

Abstract
Background
Primary coenzyme Q10 (CoQ10) deficiencies are clinically and genetically heterogeneous group of disorders associated with defects of genes involved in the CoQ10 biosynthesis pathway. COQ7‐associated CoQ10 deficiency is very rare and only two cases have been reported.
Methods and Results
We report a patient with encephalo‐myo‐nephro‐cardiopathy, persistent lactic acidosis, and basal ganglia lesions resulting in early infantile death. Using whole exome sequencing, we identified compound heterozygous variants in the COQ7 gene consisting of a deletion insertion resulting in frameshift [c.599_600delinsTAATGCATC, p.(Lys200Ilefs*56)] and a missense substitution [c.319C>T, p.(Arg107Trp), NM_016138.4]. Skin fibroblast studies showed decreased combined complex II + III activity and reduction in CoQ10 level.
Conclusion
This third patient presenting with lethal encephalo‐myo‐nephro‐cardiopathy represents the severe end of this ultra‐rare mitochondrial disease caused by biallelic COQ7 mutations. The response to CoQ10 supplement is poor and alternative treatment strategies should be developed for a more effective management of this disorder.
Keywords: coenzyme Q10, CoQ10, CoQ10 supplementation, COQ7, encephalo‐myo‐nephro‐cardiopathy, mitochondrial disease
1. INTRODUCTION
Coenzyme Q10 (CoQ10), known as ubiquinone, serves as a mitochondrial respiratory chain electron carrier shuttling electrons from NADH:ubiquinone oxidoreductase (complex I) or succinate dehydrogenase (complex II) to ubiquinol cytochrome c reductase (complex III) in the inner mitochondrial membrane.1 Besides, CoQ10 functions as an antioxidant to provide protection against lipid peroxidation as well as DNA and protein oxidation in animal cells.1 CoQ10 consists of a polar benzoquinone ring for redox reaction and a hydrophobic isoprenyl tail for diffusion in lipid bilayer and interaction with redox enzymes.2 In eukaryotes, at least 16 enzymes have been identified or proposed for CoQ1010 biosynthesis.3
Primary CoQ10 deficiency is clinically and genetically heterogeneous with an extremely wide spectrum of clinical manifestations. A recent review classified the phenotypes according to the genetic defects of the CoQ10 biosynthesis pathway: (a) glomerular renal involvement manifested as steroid resistant nephrotic syndrome (SRNS) associated with the defects of PDSS2 (COQ1 subunit 2), COQ2, COQ6, or COQ8B; (b) encephalomyopathy with COQ4, COQ7, or COQ9 defects involving hypertrophic cardiomyopathy, lactic acidosis and tubulopathy; and (c) predominant cerebellar ataxia involving only COQ8A.4 The genes associated with CoQ10 deficiency were suggested to have additional roles in mitochondrial homeostasis in addition to CoQ10 biosynthesis, which may account for the clinical heterogeneity.4 The biological mechanisms regulating various clinical phenotypes associated with different genetic defects are still unclear.
Primary CoQ10 deficiency caused by defect of the COQ7 gene is the most rarely reported. COQ7 encodes for 5‐demethoxyubiquinone hydroxylase that catalyzes the hydroxylation of 2‐polyprenyl‐3‐methyl‐6‐methoxy‐1,4‐benzoquinol (DMQH2), a critical step in CoQ10 biosynthesis. In addition to CoQ10 biosynthesis, a previous study identified a nuclear form of COQ7 which was increased in response to reactive oxygen species (ROS) and functioned independently to regulate ROS metabolism, stress responses and longevity.5 The first case reported was a 9‐year‐old Syrian boy with mildly progressive encephalo‐neuro‐nephro‐cardiopathy which was stabilized by CoQ10 treatment.6 The second case reported recently was a 6‐year‐old girl presented with spasticity and bilateral sensorineural hearing loss.7 Here, we report a Chinese boy with compound heterozygous COQ7 variants, presenting with a phenotype of mitochondrial encephalo‐myo‐nephro‐cardiopathy, persistent lactic acidosis, basal ganglia lesions and decrease in CoQ10 level in the skin fibroblasts. This is the third reported case of COQ7 defect associated with primary CoQ10 deficiency. The discovery of the present case has led to a wider clinical phenotypic spectrum of COQ7‐associated CoQ10 deficiency ranging from spasticity or mildly progressive encephalo‐neuro‐nephro‐cardiopathy to a fatal multisystemic disorder.
