

Abstract
Background
Non-opioid and opioid analgesics, as over-the-counter or prescribed medications, are widely used for the management of a diverse array of pathophysiological conditions. Previous studies have demonstrated the involvement of human cytosolic sulfotransferase (SULT) SULT1A1 in the sulfation of acetaminophen, O-desmethylnaproxen (O-DMN), and tapentadol. The current study was designed to investigate the impact of single nucleotide polymorphisms (SNPs) of the human SULT1A1 gene on the sulfation of these analgesic compounds by SULT1A1 allozymes.
Methods
Human SULT1A1 genotypes were identified by database search. cDNAs corresponding to nine SULT1A1 nonsynonymous missense coding SNPs (cSNPs) were generated by site-directed mutagenesis. Recombinant wild-type and SULT1A1 allozymes were bacterially expressed and affinity-purified. Purified SULT1A1 allozymes were analyzed for sulfation activity using an established assay procedure.
Results
Compared with the wild-type enzyme, SULT1A1 allozymes were shown to display differential sulfating activities toward three analgesic compounds, acetaminophen, O-desmethylnaproxen (O-DMN), and tapentadol, as well as the prototype substrate 4NP.
Conclusion
Results obtained indicated clearly the impact of genetic polymorphisms on the drug-sulfation activity of SULT1A1 allozymes. Such information may contribute to a better understanding about the differential metabolism of acetaminophen, O-DMN, and tapentadol in individuals with different SULT1A1 genotypes.
Introduction
The non-opiate analgesics, including acetaminophen and nonsteroidal anti-inflammatory drugs (NSAIDs) such as naproxen, are among the most commonly used analgesics [1]. These analgesics along with opioids such as tapentadol are widely used in the management of different levels of pain with a stepwise progression based on pain intensity [2–4]. Combinations of analgesic drugs are commonly used for the clinical management of acute and chronic pain either as over the counter or prescribed medications [5]. Acetaminophen occupies a unique position among analgesic drugs, both for the type of pain relieved and the associated adverse effects [1]. Among the NSAIDs, naproxen is one of the oldest and most widely used drug due to its antipyretic, anti-inflammatory, and analgesic properties [2]. Tapentadol, on the other hand, is a novel and efficient centrally-acting analgesic, prescribed mostly for the management of moderate to severe chronic pain [3].
In adults, the primary metabolic pathways of acetaminophen are the phase II glucuronidation and sulfoconjugation reactions, producing inactive and harmless metabolites. In premature infants, newborns, and young infants, the primary metabolic pathway of acetaminophen has been shown to be the sulfoconjugation due to the low levels of UDP-glucuronosyltransferases enzymes (UGTs) [1, 6]. Sulfoconjugation has also been reported to play a quantitatively more important role in tapentadol metabolism during the prenatal and neonatal stages than in adults [7]. Both glucuronide and sulfate conjugates of tapentadol have been shown to lack the analgesic activity [8, 9]. For naproxen, O-desmethylnaproxen (O-DMN) has been shown to be a major phase I metabolite, which is further biotransformed by phase II glucuronidation and sulfoconjugation reactions [10]. These latter conjugates are primarily excreted in the urine as inactive metabolites [11].
Sulfation, as mediated by the cytosolic sulfotransferase (SULT), is increasingly being recognized as a metabolic reaction which is of fundamental importance in the modulation of endogenous compounds as well as the bioinactivation of a variety of xenobiotics [12–15]. Among the thirteen known human SULTs, SULT1A1, originally referred to as the thermostable phenol sulfotransferase, has been shown to be the primary enzyme responsible for the sulfation of acetaminophen [16, 17] and O-DMN [18, 19]. SULT1A1 has also been shown to be a major enzyme responsible for the sulfation of tapentadol [20]. The gene coding for SULT1A1 has been shown to be located in the short arm of chromosome 16 [21, 22,23–25]. Genomic studies have revealed single nucleotide polymorphisms (SNPs) of SULT1A1 [22–24, 26]. Such SNPs might result in amino acid substitutions that have the potential to dramatically influence protein function [26]. It is an intriguing question whether the missense coding SNPs (cSNPs) of SULT1A1 may lead to SULT1A1 allozymes with differential sulfation activity toward acetaminophen, O-DMN, and tapentadol, thereby impacting their pharmacokinetics and thus their efficacy as well as adverse effects in individuals with distinct SULT1A1 genotypes.
In this communication, we report a systematic survey of SULT1A1 SNPs deposited in several major SNP databases. Nine missense cSNPs were selected, and the corresponding SULT1A1 allozymes were generated, expressed and purified. Purified SULT1A1 allozymes were enzymatically characterized using acetaminophen, O-DMN, and tapentadol as substrates.
Materials and Methods
Materials
Acetaminophen (≥ 99% in purity), 4-nitrophenol (4NP) (≥ 99% in purity), dimethyl sulfoxide (DMSO), isopropyl β-D-1-thiogalactopyranoside (IPTG), adenosine 5′-triphosphate (ATP), N-2-hydroxylpiperazine-N′-2-ethanesulfonic acid (HEPES), and dithiothreitol (DTT) were products of Sigma-Aldrich (St. Louis, MO, USA). Tapentadol (≥ 98% in purity) was purchased from Cayman Chemical Company (Ann Arbor, MI, USA). O-DMN (≥ 98% in purity) was a product of Toronto Research Chemicals (Toronto, ON, Canada). Carrier-free sodium [35S] sulfate was from American Radiolabeled Chemicals (St. Louis, MO, USA). Recombinant human bifunctional PAPS synthase was prepared as described previously [27]. Polygram® Cellulose 300 thin-layer chromatography (TLC) plates were from Macherey-Nagel GmbH and Co. KG (Düren, Germany). PrimeSTAR® Max DNA polymerase was a product of Takara Bio (Mountain View, CA, USA). Protein molecular weight markers were from Bioland Scientific LLC (Paramount, CA, USA). Ecolume scintillation cocktail was purchased from MP Biomedicals, LLC (Irvine, CA, USA). X-Ray films were products of FUJIFILM Corporation (Tokyo, Japan). Oligonucleotide primers were custom synthesized by Eurofins Genomics (Louisville, KY, USA). QIAprep® Spin Miniprep Kit was a product of QIAGEN (Düren, Germany). All other chemicals were of the highest standards commercially available.
Identification and analysis of human SULT1A1 SNPs
A comprehensive search was performed for SULT1A1 SNPs deposited in three online databases located at the websites of, respectively, UniProt Knowledgebase (UniProtKB), the U.S. National Center for Biotechnology Information (NCBI), and the Ensembl Variation database. Additionally, the NCBI PubMed was searched for previous studies describing SULT1A1 SNPs.
Generation of cDNAs encoding selected human SULT1A1 allozymes
Site-directed mutagenesis was performed to generate specific “mutations” in the nucleotide sequence of SULT1A1 cDNA based on a previously described procedure [28]. Briefly, wild-type SULT1A1 cDNA packaged in pGEX-2TK prokaryotic expression vector was utilized as the template in conjunction with specific mutagenic primers (cf. Table 1) to amplify “mutated” SULT1A1 cDNAs coding for different SULT1A1 allozymes. The PCR amplification conditions were 12 cycles of 30 s at 94°C, 1 min at 55°C, and 7 min at 72°C. Subsequently, the plasmids that carry the mutated SULT1A1 cDNAs were individually transformed into NEB 5-α E. coli competent cells. Colony PCR followed by agarose gel electrophoresis was carried out to identify positive colonies. Positive colonies were grown for plasmid extraction utilizing a Miniprep Kit. Nucleotide sequencing was performed to verify the sequences of the “mutated” SULT1A1 cDNAs [29] before using them for protein expression.
Table 1: List of selected single nucleotide polymorphisms with their minor allele frequencies and designed mutagenic primers of the human SULT1A1 genes.
