

Abstract
Ubiquitination is an essential protein modification that influences eukaryotic processes ranging from substrate degradation to nonproteolytic pathway alterations, including DNA repair and endocytosis. Previous attempts to analyze substrates via affinity purification approach in which ubiquitin ligases are fused to a polyubiquitin-binding domain, which allows the isolation ubiquitin. By using this protocol, ubiquitinated substrates that are specific for a given ligase can be isolated for mass spectrometry or western blot analysis. After cells have been collected, the described protocol can be completed in 2–3 d.
INTRODUCTION
Ubiquitin is a highly conserved, small polypeptide that is covalently linked to protein substrates targeted for intracellular modification. Conjugation by ubiquitin alters protein function and stability with important roles in various biological processes, such as regulation of the cell cycle, response to DNA damage, intracellular trafficking and surveillance of protein quality. A sequential enzymatic cascade transfers ubiquitin to its target, with an E3 ligase catalyzing the final step: a covalent linkage to the e-amino group of a lysine residue or an N-terminal methionine of the substrate1–4. Despite considerable efforts, the identification of substrates for specific ubiquitin ligases remains a challenge.
Past limitations of identifying ubiquitinated substrates
Approaches to identifying ubiquitinated substrates in vivo generally consist of two different types of experiments. The global protein stability profiling technique and related methods involve comparing changes in steady-state levels of total protein in the presence or absence of a given ligase using GFP-fused potential substrates5–8. Although they are successful in identifying targets of proteolysis, these techniques do not allow for the detection of nondegradative ubiquitination events or degradation of minor subpopulations. In addition, the lack of an E3 ligase can have detrimental effects on cellular physiology, thus perturbing the ubiquitin proteome indirectly9,10. Moreover, some substrates are targeted by more than one ligase; under these conditions, the absence of a single ligase may fail to substantially stabilize the substrate. For example, the yeast G1 cyclin Cln3 is targeted by the F-box proteins Grrl and Cdc4, depending on its subcellular localization, whereas human p53 turns over even in the absence of its well-studied ligase Mdm2, possibly owing to targeting by other ligases, such as Trim24, Pirh2, Copl and ARF-BP1 (refs. 11,12). Other approaches to identifying ligase targets involve the immunoprecipitation of ligase-substrate complexes followed by mass spectrometric (MS) analysis of the isolated peptides13–15. The main drawback of affinity-based methods is that ligase-substrate interactions may be too weak for co-purification of the target protein; certain ubiquitin ligases dissociate from their substrates on the order of seconds16. Although some groups have used in vivo cross-linking to overcome this challenge17,18, the weak binding of ubiquitin ligases to their substrates still remains a substantial barrier to the identification of new substrates.
Protocol overview
Here we describe a protocol that uses ubiquitin ligases fused to polyubiquitin-binding domains (ligase traps) to identify ligase substrates in yeast and mammalian cells with greater efficacy. The presence of a polyubiquitin-binding domain increases the binding affinity of a ligase to its ubiquitinated substrates. To validate our approach, we generated ligase traps using the UBA (ubiquitin-associated) domains from the soluble ubiquitin receptor proteins Rad23 and Dsk2, which deliver ubiquitinated substrates to the 26S proteasome19,20. We chose the UBA class of polyubiquitin-binding domains because they exhibit high affinity for polyubiquitinated polypeptide chains. The Rad23 UBA domain binds both K48-and K63-linked polyubiquitin, but it exhibits an approximately fourfold preference for K48-linked polyubiquitin, whereas the Dsk2 UBA domain can recognize monoubiquitin, K48-and K63-linked polyubiquitin21. To increase our ability to unambiguously identify substrates captured by the ligase trap, we adopted a two-step tandem affinity purification protocol using hexahistidine (6×His)-tagged ubiquitin to isolate the ubiquitinated species selectively (Fig. 1). First, we perform a FLAG–specific immuno-precipitation under native conditions to enrich for the ligase trap and its interacting proteins. A subsequent Ni-NTA pulldown under denaturing conditions selectively captures proteins conjugated with polyhistidine-tagged ubiquitin. This two-step purification allows for the enrichment of ubiquitin-conjugated substrates, including those that constitute only a small fraction of the total cellular protein. We have used this technique to isolate substrates of the Skp1–Cullin–F-box (SCF) family of ubiquitin ligases in both budding yeast and mammalian cells. Purified substrates are subjected to MS analysis.