2. CASE REPORT
2.1. Clinical history
The index case was born of a nonconsanguineous Chinese couple as the second twin of a dichorionic diamniotic twin pregnancy. The first twin was healthy and unaffected. Our index patient was noted to have intrauterine growth restriction, cardiomegaly and tricuspid regurgitation since antenatal period. He was born at 33 weeks of gestation by emergency cesarean section due to oligohydramnios and abnormal Doppler signals. His birth weight was 1.6 kg (10th percentile) with satisfactory Apgar scores (Table 1). Surfactant was given shortly after birth.
Table 1.
Clinical features of the three cases of COQ7 pathogenic variants reported in literature
| Freyer et al | Wang et al | Index patient | |
|---|---|---|---|
| Ancestry | Syrian | – | Chinese |
| Parents | Consanguineous | Consanguineous | Nonconsanguineous |
| Antenatal | Oligohydramnios, fetal lung hypoplasia, growth retardation | Gestational diabetes | Oligohydramnios, growth retardation, fetal cardiomegaly |
| Gestational age | Full term | 37 wk | 33 wk |
| Respiratory | Lung hypoplasia with persistent pulmonary hypertension of newborn | – | Central hypoventilation |
| Renal | Renal dysfunction with small dysplastic kidneys with impaired cortical differentiation (resolved upon follow‐up) | – | Multiple renal cysts and diffuse increase in renal parenchymal echogenicity with accentuation of cortico‐medullary differentiation |
| Cardiovascular | Left ventricular cardiac hypertrophy (with subsequent regression), systemic hypertension | – | Severe hypertrophic cardiomyopathy with pericardial effusion, moderate tricuspid regurgitation |
| Growth and feeding | Postnatal growth retardation with oromotor dysfunction requiring gastrostomy | Normal | Postnatal growth retardation with oromotor dysfunction requiring tube feeding |
| Neurology and developmental outcome | Normal MRI brain | Normal MRI brain | MRI brain showed subdural hematoma, basal ganglial and thalami hypodensities with abnormal lactate peak |
| Distal contractures since birth with progressive peripheral sensorimotor polyneuropathy, axonal, and demyelinating type | Generalized muscle wasting, more prominent in the legs, also affecting temporalis muscle | Generalized hypotonia with progressive myopathy clinically | |
| Mild learning difficulties at 9 y old, never learned to stand or walk independently | Normal early developmental milestones, followed by language delay since 14 mo, progressive motor regression since second year and became wheelchair bound at 3 | Global developmental delay with developmental age below 3 mo across all domains at 1 y | |
| Hearing | Combined sensorineural and conduction hearing impairment | Bilateral low frequency sensorineural hearing loss | Bilateral profound hearing impairment in range of 2‐4 Hz |
| Vision | Visual dysfunction | – | Lack of visual following |
| Respiratory chain enzyme activities | Complex I + III and IV deficiency | – | Complex II + III deficiency with normal isolated activity of complex II and complex III |
| COQ7 variants identified | p.(Val141Glu) | p.(Leu111Pro) | p.(Lys200Ilefs*56), p.(Arg107Trp) |
| Response to treatment | Dosage of CoQ10 not available | CoQ10 11.4 mg/kg twice daily | CoQ10 4 mg/kg/day since 2 mo |
| Regression stalled | No obvious improvement | Stepped up to 20 mg/kg/day at 1 y | |
| Significant reduction in neuromuscular pain | No deterioration or worsening spasticity | No obvious response to treatment |
Abbreviation: MRI, magnetic resonance imaging; y, years; mo, months.
