

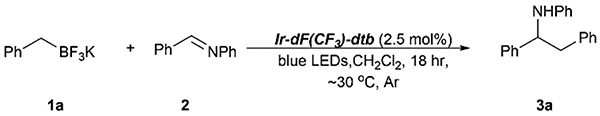
Abstract
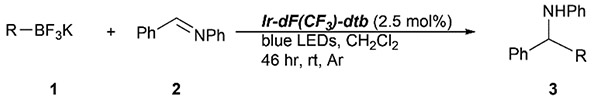
A mild, redox-neutral, alkylation of imines with potassium alkyltrifluoroborates is described. The reaction proceeds under photoredox conditions at ~30 °C with primary, secondary, and tertiary alkyltrifluoroborates, leading to alkylation products in moderate to good yield in most cases. Aryl-, vinyl-, and cyclopropyl-trifluoroborates failed to react under the reported conditions.
Graphical Abstract

Addition of organometallic reagents to imines is a general, well-established method for the synthesis of α-alkylated and arylated amines.1 Despite the attractiveness of this method, there are some drawbacks. For example, many organometallic compounds require special handling methods due to their water- and/or air-sensitivity,2 and do not exhibit sufficient tolerance toward most functional groups. Methods based on radical addition to imines circumvent many of these issues, but often require stoichiometric amounts of strong oxidants or reductants.3
Recently, the use of photoredox catalysis to promote key bond-forming steps in organic synthesis has emerged as an alternative to classical radical chemistry, and has become a practical means to achieve a number of important synthetic transformations.4 This approach relies on a catalyst that absorbs light in the visible range; the photoexcited catalyst subsequently mediates the formation of a radical from an organic substrate through a single-electron-transfer (SET) event (i.e., oxidative or reductive quenching of the photocatalyst). The resulting species may then participate in an additional SET event to regenerate the ground state photocatalyst and close the catalytic cycle. The organic radicals thus generated can engage in downstream reactions leading to the final product(s). The photocatalysts involved are often ruthenium or iridium polypyridyl complexes,4 but may also be organic molecules.5
Potassium alkyltrifluoroborates have been shown to be viable radical precursors in several visible light-promoted protocols.6 For example, Akita’s laboratory has reported radical couplings with TEMPO,7 as well as alkylations7,8 and thioalkylations9 of α,β-unsaturated carbonyls and related compounds, and Molander’s group has developed a clever single-electron transmetalation approach to Csp2–Csp3 cross-coupling,10 both using alkyl radicals derived from photo-oxidation of potassium alkyltrifluoroborates. Given their advantages as synthetic reagents, such as their stability, availability, ease of preparation, and functional group tolerance,11 our research group became interested in exploring their utility in visible light-promoted alkylations of C=N bonds. Non-photoredox additions of potassium aryl- and allyltrifluoroborates to imines12 are well-known, but there seem to be no accounts involving the direct addition of ordinary primary, secondary, or tertiary alkyl trifluoroborates to these substrates. And while there appear to be no reports to date of photoredox alkylations of imines using potassium organotrifluoroborates as alkyl radical precursors, reports of similar approaches employing alkyl bis(catecholato) silicates13 and 4-alkyl-1,4-dihydropyridines14 as alkyl radical precursors have appeared recently. In this communication, we report the results of a preliminary study employing potassium organotrifluoroborates as alkyl radical precursors in a mild, visible light-promoted, redox-neutral alkylation of imines.
