

Abstract
Porcine circovirus 3 (PCV3) was recently discovered and is a new species of the genus circovirus. Clinically, it is associated with absence of symptoms or with different clinical syndromes. It has been reported in different countries of America, Europe and Asia. Last year, in Colombia, some farms have reported symptoms similar to those caused by PCV2. Samples were taken from two farms located in the centre of the country, and the presence of PCV3 was determined by PCR in two samples, one from a pool of sera and another from mesenteric lymph node. The strains were fully sequenced (GenBank accession numbers MH327784 and MH327785) and classified into subgroups a1 and a2. According to this classification and its analysis, strain a2 is located within the group called “Linker” that may be evolving towards group “b”. In addition to the above, the two Colombian strains were compared with 104 strains reported in the GenBank database. The phylogenetic tree obtained grouped according to the classification of subgroups a1, a2, b1 and b2. It was found that subgroups a1 and a2 were well grouped when comparing whole genomes, but the same was not observed with the strains of group “b”. In the latter, no subgroups were evidenced when comparing complete genomes. It is suggested that a new classification of PCV3 subgroups should be proposed, based on whole genome sequences. This is the first report of PCV3 in Colombia and its complete genome sequence.
Keywords: Porcine Circovirus, PCV3, full genome, PCV3 subgroups
Introduction
Circoviruses are single‐stranded DNA viruses belonging to the family Circoviridae. Members of this family are classified into two genera, Circovirus and Cyclovirus, which are distinguished by the position of the origin of replication relative to the coding regions and the length of the intergenic regions (Breitbart et al. 2017; Rosario et al. 2017). Until 2016, the genus circovirus was classified into two types (PCV1 and PCV2) and, in 2017, the International Committee on Taxonomy of Viruses (ICTV) included a new type called PCV3 (Lefkowitz et al. 2018). Phylogeny‐based studies searching for the origin of PCV3 show the oldest trace of origin around 1966 from bats circovirus (Fu et al. 2017). In pigs, the presence of PCV3 has been verified since 1996 in Spain, where it was also shown that there have not been many changes since then (Klaumann et al. 2018) and its origin has been suggested as a recombination between mammalian and avian circoviruses (Franzo et al. 2018b). PCV1 is considered non‐pathogenic, whereas PCV2 is associated with the presence of a wide range of associated clinical manifestations under the term PCVAD (porcine circovirus‐associated disease). PCV3 has been found in asymptomatic pigs (Zheng et al. 2017) and in pigs with clinical symptoms similar to those caused by PCV2, such as porcine dermatitis and nephropathy syndrome (PDNS), respiratory disorders and reproductive failures (Palinski et al. 2017). PCV3 has been reported in China (Zheng et al. 2017), South Korea (Kwon et al. 2017), Japan (Hayashi et al. 2018) Poland (Stadejek et al. 2017), Spain, Denmark, Italy (Franzo et al. 2018a,b), Sweden (Ye et al. 2018), the United Kingdom (Collins et al. 2017) and Russia (Yuzhakov et al. 2018). In America, there are reports of the presence of PCV3 in the United States (Palinski et al. 2017) and Brazil (Tochetto et al. 2018). Analysis of complete genomes of PCV3 has led to propose a classification into three groups called PCV3a, PCV3b and PCV3d, based on the nucleotide sequence (Fu et al. 2017). In addition, a classification into two major groups named “a” and “b” has also been proposed based on amino acid motifs in ORF1, ORF2 and ORF3; in turn, each of these groups is subdivided into subgroups named a1, a2 and b1, b2 (Fux et al. 2018).
In this study, the presence of PCV3 in Colombian farms is reported for the first time from field samples from pigs with clinical symptoms compatible with PCV2. Phylogenetic analyses of whole genome sequences show the presence of PCV3 variants in the country. Additionally, sequence comparison analyses to other complete genomes reported in GenBank lead us to propose that a new classification is necessary for the subgroups of PCV3.