The bold underlined letters indicate altered/mutated nucleotides (SNPs).
| hSULT1A 1 allozyme | Mutagenic primer | MAF* |
|---|---|---|
| hSULT1A1 - R37Q | 5′-TGCAGAGCTTCCAGGCCCAGCCTGATGACCTGCTCAT-3′ 5′-AT GAGCAGGTCATCAGGCTGGGCCTGGAAGCTCTGCA-3′ |
0.0004 |
| hSULT1A1-P47S | 5′-CTGCTCATCAGCACCTACTCCAAGTCCGGCACTACCT-3′ 5′-AGGT AGT GCCGGACTTGGAGT AGGT GCTGATGAGCAG-3′ |
0.0002 |
| hSULT1A1-M77I | 5′-CCGAGCTCCCATCTTCATACGGGTGCCCTTCCTTGAG-3′ 5′-CTCAAGGAAGGGCACCCGTATGAAGATGGGAGCTCGG-3′ |
0.000008 |
| hSULT1A1-H149Y | 5′-TACCACATGGCCAAGGTGTACCCTGAGCCTGGGACCT-3′ 5′-AGGTCCCAGGCTCAGGGTACACCTTGGCCATGTGGTA-3′ |
0.000008 |
| hSULT1A1-Y169D | 5′-ATGGTCGGAGAAGTGTCCGACGGATCCTGGTACCAGC-3′ 5′-GCTGGTACCAGGATCCGTCGGACACTTCTCCGACCAT-3′ |
0.000008 |
| hSULT1A1 - R213H | 5′-TCCTGGAGTTTGTGGGGCACTCCCTGCCAGAGGAGAC-3′ 5′-GTCTCCTCTGGCAGGGAGTGCCCCACAAACTCCAGGA-3′ |
0.2401 |
| hSULT1A1-T227P | 5′-GACTTCGTGGTTCAGCACCCGTCGTTCAAGGAGATGA-3′ 5′-TCATCTCCTTGAACGACGGGTGCTGAACCACGAAGTC-3′ |
0.0006 |
| hSULT1A1-V243D | 5′-TGACCAACTACACCACCGACCCCCAGGAGTTCATGGA-3′ 5′-TCCATGAACTCCTGGGGGTCGGTGGTGTAGTTGGTCA-3′ |
0.00002 |
| hSULT1A1 - F247L | 5′-ACCACCGTCCCCCAGGAGCTCATGGACCACAGCATCT-3′ 5′-AGATGCTGTGGTCCATGAGCTCCTGGGGGACGGTGGT-3′ |
0.0252 |
Minor allele frequencies as mentioned in NCBI SNP database.
Expression and purification of recombinant SULT1A1 allozymes
pGEX-2TK vector harboring individual mutated SULT1A1 cDNA was transformed into BL21 E. coli competent cells for protein expression. Transformed BL21 cells were induced with IPTG for recombinant protein expression. Induced cells were collected and homogenized using an Aminco French press. Affinity chromatography using glutathione-Sepharose was performed, based on a previously established procedure [30], followed by thrombin digestion to release the recombinant SULT1A1 allozyme. The purity of the SULT1A1 allozyme thus prepared was examined using SDS-polyacrylamide gel electrophoresis (SDS-PAGE) [31, 32]. The protein concentration of the purified SULT1A1 allozymes was determined based on Bradford protein assay [33].
Sulfotransferase assay
The sulfation activity of purified recombinant human SULT1A1 allozymes toward 4NP, acetaminophen, O-DMN, and tapentadol was assayed based on a previously established protocol, with radioactive PAP[35S] as the sulfate donor [34]. Briefly, the enzymatic assay was conducted at pH 7.4 and incubated at 37°C. After a 10 min reaction, the assay mixture was terminated by heating for 3 min at 100°C, followed by TLC using a solvent system containing n-butanol: isopropanol: formic acid: water in a ratio of 3:1:1:1 (by volume) for sulfated 4NP, acetaminophen, and O-DMN, or 2:1:1:2 (by volume) for sulfated tapentadol. Upon completion of TLC, autoradiography was performed using an X-ray film to locate the radioactive spots corresponding to [35S] sulfated products. The radioactive spots located were cut out and eluted for the quantitative measurement of [35S]-radioactivity using a liquid scintillation counter. The specific activity of the SULT1A1 allozymes was calculated in the unit of nmol of sulfated product produced/min/mg of enzyme. For the kinetic studies, varying substrate concentrations, ranging from 0-100 μM for 4NP, 0-4000 μM for acetaminophen, 0-400 μM for O-DMN, and 0-500 μM for tapentadol, were tested. All assays were performed in triplicates and the data obtained were processed as described below.
Data analysis
The data obtained from the kinetic experiments were analyzed based on the non-linear regression of the Michaelis-Menten equation using the GraphPad Prism® 7 software. One-way ANOVA was performed in conjunction with Dunnett’s multiple comparison test to find data means that are significantly different from SULT1A1 wild-type enzyme.
Results
Identification and categorization of human SULT1A1 SNPs
Three online databases including the NCBI, the Ensembl Variation database, and the UniProt database, as well as relevant research articles, were searched for SULT1A1 SNPs. In total, 5765 distinct SULT1A1 SNPs were identified. The identified SULT1A1 SNPs were analyzed and categorized into coding including (synonymous, non-synonymous (missense), and nonsense SNPs) and non-coding SNPs including (introns, 3’-untranslated regions (3’UTR), and 5′-untranslated regions (5′UTR) SNPs) (cf. Table 2). The missense coding SNPs (cSNPs) identified were analyzed for locations of the amino acid changes being in the phosphosulfate binding (PSB) loop or PAPS-binding sites, substrate and/or catalysis binding sites [25, 35], and dimerization motif [36, 37]. Additionally, the impact in terms of the chemical properties of the altered amino acid residues, such as non-turn-inducing to (or from) turn-inducing residues, charged to (or from) uncharged residues, basic to (or from) acidic residues), was scrutinized. Based on these criteria, nine missense SULT1A1 cSNPs were chosen for further studies. The designated names and SNP ID numbers of these nine cSNPs are: SULT1A1-R37Q (Reference SNP (rs) rs72547527), SULT1A1-P47S (rs544820732), SULT1A1-M77I (rs765399160), SULT1A1-H149Y (rs1042008), SULT1A1-Y169D (rs767487725), SULT1A1-R213H (rs9282861), SULT1A1-T227P (rs552524124), SULT1A1-V243D (rs758145522), and SULT1A1-F247L (rs28374453). Table 1 shows these nine SULT1A1 cSNPs, their amino acid changes and locations, the documented allele frequency of individual SULT1A1 genotypes, as well as sense and antisense mutagenic primers designed for use in the PCR-amplification of the corresponding SULT1A1 cDNAs. The previously reported amino acids sequences/residues that are involved in the PAPS-binding, substrate-binding, catalysis, and dimerization, together with the locations of amino acid variations associated with the selected SULT1A1 cSNPs are depicted in Figure 1. Moreover, the locations of amino acid variations associated with above-mentioned cSNPs are illustrated in the crystal structures of SULT1A3 (Protein Data Bank code: 1LS6 [35]) (Figure S1). The cDNAs corresponding to the nine selected SULT1A1 missense cSNPs were generated and the recombinant SULT1A1 enzymes were expressed and purified as described in the Materials and Methods section. As shown in Figure 2, the SDS-PAGE electrophoretic pattern of the purified SULT1A1 allozymes revealed that the apparent molecular weights of all nine SULT1A1 allozymes were comparable to the predicted molecular weight (34.165 kDa) of the wild-type SULT1A1.
Table 2. List of single nucleotide polymorphisms in human SULT1A1 genes.
| SNP1 location | SULT1A1 |
|---|---|
| 3′-untranslated regions (3′-UTR) | 85 |
| 5′-untranslated regions (5′-UTR) | 527 |
| Intron regions | 4510 |
| Synonymous coding SNPs | 198 |
| Nonsense coding SNP | 22 |
| Missense coding SNPs | 423 |
| Total | 5765 |
SNP refers to single nucleotide polymorphism.
Figure 1.