We began our study by examining the reaction of potassium benzyltrifluoroborate (1a) with benzalaniline (2). Based on a survey of reduction potentials for potential photocatalysts,4b we chose to employ [4,4′-bis(tert-butyl)-2,2'-bipyridine]bis[3,5-difluoro-2-[5-(trifluoromethyl)-2-pyridinyl]phenyl]iridium(iii) hexafluorophosphate (Ir-dF(CF3)-dtb) in our experiments, since, in its excited state, it is likely to be strong enough (E1/2(Ir*/Ir−) = +1.21 V vs. SCE15,16) to generate the alkyl radical from a variety of potassium alkyltrifluoroborates. While the reduction potential of the excited photocatalyst is clearly sufficient to oxidize benzyl and allyl trifluoroborates (~+1 V (ref. 7)), primary and secondary substrates were seen as potentially problematic due to their higher reduction potentials (~+1.5 V for c-C6H11BF3K and ~+1.8 V for PhCH2CH2BF3K).7 In spite of this, encouraging results from Molander’s group have demonstrated that thermodynamically unfavorable single-electron oxidation, driven by irreversible C–B bond cleavage, is possible in these cases.10
We quickly found that irradiation of an argon-sparged dichloromethane solution of 1a, 2, and Ir-dF(CF3)-dtb, mounted between 2 16 W 450 nm LED floodlights for 18 h resulted in a 70% (isolated) yield of 3a (reactor assembly and LED emission spectrum shown in Fig. S1 and S2 of the ESI†). GC/MS of the reaction mixture showed that 2 was completely consumed, and only a small amount bibenzyl, resulting from homocoupling of the benzyl radical generated from 1a, was detected. Essentially no 1,2-bis(phenylamino)-1,2-diphenylethane, resulting from a potential α-amino radical derived from 2,17 was detected. Control experiments established the need for both catalyst and light; these results are shown in Table 1.
Table 1.
Visible light-promoted reaction of potassium benzyltrifluoroborate (1a) with benzalaniline (2a)a
 | |||
|---|---|---|---|
| Entry | Catalyst (Y/N) | Light (Y/N) | Yieldb(%) |
| 1 | Y | Y | 70 |
| 2 | N | Y | Trace |
| 3 | Y | N | 0 |
Reaction conditions: 1a (0.38 mmol), 2 (0.25 mmol), Ir-dF(CF3)-dtb (0.0063 mmol), CH2Cl2 (5 mL), blue LEDs, rt, Ar, 46 h.
Isolated yield.
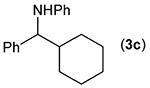
Building on these initial results, we screened a number of potassium organotrifluoroborates for suitability in this protocol. As shown in Table 2, use of a wide range of potassium alkyltrifluoroborates led to moderate to good yields when reacted with 2; addition of a proton donor (entry 1) gave a slightly better yield of product. As expected, product yields seem to be related to trifluoroborate reduction potential; thus, organotrifluoroborates that have lower reduction potentials (i.e., those that form more stable radicals and thus are more easily oxidized) result in higher yields of product (entries 1–3 and 7). Given the much higher reduction potential of primary alkyltrifluoroborates, it was somewhat surprising to find that potassium phenethyltrifluoroborate (Ered = +1.83 V (ref. 7)) was a competent radical source, although the reaction proceeded much more slowly. As shown in entry 6, only a 17% yield of amine 3f was produced after 46 h, but GC/MS analysis of the reaction mixture showed that only about 30% of 2 had been consumed during this time. Extending the reaction time to 1 week (168 h) led to complete consumption of 2 and a respectable 41% yield of 3f. Potassium α-benzyloxymethyltrifluoroborate (1h) also reacted smoothly in the protocol (entry 8), leading to a reasonable yield of the vicinal amino ether 3h. Aryl- and vinyltrifluoroborates did not react (entries 9 and 10); potassium cyclopropyltrifluoroborate (entry 4) was also not suitable in this protocol. These failures can be rationalized in terms of relative radical stabilities, which are related to R–H bond dissociation energies (BDEs).18 Vinyl-H, aryl-H, and cyclopropyl-H BDEs are generally >440 kJ mol−1, while alkyl-H BDEs are generally <420 kJ mol−1,19 reflecting the greater stability of alkyl radicals as compared to vinyl, aryl, or cyclopropyl radicals. Consistent with this trend, potassium cyclobutyltrifluoroborate (1e) could be used in the protocol (cyclobutyl-H BDE = 404 kJ mol−1),19 leading to an acceptable yield of 3e (entry 5). Unfortunately, 3e could only be isolated as an inseparable mixture with a second product in a 12 :1 molar ratio. The chemical shifts of the impurity signals, along with the HRMS of the second product seem consistent with compound 4 below, apparently derived from reaction of 2 with the dichloromethane solvent, probably via generation of the dichloromethyl radical.20
Table 2.