However, he developed heart failure with respiratory distress since day 4 of life, culminating in a re‐intubation at day 24 of life. Repeated echocardiography at 26 days old showed severe hypertrophic cardiomyopathy with moderately impaired right systolic function, moderate tricuspid regurgitation and pericardial effusion. There was significant bi‐atrial enlargement suggestive of diastolic dysfunction, with mild outflow obstruction at both ventricles. Heart failure symptoms persisted despite optimal medical therapy.
Respiratory‐wise, he was ventilator‐dependent for 7 months due to heart failure, with subsequent extubation to nasal cannula. Since 10 months, he was started on noninvasive ventilation due to central hypoventilation.
Neurologically, he was noted to have generalized hypotonia, ptosis, bilateral severe visual impairment, profound hearing impairment with progressive loss of muscle bulk and muscle weakness especially over bilateral lower limbs despite preserved jerks. Soft dysmorphic features were also noted with frontal bossing, low nasal bridge and sparse hair. He also developed infantile spasms since 10 months of life, which were responsive to phenobarbitone and vigabatrin. Magnetic resonance imaging (MRI) of the brain at 10 months of age showed multiple T2W hyperintense cystic changes involving bilateral corona radiata, basal ganglia and thalami, compatible with old lacunar infarcts, cerebral atrophy with encephalomalacic changes in bilateral frontal lobes, features of periventricular leukomalacia as well as doublet lactate peaks on magnetic resonance spectroscopy (see Figure 1A and B).
Figure 1.
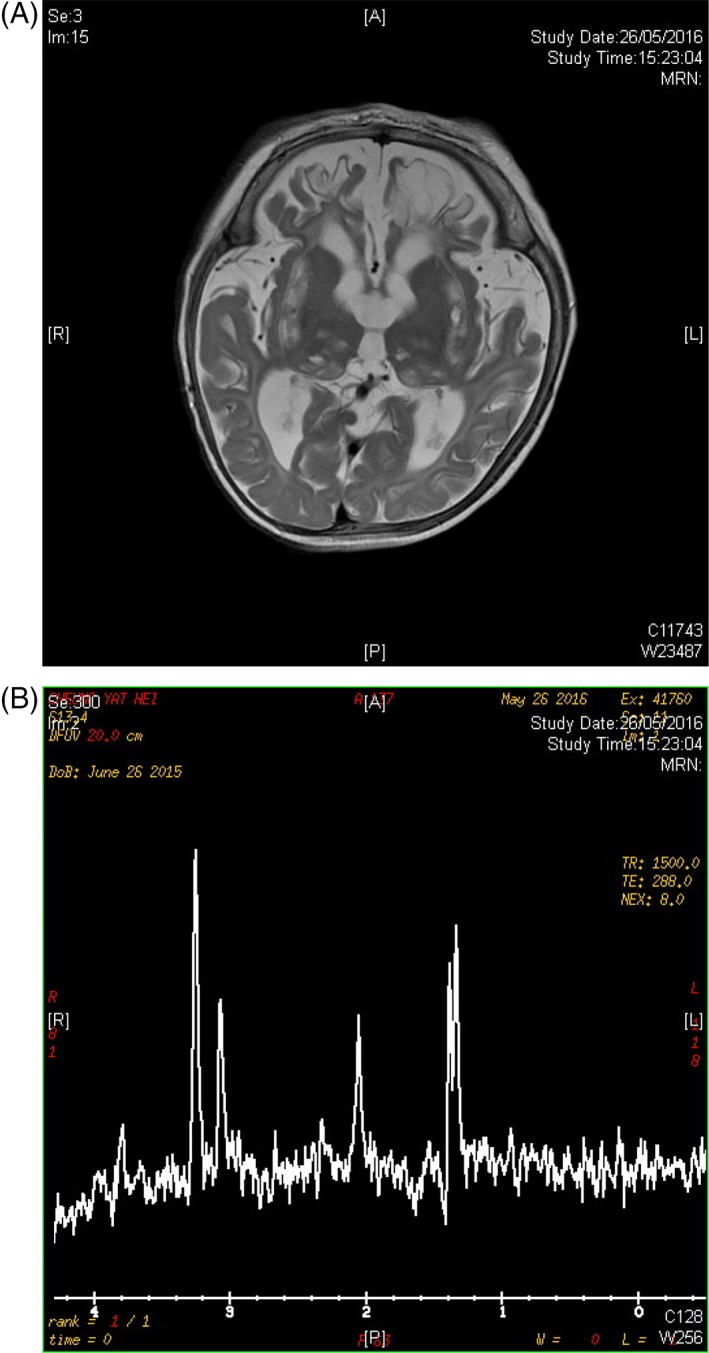
Magnetic resonance imaging brain images of the index subject. A, T2W hyperintense cystic changes involving bilateral corona radiata, basal ganglia and thalami, compatible with old lacunar infarcts, cerebral atrophy with encephalomalacic changes in bilateral frontal lobes. B, Doublet lactate peaks on magnetic resonance spectroscopy
Abbreviation: MRI, magnetic resonance imaging
Ultrasound of the urinary system showed multiple renal cysts and a diffuse increase in renal parenchymal echogenicity with accentuation of cortico‐medullary differentiation. Renal function was unremarkable.
Metabolic workup showed persistently elevated lactate up to 17 mmol/L with a raised lactate/pyruvate ratio of 50. His alanine level was up to 463 μmol/L (reference: 143‐439 μmol/L). Urine for organic acids showed increased lactate, pyruvate, 3‐hydroxybutyrate, dicarboxylic aciduria and increased excretions of Kreb cycle intermediates. These, together with the neuroimaging findings were highly suggestive of a mitochondrial disorder. He was started on CoQ10 at 2 months of life and the dose was further stepped up to 20 mg/kg/day at 12 months of life. Around the same time, he had progressive cardiorespiratory deterioration and was eventually succumbed during an episode of deterioration due to sepsis.
2.2. Genetic analysis
