

Abstract
Unrelated ligands, often found in drug discovery campaigns, can bind to the same receptor, even with the same protein residues. To investigate how this might occur, and whether it might be typically possible to find unrelated ligands for the same drug target, we sought examples of topologically unrelated ligands that bound to the same protein in the same site. Seventy-six pairs of ligands, each bound to the same protein (152 complexes total), were considered, classified into three groups. In the first (31 pairs of complexes), unrelated ligands interacted largely with the same pocket residues through different functional groups. In the second group (39 pairs), the unrelated ligand in each pair engaged different residues, though still within the same pocket. The smallest group (6 pairs) contained ligands with different scaffolds but with shared functional groups interacting with the same residues. We found that there are multiple chemically unrelated but physically similar functional groups that can complement any given local protein pocket; when these functional group substitutions are combined within a single molecule, they lead to topologically unrelated ligands that can each well-complement a site. It may be that many active and orthosteric sites can recognize topologically unrelated ligands.
Graphical Abstract

INTRODUCTION
Chemical biology and drug discovery often seek new chemical scaffolds for receptor targets, unrelated to established ligand series. Such new chemotypes can illuminate previously unknown signaling,1,2 engage new regulatory networks,3 overcome key bioavailability limits for a target,4 or confer new selectivity among target family members.5 Whereas one might expect a genuinely novel ligand to occupy a new binding site in receptor, such as an allosteric site,6 it is often the main enzyme active or receptor orthosteric sites that are the most preorganized7,8 for binding and to which the novel ligands bind. By classical structure−activity or “similar properties principle”9 medicinal chemistry, this can be surprising.
Biophysically, of course, there is little to preclude the favorable binding of unrelated chemotypes in the same protein site.10 For any given protein subsite, several functional groups can be found to complement it well, and these may be combined in multiple unrelated ligand topologies to satisfy overall receptor complementarity. This was shown in a study of protein−ligand pockets by Gao and Skolnick,11 who found that many pockets bound unrelated ligands, characterizing four examples in detail. Also, many of the structurally distinct ligands from library screening bind to the main active or orthosteric sites of the target proteins.3,5,12–14 Our interest here is how unrelated ligands are recognized in the same site, and whether one might expect it to be common or rare. We will make no distinction here between endogenous and synthetic ligands, nor will we consider the evolution of protein−ligand recognition or its promiscuity, although this is a rich topic to which the biophysics of recognition relates.14
Here, we investigate 76 pairs of unrelated ligands binding to the same target site for 71 proteins−over 150 complexes overall. While we do not pretend that this is comprehensive, this set covers most major drug target families, including G-protein coupled receptors (GPCRs), kinases, enzymes, nuclear hormone receptors (NHRs), and ion channels. We will consider whether these complexes, in the context of the biophysical considerations sketched above, support the like-lihood that many proteins may recognize, and therefore may be targeted for, diverse, unrelated ligands, even in the same binding site. Said another way, if it is theoretically reasonable that a binding site can recognize unrelated ligand chemotypes, and if there are multiple convincing examples of such recognition among extant protein−ligand complexes, then we might expect such recognition to be plausible for many drug targets.
RESULTS AND DISCUSSION
The 76 proteins pairs were taken from the Protein Data Bank (PDB)15 (see Tables SI-1–SI-3 in the Supporting Information). In each pair, the same protein was crystallized with topologically unrelated ligands; overall, 71 different proteins are represented. We excluded as ligands metal ions and crystallizing agents. Each pair of complexes was visualized and retained if the binding sites had few conformational differences and if the poses of the two ligands overlapped. The ligand pairs were chosen for each target based on their pairwise dissimilarity; each ligand pair was compared by the widely used extended connectivity fingerprint (ECFP4)16 and considered if the Tanimoto coefficient17 (Tc) was below 0.15. Briefly, in ECFP4 fingerprints ligands are converted to bit strings that represent the chemical environments within a four-bond diameter of each atom. The Tc represents the ratio of matching features to all possible features between two ligands and ranges between a value of 1 (identical molecules) and 0 (no features in common). For ECFP4 fingerprints, random pairs of molecules drawn from ChEMBL have average Tc values of ~0.20. In a few cases, the stringent Tc filter of 0.15 was relaxed to include protein families that are under-represented in the PDB or in our set (all Tc values are reported in Tables SI-1–SI-3). The 76 pairs of protein−ligand complexes were selected to represent most drug-relevant protein families, with at least three examples of each class (Figure 1).
Figure 1.
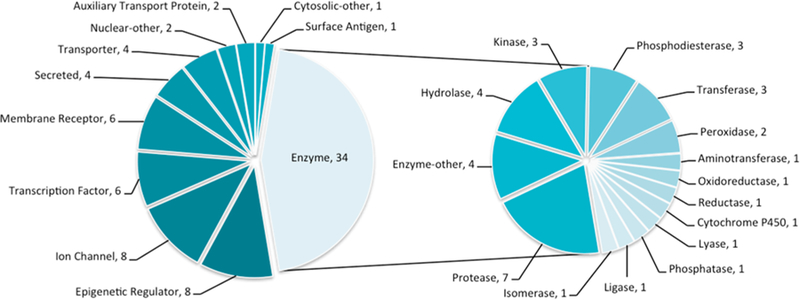
Protein families represented in this study. (Left) Distribution of major drug-relevant protein families within the 76 proteins considered (count after the comma). (Right) Distribution of enzyme subfamilies. All family names are ChEMBL target classifications.18
We categorized the pairs into three classes (Table 1): in class I, the same protein residues, or a subset of them, engage different ligand functional groups, sometimes reflecting intentional replacement of one functional group with a bioisostere19,20 (31 pairs, Table SI-1); in class II, different protein residues within the same binding site engage different ligand functional groups (39 pairs, Table SI-2); and in class III, the same protein residues engage the same ligand functional groups, but these groups emerge from different core ligand scaffolds (6 pairs, Table SI-3). With each complex pair superposed, the average root-mean-square deviation (RMSD) for all binding site atoms was of 0.41 Å from the 76 proteins, confirming that unrelated ligand binding did not reflect large conformational changes on the part of the protein (Figure 2A). Ligands in classes I, II, and III had average volume overlaps of 56.1%, 52.0%, and 64.9% with the complexes superposed, confirming they bound in the same site (Figure 2B). In only one pair of complexes, the class II complexes of α-amylase with myricetin and with D-gluconhydroximo-1,5-lactam, was the overlap of <30% of each ligand’s volume. In >70% of the complex pairs in classes I and III, 60% of at least one ligand’s respective volume was overlapped (~54% of the pairs in class II meet this criterion). Ligand volumes ranged from 65 Å3 to 825 Å3, and the average volume difference in the ligand pairs ranges between 80 Å3 and 100 Å3 in each class (see Tables SI-1–SI-3). Overall, each of the 76 pairs of ligands occupies the same space in almost identical binding sites.
Table 1.