Figure 1 |.
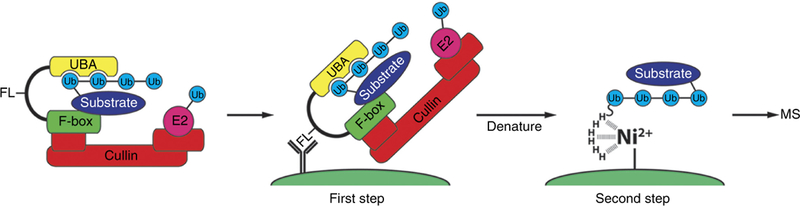
Overview of the ligase trapping procedure. F-box-UBA domain fusion proteins (i.e., ligase traps) are expressed in cells at physiological levels along with overexpression of a single copy of the ubiquitin gene containing an N-terminal hexahistidine epitope tag. The UBA of the ligase trap interacts with the nascent ubiquitin chain on endogenous SCF substrates, thereby delaying their release (left). Cells are then lysed and subjected to an anti-FLAG coimmunoprecipitation under native conditions to isolate ligase trap complexes (center). FLAG eluates are collected and a second purification is performed using Ni-NTA agarose beads under denaturing conditions to capture ubiquitinated substrates (right). This second step eliminates interactors associated with the ubiquitinated species that are not themselves substrates. FL, FLAG; H, histidine.
Advantages and applications of ligase traps
Ligase trapping allows for the reliable identification of protein ubiquitination in vivo. The protocol is fast and relatively straightforward, and the purification scheme takes ~2–3 d to complete. The efficiency and reliability of this method make this procedure generally accessible to most laboratories. This technique can be easily applied to many ubiquitin ligases, and it will prove useful in the identification of novel substrates. One advantage of ligase trapping is its high specificity and low background. Indeed, our MS analysis detected zero peptides for most off-target ligases in our yeast purifications22. Another advantage lies in the ability to use ligase trapping to validate substrates in follow-up experiments. For example, smaller-scale cultures can be used to visualize substrate ubiquitination via western blot analysis. Finally, ligase trapping can be performed under various perturbations, such as environmental stress or chemical agents, which may alter the repertoire of substrates for certain ligases.
Limitations of ligase traps
As with any biochemical method, there are a number of caveats that must be considered when you are using ligase traps. First, a ligase may not be amenable to protein fusion at either terminus (i.e., yeast Hrdl), and doing so may disrupt its ability to target its biological substrates. Second, ligase trapping is a stoichiometric procedure, and lower-affinity substrates will be identified with fewer spectral counts, although one spectral count is sufficient for identification. Furthermore, the UBA domains described here have not been well characterized for their ability to bind atypical polyubiquitin chains, such as Kll- or K33-linked ubiquitin, so we cannot attest to their success in capturing substrates modified with these chain types. Ligase trapping is optimal for the identification of substrates of specific ligases and, unlike more global approaches, it is not designed for analysis of the total ubiquitinated proteome. The limiting factor for scaling up this method is the workload required to generate additional UBA-fusion constructs for each ligase to be screened.
Comparison with alternative protocols
Numerous protocols have been developed that rely on tandem affinity purifications to capture physical interactors of ubiquitin ligases23–25. Strategies have been developed independently for the isolation of ubiquitinated proteins from cells based on the overexpression of tagged ubiquitin combined with at least one denaturing purification step to reduce nonubiquitinated interactors26–33. Alternatively, studies have used immobilized poly-ubiquitin-binding domains (poly-UBDs) to isolate endogenously ubiquitinated proteins34–39. Our ligase trap method combines elements from each of these protocols, namely the enrichment capacity of tandem affinity ligase purification, with the added selectivity for polyubiquitinated proteins via 6×Hisubiquitin denaturing purifications and fusion with UBDs.
Other MS-based approaches that enable the identification of ubiquitinated substrates of specific ligases have been published. Most of these exploit the physical association of substrates with their respective ligases, whereas some take a more global approach to identifying ubiquitinated peptides.