Whole exome sequencing (WES) and bioinformatics analyses were performed as described previously.8, 9 The variants called were annotated by Oncotator version 1.8.0.0, filtered and selected using gene panels associated with mitochondrial diseases and with strong support of mitochondrial localization suggested in MitoCarta 2.0. Compound heterozygous variants were identified in the COQ7 gene. The variants were further confirmed by Sanger sequencing. One of the variants was a deletion insertion resulting in frameshift [c.599_600delinsTAATGCATC, p.(Lys200Ilefs*56), NM_016138.4] in exon 6 which formed the stop codon 37 amino acid downstream to the wildtype stop codon. Another variant was a missense one [c.319C>T, p.(Arg107Trp), NM_016138.4] in exon 3. Both variants were not found in East Asian population according to The Genome Aggregation Database (gnomAD).
The amino acid residue of p.(Arg107Trp) is located in a highly evolutionary conserved region by multiple alignments of 100 vertebrates shown in UCSC Genome Browser with a high GERP score of 5.91 for mammalian alignment. In silico analysis including SIFT, Polyphen‐2, and Mutation Taster also predicted that the residue was located in highly conserved region and p.(Arg107Trp) was predicted to be damaging to the protein structure and function. Using the 3D modeling by STRUM, the p.(Arg107Trp) variant results a delta‐delta G value of 0.57. A positive delta‐delta G value implies the variant is responsible for COQ7 protein fold stabilization.10
Sanger sequencing of the identified variants in parental DNA was performed and showed that the variants were segregated between the parents. Father is the carrier of frameshift variant and mother is the carrier of missense variant. According to ACMG classification,11 the frameshift variant falls into the tier of “Likely pathogenic.” It was predicted to undergo nonsense mediated decay with the exon present in the biologically‐relevant transcript.12 The missense variant falls into the tier of “Uncertain significance” with a Post_P value 0.5 using the Bayesian classification framework.13
2.3. Reverse transcription polymerase chain reaction
RNA was extracted from patient's fibroblasts and semi‐quantitative reverse transcription polymerase chain reaction was performed to amplify the COQ7 complementary DNA (cDNA) consisting of the region with the two variants found. Sanger sequencing of the cDNA revealed that only the allele with the missense variant was expressed while cDNA with the frameshift variant was not identified. This finding illustrated that the mRNA with the frameshift variant could be eliminated by nonstop decay pathway which targets transcripts that do not have an in‐frame stop codon.14
2.4. Respiratory chain enzymologies analysis
Measurement of the respiratory chain succinate: cytochrome c oxidoreductase activity in skin fibroblasts revealed a significant decrease of combined complex II + III (114 mU/UCOX; reference: 325‐649) while the isolated activity of complex II and complex III were within the normal range. Fibroblast CoQ10 quantification showed a reduced level to 0.29 nmol/UCOX (reference: 1.64‐3.32). The result of these analyses indicated that this patient has CoQ10 deficiency.