Interactions within the Different Classes of Protein−Ligand Complex Pairs Used Herea
| class | protein residues | ligand functional groups |
|---|---|---|
| I | same | different |
| II | different | different |
| III | same | same |
The term “same” indicates that interacting residues or ligand functional groups are conserved between complex pairs.
Figure 2.

Statistics for binding site residue overlap, ligand volume overlap, and ligand similarity for the 76 complex pairs. (A) Binding site residue overlap (all heavy atoms) of complex pairs in each of the three classes and totaled overall. (B) Ligand volume overlap in each complex pair, separated by class and summed overall. Most pairs contain one ligand with 50% of its volume overlapped, indicating ligands are in the same binding pocket. (C) Dissimilarity of ligands in complex pairs for all three classes, by three different metrics: ECFP4 fingerprints, ROCS shape, and ROCS TanimotoCombo. Most are below common values used to distinguish chemically dissimilar scaffolds (dashed lines with corresponding colors).
Ligand Dissimilarity.
The average Tc across all 76 pairs is 0.10 (Figure 2C), which is a level of dissimilarity far greater than that characteristic of most scaffold hops.21 Since the shape and structural dissimilarity can be uncorrelated with topological dissimilarity, we compared the pairs of ligands by Rapid Overlay of Chemical Structures (ROCS) shape similarity22 and by TanimotoCombo, which incorporates the ROCS color score. The color score is akin to aligning pharmacophores, measuring the overlap of hydrogen bonds donors and acceptors, cations and anions, and hydrophobic groups.22 The average ROCS shape similarity was 0.52; only seven pairs were above the threshold of 0.75, where ligands are typically considered shape-similar.23 Meanwhile, the TanimotoCombo average was 0.68 and 75 pairs were below 1.15, which is substantially lower than the threshold of 1.4 for shape and chemical similarity.24 These levels of dissimilarity support the idea that the ligand pairs represent distant chemotypes.
Class I Complexes.
We identified 31 pairs of complexes where the same protein residues, or a shared subset of them, interacted with unrelated functional groups from the paired ligands (see Tables SI-1 and SI-4 in the Supporting Information). We illustrate three examples here.
Retinol-binding protein 4 (RBP4) binds a benzoic urea 625 (PDB: 3FMZ) and a pyrimido-thiophene tricycle 726 (PDB: 4O9S) that share a Tc of 0.24 (Figure 3A). The ligand volumes overlap by 74% and 65%, respectively, in binding sites whose residues have an RMSD of 0.56 Å. Aromatic groups of each ligand occupy a deep hydrophobic pocket; a trifluorotoluene of compound 6 makes fewer contacts than the larger pyrimido-thiophene tricycle of 7, which makes π-interactions with Phe77 and His104. The urea linker of compound 6 accepts a hydrogen bond from the backbone nitrogen of Leu37, and its benzoic acid forms a salt bridge with Arg121. Although the amide of compound 7 is positioned similarly to the urea of 6, it instead rotates to hydrogen-bond with Arg121. The benzoic acid of 6 also makes an edge-to-face π-interaction with Phe96, complemented by polar contacts with Tyr90 and Gln98, whereas a terminal trifluorotoluene of 7 makes a face-to-face π-interaction with Phe96. Despite the different combination of electrostatic and nonpolar interactions in the two complexes, the two ligands have IC50 values of 90 and 280 nM, respectively.26
Figure 3.

Examples of class I complexes, where the same protein residues interact with different ligand functional groups (see Table SI-1 and Figure SI-4 in the Supporting Information). The structure overlap, binding site of each complex, 2D outline of protein−ligand interactions (blue dots are water, red dashed lines hydrogen bonds, orange lines represent hydrophobic contact; blue residue labels are common to both), and similarity metrics are shown. (A) Retinol-binding protein 4 bound to compound 6 and to compound 7. (B) M2 muscarinic receptor bound to Iperoxo (8) and to QNB (9). (C) Tankyrase-1 bound to compounds 10 and 11.
A second example is the recognition of the agonist Iperoxo (8)27 (PDB: 4MQS) and the antagonist QNB (9)28 (PDB: 3UON) by the M2 muscarinic G-protein coupled receptor (GPCR) (Figure 3B). In both ligands, a cationic tertiary or quaternary amine ion pairs with Asp103. However, this charged amine is the only shared feature between these ligands, and they have a Tc of merely 0.05. A terminal dihydroisoxazole of Iperoxo accepts a hydrogen bond from Asn404, whereas an α-hydroxy ester of QNB both accepts and donates a hydrogen bond to the same Asn. The dihydroisox-azole of Iperoxo also superposes with a phenyl ring of QNB, making more-distant π-interactions with surrounding Trp155 and Trp400. The π-system of the alkyne linker of Iperoxo is sandwiched by Tyr104 and Trp400, whereas these residues T-stack with the phenyl rings of QNB that extend opposite the cationic nitrogen; the terminal Trp400 torsion angle is rotated nearly 60° to optimize these interactions. More generally, the binding sites shift somewhat more than is typical for this set, superposing with a 1.54 Å RMSD. The position of the ligands also overlaps slightly less, at 57% and 35% of their respective volumes. Notwithstanding their chemical, functional, and spatial differences, both ligands bind in the mid-pM range.27,29
Finally, two relatively rigid, aromatic ligands 1030 (PDB: 4OA7) and 1131 (PDB: 3UDD) inhibit tankyrase-1 (TNKS) by targeting the adenosine subsite of the NAD binding pocket (Figure 3C). Both ligands share ~66% volume overlap, but have a Tc of only 0.11. At one end, a quinolone of compound 10 and a methoxyphenyl of compound 11 both π-stack with His1201. Both ligands also hydrogen-bond with the backbone nitrogen of Asp1198; as a hydrogen bond acceptor, the amide oxygen of 10 mimics the 2-nitrogen of the 1,2,4-oxadiazole in 11. Similarly, the backbone nitrogen of Tyr1213 donates a hydrogen bond to each ligand; again, the ligand acceptors are a carbonyl oxygen in 10−now part of a succinamide moiety−in contrast to an aromatic nitrogen in 11−now part of a triazole ring. The other carbonyl oxygen of the succinamide in 10 points directly at the face of Tyr1203, whereas this residue makes a π-contact with a methoxyphenyl in 11. The methoxyphenyl also makes an edge-to-face π-interaction with Phe1188. Although Tyr1203 rearranges to accommodate the methoxyphenyl of 11, the binding sites superpose to an RMSD of 0.88 Å. These chemically different ligands are similarly situated, contact similar residues, and inhibit TNKS with similar IC50 values of 240 nM and 33 nM, respectively.30,31
Summary of Class I Complexes.