Materials and methods
Porcine clinical samples
At the beginning of 2018, clinical cases associated with PCVAD, particularly porcine dermatitis and nephropathy syndrome (PDNS), were reported in pigs from at least four farms located in the centre of the country (region of Cundinamarca). Two of those farms (A and B) submitted samples to our laboratory to determine PCV2 by real time PCR (qPCR). Farm A submitted tissues (two lungs and two mesenteric ganglia) with morphological changes at necropsy as superficial dermatitis, lungs with collapse and failure, lobular pneumonia and lymph nodes enlarged. Farm B submited six serum pools from pigs at 3, 7, 10, 13, 16, 20 weeks of age. Each pool corresponded to three pigs of the same age.
DNA extraction and PCR detection of PCV3
DNA from tissues and from each serum pool was extracted using the DNA Qiamp Kit (QIAGEN), according to the manufacturer′s instructions. To demonstrate the presence of PCV2 DNA, qPCR assays were performed using specific primers designed and routinely employed in the diagnostic service of our laboratory (ABF 5′CCAGAATTCAACCTTMACYTTYC 3′) position 1416 ‐ 1439 and (ABR 5′ GCGGTGGACATGMTGAGATT) position 1515‐1534 generating an amplicon of 118 bp (unpublished data). The lower limit of detection (or cutoff) was 5.4 × 10−4 copies per ml. This cut‐off point corresponded to the threshold (Ct) of 34.11, therefore, samples with Ct values lower than this value were considered as negative. For PCV3 DNA detection in tissues and serum samples, conventional PCR assays were performed using a set of PCV3‐specific primers reported by Chen et al. (2017). Reactions were performed in a total volume of 25 μL containing 0.25 μL of Taq polymerase (5 U μL−1) (Go taq flexi ‐ Promega®), 5x Taq buffer (2.5 μL), 2 mmol L−1 MgCl2, 0.5 mmol L−1 dNTP, 1 μL of each primer (20 μmol L−1) and 2 μL of extracted DNA. The PCR reactions were performed on a Biorad® ‐ DNA thermocycler using a protocol consisting of denaturation at 94°C for 5 min, followed by 35 cycles including a denaturation at 94°C for 30 s, annealing at 58°C for 30 s and extension at 72°C for 45 s, with a final extension at 72°C for 5 min. The reaction generated a 267 bp PCR product.
Sequencing of the complete genome of PCV3
The PCV3 full genome of two samples was amplified using four sets of specific sequencing primers reported by Palinski et al. (2017). Reactions were performed in a total volume of 25 μL containing 0.25 μL of AccuPrime Taq (5 U μL−1) (Thermofisher®), 1x AccuPrime PCR Buffer I (2.5 μL), 1 μL of each primer (20 μmol L−1) and 2 μL of extracted DNA. The PCR reactions were performed on a Biorad® ‐ DNA thermocycler using a protocol consisting of denaturation at 94°C for 2 min, followed by 35 cycles including a denaturation at 94°C for 30 s, annealing at 55°C for 30 s and extension at 68°C for 1 min. PCR products were directly sequenced in both directions at the commercial sequencing facility SSiGMol (Servicio de Secuenciación y Análisis Molecular, Instituto de Genética, Universidad Nacional de Colombia). Genome assembly and annotation were done using Geneious version 9.1.8. The complete genome sequence of PCV‐3 has been deposited in GenBank under the accession numbers MH327784 and MH327785.
Phylogenetic analysis
The two full‐length PCV3 genomes from this study were aligned with all complete genome sequences of PCV‐3 strains obtained from GenBank to date. The evolutionary history was inferred using the Neighbor‐Joining method (Saitou & Nei 1987). The optimal tree with the sum of branch length = 0.27490272 is shown. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (2000 replicates) are shown next to the branches (Felsenstein 1985). The evolutionary distances were computed using the Kimura 2‐parameter method (Kimura 1980) and are in the units of the number of base substitutions per site. The analysis involved 106 nucleotide sequences. All positions containing gaps and missing data were eliminated. There were a total of 1992 positions in the final dataset. Evolutionary analyses were conducted in MEGA7 (Kumar et al. 2016).