3. DISCUSSION
3.1. Third reported case of pathogenic variant in COQ7
In this study, we reported the third case of COQ7 defect associated with primary CoQ10 deficiency through WES. This again demonstrated that WES is an important molecular diagnostic test for mitochondrial disorders as a similar clinical phenotype (such as multiple organ involvement) can be resulted from mutations of different mitochondrial or nuclear genes.9 We have chosen skin biopsy as a less invasive investigation for complex activity and CoQ10 level because a previous study illustrated that analysis of fibroblasts was useful to demonstrate a deficiency even though the muscle showed false negative results.15 However, a normal CoQ10 level in fibroblasts did not exclude CoQ10 deficiency in some cases.16, 17 Tissue specificity suggested that the choice of appropriate tissues is important and study of different tissues in case of a negative result is necessary to confirm CoQ10 deficiency.
3.2. Comparison with previous cases of COQ7 and COQ9 deficiency
Our subject presented with encephalo‐myo‐nephro‐cardiopathy, persistent lactic acidosis and basal ganglia lesions. The clinical presentation of our patient was comparable to one of reported classical phenotypes of CoQ10 deficiency as encephalomyopathy with hypertrophic cardiomyopathy, lactic acidosis and tubulopathy4 and this verified that COQ7 deficiency could cause multiple organ involvement. There were many similarities between our index subject and that of the previously reported cases of COQ7 deficiency. All three subjects had hearing impairment and global developmental delay or intellectual disability, with involvement of the peripheral nervous system. Despite the similarities, there were also many notable differences among the three cases. Our index subject had a fatally progressive multisystemic involvement with cardiac failure and clinically significant tubulopathy. The MRI brain showed typical features of a mitochondrial disorder which were absent in the previously reported cases. What remains to be better ascertained, is the response to CoQ10 supplements. A comparison of the three patients is available in Table 1.
Different types and positions of the COQ7 variants could alter the function of COQ7 protein in different ways and affect the presenting phenotypes of the patients. The second case study demonstrated that the variant p.(Leu111Pro) showed a milder reduction in CoQ10 level than the p.(Val141Glu) variant in the first case and so the phenotype of the second case is milder.7 The p.(Val141Glu) variant next to p.Glu142 residue is predicted to be part of the di‐iron motif and was suggested to impair iron binding for hydroxylation.18 The p.(Arg107Trp) variant in the present study was located in another highly conserved region but the biological significance of that region is not well‐studied. The 3D modeling by STRUM implies that the p.(Arg107Trp) variant is responsible for COQ7 protein fold stabilization.10 Further, mutations at the COQ7 Arg107 residue is predicted to affect the interaction with COQ9, disrupting the CoQ10 biosynthesis.19 A change from Arg to Trp results in an increased size, increased in hydrophobicity and a loss in charge, suggesting a loss of interactions with other molecules. Further in‐vitro investigation on COQ7 structure and function can provide more insights to the phenotype‐genotype relationship.