The ligands in these complexes are unrelated topologically and use different functional groups to interact with the same protein residues (for all 31 complexes, see Table SI-1 and Figure SI-3 in the Supporting Information); most pairs of ligands nevertheless bind with similar affinities. Binding site lysines and arginines may complement anions in one ligand and neutral hydrogen bond acceptors in another (as with retinol binding protein, above, or with the demethylase KDM4a; see Table SI-1 and Figure SI-3). The same aromatic residues of a binding site may complement an aryl ring from one ligand or an alkyne linker in another, as in the muscarinic receptor (above), while in other targets, the same aromatic residues may recognize unrelated ligand proton donors, such as a guanidinium or a pyrazole (as in M1-aminopeptidase; see Table SI-1 and Figure SI-3). A receptor backbone amide may hydrogen-bond with one ligand and participate in a dipole−quadrupole interaction with another ligand, such as tankyrase. Not discussed in our three examples are the many examples of nonpolar protein residues that complement a wide range of topologically unrelated hydrophobic ligand side chains, as in the ditrifluoromethyl and steroid groups bound to the glucocorticoid receptor (NR3C1, Table SI-1 and Figure SI-3). In class I complexes, no single type of interaction or driving force can explain the binding of the unrelated ligands to their shared binding sites.
Class II Complexes.
Class II are those complex pairs where two chemically different ligands interact with mostly different protein residues, yet still overlap in the same binding site. We categorized 39 pairs as class II, and again describe three examples, now emphasizing protein−ligand interactions unique to each ligand (a full list is given in Tables SI-2 and SI-5 in the Supporting Information).
Coagulation factor Xa recognizes DX-9056a (12)32 (PDB: 1FAX), which has long been a characteristic inhibitor, and the novel chloroindole pyridine compound 134 (PDB: 1WU1) (Figure 4A). Whereas the IC50 values of the ligands are within one logarithmic unit, at 85 nM and 18 nM, respectively, they share little chemical similarity with a Tc of 0.16, especially in their warheads. Still, at a gross level, each adopts an L-shape and overlap 63% of their volumes in binding sites that superpose with an RMSD of 0.45 Å. The S1 pocket, lined by Asp189, Ser195 and Tyr228, was historically targeted with a basic amine, such as the amidinium in DX-9056a.32 It was thus astonishing that the chloroindole in 13 could target the same pocket by making a halogen−π interaction with Tyr228; the ability to make such halogen-bonds in coagulation-cascade proteases has enabled drugs such as rivaroxaban33 to overcome the pharmacokinetic problems of the cationic inhibitors. A tertiary carbon with a carboxylic acid substituent in DX-9056a makes the L-turn, whereas the shape is defined by a sulfonylpiperazine in 13. The S4 aryl-binding pocket, made up by Tyr99, Phe174, and Trp215 is the other focal point for ligand design. DX-9056a features a phenyl ring and a terminal acetimidoylpyrrolidine; the ligand’s second amidinium simultaneously hydrogen-bonds to the backbone carbonyl of Glu97 and forms a salt bridge with its side-chain carboxylic acid. In contrast, compound 13 features a 5-pyridylpyrimidine that makes no polar or ionic interactions in S4; the two linked aromatic rings π−π or T-stack with Tyr99, Phe174, and Trp215.
Figure 4.

Examples of class II complexes where different protein residues engage different ligand functional groups (see Table SI-2 and Figure SI-5 in the Supporting Information). The 3D crystal structure overlap, enlarged binding site of each complex, 2D outline of protein−ligand interactions (colored as in Figure 3), and similarity metrics are shown. (A) Coagulation factor Xa bound to DX-9056a (12) and to compound 13. (B) Progesterone receptor bound to ulipristal acetate (14) and to compound 15. (C) Glucose transporter bound to cytochalasin B (16) and to compound 17.
In a second class II pair, the progesterone receptor binds both the antagonistic steroid-analogue ulipristal acetate (14)34 (PDB: 4OAR) and the agonist isoxazole sulfonamide 1535 (PDB: 4APU) (Figure 4B). The two molecules are unrelated topologically, with a Tc of 0.10, but overlap in the binding site by 52% and 67% of their respective volumes. Ulipristal acetate hydrogen-bonds to Thr894 through its terminal ester and makes three additional hydrogen bonds from its cyclic ketone to Gln725, Arg766, and to an ordered water molecule. Conversely, compound 15 only hydrogen bonds once to the receptor, and to a different residue, Asn719. The isoxazole of 15 does make a polar interaction with the same ordered water molecule as ulipristal acetate, bridging to Gln725 and Arg766. While both ligands make nonpolar interactions with the receptor, the specific residues with which they interact differ. For instance, ulipristal acetate makes π-contacts with Phe778 and Trp755, whereas compound 15 does so with Phe778 and Tyr890. Both ligands are potent, with an IC50 value of 0.2 nM (14) and an EC50 value of 8.6 nM (15). Aside from a conformational change in Met909, the binding site residues superpose with an RMSD of 0.58 Å.
A final class II example is the glucose transporter (SLC2A1), which binds the multicyclic cytochalasin B (16)36 (PDB: 5EQI), and a more linear phenylalanine derivative (17)36 (PDB: 5EQG) (see Figure 4C). Again, the IC50 values of each ligand are similar at 110 nM and 267 nM, respectively, yet the compounds share little topologically with a Tc of 0.15. Nevertheless, both ligands overlap 53% and 60% of their respective volumes in structures that superpose with an RMSD of 0.36 Å. Cytochalasin B hydrogen-bonds to Thr137 and Trp388, and otherwise makes apolar contacts with Asn411 and Trp412, which are characteristic of inhibitors of this transporter. Compound 17 similarly interacts with Asn411 and Trp412, but instead of hydrogen-bonding with Trp388, it forms π−π interactions with this residue, and in addition with His160, and Phe379.
Summary of Class II Complexes.
The ligands in these complexes, like those in class I, are unrelated topologically, but here they more often engage with different protein residues, and less so with shared ones. Nevertheless, they bind in the same protein site, often with similar affinities (all 39 complexes are summarized in Tables SI-2 and SI-5). A mix of different driving interactions may be seen between the different ligand pairs in any given complex, such as the recognition of an amidinium or a halogen bond−by different residues−by the factor Xa, or the swapping of a π−π interaction in the SLC2A1 transporter for a tryptophan-donated hydrogen bond. In this sense, the class II complexes resemble the diverse forms of alternating interactions observed in the class I complexes. Still, he class II complexes more often swap nonpolar interactions among different hydrophobic protein residues than seemed to be true for the class I complexes.
Class III Complexes.
In six pairs of complexes that we analyzed, the protein recognizes functional groups that are shared between the paired ligands (i.e., the ligand “warheads”), while the core ligand scaffolds that present these shared side chains differ. We consider three examples (a full list is presented in Tables SI-3 and SI-6 in the Supporting Information).