Results and discussion
All serum pools and tissues were found negative for PCV2, while only one mesenteric ganglion from farm A and one serum pool from pigs of 19 weeks of age from farm B were positive for PCV3. The complete genome of PCV3 was amplified from both samples, and sequences were designated as PCV3/COL/cundinamarca1/2018 (COL1) and PCV3/COL/Cundinamarca2/2018 (COL2) and deposited in GenBank under accession numbers MH327784 and MH327785. The length of the genomes was 2000 nt and these displayed an ambisense genomic organization, with two inversely arranged major open reading frames (ORFs), encoding the replication associated protein (Rep) and the capsid protein (Cap). The NJ tree based on PCV3 complete genome sequences, including COL1, COL2 and 104 reported complete genomes is shown in Figure 1. The sequence identity between the Colombian sequences was 99.60%, and both Colombian strains showed a range of identity between 99.6% and 99.9% compared with isolates from South Korea (KY996341 and KY 996343), China (MF069116), Brazil (MF079254) and Italy (MF162299). All of these strains were grouped into PCV3 group 2, according to (Ku et al. 2017), and into group “a”, according to (Fux et al. 2018).
Figure 1.
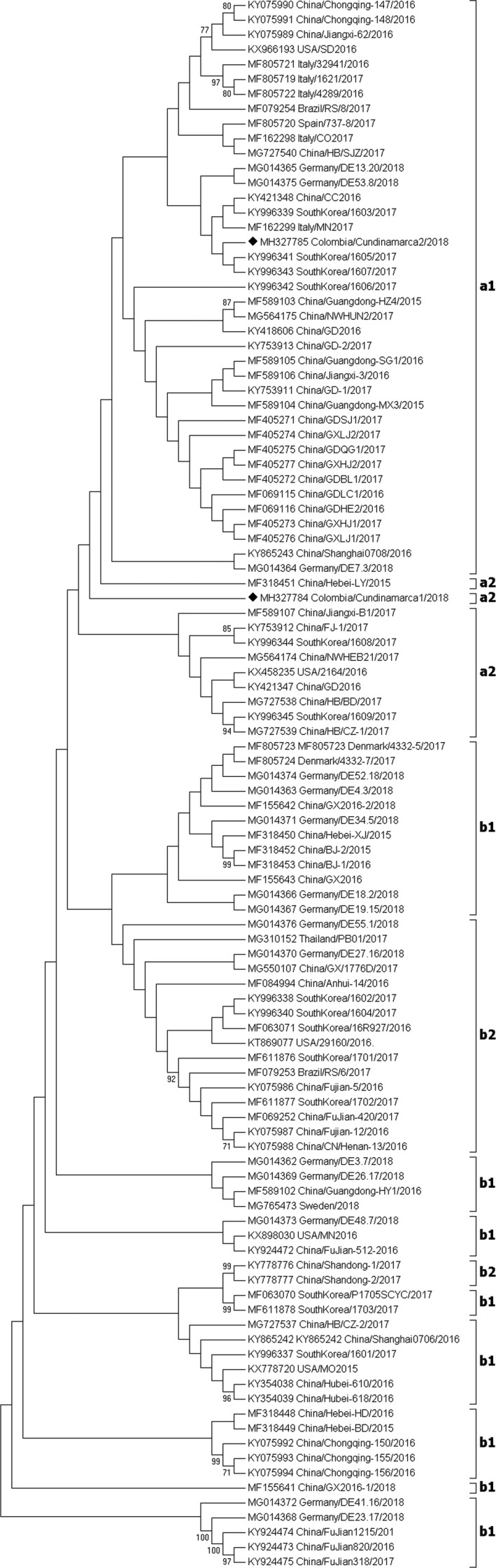
Phylogenetic analysis based on the complete genome sequence of two Colombian strains (♦) and 104 reference strains (GenBank accession number, country and year of collection). The tree was inferred using the Neighbor‐Joining method. Bootstrap values greater than 70% from 2000 replicates are indicated at the nodes. a1, a2, b1 and b2 correspond to the location of the sequences, according to the classification by subgroups for PCV3 proposed by Fux et al. (2018).