The Parallel Adapter Capture (PAC) technology developed by Harper and colleagues40 uses comparative MS to analyze immunopurified ligase adapter proteins in the presence or absence of proteasome or NEDD8 inhibition. Compared with ligase trapping, this method is similar in that it also enables the identification of substrates for a specific ligase adapter, as opposed to a family of adapters or ligases. Similarly, it can identify substrates of low abundance, as a single peptide is sufficient for identification. Both protocols rely on the identification of interactors that are specific to a particular ligase by comparing MS results for many different ligases, either using the Comparative Proteomics Analysis Software Suite (CompPASS), for PAC41,42, or a similar method that compares each ligase trap IP with all other ligase trap IPs done in the same organism within our laboratory22,43. A notable difference between the two protocols is that ligase trapping greatly enriches for substrates, whereas PAC does not distinguish substrates from other stably interacting proteins. Further, ligase trapping provides a way to validate candidate substrates, as ubiquitinated species of substrate proteins can be purified by the ligase trap construct and visualized by western blot analysis. A comparison of our recent analysis of substrates of the human F-box β-transducing-repeat-containing protein (β-TrCP)43 with a similar study that used PAC44 shows the relative strengths of these techniques. For the well-studied F-box protein β-TrCP, ligase trapping identified 28 unique interactors, of which l2 were known substrates. We attempted to validate 14 of the novel substrates, and we successfully purified ubiquitinated species of 11 of these. By using PAC to study the same ligase, Kim et al.44 identified 151 interactors, of which 16 were previously known substrates. They tested whether nine of the novel interactors that they identified were actually β-TrCP substrates, and the showed that three were stabilized by β-TrCP knockdown and that two additional substrates bound to β-TrCP only in the presence of the proteasome inhibitor MG132, and thus they are probable substrates. We used ~109 cells for each MS experiment, whereas a typical PAC protocol uses 107 cells40. Thus, experiments published so far suggest that a far higher percentage of the interactors discovered by ligase trapping are bona fide substrates, whereas PAC may discover a slightly higher number of true substrates and requires substantially less material.
Ubiquitinated peptides can be identified via MS detection of Gly-Gly (di-GLY) residues that arise after trypsinization of ubiquitinated samples26,45–47. Recently, this technology has been adapted to identify ligase substrates by comparing the repertoire of diGLY-modified peptides after genetic or chemical perturbation of a particular ligase48–50. These studies identified comparable numbers of substrates as ligase trapping. However, this approach suffers reproducibility issues for low-abundance targets, because of stochastic sampling51, and, as mentioned earlier, altering the function or abundance of a given ligase may lead to nonphysiologic results.
Experimental design
Generation and growth of ligase trap yeast strains.
This protocol requires a yeast strain that expresses both (ii) galactose-inducible 6×His-tagged ubiquitin and (ii) an F-box protein fused to a UBA domain via a 3×FLAG linker sequence. To generate the latter construct, we used the integrating vector pRS306 to clone the DNA in the following order: a partial C terminus of an F-box protein, a 3×FLAG linker sequence and either the two C-terminal UBA domains of the RAD23 gene (codons 143–397) or the single UBA domain of the DSK2 gene (codons 327–373). To integrate the ligase trap into its endogenous locus, the plasmid was linearized at a unique restriction site in the DNA encoding the F-box protein and transformed into yeast cells. Ligase traps with the UBA domain fused to the N terminus of the F-box protein were also created (promoter-UBA-3×FLAG-F-box). To overexpress 6×His-ubiquitin, we inserted the His3MX-GAL1–6×His cassette upstream of the last ubiquitin sequence in the UBI4 locus (which contains five ubiquitin sequences in tandem), thus deleting the UBI4 promoter. Colonies were checked by PCR, and we selected a strain containing only a single remaining copy of ubiquitin tagged with 6×His under the GAL1 promoter. The tagged ubiquitin is expressed at a high level upon growth in galactose, such that at least 50% of the total ubiquitin in the cell is tagged. To express sufficient levels of 6×His-ubiquitin, yeast cell lines in log phase are grown in galactose for slightly less than two doubling times before collection, lysis and two-step immunoprecipitation.
Generation and growth of ligase trap–stable mammalian cell lines.