Amino acid sequence of human SULT1A1 marked with residues/regions reported to be critical to PAPS-binding, substrate-binding, dimerization, and/or catalysis. Residues circled with a white background are those involved in PAPS-binding. Residues enclosed in diamond symbols are those involved in catalysis. Residues circled with light gray background refer to the dimerization-motif. Residues enclosed in square symbols are those involved in substrate-binding. Residues Met77, His149, Tyr169, Val243, and Phe247 are part of the L-shaped substrate-binding pocket. Residues circled with black background refer to the locations of amino acid substitutions associated with the SULT1A1 allozymes analyzed. Residues circled with dark gray background represent the substituting amino acid residues. The figure was generated using Protter, a web tool for interactive protein feature visualization [54].
Figure 2.
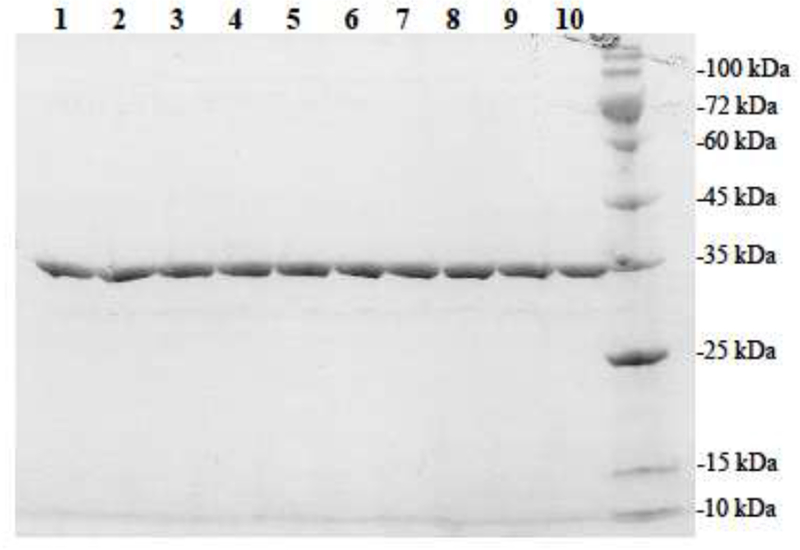
SDS gel electrophoretic pattern of the purified human SULT1A1 allozymes. SDS-PAGE was performed on a 12% gel, followed by Coomassie Blue staining. Samples analyzed in lanes 1 through 10 correspond to SULT1A1-wild-type, SULT1A1-P47S, SULT1A1-M77I, SULT1A1-H149Y, SULT1A1-Y169D, SULT1A1-T227P, SULT1A1-V243D, SULT1A1-R37Q, SULT1A1-R213H, and SULT1A1-F247L. Positions of protein molecular weight markers co-electrophoresed are indicated on the right.
Sulfation of 4NP, acetaminophen, O-DMN, and tapentadol by the wild-type SULT1A1
Sulfating properties of the wild-type SULT1A1 were first analyzed using 4NP, acetaminophen, O-DMN, and tapentadol as substrates. Figure 3 shows the concentration-dependent sulfation of each of the four substrates by the wild-type SULT1A1. Data obtained were processed using GraphPad Prism® 7 software for the determination of kinetic constants, Km, Vmax, and Vmax/Km. As shown in Table 3, the Km value (2.67 ± 0.12 μM) determined for the wild-type SULT1A1 enzyme with the prototypic substrate 4NP was lower than those (394.20 ± 17.82, 8.25 ± 0.67 and 92.54 ± 4.13 μM) determined with acetaminophen, O-DMN, and tapentadol. The Km with O-DMN (8.25 ± 0.67 μM) was more than three times higher than that with 4NP. Much higher Km values (394.20 ± 17.82 and 92.54 ± 4.13 μM) were found with acetaminophen and tapentadol, being approximately 147 and 34 times that of 4NP. Regarding the Vmax, the value determined with acetaminophen (21.58 ± 0.29) was found to be comparable with that with 4NP (21.76±0.83 nmol/min/mg). A slightly higher Vmax value (27.73 ± 0.51 nmol/min/mg) was found with O-DMN, whereas a more than 90% lower Vmax value (1.14 ± 0.01 nmol/min/mg) was found with tapentadol. Based on these data, the catalytic efficiency as reflected by Vmax/Km appeared to be lower with acetaminophen, O-DMN, and tapentadol than with 4NP.
Figure 3.
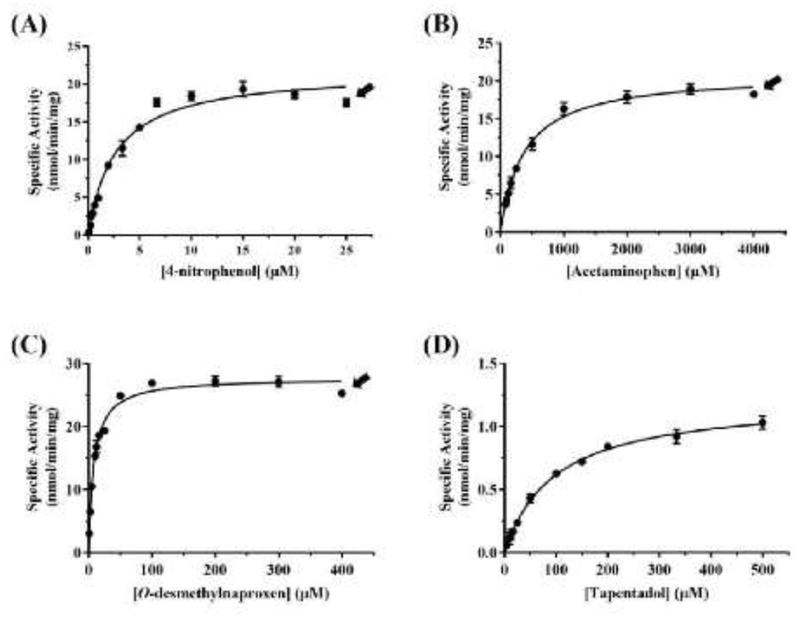
Kinetic analysis of the sulfation of the tested substrates by wild-type human SULT1A1.(A), (B), (C), and (D) show the nonlinear Michaelis–Menten curves for the sulfation of, respectively, 4NP, acetaminophen, O-DMN, and tapentadol. The nonlinear curves were generated using GraphPad Prism® 7 software. Arrow signs indicate the concentrations at which substrate inhibition started taking place. Data shown represent calculated mean ± standard deviation derived from three experiments.
Table 3.
Kinetic parameters of the human SULT1A1 wild type with 4NP, acetaminophen, O-DMN, and tapentadol as substrates.
| Substrate | Km (μM) | Vmax (nmol/min/mg) | Vmax/Km (ml/min/mg) |
|---|---|---|---|
| 4NP | 2.67 ± 0.12 | 21.76 ± 0.83 | 8.15 |
| Acetaminophen | 394.20 ± 17.82 | 21.58 ± 0.29 | 0.05 |
| O-DMN | 8.25 ± 0.67 | 27.73 ± 0.51 | 3.36 |
| Tapentadol | 92.54 ± 4.13 | 1.14 ± 0.01 | 0.01 |
Enzymatic characterization of the SULT1A1 allozymes
Purified SULT1A1 allozymes were analyzed, in comparison with the wild-type enzyme, for their sulfation activity with 4NP, acetaminophen, O-DMN, and tapentadol as substrates. Based on the results from the above-mentioned studies with the wild-type SULT1A1, three different concentrations (one much below Km, one close to Km, and one well above Km) of each of the four substrates were tested. Figures 4–7 show the results obtained from these comparative studies.
Figure 4.