As COQ9 was suggested to interact with COQ7 physically and functionally in CoQ10 biosynthesis,20 the clinical phenotypes were compared. Up till now, six patients from three families were reported to have COQ9 deficiency.21, 22, 23 Both COQ7 and COQ9 deficiency may cause a multisystemic disorder with encephalopathy, lactic acidosis and a variable combination of cardiopathy and nephropathy. However, patients with biallelic COQ9 mutations all had severely intractable symptoms causing neonatal or infantile death and a lack of response to CoQ10 supplementation, similar to our index patient. Intriguingly, the other two patients with biallelic COQ7 mutations as mentioned above had a much milder phenotype with variable response to CoQ10. Understanding the precise role of COQ9 in CoQ10 biosynthesis would be necessary to explain the phenotypic difference between COQ7 and COQ9 deficiency.
3.3. Role of CoQ10 supplements in mitochondrial disorders
In contrast to the first case with the clinical progression being stabilized by CoQ10 treatment, our patient had no response to CoQ10 supplementation at a recommended dose of 20 mg/kg/day.24 However, a higher dose of 30 mg/kg/day could have been tried.25 CoQ10 deficiency is potentially treatable by CoQ10 supplementation but the outcomes are still variable. Different tissue involvements in different cases may influence the responsiveness to CoQ10 treatments. In a previous report, CoQ10 supplementation is effective in patient with COQ2 defect manifested with neurological signs, nephrotic syndrome or stroke‐like episode involving vascular structures.26 Besides, improvements including better coordination, decrease in muscle weakness, better speech articulation, etc. were reviewed.27 However, the responses of central nervous system are poor for some patients such as Leigh syndrome caused by PDSS2 defects and refractory seizures resulting from COQ9 mutations.22, 28 Other neurological symptoms including developmental delay were also not improved.27 Possible contributing factors are poor bioavailability of CoQ10 leading to poor penetration across the blood‐brain barrier and presence of irreversible brain damage before CoQ10 treatment.29 These can be the reasons for poor CoQ10 responsiveness in our patient, owing to the early progressive and severe neurological damages.
As demonstrated by this first report,6 2,4‐dihydroxybensoic acid (2,4DHB), a CoQ10 biosynthetic precursor with an additional hydroxyl group which is normally added by COQ7, was able to rescue the CoQ10 deficiency in patient fibroblasts by bypassing the need for COQ7 and this finding was supported by another in vivo study.30 On the other hand, the second case report demonstrated that 2,4DHB, at the same time, can inhibit native CoQ10 biosynthesis.7 In Caenorhabditis elegans model, overexpression of the CLD1 gene, encoding a phospholipase A, restored CoQ10 wildtype levels, suggesting a recovery of COQ7 function by structural remodeling.31 All these studies highlighted nonlinearity of CoQ10 biosynthesis and opened up alternative treatment strategies for CoQ10 deficiency.
In conclusion, the present study reported a severe case of COQ7‐associated primary CoQ10 deficiency with progressive and fatal encephalo‐myo‐nephro‐cardiopathy not responding to CoQ10 treatment. Alternative treatment strategies should be developed for a more effective management of this disorder.
CONFLICT OF INTERESTS
A.K.Y.K., A.T.G.C., M.H.Y.T., K.‐S.L., R.J.T.R., B.H.Y.C., and C.W.F. declare that they have no conflict of interest. J.S. is the CEO of Khondrion, a pharmaceutical company developing compounds to potentially treat mitochondrial disease.
AUTHOR CONTRIBUTIONS
Conception and design of study: A.K.Y.K., C.W.F., and B.H.Y.C. Drafting the manuscript: A.K.Y.K., C.W.F., and A.T.G.C. Evaluation of manuscript for content: A.K.Y.K., C.W.F., B.H.Y.C., T.M.H.Y., K.S.L., and J.S. Data analysis and interpretation: A.K.Y.K., A.T.G.C., T.M.H.Y., and R.J.T.R.
ETHICAL APPROVAL STATEMENT
Ethical approval had been obtained from the Institutional Review Board (IRB) of the University of Hong Kong‐Hong Kong West Cluster (IRB Ref. No.: UW 11‐190). Written consent was obtained from the parents of the patient.