Scope of the reaction employing various potassium organotrifluoroboratesa
 | |||
|---|---|---|---|
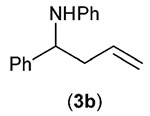
| Entry | RBF3K (1) | Product (3) | Yieldb (%) |
| 1 |  |
 |
70 (76c) |
| 2 | 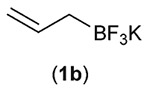 |
 |
70 (0d) (0e) |
| 3 | 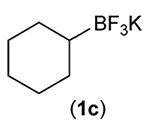 |
 |
73 |
| 4 | 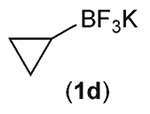 |
 |
0 |
| 5 | 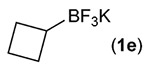 |
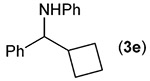 |
48 f,g |
| 6 | 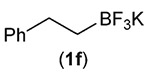 |
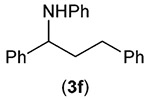 |
17 (41h) |
| 7 | 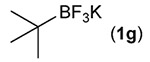 |
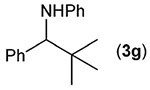 |
60 |
| 8 |  |
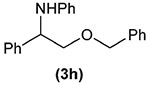 |
61 |
| 9 |  |
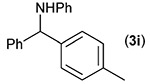 |
0 |
| 10 | 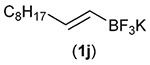 |
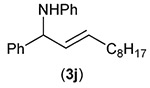 |
0 |
Reaction conditions: 1 (0.38 mmol), 2 (0.25 mmol), Ir-dF(CF3)-dtb (0.0063 mmol), CH2Cl2 (5 mL), blue LEDs, rt, Ar. 46 h.
Isolated yields.
(PhO)2PO2H (0.38 mmol) added to the reaction mixture.
No catalyst.
No light.
Isolated as an inseparable mixture with 4 (3e : 4 = 10: 1 by NMR).
Calculated yield of 3e based on NMR purity.
168 h.

Since addition of potassium allyltrifluoroborate (1b) to certain imines previously had been shown to occur in the presence of Lewis acids,12b the reaction of 1b was also examined in the absence of both catalyst and light (entry 2); no product was produced in either case, further validating the need for both in the protocol.
Based on literature precedent, thermodynamic considerations and the absence of homocoupling products derived from benzalaniline, we postulate that this reaction proceeds by a closed-cycle radical addition mechanism as shown in Scheme 1. Irradiation of the iridium catalyst should give an excited-state species which is able to oxidize an organotrifluoroborate to radical intermediate A. Radical addition of A to the imine should produce N-centered radical B. Ir-dF(CF3)-dtb, in its reduced state (E1/2(IrIII/IrII) = −1.37 V (ref. 15)), will likely be strong enough to convert B to amide ion C,21 thus closing the catalytic cycle. A similar mechanism has recently been proposed for radical alkylations of imines with alkyl silicates.13b
Scheme 1.

Proposed mechanism for the visible light-promoted alkylation of imines.
However, many visible-light photoredox reactions which are postulated as closed-cycle mechanisms may actually proceed via radical chain processes, since a group transfer event in a radical chain process generates the same intermediate as an electron transfer event.22 To screen for this possibility, we performed a “light–dark” experiment, alternating between irradiation and non-irradiation, and analyzing the extent of product formation after each period. These results are shown in Fig. 1. Based on these data, it appears that product formation only occurs during periods of illumination, and does not occur to any appreciable extent during periods of no illumination. It should be noted that after about 60% conversion to product, the reaction mixture was left in the dark overnight (~16 h), during which time no additional product formation was observed. After that period, product formation resumed upon further illumination. These results are consistent with a closed cycle mechanism, although further study (i.e., determination of the quantum yield) is necessary to completely rule out a radical chain process.23
Fig. 1.

Light–dark experiment results. Top: Time course for the appearance of 3a, measured as GC area ratio of 3a to that of an internal standard (decane). Bottom: Illumination of reaction mixture vs. time. Experimental details can be found in the ESI.†
In summary, we have developed a mild, visible light-promoted protocol for the alkylation of imines using potassium organotrifluoroborates. Moderate to good yields of amine products are obtained with a wide variety of these substrates, including ordinary primary, secondary, and tertiary alkyl examples. Further optimization of the methodology, as well as investigations into the scope and mechanism of the transformation are ongoing in our laboratory, and the results will be reported in due course.