The β2-adrenergic receptor binds the inverse agonist, timolol (18)37 (PDB: 3D4S) and the agonist BI167107 (19)38 (PDB: 4LDE) (Figure 5A). Both ligands make conserved interactions through a cationic nitrogen and an alcohol to Asp113, Asn312, Tyr316. The common anchor causes substantial volume overlap of the ligands: 75% and 62%, respectively. Substituted morpholines in both scaffolds make a crucial polar interaction with Asn293 deep in the orthosteric pocket and aromatic ligand groups T-stack with Phe290. Apart from the morpholine, the molecular scaffolds differ, and the overall Tc of the two molecules is 0.17. An additional tolyl on BI167107, which interacts with Trp109, and different polar groups are responsible for the topological dissimilarity between the two ligands. A thiadiazole of timolol hydrogen bonds to Thr118, while BI167107 makes two additional hydrogen bonds to Ser203 and Ser207. The binding site residues of the two complexes overlap with an RMSD of 0.64 Å; both ligands bind the β2-adrenergic receptor with subnanomolar affinity.
Figure 5.

Examples of class III complexes where the same protein residues interact with similar functional groups on the ligands (also see Table SI-3 and SI-6). The structure overlap, enlarged binding site of each complex, 2D outline of protein−ligand interactions (colored as in Figure 3), and similarity metrics are shown: (A) β2-adrenergic receptor bound to timolol (18) and to BI167107 (19); (B) FABP4 bound to compound 20 and to ibuprofen (21); and (C) adenosine receptor A2a bound to adenosine (22) and to compound 23.
A second characteristic class III pair involves fatty acid binding protein 4 (FABP4), which binds both indole compound 2039 (PDB: 5D45) and ibuprofen (21)40 (PDB: 3P6H) (Figure 5B). The carboxylic acid of each ligand makes a salt bridge and hydrogen bonds with Arg126, Tyr128, and an ordered water molecule bridging to Arg106. The rest of the recognition elements in the site appear to be hydrophobic and it is perhaps unsurprising that ligands have found several topologies to complement these residues, resulting in a pairwise Tc of 0.14. Compound 20 is much larger, with two additional phenyl rings that displace members of a water network around ibuprofen. The size difference between the two ligands is reflected in only 44% of the larger compound 20 being overlapped, compared to 80% of ibuprofen. The binding sites of each complex superpose with an RMSD of 0.29 Å and both ligands bind with Ki values of ~100 nM.39,40
A final example of class III complexes is shown with the A2A adenosine receptor, a GPCR, which binds adenosine (22)41 (PDB: 2YDO) and a triazine-and chlorophenol-containing antagonist 2342 (PDB: 3UZC) (Figure 5C). Both ligands hydrogen bond to Asn253 and His278 through an extracyclic nitrogen on nitrogen-rich heterocycles and hydroxyl groups, respectively. Although the agonist and antagonist share these polar contacts and overlap 65% and 54% of their respective volumes in the binding site, they differ chemically, with a Tc of only 0.09. Whereas adenosine makes several hydrogen bonds with Glu169, Ser277, and a bridging water molecule to His250, compound 23 makes a halogen−π interaction with His278 and Both ligands bind well, with Ki values of 30.9 nM and 1.41 nM, respectively.41,42 The binding site residues superpose with an RMSD of 0.98 Å.
Summary of Class III Complexes.
As in the class I and class II complexes, the class III ligand pairs are unrelated topologically. Unlike class I and II, the differences in class III ligands occur in the core scaffolds, not the ligand side chains most involved in interacting with the proteins. Accordingly, there can be more similarity in the interactions between the unrelated ligands. Still, even among these complexes, one observes the same switching of driving interactions that prevails in the class I and, to a lessor extent, the class II complexes: the atom types of donors or acceptors can change, hydrogen bonds themselves may be swapped for dipole−quadrupole interactions, and there is great plasticity in the nonpolar interactions. A theme that emerges in all three classes is that there are multiple topologically unrelated ligand groups that can recognize most receptor side chains and binding site environments.
Water Molecules Mediating Ligand Binding.
Water molecules can mediate interactions between ligand and protein43,44 to enable more diverse ligands to bind in the same site. Of the 76 pairs studied, 45 had at least one water molecule; 13 of them bridged one ligand to a residue that was directly engaged by the other ligand in the pair. We illustrate two examples.
Pteridine reductase binds both the benzimidazole 2445 and the flavanol 2546 (Figure 6A). The chemically unrelated ligands have a Tc of 0.05 and overlap well in the biopterin pocket (80% and 39% of each ligand’s volume). Compound 25 hydrogen bonds directly to Asp161 through its 3′ phenolic oxygen, which also engages two ordered water molecules, forming a hydrogen bond network with Cys168 and Asn175. This phenolic oxygen is 1 Å from a superposed water molecule from the cocrystal of 24. In this second complex, the imidazole nitrogen of compound 24 hydrogen-bonds to this water molecule, which connects to the water network with the same residues, Asp161, Cys168, and Asn175. The 7-OH of 25 hydrogen-bonds to Ser95 and to the NADP cofactor, whereas a water molecule bridges the exocyclic nitrogen of 24 to the NADP cofactor; the water molecule is again <1 Å from the superposed 7-OH of 25. The two binding sites superpose with an RMSD of 0.18 Å; the Ki of 24 is 288 μM, while the IC50 of 25 is 104 μM.
Figure 6.

Complex pairs where water mediates interactions with one ligand whereas the second ligand makes these interactions directly. The structure overlap, binding site of each complex, 2D outline of protein−ligand interactions (colored as in Figure 3), and similarity metrics are shown. (A) Pteridine reductase co-crystallized with compound 24 and with compound 25. (B) Renin co-crystallized with compound 26 and with compound 27.
A second example of the role of water is shown in the complexes of renin with the flexible polyhydroxy 2647 (PDB: 1HRN) and with the more-rigid diaminopyrimidine benzox-azine 2748 (PDB: 2G1Y) (Figure 6B). Here, too, the ligands are chemically unrelated with a pairwise Tc of 0.06. Nevertheless, 43% and 58% of their respective volumes overlap in the site, and they each interact with several of the same residues, which superpose with an RMSD of 0.26 Å. This is partly explained by bridging water molecules, one of which bridges the terminal diaminopyrimidine of 27 and Ser76, while a hydroxyl of 26 displaces this water and hydrogen-bonds directly to the same Ser76. Similarly, in the S3 subpocket, a water network bridges a hydroxyl in 26 to the backbone of Tyr14, while compound 27 hydrogen-bonds directly to the same Tyr14. Meanwhile, the IC50 values of the two compounds are within one logarithmic order at 9 and 90 nM, respectively.