To produce an analogous stable mammalian cell line, we integrated three constructs into 293 FlpIn TRex cells. 293 FlpIn TRex cells already express the tet repressor; therefore, genes whose promoter sequences contain the tet operator are expressed only upon the addition of doxycycline. Into these cells, we first tranfected a linearized plasmid encoding doxycycline-inducible 6×His-ubiquitin marked with neoR, selected clonal stable cell lines and screened for those with the highest doxycycline-inducible 6×His-ubiquitin expression, such that tagged ubiquitin represents at least one-quarter of the total ubiquitin pool upon doxycycline treatment. Next, we transfected this stable cell line with a linearized plasmid encoding both an shRNA against the endogenous ubiquitin ligase of interest and an shRNA-resistant version of this ligase fused to 3×FLAG and the C terminus of RAD23B (codons 185–409), which contains two UBA domains; this plasmid was marked with hygromycin resistance. We selected hygromycin-resistant clonal cell lines and screened for those that both repressed the endogenous ligase (where this was possible to ascertain) and expressed the ligase trap at nearendogenous levels, as measured by western blotting. For ligases for which no antibody was available, we compared the expression of the ligase trap with the expression of another ligase trap for which an antibody was available, and we calculated the appropriate expression level on the basis of the relative message levels of the two ligases in HEK293 cells (see Sultan et al.52).
Efficient identification of ubiquitin ligase substrates using this technique requires a large number of cells: 63 just-subconfluent 245 × 245 mm dishes. As mentioned earlier, this is substantially more cells than are used in similar techniques: we use ~109 cells, whereas Harper and colleagues’ PAC protocol uses 107 cells40. The larger initial input in our protocol is probably required because our two-step purification only captures the small pool of any sub-strate that is ubiquitinated at the moment of cell lysis. However, two repeats of each purification are typically sufficient to identify substrates. Cells are treated with MG132 for 4 h before collection, lysis and immunoprecipitation.
Proteomic conditions.
As with all proteomics, care should be taken to avoid keratin contamination of samples, as this will interfere with the detection of substrate peptides. For reagent preparation, we recommend the following precautions:
Purchase HPLC-grade water for all solutions.
Use polypropylene supplies for all purification procedures. If you must use durable plastic or glassware, be sure to wash them well with Milli-Q water.
Use dedicated reagents and supplies for MS analysis, if possible. Minimize handling. (Autoclaving does not remove keratin.)
Sterilize the buffers by filtration through a 0.2-μm syringe filter.
Use sterile, disposable consumables, tubes and bottles for reagent storage.
Wear a lab coat and change gloves often.
Wear disposable hair bonnets, if possible.
Use barrier pipette tips.
Sample monitoring.
Before MS analysis, it is useful to collect a small quantity of each eluate for western blotting and silver staining to estimate the efficiency of purification. This is noted in Steps 1A and 1B(x, xiii,xvi,xvii,xix).
Timing.
To produce a protein sample for MS analysis from saturated liquid yeast cultures or confluent tissue culture plates, it takes ~2.5 or 5 d, respectively. After data collection, processing of the results obtained can take markedly longer.
Future directions.
Ligase trapping works very well for some ubiquitin ligases (e.g., Grr1 and β-TrCP), and it works less well for others, such as Hrd1, Cdc20 and Fbw7). The identification of substrates for some ligases may be improved by optimization of the ligase trap. Ubiquitin ligases may be fused to the UBA domain on the opposite end, fused to a different UBA domain or expressed at a higher level. We will determine whether the efficacy of this protocol is increased by the use of UBA domains with increased affinities for polyubiquitin chains, such as tandem ubiquitin-binding entities37, or by the use of linkage-specific polyubiquitin-binding domains (i.e., TAB2 NZF for K63, NEMO UBAN for linear)53,54. We are currently rebuilding ligase traps with 4–6 copies of the UBA. Although we were initially worried that high-affinity UBAs might nonspecifically pull down poly-ubiquitinated proteins, this does not appear to be a problem when ligases are expressed at endogenous levels.
This protocol describes the use of UBA-ligase fusion proteins to identify substrates of the SCF family of ubiquitin ligases, but this work could be expanded to other families of ubiquitin ligases. In addition, other ubiquitin-like modifiers, such as SUMO or LC3, also have specific binding domains55,56, and they can be used to generate an analogous enzyme ‘trap’ to search for target substrates.