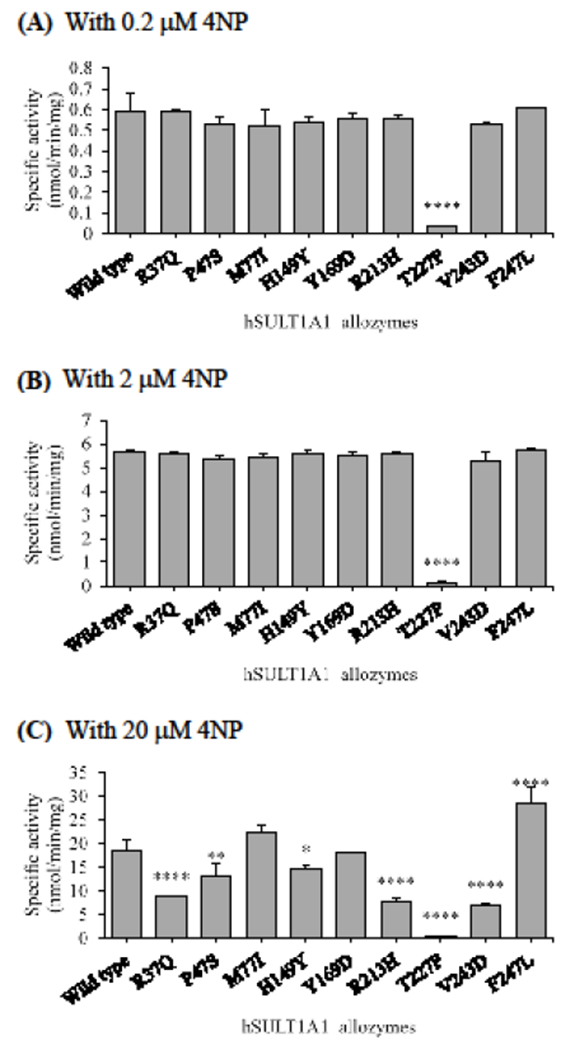
Specific activities of the sulfation of 4NP by human SULT1A1 allozymes. The concentrations of 4NP used in the enzymatic assays were 0.2 μM (A), 2 μM (B), and 20 μM (C). Data shown represent mean ± standard deviation derived from three determinations. One-way ANOVA was performed in combination with Dunnett’s multiple comparison test (statistically significant from SULTlAl-wild-type: *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001).
Figure 7.
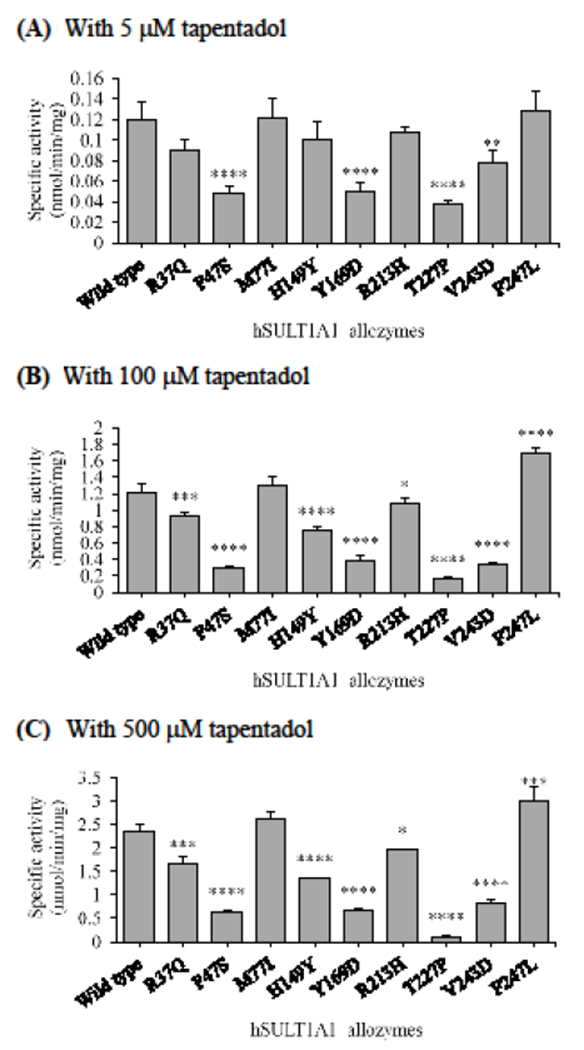
Specific activities of the sulfation of tapentadol by human SULT1A1 allozymes. The concentrations of tapentadol used in the enzymatic assays were 5 μM (A), 100 μM (B), and 500 μM (C). Data shown represent mean ± standard deviation derived from three determinations. One-way ANOVA was performed in combination with Dunnett’s multiple comparison test (statistically significant from SULTlAl-wild-type: *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001).
4NP as the substrate
As shown in Figure 4A and 4B, at low substrate concentrations (0.2 and 2 μM), all SULT1A1 allozymes displayed sulfating activities comparable to that of the wild-type enzyme, except SULT1A1-T227P which exhibited a dramatically lower activity (less than 5% of that of wild-type SULT1A1). At high substrate concentration (20 μM), SULT1A1-Y169D showed a sulfation activity comparable to that of the wild-type enzyme, while SULT1A1-M77I and SULT1A1-F247L displayed higher sulfating activities than that of the wild-type. In contrast, the other six SULT1A1 allozymes (SULT-R37Q, SULT-P47S, SULT-H149Y, SULT-R213H, SULT-T227P, and SULT-V243D) all showed significantly lower sulfating activities, with SULT1A1-T227P again displaying the lowest specific activity that was only about 2.8% of that of the wild-type enzyme (Figure 4C).
Acetaminophen as the substrate
At all three substrate concentrations (40, 400, and 1000 μM), similar patterns of differential acetaminophen-sulfating activities were found for the SULT1A1 allozymes tested (Figure 5). Of the nine human SULT1A1 allozymes, seven (SULT1A1-R37Q, SULT1A1-P47S, SULT1A1-H149Y, SULT1A1-Y169D, SULT1A1-R213H, SULT1A1-T227P, and SULT1A1-V243D) consistently exhibited lower sulfating activities than the wild-type enzyme. Among them, SULT1A1-T227P allozyme displayed barely detectable activities with all the three substrates concentrations tested. Of the remaining six allozymes, two (SULT1A1-R37Q and SULT1A1-R213H) showed sulfating activities that were more than 13% lower and four (SULT1A1-P47S, SULT1A1-H149Y, SULT1A1-Y169D, and SULT1A1-V243D) showed sulfating activities that were greater than 77% lower than the wild-type enzyme. In contrast, SULT1A1-M77I and SULT1A1-F247L displayed sulfating activities significantly higher that of the wild-type enzyme.
Figure 5.
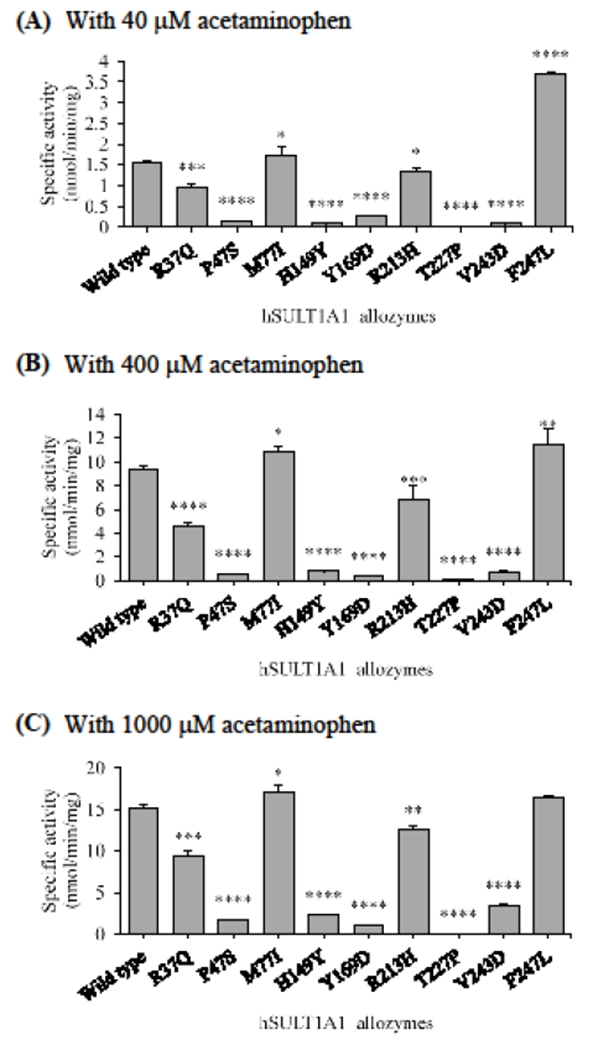
Specific activities of the sulfation of acetaminohen by human SULT1A1 allozymes. The concentrations of acetaminophen used in the enzymatic assays were 40 μM (A), 400 μM (A), and 1000 μM (C). Data shown represent mean ± standard deviation derived from three determinations. One-way ANOVA was performed in combination with Dunnett’s multiple comparison test (statistically significant from SULT1A1-wild-type: *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001).