An intra‐specific classification for PCV3 has been defined by searching for amino acid motifs. Fux et al. (2018) proposed codon 122 of ORF1 and codons 24, 27, 77 and 150 of ORF2 for defining groups (Table 1). Motif AVKSI defines group “a”, while motif SARSI defines group “b”. Additionally, variations found within each motif serve to determine subgroups. COL1 strain displays motif SAKSI, which together with the strains containing the SVKSI motif constitute subgroup a2. On the other hand, COL2 strain has motif AVKSI corresponding to subgroup a1 (Table 2). Interestingly, COL2 strain also has a change in codon 19 of ORF1, corresponding to Q in the site where P is common. This change is only associated with isolates from South Korea (KY996341, KY996342, KY996343). The implications of this change in both the evolution and the characteristics of the virus are unknown. On the other hand, according to the proposal by (Fux et al. 2018) in terms of the intra‐specific classification for PCV3, COL1 strain can be located within the strains that evolutionarily represent a transition (linker) between group “a” and group “b”, because it has an intermediate motif (S/A A K S I) found on codon 122 of ORF1. Additionally, the analysis of codons 1, 4 and 227 of ORF3 can also be used for the classification of subgroups “a” and “b”. Motif FDG defines group a1, motif F G/D V corresponds to group a2, and motif SGV to group b1 or b2 (Table 1). In COL1 strain, motif FGV was found, which places it in subgroup a2, while COL2 strain presented motif FDG that places it in group a1 (Table 2). The analysis of ORF1, ORF2 and ORF3 motifs located COL1 strain as PCV3a2 and COL2 strain as PVC3a1. These results could indicate the probable presence of at least two groups of PCV3 in Colombia or the presence of strains of group a in an evolutionary process, for which it is necessary to isolate and sequence more genomes of representative strains to understand the evolutionary processes between PCV‐3 and host swine in Colombia and worldwide.
Table 1.
Motifs patterns to PCV‐3 group specific
| Group specific motifs | ORF1 | ORF2 | ORF3 | |||||
|---|---|---|---|---|---|---|---|---|
| 122 | 24 | 27 | 77 | 150 | 1 | 4 | 227 | |
| a1 | A | V | K | S/N | I | F | D | G |
| a2 | S | V/A | K | S | I/L | F | D/G | V |
| b1 | S/A | A/V | R | S | I/L | S | G/D | V/G |
| b2 | S | A | R | T | L/I | S | G | V |
Table 2.
Group specific motifs of the some PCV‐3 genomes compared with Colombia PCV‐3 genomes
| Genbank accession number | Country | Group specific motifs | ORF1 | ORF2 | ORF3 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| 122 | 24 | 27 | 77 | 150 | 1 | 4 | 227 | |||
| MH327785 | Colombia 2 | a1 | A | V | K | S | I | F | D | G |
| KY996341 | South Korea | a1 | A | V | K | S | I | F | D | G |
| KY 996342 | South Korea | a1 | A | V | K | S | I | F | D | G |
| KY 996343 | South Korea | a1 | A | V | K | S | I | F | D | G |
| MF069116 | China | a1 | A | V | K | S | I | F | D | G |
| MF079254 | Brazil | a1 | A | V | K | S | I | F | D | G |
| MF162299 | Italy | a1 | A | V | K | S | I | F | D | G |
| MH327784 | Colombia 1 | a2 | S | A | K | S | I | F | G | V |
We carried out a sequence analysis of 106 complete genome sequences for PCV3, including two reported here and the remaining reported in GenBank. A classification was made according to the proposal by Fux et al. (2018), who analysed ORFs 1, 2 and 3 of 50 sequences. Based on this classification, it was found that 39/106 sequences were located within subgroup a1; these are very homogeneous in the three ORFs analysed, and only one sequence (KY865243) showed a difference in one codon (77 of ORF2). In addition, 11/106 sequences were located in subgroup a2, which are not as homogeneous among them as compared with subgroup a1. Accordingly, the greatest variation was found in codon 24 of ORF2, where A was found in six sequences, while V was found in five sequences. The remainder were identical, except for sequence MF318451 where L was found instead of I at codon 150 of ORF2. For subgroup b1, 38 of 106 sequences were found in this subgroup, which showed great stability at codons 27 (R) and 77 (S) of ORF2 and codon 1 (S) of ORF3. A single variant was found in codon 24 of ORF2 (V/A) (KY996337), codon 4 of ORF3 (D/G) (KY996337), and codon 227 of ORF3 (F/V). A group of five sequences (KX778720, KX778720, KY354039, MF155641, MG727537) showed variation in codon 122 of ORF1 (A/S) and codon 227 of ORF3 (G/V). MG727537 showed three differences compared with the common pattern (SARSISGV instead of AARSLSGG). Subgroup b2 contained 18/106 sequences that were very stable in general. Only one sequence (MG014376) showed a difference in codon 150 of ORF2 (I/L) (Data S1).