CONCLUSION
A key observation from this study is that many proteins can bind unrelated ligands in the same binding site. This is true for all major target families, including GPCRs, transporters, ion channels, nuclear hormone receptors, and enzymes. Often, the more deeply that a target is studied, the more chemotypes one can find for it. Consider the long-studied μ-opioid49 and estrogen receptors, for which at least 22 and 21 unrelated chemotypes, respectively, may be distinguished from the known ligands with <1 nM affinity in the ChEMBL database18 (using an ECFP4 Tc ≤ 0.35, suggesting scaffold hops21). The recognition of different ligands that overlap in the same site can occur in several ways: wholly unrelated ligands can interact with largely the same residues (our class I), they can bind in the same site but engage different residues within it (our class II), or unrelated scaffolds might project similar functional groups to interact with the same protein residues (our class III). No single type of driving force seems to explain this plasticity, with multiple types of functional group recognition occurring in many of the pairs of ligands (salt-bridges exchanged for ion-dipole interactions, one type of hydrogen-bond donor or acceptors swapped for another, hydrogen bonds replaced with dipole quadrupole interactions, and great plasticity among nonpolar side-chain interactions). An encouraging inference of this study is that structural complementarity allows for the discovery of entirely new chemotypes, even for heavily studied targets.5,14
At first glance, the recognition of diverse ligands by the same site may seem unsurprising. For many targets, multiple chemotypes will be well-known to specialists; for instance, opioid researchers are conscious of morphine, fentanyl, oliceridine,50 and others,14 where pairwise ECFP4 Tc values range from 0.12 to 0.19. The ability of unrelated proteins to bind the same ligands−the logical complement of this study−has also been shown both for substrates−leading to enzyme moonlighting and perhaps evolution51−metabolites52 and for synthetic molecules.11,53,54 Meanwhile, medicinal chemists have long used bioisosteres to exchange topologically unrelated functional groups. Well-known examples include the replacement of carboxylates with tetrazoles, hydroxamic acids, and isoxazoles,55 the replacement of benzene with thiophene,19 the replacement of catechols with benzimidazoles,56 and the replacement of phosphates with salicylates or isothiazolidinones.20 Of the 76 pairs of protein−ligand complexes studied (152 total complexes), 32 reflected such bioisosteric swaps. More surprising, but just as common in this set, are cases where the same protein residue interacts with pairs of ligand functionalities that are not classically bioisosteric, including the hydrogen bonding or π-stacking of a tryptophan with either a ligand lactam or aryl group (as in 16 and 17 interacting Trp388 of the glucose transporter, Figure 4C) or the interaction of a cation or a halogen in the S1 pocket of factor Xa (Figure 4A). From a biophysics standpoint, we are taught to consider energies of interacting groups, not their topologies. From this perspective, two ligands that both present an anion to interact with a threonine will likely capture similar interaction energies, after correcting for pKa differences, regardless if one is an isoxazole phenolate and the other is a carboxylate, as in glutamate and AMPA binding to the ionotropic glutamate receptor (seeTables SI-1 and SI-4).
Even nodding to this biophysical understanding, what emerge from this study are the wide range of targets for which unrelated ligands can bind, and the profound nature of the dissimilarity. Thus, classic bioisosteric swaps are a minority of our cases, and even where they can be detected they rarely represent the largest change. Especially in classes I and II, no single chemical switch links most of the ligand pairs. None of this breaks the laws of biophysics, of course, but that is one of the key points of this study.
Certain caveats merit airing. We do not pretend to be comprehensive in this study: there are likely many other pairs of dissimilar ligands binding to the same site. Conversely, and more generally, unrelated ligands binding to the same site remain exemplary exceptions, not the rule; molecules falling into related families dominate most ligand−protein complexes, as this reflects the history of the medicinal chemistry undertaken against them. Consequently, the ratios of our classes I, II, and III are not meaningful in themselves, nor is the ratio of complexes with unrelated ligands meaningful to those with related ligands. Nor do we pretend that even our classes are definitive others might easily be used. Even among our classes, choosing the class into which a pair of complexes fell was often a judgment call; we had no fully quantitative metric for assigning a pair of complexes to one class or the other, although often the choice seemed clear. Although we sought examples from multiple protein families, we cannot confidently write that all proteins will recognize unrelated chemotypes. Although the binding sites of each complex pair overlap by crystallography, they may differ sufficiently to change the energy landscape.57 Finally, using any single metric, such as topology-based Tanimoto coefficients, to characterize chemical similarity can mislead, especially when one molecule is much larger than the other.
These caveats notwithstanding, it seems clear that many proteins can bind−and many others should be able to bind−unrelated chemotypes in the same site. While the study was not comprehensive, it was extensive, represented many families of drug targets, and it was unbiased in the selection of complexes. Crucially, biophysical considerations, such as the equivalence of topologically unrelated groups, and the size and diversity of the chemical space from which such groups might be drawn, bolster the empirical observations described here. Thus, while we make no statistical argument as to the relative occurrence of unrelated and related ligands binding to the same site−the latter dominate, because of the structure−activity focus of the field‒there is both a theoretical expectation that the same binding site can bind unrelated ligands, and extensive empirical evidence to support it. The ability of most binding sites to recognize diverse and even unrelated chemotypes supports the community’s longstanding program of structure-based novel ligand discovery, and the new biology that they promise.49
METHODS
Protein−ligand complexes were selected in two ways. First, a list of PDB structures containing ligands, solved by X-ray crystallography to a resolution of better than 2.5 Å, was compiled using the “Advanced Search” interface in RCSB.org. All entries with Uniprot identifiers were processed using the KNIME analytics platform (https://www.knime.com). Of 70879 PDB structures with 17892 ligands, we excluded the ~11% that were crystallized with only one ligand, and another ~6% with frequent ligands such as buffer components and ions. Only entries with at least 20 unique ligands were selected, thereby reducing this sizable list to 188 Uniprot IDs with up to 389 ligands for a single target. Using the RDKit Fingerprint module within KNIME, we calculated pairwise ECFP4-based Tcs, and generated similarity matrices between all ligands for each Uniprot ID. Using these matrices, we prioritized complexes with maximally dissimilar ligands; these were visually inspected in PyMOL and in DiscoveryStudio 4.1 (Biovia, Inc.).
Second, to account for under-represented protein classes, the list of complexes was complemented by a more lenient curation that did not require 20 unique ligands. After removing buffers, ions and other crystallizing agents, this left 15 120 complexes, corresponding to 1306 unique Uniprot IDs. Again, a pairwise ECFP4-based Tc was calculated for all ligands within each Uniprot ID. The lowest Tc pairs from randomly selected protein structures were visualized until each protein major class was represented. If the lowest Tc-pair seemed inappropriate for instance, if there were large domain shifts in the protein, or if the ligands were not in the same binding pocket then other pairs of ligands were visualized.
Initially, 365 unique PDB pairs were visualized in PyMOL, using a script that was optimized to superpose the structures based on their fold. A maximum of 10 ligand−ligand receptor pairs were visualized for each protein; these were the lowest pairs for that protein, as determined by pairwise Tc similarity of their ligands. For each protein, we selected the first appropriate complex that we visualized, based on the evaluation of hydrogen bond networks, hydrophobic stacking interactions, water-mediated interactions (where applicable), interaction with individual amino acids, and superposition of ligand volumes. Ultimately, 76 protein−ligand complex pairs were for 71 unique proteins and 151 unique ligands; five proteins are duplicated, as are two ligands.