O-DMN as the substrate
Like with acetaminophen, similar patterns of differential O-DMN-sulfating activities were found for the SULT1A1 allozymes tested at all three substrate concentrations (Figure 6). Among the nine allozymes, SULT1A1-F247L displayed higher sulfation activity than the wild-type enzyme by about 33%. SULT1A1-M77I, on the other hand, exhibited sulfating activities that were moderately higher (with 4%, 8%, and 0.12% increase at, respectively, 1, 10, and 100 μM) than the wild-type. The other seven SULT1A1 allozymes (SULT1A1-R37Q, SULT1A1-P47S, SULT1A1-H149Y, SULT1A1-Y169D, SULT1A1-R213H, SULT1A1-T227P, and SULT1A1-V243D) all showed lower sulfation activities than with the wild-type enzyme. Of these seven allozymes, SULT1A1-R37Q and SULT1A1-R213H showed approximately a 20% decrease in O-DMN-sulfation activity, while SULT1A1-H149Y and SULT1A1-V243D showed approximately a 60% decrease in O-DMN-sulfation activity, compared with the wild-type SULT1A1. SULT1A1-P47S and SULT1A1-Y169D, on the other hand, displayed approximately an 80% decrease in sulfation activity than the wild-type enzyme. The lowest activity was detected for SULT1A1-T227P, which showed a greater than 97% decrease in O-DMN-sulfation activity.
Figure 6.
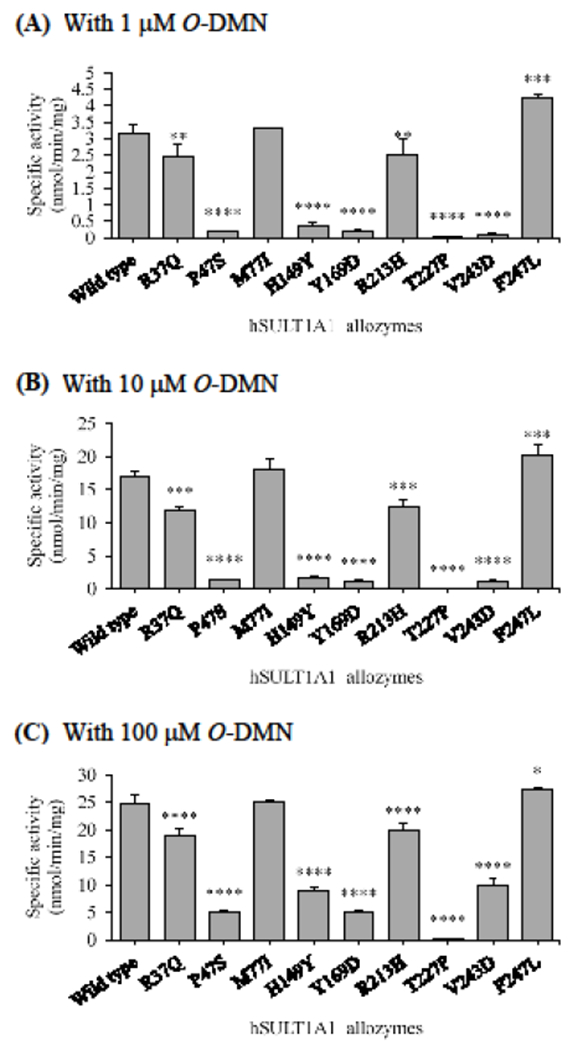
Specific activities of the sulfation of O-DMN by human SULT1A1 allozymes. The concentrations of O-DMN used in the enzymatic assays were 1 μM (A), 10 μM (B), and 100 μM (C). Data shown represent mean ± standard deviation derived from three determinations. One-way ANOVA was performed in combination with Dunnett’s multiple comparison test (statistically significant from SULTlAl-wild-type: *p<0,05, **p<0.01, ***p<0.001, and ****p<0.0001).
Tapentadol as the substrate
With 5 μM tapentadol as a substrate (Figure 7A), two allozymes, SULT1A1-M77I and SULT1A1-F247L, displayed sulfating activities comparable to that of the wild-type enzyme, while three allozymes, SULT1A1-R37Q, SULT1A1-H149Y, and SULT1A1-R213H, exhibited slightly lower (75%, 84%, and 90%, respectively, that of the wild-type) sulfating activities. The remaining four SULT1A1 allozymes exhibited much lower sulfating activities. Of these four allozymes, SULT1A1-T227D showed the lowest sulfation activity, being only 31% of that of the wild-type enzyme. At mid and high substrate concentrations (100 and 500 μM, respectively), similar patterns of tapentadol-sulfating activities were found for the SULT1A1 allozymes analyzed (Figure 7B and C). Seven of them, SULT1A1-R37Q, SULT1A1-P47S, SULT1A1-H149Y, SULT1A1-Y169D, SULT1A1-R213H, SULT1A1-T227P, and SULT1A1-V243D, displayed significantly lower sulfation activities than the wild-type enzyme. Of the seven, SULT1A1-T227P displayed the lowest tapentadol-sulfation activity, being only 13% and 5% (at, respectively, 100 μM and 500 μM substrate concentration) of that of the wild-type enzyme. Of the other six allozymes, the sulfation activities of three (SULT1A1-R37Q, SULT1A1-H149Y, and SULT1A1-R213H) were approximately 12% lower than that of the wild-type, while the sulfation activities of the other three (SULT1A1-P47S, SULT1A1-Y169D, and SULT1A1-V243D) were approximately 24%, 32%, and 28% at 100 μM, respectively, and 27%, 28%, and 36% at 500 μM, respectively, of that for the wild-type enzyme. In contrast, SULT1A1-M77I allozyme showed slightly higher sulfation activities, being 7% and 11% higher at, respectively, 100 μM and 500 μM than that of the wild-type enzyme, while SULT1A1-F247L displayed higher tapentadol-sulfation activities, being 39% and 27%, respectively, higher at 100 μM and 500 μM than that of the wild-type enzyme.
In silico analyses of the effect of the substitution on the SULT1A1 structure
Rotamer analyses of the substituted-residues were carried out using the Dunbrack backbone-dependent rotamer library [38]. Top-ranked rotamers of the substituted residues, with 0.30, 0.50, 0.82, 0.70, 0.42, 0.16, 0.99, 0.41, 0.57 probabilities for Q37, S47, T77, Y149, D169, H213, P227, D243, and L247, respectively, are shown in Figure S2. Additional in silico analyses showed the effect of substitutions on the structural interactions between the substituted residues and the surrounding residues. Among the substitutions, H149Y, V243D, and F247L appeared to change the interaction with the substrate and T227P appeared to change the interaction with PAP (PAPS). While P47S and Y169D appeared to affect the substrate-binding pockets by altering the interaction between the substrate and enzyme, M77T seemed to have smaller effect on the substrate-binding pocket. R37Q and R213H, on the other hand, appeared to affect the surface structures, which may change the overall structure of SULT1A1.