The phylogenetic analysis of the 106 complete genome sequences confirms the classification based on ORF1, 2 and 3 sequences classified as a1 and a2, because the complete sequences are distributed in blocks in the tree (Fig. 1). On the contrary, the classification of strains belonging to subgroups b1 and b2 is not clearly shown when analysing the complete sequences. As can be seen in Figure 1, strains b1 and b2 are mixed in alternating blocks. Therefore, it is proposed that a new classification of PCV3 should be performed according to phylogenetic analyses based on the complete genome sequences. Additionally, a new classification could be more appropriate to explain the geographic changes by phylogenetic distribution.
This study is the first report of the presence of PCV3 in Colombia. Due to the existence of farms that report symptoms associated with PCV2, it is necessary to carry out studies that demonstrate whether PCV3 is the cause of this symptomatology and the consequences of this on the productive system. Likewise, the sequence variation observed among the Colombian PCV3 genomes reported here suggests that the virus varies among farms and this may also be the case within farms. Additionally, a new classification of the PCV3 subgroups based on the phylogenetic distribution of the complete genome sequences is proposed.
Source of funding
This research was supported by the Facultad de Medicina Veterinaria y de Zootecnia, Universidad Nacional de Colombia and by the corresponding author's own resources.
Conflict of interest
The authors declare no conflict of interest.
Ethical statement
The authors confirm that the ethical policies of the journal, as noted on the journal's author guidelines page, have been adhered to and the appropriate ethical review committee approval has been received. The US National Research Council's guidelines for the Care and Use of Laboratory Animals were followed.’
Contributions
DSVB conceived and designed the study, performed experiments, analysed data and wrote the paper. FSC analysed data, designed figures and wrote the paper. LB performed experiments. DM designed the study and contributed samples. JJ provided financial support, conceived and designed the study, analysed data and wrote the paper. All authors read and approved the final manuscript.
Supporting information
Data S1. Amino acid alignments of the putative ORFs 1, 2 and 3 used to identify group specific motifs by Fux et al., 2018.
Acknowledgements
The Authors thank the veterinarians who facilitated the samples and who collaborated with data on the health status of the farms. To Andrea González for revising the manuscript.
References
- Breitbart M., Delwart E., Rosario K., Segalés J., Varsani A.; Ictv Report Consortium (2017) ICTV virus taxonomy profile: circoviridae. The Journal of General Virology 98, 1997–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G.H., Mai K.J., Zhou L., Wu R.T., Tang X.Y., Wu J.L. et al (2017) Detection and genome sequencing of porcine circovirus 3 in neonatal pigs with congenital tremors in South China. Transboundary and Emerging Diseases 64, 1650–1654. [DOI] [PubMed] [Google Scholar]
- Collins P.J., McKillen J. & Allan G. (2017) Porcine circovirus type 3 in the UK. The Veterinary Record 181, 599. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39, 783–791. [DOI] [PubMed] [Google Scholar]
- Franzo G., Legnardi M., Hjulsager C.K., Klaumann F., Larsen L.E., Segales J. & Drigo M. (2018a) Full‐genome sequencing of porcine circovirus 3 field strains from Denmark, Italy and Spain demonstrates a high within‐Europe genetic heterogeneity. Transboundary and Emerging Diseases 65, 602–606. [DOI] [PubMed] [Google Scholar]
- Franzo G., Segales J., Tucciarone C.M., Cecchinato M. & Drigo M. (2018b) The analysis of genome composition and codon bias reveals distinctive patterns between avian and mammalian circoviruses which suggest a potential recombinant origin for Porcine circovirus 3. PLoS ONE 13, e0199950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X., Fang B., Ma J., Liu Y., Bu D., Zhou P. et al (2017). Insights into the epidemic characteristics and evolutionary history of the novel porcine circovirus type 3 in southern China. Transboundary and Emerging Diseases, 65, e296–e303. [DOI] [PubMed] [Google Scholar]