ACKNOWLEDGMENTS
We would like to acknowledge the Society for the Relief of Disabled Children and the Joshua Hellmann Foundation for Orphan Disease for donations to financially support this study. Besides, we also thank the Centre of Genomic Sciences of the University of Hong Kong for providing bioinformatics services.
Kwong AK‐Y, Chiu AT‐G, Tsang MH‐Y, et al. A fatal case of COQ7‐associated primary coenzyme Q10 deficiency. JIMD Reports. 2019;47:23–29. 10.1002/jmd2.12032
Communicating Editor: Saskia Brigitte Wortmann
Funding information Joshua Hellmann Foundation for Orphan Disease; Society for the Relief of Disabled Children
Contributor Information
Brian H.‐Y. Chung, Email: bhychung@hku.hk
Cheuk‐Wing Fung, Email: fcw1209m@hku.hk.
REFERENCES
- 1. Ernster L, Dallner G. Biochemical, physiological and medical aspects of ubiquinone function. Biochim Biophys Acta. 1995;1271(1):195‐204. [DOI] [PubMed] [Google Scholar]
- 2. Sakamoto K, Miyoshi H, Ohshima M, et al. Role of the Isoprenyl tail of ubiquinone in reaction with respiratory enzymes: studies with bovine heart mitochondrial complex I and Escherichia coli bo‐type ubiquinol oxidase. Biochemistry. 1998;37(43):15106‐15113. [DOI] [PubMed] [Google Scholar]
- 3. Stefely JA, Pagliarini DJ. Biochemistry of mitochondrial coenzyme Q biosynthesis. Trends Biochem Sci. 2017;42(10):824‐843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Acosta MJS, Fonseca LV, Desbats MA, et al. Coenzyme Q biosynthesis in health and disease. BBA‐Bioenergetics. 2016;1857(8):1079‐1085. [DOI] [PubMed] [Google Scholar]
- 5. Monaghan RM, Barnes RG, Fisher K, et al. A nuclear role for the respiratory enzyme CLK‐1 in regulating mitochondrial stress responses and longevity. Nat Cell Biol. 2015;17(6):782‐792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Freyer C, Stranneheim H, Naess K, et al. Rescue of primary ubiquinone deficiency due to a novel COQ7 defect using 2,4‐dihydroxybensoic acid. J Med Genet. 2015;52(11):779‐783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang Y, Smith C, Parboosingh JS, Khan A, Innes M, Hekimi S. Pathogenicity of two COQ7 mutations and responses to 2,4‐dihydroxybenzoate bypass treatment. J Cell Mol Med. 2017;21(10):2329‐2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Neveling K, Feenstra I, Gilissen C, et al. A post‐hoc comparison of the utility of sanger sequencing and exome sequencing for the diagnosis of heterogeneous diseases. Hum Mutat. 2013;34(12):1721‐1726. [DOI] [PubMed] [Google Scholar]
- 9. Wortmann SB, Koolen DA, Smeitink JA, van den Heuvel L, Rodenburg RJ. Whole exome sequencing of suspected mitochondrial patients in clinical practice. J Inherit Metab Dis. 2015;38(3):437‐443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Quan L, Lv Q, Zhang Y. STRUM: structure‐based prediction of protein stability changes upon single‐point mutation. Bioinformatics. 2016;32(19):2936‐2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the association for molecular pathology. Genet Med. 2015;17(5):405‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Abou Tayoun AN, Pesaran T, DiStefano MT, et al. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum Mutat. 2018;39(11):1517‐1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tavtigian SV, Greenblatt MS, Harrison SM, et al. Modeling the ACMG/AMP variant classification guidelines as a Bayesian classification framework. Genet Med. 2018;20(9):1054‐1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Klauer AA, Van Hoof A. Degradation of mRNAs that lack a stop codon: a decade of nonstop progress. Wiley Interdiscip Rev RNA. 2012;3:649‐660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Montero R, Sánchez‐Alcázar JA, Briones P, et al. Analysis of coenzyme Q10 in muscle and fibroblasts for the diagnosis of CoQ10 deficiency syndromes. Clin Biochem. 2008;41(9):697‐700. [DOI] [PubMed] [Google Scholar]