To calculate the ligand volume overlap for each complex pair, the two PDB structures were superposed using the MatchMaker function in Chimera, and the overlapping ligands saved. The volume of each ligand was calculated with Python scripts that computes van der Waals spheres for each atom. The two ligands are virtually combined and their joint volume is calculated. The volume overlap of two ligands is found by adding the individual volumes and subtracting the joint volume. ROCS shape-based overlay values were computed using ROCS Release 3.2.0.4 from OpenEye. The same ligand input was used for the volume ROCS calculations.
Three-dimensional structural images were generated with PyMOL.58 Two-dimensional ligand−receptor interactions were generated using LigPlot59 and Pose view (a part of the ProteinsPlus web service);60 the interactions were manually merged in ChemDraw Prime 15.1, to ensure same ligand site orientation.
Supplementary Material
ACKNOWLEDGMENTS
We thank R. Stein and J. Irwin for critiquing this manuscript, T. Balius for volume calculation scripts, J. Irwin for help with clustering and with chemoinformatics, and OpenEye Software for free use of ROCS.
Funding
This work was supported by NIH Grant Nos. R35GM122481 (from the grant sponsor, National Institute of General Medical Sciences, to B.K.S.) and in part by 5F32GM122191 (to J.P.) and ALSAC (to M.F.).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschem-bio.8b00443.
Full list of complex pairs from classes I, II, and III, including binding site RMSD, ligand similarity metrics, volume overlap, and 3D comparisons (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Frimurer TM, Mende F, Graae A-S, Engelstoft MS, Egerod KL, Nygaard R, Gerlach L-O, Hansen JB, Schwartz TW, and Holst B (2017) Model-Based Discovery of Synthetic Agonists for the Zn2+-Sensing G-Protein-Coupled Receptor 39 (GPR39) Reveals Novel Biological Functions. J. Med. Chem 60, 886–898. [DOI] [PubMed] [Google Scholar]
- (2).Huang X-P, Karpiak J, Kroeze WK, Zhu H, Chen X, Moy SS, Saddoris KA, Nikolova VD, Farrell MS, Wang S, Mangano TJ, Deshpande DA, Jiang A, Penn RB, Jin J, Koller BH, Kenakin T, Shoichet BK, and Roth BL (2015) Allosteric ligands for the pharmacologically dark receptors GPR68 and GPR65. Nature 527, 477–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Powers RA, Morandi F, and Shoichet BK (2002) Structure-Based Discovery of a Novel, Noncovalent Inhibitor of AmpC β-Lactamase. Structure 10, 1013–1023. [DOI] [PubMed] [Google Scholar]
- (4).Komoriya S, Haginoya N, Kobayashi S, Nagata T, Mochizuki A, Suzuki M, Yoshino T, Horino H, Nagahara T, Suzuki M, Isobe Y, and Furugoori T (2005) Design, synthesis, and biological activity of non-basic compounds as factor Xa inhibitors: SAR study of S1 and aryl binding sites. Bioorg. Med. Chem 13, 3927–3954. [DOI] [PubMed] [Google Scholar]
- (5).Wang S, Wacker D, Levit A, Che T, Betz RM, McCorvy JD, Venkatakrishnan AJ, Huang X-P, Dror RO, Shoichet BK, and Roth BL (2017) D4 dopamine receptor high-resolution structures enable the discovery of selective agonists. Science 358, 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Wenthur CJ, Gentry PR, Mathews TP, and Lindsley CW (2014) Drugs for Allosteric Sites on Receptors. Annu. Rev. Pharmacol. Toxicol 54, 165–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Warshel A (1998) Electrostatic Origin of the Catalytic Power of Enzymes and the Role of Preorganized Active Sites. J. Biol. Chem 273, 27035–27038. [DOI] [PubMed] [Google Scholar]
- (8).Shoichet BK, Baase WA, Kuroki R, and Matthews BW (1995) A relationship between protein stability and protein function. Proc. Natl. Acad. Sci. U. S. A 92, 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Johnson MA, and Maggiora GM (1990) Concepts and Applications of Molecular Similarity; Wiley: New York. [Google Scholar]
- (10).Nobeli I, Favia AD, and Thornton JM (2009) Protein promiscuity and its implications for biotechnology. Nat. Biotechnol 27, 157–167. [DOI] [PubMed] [Google Scholar]
- (11).Gao M, and Skolnick J (2013) A Comprehensive Survey of Small-Molecule Binding Pockets in Proteins. PLoS Comput. Biol 9, e1003302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Wilhelm S, Carter C, Lynch M, Lowinger T, Dumas J, Smith RA, Schwartz B, Simantov R, and Kelley S (2006) Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat. Rev. Drug Discovery 5, 835. [DOI] [PubMed] [Google Scholar]
- (13).Dorr P, Westby M, Dobbs S, Griffin P, Irvine B, Macartney M, Mori J, Rickett G, Smith-Burchnell C, Napier C, Webster R, Armour D, Price D, Stammen B, Wood A, and Perros M (2005) Maraviroc (UK-427,857), a Potent, Orally Bioavailable, and Selective Small-Molecule Inhibitor of Chemokine Receptor CCR5 with Broad-Spectrum Anti-Human Immunodeficiency Virus Type 1 Activity. Antimicrob. Agents Chemother 49, 4721−4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Manglik A, Lin H, Aryal DK, McCorvy JD, Dengler D, Corder G, Levit A, Kling RC, Bernat V, Hübner H, Huang X-P, Sassano MF, Giguère PM, Löber S, Duan D, Scherrer G, Kobilka BK, Gmeiner P, Roth BL, and Shoichet BK (2016) Structure-based discovery of opioid analgesics with reduced side 687 effects. Nature 537, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, and Bourne PE (2000) The Protein Data Bank. Nucleic Acids Res 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Rogers D, and Hahn M (2010) Extended-Connectivity Fingerprints. J. Chem. Inf. Model 50, 742–754. [DOI] [PubMed] [Google Scholar]
- (17).Rogers DJ, and Tanimoto TT (1960) A computer program for classifying plants. Science 132, 1115–1118. [DOI] [PubMed] [Google Scholar]
- (18).Bento AP, Gaulton A, Hersey A, Bellis LJ, Chambers J, Davies M, Krüger FA, Light Y, Mak L, McGlinchey S, Nowotka M, Papadatos G, Santos R, and Overington JP (2014) The ChEMBL bioactivity database: An update. Nucleic Acids Res 42, D1083–D1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Patani GA, and LaVoie EJ (1996) Bioisosterism: A Rational Approach in Drug Design. Chem. Rev 96, 3147–3176. [DOI] [PubMed] [Google Scholar]