Discussion
The nonopioid and opioid analgesic drugs, such as acetaminophen, O-DMN, and tapentadol, are widely used in the clinical management of a variety of diseases and symptoms in various age groups [1–3]. Therefore, it is important to understand better the possible impact of genetic polymorphism on the metabolism hence the efficacy and toxicity of these drug compounds in different individuals. Members of the SULT enzyme family catalyze the sulfation of a multitude of endogenous and exogenous compounds [14]. Among the known human SULT enzymes, SULT1A1 is known to be a major SULT in xenobiotic metabolism [25], due in part to its broad substrate range and its wide distribution throughout the body including the brain, gastrointestinal tract, liver, lung, skin, platelets, and kidney [14, 25]. The enzyme is also important from a developmental biology perspective, since SULT1A1 has been reported to be extensively expressed at fetal stages [39–41]. As such, SULT1A1 plays an essential role in the chemical defense during fetal development, through neonatl/child development, onto adulthood [41].
Recent studies have demonstrated the involvement of SULT1A1 in the sulfation of acetaminophen [17], O-DMN [19], and tapentadol [20]. Studies have shown that genetic variability in the genes encoding enzymes, including SULTs, responsible for the metabolism of acetaminophen may be associated with risk of certain individuals for acetaminophen-induced toxicity [16, 42, 43]. It is noted that single nucleotide polymorphisms (SNPs) of the SULT1A1 gene have been reported [22, 23]. Earlier studies using platelet samples collected from human subjects with different SULT1A1 genotypes showed greater than 50-fold variations in their sulfation activity toward 4-nitrophenol, a prototype substrate for SULT1A1 [22]. In another study [44], two SULT1A1 allozymes expressed in cultured cells were found to display significantly different activity with 3,3′,5-triiodothyronine as the substrate. In the current study, a systematic database search was performed to identify the missense cSNPs of human SULT1A1 genes. Nine SULT1A1 missense cSNPs were selected, and corresponding SULT1A1 allozymes were expressed, purified, and analyzed in regard to their sulfation activity toward acetaminophen, O-DMN, and tapentadol, as well as 4NP as a prototype substrate.
Our initial study on the sulfation of 4NP, a prototype substrate, and the three tested analgesic compounds showed that the wild-type SULT1A1 displayed kinetic constants similar to those previously reported [17, 19, 20, 45–47; cf Table 2]. Based on the Km values determined with each of the four compounds, three concentrations each of these four substrates, one well below Km, one around Km, and one well above Km, were used to evaluate the effects of amino acid variations on the sulfation activity of different SULT1A1 allozymes. Activity data shown in Figures 4–7 indicated clearly the differential sulfation activity of different SULT1A1 allozymes. Several crystal structures of SULT1A1 have been reported [35, 48, 49]. Some of the structural elements involved in the functioning of the enzyme include the phosphosulfate binding loop (PSB) and other PAPS-binding regions/residues (residues 45TYPKSGTT51, Arg130, Ser138, and 257RKG259), as well as the substrate-binding residues (Ile-21, Phe24, Met77, Pro90, Ala146, His149, Tyr169, Phe247, and Val243) [25, 35, 37]. It is interesting to note that most of the SULT1A1 cSNPs examined involve amino acid changes either within or close to these important structural elements. For example, in the case of SULT1A1-P47S which showed a decrease in sulfation activity, Pro47 has been reported to be part of the PSB loop that is important for orienting the cofactor, PAPS, for in-line sulfuryl transfer to the acceptor substrate [25, 50]. The replacement of Pro (a non-polar, turning inducing amino acid residue) with Ser (a polar, non-turning inducing residue) might result in conformational changes that affect the role of the PSB loop in the sulfation reaction, and ultimately render the decrease in catalytic activity of this allozyme. In addition, in silico analyses showed that the replacement of Pro with Ser may affect the structure of substrate-binding pocket (Figure S2). Similarly, for SULT1A1-T227P, the replacement of Thr with Pro resulted in the near abolishment of sulfation activity. In SULT1A1-T227P, Pro as a turn-inducing amino acid may compel the local structure to adopt a more restricted conformation. Furthermore, the PAPS adenine ring has been shown to interact with the conserved residues Trp53 and Phe229. These interactions are stabilized by hydrogen bond interactions which are determined by the side chains of residues Thr227 and Tyr193 [25, 35]. In silico analysis demonstrated indeed that the replacement of Pro with Ser may affect the interaction between Thr227 and Trp53 and PAP, which may disrupt the stability of PAPS binding (Figure S2). In SULT1A1-T227P, these interactions are likely weakened/disrupted, thereby destabilizing the PAPS binding and leading to the dramatic decrease in sulfation activity toward all four tested substrates.
It was noted that compared to the wild-type enzyme, only slight changes in sulfation activity were detected for all SULT1A1 allozymes tested with low and mid 4NP concentrations, except SULT1A1-T227P which showed a dramatic decrease (Figure 4). In contrast, at high substrate concentration, a wide variation in 4NP-sulfation activity was detected among the nine SULT1A1 allozymes. It has been reported that the active site of SULT1A1 is capable of binding two molecules of 4NP simultaneously (cf. Figure S1) and that decreased enzyme activity at high substrate concentrations is caused by impeded catalysis when both binding sites are occupied [35]. Therefore, the differential sulfation activities of SULT1A1 allozymes at high substrate concentration might reflect their differential binding affinities for 4NP. It is noted that substrate inhibition has been reported previously for SULT1A1 [46, 51].
SULT1A1-H149Y allozyme exhibited lower sulfating activities than the wild-type enzyme toward the four substrates tested. The positively charged/basic amino acid His149 residue has been shown to be part of the substrate binding pocket in the structure of SULT1A1 [49; cf Figure S2], and is critical to the determination of the enzyme substrate specificity [35, 49]. The side chain of His149 may play a role in the stabilization of the substrate in the substrate binding pocket through the formation of electrostatic interactions with the negatively charged nitro group of the substrate, 4NP [49]. It is therefore not surprising that the replacement of His149 with Tyr resulted in dramatically lower sulfating activities, compared with the wild-type enzyme. SULT1A1-Y169D and SULT1A1-V243D also displayed decrease sulfating activities toward the tested substrates compared with the wild-type enzyme. These two allozymes involve amino acid changes to a negatively charged/acidic residue, Asp and may dramatically alter the structure of substrate-binding pocket as indicated by in silico analyses (Figure S2). Tyr169 residue has been shown to be a component of the substrate binding pocket of SULT1A1 [35, 51; cf. Figure S2]. In silico analyses suggested that replacement of Tyr169 with Asp may eliminate the hydrogen bonding between Tyr169 and Glu83 in the loop-1, leading to the conformational change of loop-2. Replacement of Tyr169 with the negatively charged Asp thereby disrupting the substrate binding pocket may underscore the 26-fold decrease in sulfation activity with acetaminophen as a substrate, compared with the wild-type enzyme (Figure 5). Val243, as part of the eight aliphatic residues in the L-shaped substrate binding pocket of the SULT1A1 structure, has been shown to be involved in the catalytic activity of SULT1A1 [35, 37, 51]. Moreover, as shown in Figure S2, replacement with Asp may cause the alteration of the conformation of loop-3. Substitution of this amino acid with the negatively charged Asp also exhibited significant lower sulfating activities with all four tested substrates (Figure 4–7).