- Fux R., Söckler C., Link E.K., Renken C., Krejci R., Sutter G. et al (2018) Full genome characterization of porcine circovirus type 3 isolates reveals the existence of two distinct groups of virus strains. Virology Journal 15, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi S., Ohshima Y., Furuya Y., Nagao A., Oroku K., Tsutsumi N. et al (2018) First detection of porcine circovirus type 3 in Japan. The Journal of Veterinary Medical Science 80, 1468–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. Journal of Molecular Evolution 16, 111–120. [DOI] [PubMed] [Google Scholar]
- Klaumann F., Franzo G., Sohrmann M., Correa‐Fiz F., Drigo M., Núñez J. I. et al (2018). Retrospective detection of Porcine circovirus 3 (PCV‐3) in pig serum samples from Spain. Transboundary and Emerging Diseases 65, 1290–1296. [DOI] [PubMed] [Google Scholar]
- Ku X., Chen F., Li P., Wang Y., Yu X., Fan S. et al (2017) Identification and genetic characterization of porcine circovirus type 3 in China. Transboundary and Emerging Diseases 64, 703–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S., Stecher G., Tamura K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Molecular Biology and Evolution, 33, 1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon T., Yoo S.J., Park C.‐K. & Lyoo Y.S. (2017) Prevalence of novel porcine circovirus 3 in Korean pig populations. Veterinary Microbiology 207, 178–180. [DOI] [PubMed] [Google Scholar]
- Lefkowitz E.J., Dempsey D.M., Hendrickson R.C., Orton R.J., Siddell S.G. & Smith D.B. (2018) Virus taxonomy: the database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Research 46(D1), D708–D717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palinski R., Piñeyro P., Shang P., Yuan F., Guo R., Fang Y. et al (2017). A novel porcine circovirus distantly related to known circoviruses is associated with porcine dermatitis and nephropathy syndrome and reproductive failure. Journal of Virology, 91, e01879–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosario K., Breitbart M., Harrach B., Segalés J., Delwart E., Biagini P. & Varsani A. (2017) Revisiting the taxonomy of the family Circoviridae: establishment of the genus Cyclovirus and removal of the genus Gyrovirus. Archives of Virology 162, 1447–1463. [DOI] [PubMed] [Google Scholar]
- Saitou N. & Nei M. (1987) The neighbor‐joining method: a new method for reconstructing phylogenetic trees. Molecular Biology and Evolution 4, 406–425. [DOI] [PubMed] [Google Scholar]
- Stadejek T., Woźniak A., Miłek D. & Biernacka K. (2017) First detection of porcine circovirus type 3 on commercial pig farms in Poland. Transboundary and Emerging Diseases 64, 1350–1353. [DOI] [PubMed] [Google Scholar]
- Tochetto C., Lima D.A., Varela A.P.M., Loiko M.R., Paim W.P., Scheffer C.M. & Roehe P.M. (2018) Full‐Genome Sequence of Porcine Circovirus type 3 recovered from serum of sows with stillbirths in Brazil. Transboundary and Emerging Diseases 65, 5–9. [DOI] [PubMed] [Google Scholar]
- Ye X., Berg M., Fossum C., Wallgren P. & Blomström A.‐L. (2018) Detection and genetic characterisation of porcine circovirus 3 from pigs in Sweden. Virus Genes 54, 466–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzhakov A.G., Raev S.A., Alekseev K.P., Grebennikova T.V., Verkhovsky O.A., Zaberezhny A.D. & Aliper T.I. (2018) First detection and full genome sequence of porcine circovirus type 3 in Russia. Virus Genes 54, 608–611. [DOI] [PubMed] [Google Scholar]
- Zheng S., Wu X., Zhang L., Xin C., Liu Y., Shi J. et al (2017) The occurrence of porcine circovirus 3 without clinical infection signs in Shandong Province. Transboundary and Emerging Diseases 64, 1337–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Amino acid alignments of the putative ORFs 1, 2 and 3 used to identify group specific motifs by Fux et al., 2018.