- (20).Zhang Y, Borrel A, Ghemtio L, Regad L, Boije af Gennas̈ G, Camproux A-C, Yli-Kauhaluoma J, and Xhaard H (2017) Structural Isosteres of Phosphate Groups in the Protein Data Bank. J. Chem. Inf. Model 57, 499–516. [DOI] [PubMed] [Google Scholar]
- (21).Muchmore SW, Debe DA, Metz JT, Brown SP, Martin YC, and Hajduk PJ (2008) Application of Belief Theory to Similarity Data Fusion for Use in Analog Searching and Lead Hopping. J. Chem. Inf. Model 48, 941–948. [DOI] [PubMed] [Google Scholar]
- (22).Hawkins PCD, Skillman AG, and Nicholls A (2007) Comparison of Shape-Matching and Docking as Virtual Screening Tools. J. Med. Chem 50, 74–82. 712 [DOI] [PubMed] [Google Scholar]
- (23).Rush TS, Grant JA, Mosyak L, and Nicholls A (2005) A Shape-Based 3-D Scaffold Hopping Method and Its Application to a Bacterial Protein−Protein Interaction. J. Med. Chem 48, 1489–1495. [DOI] [PubMed] [Google Scholar]
- (24).Nicholls A, McGaughey GB, Sheridan RP, Good AC, Warren G, Mathieu M, Muchmore SW, Brown SP, Grant JA, Haigh JA, Nevins N, Jain AN, and Kelley B (2010) Molecular Shape and Medicinal Chemistry: A Perspective. J. Med. Chem 53, 3862–3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Motani A, Wang Z, Conn M, Siegler K, Zhang Y, Liu Q, Johnstone S, Xu H, Thibault S, Wang Y, Fan P, Connors R, Le H, Xu G, Walker N, Shan B, and Coward P (2009) Identification and Characterization of a Non-retinoid Ligand for Retinol-binding Protein 4 Which Lowers Serum Retinol-binding Protein 4 Levels in Vivo. J. Biol. Chem 284, 7673–7680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Wang Y, Connors R, Fan P, Wang X, Wang Z, Liu J, Kayser F, Medina JC, Johnstone S, Xu H, Thibault S, Walker N, Conn M, Zhang Y, Liu Q, Grillo MP, Motani A, Coward P, and Wang Z (2014) Structure-assisted discovery of the first non-retinoid ligands for Retinol-Binding Protein 4. Bioorg. Med. Chem. Lett 24, 2885–2891. [DOI] [PubMed] [Google Scholar]
- (27).Kruse AC, Ring AM, Manglik A, Hu J, Hu K, Eitel K, Hubner H, Pardon E, Valant C, Sexton PM, Christopoulos A, Felder CC, Gmeiner P, Steyaert J, Weis WI, Garcia KC, Wess J, and Kobilka BK (2013) Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504, 101–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Haga K, Kruse AC, Asada H, Yurugi-Kobayashi T, Shiroishi M, Zhang C, Weis WI, Okada T, Kobilka BK, Haga T, and Kobayashi T (2012) Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature 482, 547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Schrage R, Holze J, Klöckner J, Balkow A, Klause AS, Schmitz A-L, De Amici M, Kostenis E, Tränkle C, Holzgrabe U, and Mohr K (2014) New insight into active muscarinic receptors with the novel radioagonist [3H]iperoxo. Biochem. Pharmacol 90, 307–319. [DOI] [PubMed] [Google Scholar]
- (30).Kulak O, Chen H, Holohan B, Wu X, He H, Borek D, Otwinowski Z, Yamaguchi K, Garofalo LA, Ma Z, Wright W, Chen C, Shay JW, Zhang X, and Lum L (2015) Disruption of Wnt/β-Catenin Signaling and Telomeric Shortening Are Inextricable Consequences of Tankyrase Inhibition in Human Cells. Mol. Cell. Biol 35, 2425–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Shultz MD, Kirby CA, Stams T, Chin DN, Blank J, Charlat O, Cheng H, Cheung A, Cong F, Feng Y, Fortin PD, Hood T, Tyagi V, Xu M, Zhang B, and Shao W (2012) [1,2,4]Triazol-3-ylsulfanylmethyl)-3-phenyl-[1,2,4]oxadiazoles: Antagonists of the Wnt Pathway That Inhibit Tankyrases 1 and 2 via Novel Adenosine Pocket Binding. J. Med. Chem 55, 1127–1136. [DOI] [PubMed] [Google Scholar]
- (32).Brandstetter H, Kühne A, Bode W, Huber R, von der Saal W, Wirthensohn K, and Engh RA (1996) X-ray Structure of Active Site-inhibited Clotting Factor Xa: IMPLICATIONS FOR DRUG DESIGN AND SUBSTRATE RECOGNITION. J. Biol. Chem 271, 29988–29992. [DOI] [PubMed] [Google Scholar]
- (33).Bissantz C, Kuhn B, and Stahl M (2010) A Medicinal Chemist’s Guide to Molecular Interactions. J. Med. Chem 53, 5061–5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Petit-Topin I, Fay M, Resche-Rigon M, Ulmann A, Gainer E, Rafestin-Oblin ME, and Fagart J (2014) Molecular determinants of the recognition of ulipristal acetate by oxo-steroid 770 receptors. J. Steroid Biochem. Mol. Biol 144, 427–435. [DOI] [PubMed] [Google Scholar]
- (35).Lusher SJ, Raaijmakers HCA, Vu-Pham D, Dechering K, Lam TW, Brown AR, Hamilton NM, Nimz O, Bosch R, McGuire R, Oubrie A, and de Vlieg J (2011) Structural Basis for Agonism and Antagonism for a Set of Chemically Related Progesterone Receptor Modulators. J. Biol. Chem 286, 35079–35086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Kapoor K, Finer-Moore JS, Pedersen BP, Caboni L, Waight A, Hillig RC, Bringmann P, Heisler I, Müller T, Siebeneicher H, and Stroud RM (2016) Mechanism of inhibition of human glucose transporter GLUT1 is conserved between cytochalasin B and phenylalanine amides. Proc. Natl. Acad. Sci. U. S. A 113, 4711–4716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Hanson MA, Cherezov V, Roth CB, Griffith MT, Jaakola V-P, Chien EYT, Velasquez J, Kuhn P, and Stevens RC (2008) A specific cholesterol binding site is established by the 2.8 Å structure of the human β(2)-adrenergic receptor in an alternate crystal form. Structure (Oxford, U. K.) 16, 897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Ring AM, Manglik A, Kruse AC, Enos MD, Weis WI, Garcia KC, and Kobilka BK (2013) Adrenaline-activated structure of the β(2)-adrenoceptor stabilized by an engineered nanobody. Nature 502, 575–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Tagami U, Takahashi K, Igarashi S, Ejima C, Yoshida T, Takeshita S, Miyanaga W, Sugiki M, Tokumasu M, Hatanaka T, Kashiwagi T, Ishikawa K, Miyano H, and Mizukoshi T (2016) Interaction Analysis of FABP4 Inhibitors by