SULT1A1-R37Q and SULT1A1-R213H, with the positively charged Arg replaced by either a Gin or His, also showed lower sulfation activities than the wild-type enzyme with all four tested substrates (Figure 4–7). Previous studies have demonstrated the importance of the Arg residues for the SULT1A1 catalytic activity [26, 35, 51]. These two SULT1A1 SNPs had been identified in earlier polymorphic studies, which showed also that the platelet samples corresponding to the SULT1A1-R213H genotype displayed much lower sulfation activity than the platelet samples corresponding to wild-type SULT1A1 genotype [22, 23, 44]. Substitution of a basic Arg residue with an uncharged Gin residue in SULT1A1-R37Q may alter the loop structure containing R37 by eliminating the hydrogen bonding with Asp39 (cf. Figure S2), which could in part explain the impairment in the sulfation activity of SULT1A1-R37Q. In SULT1A1-R213H, the substitution of Arg with His may cause altered interaction with neighboring residues, leading to changes in protein stability as well as PAPS-binding [35, 52]. While His213 in SULT1A1-R213H is not thought to contribute directly to the substrate-binding pocket, it may interact with other residues, leading to certain structural alterations in SULT1A1 protein and indirectly the substrate binding pocket [52]. Indeed, in silico analyses indicated that the replacement of Arg213 with His may dramatically alter the interaction between Arg213 and Tyr62 located in a-helix3, which may affect the interaction of PAPS and PSB-loop near the α-helix3. It is important to note that segregation analysis of data derived from family studies revealed SULT1A1-R213H genotype as a rather common polymorphism found among Caucasians, Asians, and Africans [22, 53]. This SULT1A1 genotype was found to have an allele frequency of about 30% in the Caucasians, 16.8% in the Japanese population [25, 53], and 27% in African American population [26].
To conclude, the current study aimed to systematically evaluate the effects of genetic polymorphisms on the sulfation activity of SULT1A1 allozymes towards 4NP, acetaminophen, O-DMN, and tapentadol. Enzymatic characterization of these allozymes revealed significant and sometimes dramatic differences in their catalytic activity with the tested substrates. These data obtained help provide insights into the functional consequences of polymorphic changes that may in the future aid in the interpretation of the pharmacokinetic profiles of those drugs found for individuals with different SULT1A1 genotypes.
Supplementary Material
Acknowledgments:
This work was supported in part by a grant from National Institutes of Health (Grant # R03HD071146).
Abbreviations:
- 4NP
4-nitrophenol
- O-DMN
O-desmethylnaproxen
- PAPS
3’-phosphoadenosine-5’-phosphosulfate
- SULT
cytosolic sulfotransferase
- TLC
thin-layer chromatography
- SNP
single nucleotide polymorphism
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: None declared.
References
- 1.Bertolini A, Ferrari A, Ottani A, Guerzoni S, Tacchi R, Leone S. Paracetamol: new vistas of an old drug. CNS Drug. 2006;12(3-4):250–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duggan KC, Walters MJ, Musee J, Harp JM, Kiefer JR, Oates JA, Marnett LJ. Molecular basis for cyclooxygenase inhibition by the non-steroidal anti-inflammatory drug naproxen. J Biol Chem. 2010; 5;285(45):34950–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kress HG. Tapentadol and its two mechanisms of action: is there a new pharmacological class of centrally-acting analgesics on the horizon? Eur J Pain. 2010;14(8):781–3. [DOI] [PubMed] [Google Scholar]
- 4.Constance JE, Campbell SC, Somani AA, Yellepeddi V, Owens KH, Sherwin CMT. Pharmacokinetics, pharmacodynamics, and pharmacogenetics associated with nonsteroidal anti-inflammatory drugs and opioids in pediatric cancer patients. Expert Opinion on Drug Metabolism & Toxicology. 2017;13(7):715–724. [DOI] [PubMed] [Google Scholar]
- 5.Li Wan Po A, Zhang WY. Systematic overview of co-proxamol to assess analgesic effects of addition of dextropropoxyphene to paracetamol. BMJ. 1997;13;315(7122):1565–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adjei AA, Gaedigk A, Simon SD, Weinshilboum RM, Leeder JS. Interindividual variability in acetaminophen sulfation by human fetal liver: implications for pharmacogenetic investigations of drug-induced birth defects. Birth Defects Res A Clin Mol Teratol. 2008;82(3):155–65. [DOI] [PubMed] [Google Scholar]
- 7.Terlinden R, Ossig J, Fliegert F, Lange C, Göhler K. Absorption, metabolism, and excretion of 14C-labeled tapentadol HCl in healthy male subjects. Eur J Drug Metab Pharmacokinet. 2007;32(3):163–9. [DOI] [PubMed] [Google Scholar]
- 8.Coulter C, Taruc M, Tuyay J, Moore C. Determination of tapentadol and its metabolite N-desmethyltapentadol in urine and oral fluid using liquid chromatography with tandem mass spectral detection. J Anal Toxicol. 2010;34(8):458–63. [DOI] [PubMed] [Google Scholar]
- 9.Singh DR, Nag K, Shetti AN, Krishnaveni N. Tapentadol hydrochloride: A novel analgesic. Saudi J Anaesth. 2013;7(3):322–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Segre EJ. Naproxen metabolism in man. J Clin Pharmacol. 1975;15(4 Pt. 2):316–23. [DOI] [PubMed] [Google Scholar]
- 11.Davies NM and Anderson KE. Clinical pharmacokinetics of naproxen. Clin Pharmacokinet. 1997;32(4):268–93. [DOI] [PubMed] [Google Scholar]
- 12.Mulder GJ, Jakoby WB, editors. Drug Metabolism, London: Taylor and Francis; 1990. [Google Scholar]
- 13.Weinshilboum RM, Otterness DM, Aksoy IA, Wood TC, Her C, Raftogianis RB. Sulfation and sulfotransferases 1: Sulfotransferase molecular biology: cDNAs and genes. FASEB J. 1997;11(1):3–14. [PubMed] [Google Scholar]
- 14.Coughtrie MW. Sulfation through the looking glass--recent advances in sulfotransferase research for the curious. Pharmacogenomics J. 2002;2(5):297–308. [DOI] [PubMed] [Google Scholar]
- 15.Strott CA. Sulfonation and molecular action. Endocr Rev. 2002;23(5):703–32. [DOI] [PubMed] [Google Scholar]
- 16.Zhao L, Pickering G. Paracetamol metabolism and related genetic differences. Drug Metab Rev. 2011;43(1):41–52. [DOI] [PubMed] [Google Scholar]
- 17.Yamamoto A, Liu MY, Kurogi K, Sakakibara Y, Saeki Y, Suiko M, et al. Sulphation of acetaminophen by the human cytosolic sulfotransferases: a systematic analysis. J Biochem. 2015;158(6):497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Falany CN, Ström P, Swedmark S. Sulphation of O-desmethylnaproxen and related compounds by human cytosolic sulfotransferases. Br J Clin Pharmacol 2005;60(6):632–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alherz FA, Almarghalani DA, Hussein NA, Kurogi K, Liu MC. A reappraisal of the 6-O-desmethylnaproxen-sulfating activity of the human cytosolic sulfotransferases. Can J Physiol Pharmacol. 2017;95(6):647–651. [DOI] [PubMed] [Google Scholar]
- 20.Bairam AF, Rasool MI, Kurogi K, Liu MC. On the Molecular Basis Underlying the Metabolism of Tapentadol Through Sulfation. Eur J Drug Metab Pharmacokinet. 2017;42(5):793–800. [DOI] [PubMed] [Google Scholar]
- 21.Raftogianis RB, Her C, Weinshilboum RM. Human phenol sulfotransferase pharmacogenetics: STP1 gene cloning and structural characterization. Pharmacogenetics. 1996;6(6):473–87. [DOI] [PubMed] [Google Scholar]