Supplementary Material
Acknowledgements
Acknowledgment is made to the Donors of the American Chemical Society Petroleum Research Fund (58270-UR1) for support of this research. Additional support was provided by an SC-INBRE grant from the National Institute of General Medical Sciences (P20 GM103499) and the Winthrop University Research Council. The authors would also like to thank Michael Walla of the University of South Carolina Department of Chemistry and Biochemistry for high resolution mass spectrometry analysis.
Footnotes
Conflicts of interest
There are no conflicts to declare.
Electronic supplementary information (ESI) available: Experimental procedures, and compound 1H- and 13C-NMR spectra.
Notes and references
- 1 (a).Marcantoni E and Petrini M, Lewis acid promoted addition reactions of organometallic compounds, Compr. Org. Synth, 2014, 1, 344–364, (2nd Ed.) [Google Scholar]; (b) Yamada K.-i. and Tomioka K, Copper-catalyzed asymmetric alkylation of imines with dialkylzinc and related reactions, Chem. Rev, 2008, 108, 2874–2886; [DOI] [PubMed] [Google Scholar]; (c) Bloch R, Additions of organometallic reagents to C=N Bonds: Reactivity and selectivity, Chem. Rev, 1998, 98, 1407–1438. [DOI] [PubMed] [Google Scholar]
- 2.Recently, however, addition of alkyllithiums and Grignard reagents to imines in water under air has been reported: Dilauro G, Dell’Aera M, Vitale P, Capriati V and Perena FM, Unprecedented nucleophilic additions of highly polar organometallic compounds to imines and nitriles using water as a non-innocent reaction medium, Angew. Chem., Int. Ed, 2017, 56, 10200–10203. [DOI] [PubMed] [Google Scholar]
- 3 (a).Tauber J, Imbri D and Opatz T, Radical addition to iminium ions and cationic heterocycles, Molecules, 2014, 19, 16190–16222; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cannella R, Clerici A, Pastori N, Regolini E and Porta O, One-pot four-component reaction: Aqueous TiCl3/PhN2+-mediated alkyl radical addition to imines generated in situ, Org. Lett, 2005, 7, 645–648; [DOI] [PubMed] [Google Scholar]; (c) Friestad GK, Addition of carbon-centered radicals to imines and related compounds, Tetrahedron, 2001, 57, 5461–5496. [Google Scholar]
- 4 (a).Shaw MH, Twilton J and MacMillan DW, Photoredox catalysis in organic chemistry, J. Org. Chem, 2016, 81, 6898–6926; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Prier CK, Rankic DA and MacMillan DW, Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis, Chem. Rev, 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Romero NA and Nicewicz DA, Organic photoredox catalysis, Chem. Rev, 2016, 116, 10075–10166. [DOI] [PubMed] [Google Scholar]
- 6.Chenneberg L, Lévêque C, Corcé V, Baralle A, Goddard J-P, Ollivier C and Fensterbank L, Single-Electron-Transfer Oxidation of Trifluoroborates and Silicates with Organic Reagents: A Comparative Study, Synlett, 2016, 27, 731–735. [Google Scholar]
- 7.Yasu Y, Koike T and Akita M, Visible light-induced selective generation of radicals from organoborates by photoredox catalysis, Adv. Synth. Catal, 2012, 354, 3414–3420. [Google Scholar]
- 8.Chinzei T, Miyazawa K, Yasu Y, Koike T and Akita M, Redox-economical radical generation from organoborates and carboxylic acids by organic photoredox catalysis, RSC Adv., 2015, 5, 21297–21300. [Google Scholar]
- 9.Li Y, Miyazawa K, Koike T and Akita M, Alkyl- and aryl-thioalkylation of olefins with organotrifluoroborates by photoredox catalysis, Org. Chem. Front, 2015, 2, 319–323. [Google Scholar]
- 10.Primer DN, Karakaya I, Tellis JC and Molander GA, Single-electron transmetalation: an enabling technology for secondary alkylboron cross-coupling, J. Am. Chem. Soc, 2015, 137, 2195–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11 (a).Molander GA, Organotrifluoroborates: Another branch of the mighty oak, J. Org. Chem, 2015, 80, 7837–7848; [DOI] [PubMed] [Google Scholar]; (b) Darses S and Genet J-P, Potassium trifluoro (organo)borates: New perspectives in organic chemistry, Eur. J. Org. Chem, 2003, 4313–4327. [Google Scholar]
