1338–1345. doi: 10.1038/sj.onc.1210202. [DOI] [PubMed] [Google Scholar]
- Escola JM, Kleijmeer MJ, Stoorvogel W, Griffith JM, Yoshie O, Geuze HJ. Selective enrichment of tetraspan proteins on the internal vesicles of multivesicular endosomes and on exosomes secreted by human B-lymphocytes. J Biol Chem. 1998;273:20121–20127. doi: 10.1074/jbc.273.32.20121. [DOI] [PubMed] [Google Scholar]
- Espinosa-Neira R, Mejia-Rangel J, Cortes-Reynosa P, Salazar EP. Linoleic acid induces an EMT-like process in mammary epithelial cells MCF10A. Int J Biochem Cell Biol. 2011;43:1782–1791. doi: 10.1016/j.biocel.2011.08.017. [DOI] [PubMed] [Google Scholar]
- Ferrucci L, Cherubini A, Bandinelli S, Bartali B, Corsi A, Lauretani F, Martin A, Andres-Lacueva C, Senin U, Guralnik JM. Relationship of plasma polyunsaturated fatty acids to circulating inflammatory markers. J Clin Endocrinol Metab. 2006;91:439–446. doi: 10.1210/jc.2005-1303. [DOI] [PubMed] [Google Scholar]
- Fritsche KL. Too much linoleic acid promotes inflammation-doesn't it? Prostaglandins Leukot Essent Fatty Acids. 2008;79:173–175. doi: 10.1016/j.plefa.2008.09.019. [DOI] [PubMed] [Google Scholar]
- Galindo-Hernandez O, Serna-Marquez N, Castillo-Sanchez R, Salazar EP. Extracellular vesicles from MDA-MB-231 breast cancer cells stimulated with linoleic acid promote an EMT-like process in MCF10A cells. Prostaglandins Leukot Essent Fatty Acids. 2014;91:299–310. doi: 10.1016/j.plefa.2014.09.002. [DOI] [PubMed] [Google Scholar]
- Galindo-Hernandez O, Gonzales-Vazquez C, Cortes-Reynosa P, Reyes-Uribe E, Chavez-Ocana S, Reyes-Hernandez O, Sierra-Martinez M, Salazar EP (2015) Extracellular vesicles from women with breast cancer promote an epithelial-mesenchymal transition-like process in mammary epithelial cells MCF10A. Tumour Biol 36:9649–9659 [DOI] [PubMed]
- He M, Qin H, Poon TC, Sze SC, Ding X, Co NN, Ngai SM, Chan TF, Wong N. Hepatocellular carcinoma-derived exosomes promote motility of immortalized hepatocyte through transfer of oncogenic proteins and RNAs. Carcinogenesis. 2015;36:1008–1018. doi: 10.1093/carcin/bgv081. [DOI] [PubMed] [Google Scholar]
- Huang C, Jacobson K, Schaller MD. MAP kinases and cell migration. J Cell Sci. 2004;117:4619–4628. doi: 10.1242/jcs.01481. [DOI] [PubMed] [Google Scholar]
- Iraci N, Leonardi T, Gessler F, Vega B, Pluchino S (2016) Focus on extracellular vesicles: physiological role and signalling properties of extracellular membrane vesicles. Int J Mol Sci 17:171 [DOI] [PMC free article] [PubMed]
- Irie HY, Pearline RV, Grueneberg D, Hsia M, Ravichandran P, Kothari N, Natesan S, Brugge JS. Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial-mesenchymal transition. J Cell Biol. 2005;171:1023–1034. doi: 10.1083/jcb.200505087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EH, Willett WC, Colditz GA, Hankinson SE, Stampfer MJ, Hunter DJ, Rosner B, Holmes MD. Dietary fat and risk of postmenopausal breast cancer in a 20-year follow-up. Am J Epidemiol. 2006;164:990–997. doi: 10.1093/aje/kwj309. [DOI] [PubMed] [Google Scholar]
- Kowal J, Tkach M, Thery C. Biogenesis and secretion of exosomes. Curr Opin Cell Biol. 2014;29:116–125. doi: 10.1016/j.ceb.2014.05.004. [DOI] [PubMed] [Google Scholar]
- Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8:627–644. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matheny RW, Jr, Adamo ML. Current perspectives on Akt Akt-ivation and Akt-ions. Exp Biol Med (Maywood) 2009;234:1264–1270. doi: 10.3181/0904-MR-138. [DOI] [PubMed] [Google Scholar]