- 22.Raftogianis RB, Wood TC, Otterness DM, Van Loon JA, Weinshilboum RM. Phenol sulfotransferase pharmacogenetics in humans: association of common SULT1A1 alleles with TS PST phenotype. Biochem Biophys Res Commun. 1997;239(1):298–304. [DOI] [PubMed] [Google Scholar]
- 23.Raftogianis RB, Wood TC, Weinshilboum RM. Human phenol sulfotransferases SULT1A2 and SULT1A1: genetic polymorphisms, allozyme properties, and human liver genotype-phenotype correlations. Biochem Pharmacol. 1999;58(4):605–16. [DOI] [PubMed] [Google Scholar]
- 24.Hebbring SJ, Adjei AA, Baer JL, Jenkins GD, Zhang J, Cunningham JM, et al. Human SULT1A1 gene: copy number differences and functional implications. Hum Mol Genet. 2007;16(5):463–70. [DOI] [PubMed] [Google Scholar]
- 25.Hempel N, Gamage N, Martin JL, McManus ME. Human cytosolic sulfotransferase SULT1A1. Int J Biochem Cell Biol. 2007;39(4):685–9. [DOI] [PubMed] [Google Scholar]
- 26.Hildebrandt MA, Carrington DP, Thomae BA, Eckloff BW, Schaid DJ, Yee VC, et al. Genetic diversity and function in the human cytosolic sulfotransferases. Pharmacogenomics J. 2007;7(2):133–43. [DOI] [PubMed] [Google Scholar]
- 27.Yanagisawa K, Sakakibara Y, Suiko M, Takami Y, Nakayama T, Nakajima H, et al. Cdna cloning, expression, and characterization of the human bifunctional ATP sulfurylase/adenosine 5′-phosphosulfate kinase enzyme. Biosci Biotechnol Biochem. 1998;62(5):1037–40. [DOI] [PubMed] [Google Scholar]
- 28.Hui Y, Liu MC. Sulfation of ritodrine by the human cytosolic sulfotransferases (SULTs): Effects of SULT1A3 genetic polymorphism. Eur J Pharmacol. 2015; 761:125–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 1977;74(12):5463–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harper S, Speicher DW. Purification of proteins fused to glutathione S-transferase. Methods Mol Biol. 2011; 681:259–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shapiro AL, Viñuela E, Maizel JV Jr. Molecular weight estimation of polypeptide chains by electrophoresis in SDS-polyacrylamide gels. Biochem Biophys Res Commun. 1967;28(5):815–20. [DOI] [PubMed] [Google Scholar]
- 32.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227(5259):680–5. [DOI] [PubMed] [Google Scholar]
- 33.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976; 72:248–54. [DOI] [PubMed] [Google Scholar]
- 34.Kurogi K, Chen M, Lee Y, Shi B, Yan T, Liu MY, et al. Sulfation of buprenorphine, pentazocine, and naloxone by human cytosolic sulfotransferases. Drug Metab Lett. 2012;6(2):109–15. [PubMed] [Google Scholar]
- 35.Gamage NU, Duggleby RG, Barnett AC, Tresillian M, Latham CF, Liyou NE, et al. Structure of a human carcinogen-converting enzyme, SULT1A1. Structural and kinetic implications of substrate inhibition. J Biol Chem. 2003;278(9):7655–62. [DOI] [PubMed] [Google Scholar]
- 36.Petrotchenko EV, Pedersen LC, Borchers CH, Tomer KB, Negishi M. The dimerization motif of cytosolic sulfotransferases. FEBS Lett. 2001;490(1–2):39–43. [DOI] [PubMed] [Google Scholar]
- 37.Tibbs ZE, Rohn-Glowacki KJ, Crittenden F, Guidry AL, Falany CN. Structural plasticity in the human cytosolic sulfotransferase dimer and its role in substrate selectivity and catalysis. Drug Metab Pharmacokinet. 2015;30(1):3–20. [DOI] [PubMed] [Google Scholar]
- 38.Dunbrack RL Jr. Rotamer libraries in the 21st century. Curr. Opin. Struct. Biol 2002: 2(4):431–40. [DOI] [PubMed] [Google Scholar]
- 39.Richard K, Hume R, Kaptein E, Stanley EL, Visser TJ, Coughtrie MW. Sulfation of thyroid hormone and dopamine during human development: ontogeny of phenol sulfotransferases and arylsulfatase in liver, lung, and brain. J Clin Endocrinol Metab. 2001;86(6):2734–42. [DOI] [PubMed] [Google Scholar]
- 40.Vietri M, Pietrabissa A, Mosca F, Rane A, Pacific GM. Human adult and fetal liver sulphotransferases: inhibition by mefenamic acid and salicylic acid. Xenobiotica. 2001;31(3):153–61. [DOI] [PubMed] [Google Scholar]
- 41.Stanley EL, Hume R, Coughtrie MW. Expression profiling of human fetal cytosolic sulfotransferases involved in steroid and thyroid hormone metabolism and in detoxification. Mol Cell Endocrinol. 2005;240(1–2):32–42. [DOI] [PubMed] [Google Scholar]
- 42.Patel M, Tang BK, Kalow W. Variability of acetaminophen metabolism in Caucasians and Orientals. Pharmacogenetics. 1992;2(1):38–45. [DOI] [PubMed] [Google Scholar]
- 43.Court MH, Peter I, Hazarika S, Vasiadi M, Greenblatt DJ, Lee WM. Candidate gene polymorphisms in patients with acetaminophen-induced acute liver failure. Drug Metab Dispos. 2014;42(1):28–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li X, Clemens DL, Cole JR, Anderson RJ. Characterization of human liver thermostable phenol sulfotransferase (SULT1A1) allozymes with 3,3’,5-triiodothyronine as the substrate. J Endocrinol. 2001;171(3):525–32. [DOI] [PubMed] [Google Scholar]
- 45.Tabrett CA. and Coughtrie MW. Phenol sulfotransferase 1A1 activity in human liver: kinetic properties, interindividual variation and re-evaluation of the suitability of 4-nitrophenol as a probe substrate. Biochem Pharmacol. 2003;66(11):2089–97. [DOI] [PubMed] [Google Scholar]
- 46.Wang LQ, Lehmler HJ, Robertson LW, James MO. Polychlorobiphenylols are selective inhibitors of human phenol sulfotransferase 1A1 with 4-nitrophenol as a substrate. Chem Biol Interact. 2006;159(3):235–46. [DOI] [PubMed] [Google Scholar]
- 47.Riches Z, Bloomer JC, Coughtrie MW. Comparison of 2-aminophenol and 4-nitrophenol as in vitro probe substrates for the major human hepatic sulfotransferase, SULT1A1, demonstrates improved selectivity with 2-aminophenol. Biochem Pharmacol. 2007;74(2):352–8. [DOI] [PubMed] [Google Scholar]
- 48.Gamage NU, Tsvetanov S, Duggleby RG, McManus ME, Martin JL. The structure of human SULT1A1 crystallized with estradiol. An insight into active site plasticity and substrate inhibition with multi-ring substrates. J Biol Chem. 2005;280(50):41482–6. [DOI] [PubMed] [Google Scholar]
- 49.Lu J, Li H, Zhang J, Li M, Liu MY, An X, et al. Crystal structures of SULT1A2 and SULT1A1 *3: insights into the substrate inhibition and the role of Tyr149 in SULT1A2. Biochem Biophys Res Commun. 2010;396(2):429–34. [DOI] [PubMed] [Google Scholar]
- 50.Kakuta Y, Petrotchenko EV, Pedersen LC, Negishi M. The sulfuryl transfer mechanism. Crystal structure of a vanadate complex of estrogen sulfotransferase and mutational analysis. J Biol Chem. 1998;273(42):27325–30. [DOI] [PubMed] [Google Scholar]
- 51.Barnett AC, Tsvetanov S, Gamage N, Martin JL, Duggleby RG, McManus ME. Active site mutations and substrate inhibition in human sulfotransferase 1A1 and 1A3. J Biol Chem. 2004;279(18):18799–805. [DOI] [PubMed] [Google Scholar]
- 52.Nagar S, Walther S, Blanchard RL. Sulfotransferase (SULT) 1A1 polymorphic variants *1, *2, and *3 are associated with altered enzymatic activity, cellular phenotype, and protein degradation. Mol Pharmacol. 2006;69(6):2084–92. [DOI] [PubMed] [Google Scholar]
- 53.Ozawa S, Shimizu M, Katoh T, Miyajima A, Ohno Y, Matsumoto Y, et al. Sulfating-activity and stability of cDNA-expressed allozymes of human phenol sulfotransferase, ST1A3*1 ((213) Arg) and ST1A3*2 ((213) His), both of which exist in Japanese as well as Caucasians. J Biochem. 1999;126(2):271–7. [DOI] [PubMed] [Google Scholar]
- 54.Omasits U, Ahrens CH, Müller S, Wollscheid B. Protter: interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics. 2014;30(6):884–6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.