- 16. Lagier‐Tourenne C, Tazir M, López LC, et al. ADCK3, an ancestral kinase, is mutated in a form of recessive ataxia associated with coenzyme Q10 deficiency. Am J Hum Genet. 2008;82(3):661‐672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ogasahara S, Engel AG, Frens D, Mack D. Muscle coenzyme Q deficiency in familial mitochondrial encephalomyopathy. Proc Natl Acad Sci U S A. 1989;86(7):2379‐2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stenmark P, Grunler J, Mattsson J, Sindelar PJ, Nordlund P, Berthold DA. A new member of the family of di‐iron carboxylate proteins. Coq7 (clk‐1), a membrane‐bound hydroxylase involved in ubiquinone biosynthesis. J Biol Chem. 2001;276(36):33297‐33300. [DOI] [PubMed] [Google Scholar]
- 19. Lohman DC, Aydin D, Von Bank HC, et al. An isoprene lipid‐binding protein promotes eukaryotic coenzyme Q biosynthesis. Mol Cell. 2019;73:763‐774.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lohman DC, Forouhar F, Beebe ET, et al. Mitochondrial COQ9 is a lipid‐binding protein that associates with COQ7 to enable coenzyme Q biosynthesis. Proc Natl Acad Sci U S A. 2014;111(44):E4697‐E4705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Danhauser K, Herebian D, Haack TB, et al. Fatal neonatal encephalopathy and lactic acidosis caused by a homozygous loss‐of‐function variant in COQ9. Eur J Hum Genet. 2016;24(3):450‐454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Duncan AJ, Bitner‐Glindzicz M, Meunier B, et al. A nonsense mutation in COQ9 causes autosomal‐recessive neonatal‐onset primary coenzyme Q10 deficiency: a potentially treatable form of mitochondrial disease. Am J Hum Genet. 2009;84(5):558‐566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Smith AC, Ito Y, Ahmed A, et al. A family segregating lethal neonatal coenzyme Q10 deficiency caused by mutations in COQ9. J Inherit Metab Dis. 2018;41(4):719‐729. [DOI] [PubMed] [Google Scholar]
- 24. Danhauser K, Smeitink JAM, Freisinger P, et al. Treatment options for lactic acidosis and metabolic crisis in children with mitochondrial disease. J Inherit Metab Dis. 2015;38(3):467‐475. [DOI] [PubMed] [Google Scholar]
- 25. Montini G, Malaventura C, Salviati L. Early coenzyme Q10 supplementation in primary coenzyme Q10 deficiency. N Engl J Med. 2008;358(26):2849‐2850. [DOI] [PubMed] [Google Scholar]
- 26. Rahman S, Clarke CF, Hirano M. 176th ENMC international workshop: diagnosis and treatment of coenzyme Q10 deficiency. Neurochem Res NMD. 2012;22(1):76‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rötig A, Mollet J, Rio M, Munnich A. Infantile and pediatric quinone deficiency diseases. Mitochondrion. 2007;7:S112‐S121. [DOI] [PubMed] [Google Scholar]
- 28. Lopez LC, Schuelke M, Quinzii CM, et al. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am J Hum Genet. 2006;79(6):1125‐1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Quinzii CM, DiMauro S, Hirano M. Human coenzyme Q10 deficiency. Neurochem Res. 2007;32(4–5):723‐727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang Y, Oxer D, Hekimi S. Mitochondrial function and lifespan of mice with controlled ubiquinone biosynthesis. Nat Commun. 2015;6:6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kar A, Beam H, Borror MB, Luckow M, Gao X, Rea SL. CLD1 reverses the ubiquinone insufficiency of mutant cat5/coq7 in a Saccharomyces cerevisiae model system. PLoS One. 2016;11(9):e0162165. [DOI] [PMC free article] [PubMed] [Google Scholar]