- 12 (a).Carrera DE, The acid promoted Petasis reaction of organotrifluoroborates with imines and enamines, Chem. Commun, 2017, 53, 11185–11188; [DOI] [PubMed] [Google Scholar]; (b) Lee S, Lee WL and Yun J, Rhodium-catalyzed addition of alkyltrifluoroborate salts to imines, Adv. Synth. Catal, 2015, 357, 2219–2222; [Google Scholar]; Batey R and Ramadhar T, Allylation of imines and their derivatives with organoboron reagents: Stereocontrolled synthesis of homoallylic amines, Synthesis, 2011, 1321–1346; [Google Scholar]; (c) Tian P, Dong H-Q and Lin G-Q, Rhodium-catalyzed asymmetric arylation, ACS Catal, 2012, 2, 95–119; [Google Scholar]; (d) Luo Y, Hepburn HB, Chotsaeng N and Lam HW, Enantioselective rhodium-catalyzed nucleophilic allylation of cyclic imines with allylboron reagents, Angew. Chem., Int. Ed, 2012, 51, 8309–8313; [DOI] [PubMed] [Google Scholar]; (e) Brak K and Ellman JA, Asymmetric Rh(I)-catalyzed addition of MIDA boronates to N-tert-butanesulfinyl aldimines: Development and comparison to trifluoroborates, J. Org. Chem, 2010, 75, 3147–3150. [DOI] [PubMed] [Google Scholar]
- 13 (a).Corce V, Chamoreau LM, Derat E, Goddard JP, Ollivier C and Fensterbank L, Silicates as latent alkyl radical precursors: Visible-light photocatalytic oxidation of hypervalent bis-catecholato silicon compounds, Angew. Chem., Int. Ed, 2015, 54, 11414–11418; [DOI] [PubMed] [Google Scholar]; (b) Patel NR, Kelly CB, Siegenfeld AP and Molander GA, Mild, redox-neutral alkylation of imines enabled by an organic photocatalyst, ACS Catal, 2017, 7, 1766–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang HH and Yu S, Radical alkylation of imines with 4-alkyl-1,4-dihydropyridines enabled by photoredox/Bronsted acid cocatalysis, J. Org. Chem, 2017, 82, 9995–10006. [DOI] [PubMed] [Google Scholar]
- 15.Lowry MS, Goldsmith JI, Slinker JD, Rohl R, Pascal RA, Malliaras GG and Bernhard S, Single-layer electroluminescent devices and photoinduced hydrogen production from an ionic iridium(III) complex, Chem. Mater, 2005, 17, 5712–5719. [Google Scholar]
- 16.Unless otherwise noted, reduction potentials are reported vs. the saturated calomel electrode (SCE) in acetonitrile.
- 17.Nakajima M, Fava E, Loescher S, Jiang Z and Rueping M, Photoredox-catalyzed reductive coupling of aldehydes, ketones, and imines with visible light, Angew. Chem., Int. Ed, 2015, 54, 8828–8832. [DOI] [PubMed] [Google Scholar]
- 18.Carey FA and Sundberg RJ, Advanced Organic Chemistry, Springer, New York, 4th edn, 2000, pp. 695–697. [Google Scholar]
- 19.CRC Handbook of Chemistry and Physics, ed. Lide DR, CRC Press, Boca Raton, Florida, 74th edn, 1993, pp. 9–136. [Google Scholar]
- 20.Generation of the dichloromethyl radical has been accomplished using peroxide initiators: Tian Y and Liu Z-Q, Metal-free radical cascade dichloromethylation of activated alkenes using CH2Cl2: Highly selective activation of the C–H bond, RSC Adv., 2014, 4, 64855–64859. [Google Scholar]
- 21.The reduction potential of the similar N-methylanilinyl radical has been reported to be −0.78 V. See: Jonsson M, Wayner DDM and Lusztyk J, Redox and acidity properties of alkyl- and arylamine radical cations and the corresponding aminyl radicals, J. Phys. Chem, 1996, 100, 17539–17543. [Google Scholar]
- 22.Kärkäs MD, Matsuura BS and Stephenson CRJ, Enchained by visible light–mediated photoredox catalysis, Science, 2015, 349, 1285–1286. [DOI] [PubMed] [Google Scholar]
- 23.Cismesia MA and Yoon TP, Characterizing chain processes in visible light photoredox catalysis, Chem. Sci, 2015, 6, 5426–5434. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.