X-ray Crystallography and Fragment Molecular Orbital Analysis. ACS Med. Chem. Lett 7, 435–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Gonzalez JM, and Fisher SZ (2015) Structural analysis of ibuprofen binding to human adipocyte fatty-acid binding protein (FABP4). Acta Crystallogr., Sect. F: Struct. Biol. Commun 71, 163–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Lebon G, Warne T, Edwards PC, Bennett K, Langmead CJ, Leslie AGW, and Tate CG (2011) Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 474, 521–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Congreve M, Andrews SP, Doré AS, Hollenstein K, Hurrell E, Langmead CJ, Mason JS, Ng IW, Tehan B, Zhukov A, Weir M, and Marshall FH (2012) Discovery of 1,2,4-Triazine Derivatives as Adenosine A2A Antagonists using Structure Based Drug Design. J. Med. Chem 55, 1898−1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Barillari C, Taylor J, Viner R, and Essex JW (2007) Classification of Water Molecules in Protein Binding Sites. J. Am. Chem. Soc 129, 2577–2587. [DOI] [PubMed] [Google Scholar]
- (44).Lu Y, Wang R, Yang C-Y, and Wang S (2007) Analysis of Ligand-Bound Water Molecules in High-Resolution Crystal Structures of Protein−Ligand Complexes. J. Chem. Inf. Model 47, 668–675. [DOI] [PubMed] [Google Scholar]
- (45).Mpamhanga CP, Spinks D, Tulloch LB, Shanks EJ, Robinson DA, Collie IT, Fairlamb AH, Wyatt PG, Frearson JA, Hunter WN, Gilbert IH, and Brenk R (2009) One Scaffold, Three Binding Modes: Novel and Selective Pteridine Reductase 1 Inhibitors Derived from Fragment Hits Discovered by Virtual Screening. J. Med. Chem 52, 4454–4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Borsari C, Luciani R, Pozzi C, Poehner I, Henrich S, Trande M, Cordeiro-da-Silva A, Santarem N, Baptista C, Tait A, Di Pisa F, Dello Iacono L, Landi G, Gul S, Wolf M, Kuzikov M, Ellinger B, Reinshagen J, Witt G, Gribbon P, Kohler M, Keminer O, Behrens B, Costantino L, Tejera Nevado P, Bifeld E, Eick J, Clos J, Torrado J, Jiménez-Antón MD, Corral MJ, Alunda JM, Pellati F, Wade RC, Ferrari S, Mangani S, and Costi MP (2016) Profiling of Flavonol Derivatives for the Development of Antitrypanosomatidic Drugs. J. Med. Chem 59, 7598–7616. [DOI] [PubMed] [Google Scholar]
- (47).Tong L, Pav S, Lamarre D, Pilote L, LaPlante S, Anderson PC, and Jung G (1995) High Resolution Crystal Structures of Recombinant Human Renin in Complex with Polyhydroxymonoamide Inhibitors. J. Mol. Biol 250, 211–222. [DOI] [PubMed] [Google Scholar]
- (48).Powell NA, Ciske FL, Cai C, Holsworth DD, Mennen K, Van Huis CA, Jalaie M, Day J, Mastronardi M, McConnell P, Mochalkin I, Zhang E, Ryan MJ, Bryant J, Collard W, Ferreira S, Gu C, Collins R, and Edmunds JJ (2007) Rational design of 6-(2,4-diaminopyrimidinyl)-1,4-benzoxazin-3-ones as small molecule renin inhibitors. Bioorg. Med. Chem 15, 5912–5949. [DOI] [PubMed] [Google Scholar]
- (49).Roth BL, Irwin JJ, and Shoichet BK (2017) Discovery of new GPCR ligands to illuminate new biology. Nat. Chem. Biol 13, 1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).DeWire SM, Yamashita DS, Rominger DH, Liu G, Cowan CL, Graczyk TM, Chen X-T, Pitis PM, Gotchev D, Yuan C, Koblish M, Lark MW, and Violin JD (2013) A G Protein-Biased Ligand at the μ-Opioid Receptor Is Potently Analgesic with Reduced Gastrointestinal and Respiratory Dysfunction Compared with Morphine. J. Pharmacol. Exp. Ther 344, 708–717. [DOI] [PubMed] [Google Scholar]
- (51).Khersonsky O, Roodveldt C, and Tawfik DS (2006) Enzyme promiscuity: evolutionary and mechanistic aspects. Curr. Opin. Chem. Biol 10, 498–508. [DOI] [PubMed] [Google Scholar]
- (52).Kahraman A, Morris RJ, Laskowski RA, Favia AD, and Thornton JM (2010) On the diversity of physicochemical environments experienced by identical ligands in binding pockets of unrelated proteins. Proteins: Struct., Funct., Genet 78, 1120–1136. [DOI] [PubMed] [Google Scholar]
- (53).Lin H, Sassano MF, Roth BL, and Shoichet BK (2013) A pharmacological organization of G protein-coupled receptors. Nat. Methods 10, 140–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Barelier S, Sterling T, O’Meara MJ, and Shoichet BK (2015) The Recognition of Identical Ligands by Unrelated Proteins. ACS Chem. Biol 10, 2772–2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Lassalas P, Gay B, Lasfargeas C, James MJ, Tran V, Vijayendran KG, Brunden KR, Kozlowski MC, Thomas CJ, Smith AB, Huryn DM, and Ballatore C (2016) Structure Property Relationships of Carboxylic Acid Isosteres. J. Med. Chem 59, 3183–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Arnett CD, Wright J, and Zenker N (1978) Synthesis and adrenergic activity of benzimidazole bioisosteres of norepinephrine and isoproterenol. J. Med. Chem 21, 72–78. [DOI] [PubMed] [Google Scholar]
- (57).Sigala PA, Kraut DA, Caaveiro JMM, Pybus B, Ruben EA, Ringe D, Petsko GA, and Herschlag D (2008) Testing Geometrical Discrimination within an Enzyme Active Site: Con-strained Hydrogen Bonding in the Ketosteroid Isomerase Oxyanion Hole. J. Am. Chem. Soc 130, 13696–13708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Wishart DS, Jewison T, Guo AC, Wilson M, Knox C, Liu Y, Djoumbou Y, Mandal R, Aziat F, Dong E, Bouatra S, Sinelnikov I, Arndt D, Xia J, Liu P, Yallou F, Bjorndahl T, Perez-Pineiro R, Eisner R, Allen F, Neveu V, Greiner R, and Scalbert A (2012) HMDB 3.0−The Human Metabolome Database in 2013. Nucleic Acids Res 41, D801–D807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Laskowski RA, and Swindells MB (2011) LigPlot+: Multiple Ligand−Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model 51, 2778–2786. [DOI] [PubMed] [Google Scholar]
- (60).Fährrolfes R, Bietz S, Flachsenberg F, Meyder A, Nittinger E, Otto T, Volkamer A, and Rarey M (2017) ProteinsPlus: a web portal for structure analysis of macromolecules. Nucleic Acids Res 45, W337–W343. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.