- Minciacchi VR, Freeman MR, Di Vizio D. Extracellular vesicles in cancer: exosomes, microvesicles and the emerging role of large oncosomes. Semin Cell Dev Biol. 2015;40:41–51. doi: 10.1016/j.semcdb.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muralidharan-Chari V, Clancy JW, Sedgwick A, D'Souza-Schorey C. Microvesicles: mediators of extracellular communication during cancer progression. J Cell Sci. 2010;123:1603–1611. doi: 10.1242/jcs.064386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutschelknaus L, Azimzadeh O, Heider T, Winkler K, Vetter M, Kell R, Tapio S, Merl-Pham J, Huber SM, Edalat L, Radulovic V, Anastasov N, Atkinson MJ, Moertl S. Radiation alters the cargo of exosomes released from squamous head and neck cancer cells to promote migration of recipient cells. Sci Rep. 2017;7:12423. doi: 10.1038/s41598-017-12403-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penfornis P, Vallabhaneni KC, Whitt J, Pochampally R. Extracellular vesicles as carriers of microRNA, proteins and lipids in tumor microenvironment. Int J Cancer. 2016;138:14–21. doi: 10.1002/ijc.29417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu JL, Qu XJ, Zhao MF, Teng YE, Zhang Y, Hou KZ, Jiang YH, Yang XH, Liu YP (2009) Gastric cancer exosomes promote tumour cell proliferation through PI3K/Akt and MAPK/ERK activation. Dig Liver Dis 41:875–880 [DOI] [PubMed]
- Schulz M, Hoffmann K, Weikert C, Nothlings U, Schulze MB, Boeing H. Identification of a dietary pattern characterized by high-fat food choices associated with increased risk of breast cancer: the European prospective investigation into cancer and nutrition (EPIC)-Potsdam study. Br J Nutr. 2008;100:942–946. doi: 10.1017/S0007114508966149. [DOI] [PubMed] [Google Scholar]
- Serna-Marquez N, Villegas-Comonfort S, Galindo-Hernandez O, Navarro-Tito N, Millan A, Salazar EP. Role of LOXs and COX-2 on FAK activation and cell migration induced by linoleic acid in MDA-MB-231 breast cancer cells. Cell Oncol (Dordr) 2013;36:65–77. doi: 10.1007/s13402-012-0114-4. [DOI] [PubMed] [Google Scholar]
- Serna-Marquez N, Diaz-Aragon R, Reyes-Uribe E, Cortes-Reynosa P, Salazar EP. Linoleic acid induces migration and invasion through FFAR4- and PI3K−/Akt-dependent pathway in MDA-MB-231 breast cancer cells. Med Oncol. 2017;34:111. doi: 10.1007/s12032-017-0969-3. [DOI] [PubMed] [Google Scholar]
- Simopoulos AP (2006) Evolutionary aspects of diet, the omega-6/omega-3 ratio and genetic variation: nutritional implications for chronic diseases. Biomed Pharmacother 60:502–507 [DOI] [PubMed]
- Soto-Guzman A, Navarro-Tito N, Castro-Sanchez L, Martinez-Orozco R, Salazar EP. Oleic acid promotes MMP-9 secretion and invasion in breast cancer cells. Clin Exp Metastasis. 2010;27:505–515. doi: 10.1007/s10585-010-9340-1. [DOI] [PubMed] [Google Scholar]
- Toker A (2012) Achieving specificity in Akt signaling in cancer. Adv Biol Regul 52:78–87 [DOI] [PMC free article] [PubMed]
- Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010;11:329–341. doi: 10.1038/nrm2882. [DOI] [PubMed] [Google Scholar]
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- Webber J, Yeung V, Clayton A. Extracellular vesicles as modulators of the cancer microenvironment. Semin Cell Dev Biol. 2015;40:27–34. doi: 10.1016/j.semcdb.2015.01.013. [DOI] [PubMed] [Google Scholar]
- Yang SX, Polley E, Lipkowitz S. New insights on PI3K/AKT pathway alterations and clinical outcomes in breast cancer. Cancer Treat Rev. 2016;45:87–96. doi: 10.1016/j.ctrv.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonezawa T, Katoh K, Obara Y. Existence of GPR40 functioning in a human breast cancer cell line, MCF-7. Biochem Biophys Res Commun. 2004;314:805–809. doi: 10.1016/j.bbrc.2003.12.175. [DOI] [PubMed] [Google Scholar]
- Yonezawa T, Haga S, Kobayashi Y, Katoh K, Obara Y. Unsaturated fatty acids promote proliferation via ERK1/2 and Akt pathway in bovine mammary epithelial cells. Biochem Biophys Res Commun. 2008;367:729–735. doi: 10.1016/j.bbrc.2007.12.190. [DOI] [PubMed] [Google Scholar]
- Yoon YJ, Kim DK, Yoon CM, Park J, Kim YK, Roh TY, Gho YS. Egr-1 activation by cancer-derived extracellular vesicles promotes endothelial cell migration via ERK1/2 and JNK signaling pathways. PLoS One. 2014;9:e115170. doi: 10.1371/journal.pone.0115170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Guan JL. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv Drug Deliv Rev. 2011;63:610–615. doi: 10.1016/j.addr.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PNG 421 kb)
(PNG 203 kb)