MATERIALS
REAGENTS
Materials related to budding yeast culture (Step 1A)
Yeast strain (Saccharomyces cerevisiae)—ubi4::GAL1pr-6×HisUb for the generation of ligase trap strain (available upon request)
Plasmid with F-box-UBA fusion for the generation of ligase trap strain (available upon request)
Adenine (Sigma-Aldrich, cat. no. A8626)
Uracil (Sigma-Aldrich, cat. no. U0750)
Yeast extract (Fisher, cat. no. BP1422)
Bacto peptone (BD Biosciences, cat. no. 211677)
Ammonium sulfate (Fisher, cat. no. A702)
Succinic acid (Sigma-Aldrich, cat. no. S7501)
Yeast nitrogen base without AA, carbohydrate and AS (US Biological, cat. no. Y2030)
Raffinose pentahydrate, low glucose (US Biological, cat. no. R1030)
D-(+)-Galactose (Sigma-Aldrich, cat. no. G0750)
Materials related to tissue culture (Step 1B)
Cell line 293 FlplnTRex pTB30 clone 4 (available upon request), which expresses 6×His-ubiquitin when induced with doxycycline, to generate the ligase trap cell lines. 293 FlpIn TRex cells are available from Life Technologies (cat. no. R780–07)
Ligase trap plasmids (available upon request)
DMEM, high glucose (Life Technologies, cat. no. 11965)
FBS (Sigma-Aldrich, cat. no. 12306C): choose a lot that is low in tetracycline
Large, square (245 × 245 mm) tissue-culture treated dishes (Corning, cat. no. 431110)
Trypsin-EDTA (Life Technologies, cat. no. 25200)
Doxycycline hyclate (Sigma-Aldrich, cat. no. D9891)
1× PBS (Life Technologies, cat. no. 14190)
T-150 filter cap tissue culture flasks (Cyto-One, cat. no. CC7682–4815)
Materials required for both protocols (Steps 1A and 1B)
Water, filtered, HPLC grade (Fisher Scientific, cat. no. W5)
DMSO (Sigma-Aldrich, cat. no. D8418)
HEPES (Fisher Scientific, cat. no. BP310)
Potassium acetate (Fisher Scientific, cat. no. BP364)
Magnesium chloride (Fisher Scientific, cat. no. M33)
Calcium chloride (Fisher Scientific, cat. no. C77)
Sodium fluoride (Fisher Scientific, cat. no. S299)
Protease inhibitor tablets, EDTA-free (Roche, cat. no. 04693132001)
Phosphatase inhibitor cocktail tablets, PhosSTOP (Roche, cat. no. 04906837001)
Leupeptin (Sigma-Aldrich, cat. no. L2023)
Bestatin (Sigma-Aldrich, cat. no. B8385)
Benzamidine HCl (Sigma-Aldrich, cat. no. B6506)
Pepstatin A (Sigma-Aldrich, cat. no. P5318)
PMSF (Sigma-Aldrich, cat. no. 78830) ! CAUTION PMSF is harmful; handle it with extra care.
Sodium orthovanadate (ACROS Organics, cat. no. 205330500)
β-Glycerophosphate disodium salt hydrate (Sigma-Aldrich, cat. no. G5422)
Sodium chloride (Fisher Scientific, cat. no. S640)
Potassium chloride (Fisher Scientific, cat. no. BP366)
Sodium phosphate (ACROS Organics, cat. no. 424395000)
Potassium phosphate dibasic (Fisher Scientific, cat. no. BP363)
Sodium hydroxide (Fisher Scientific, cat. no. S318) ! CAUTION Sodium hydroxide is corrosive; handle it with extra care.
DNase I from bovine pancreas (Sigma-Aldrich, cat. no. D4527)
MG132 (Sigma-Aldrich, cat. no. C2211)
Iodoacetamide (Sigma-Aldrich, cat. no. I1149)
Anti-FLAG M2 magnetic beads (Sigma-Aldrich, cat. no. M8823)
3×FLAG peptide, lyophilized powder (Sigma-Aldrich, cat. no. F4799)
Nonidet P-40 (US Biological, cat. no. N3500)
Tris base (Fisher, cat. no. BP152)
Ni-NTA agarose (Invitrogen, cat. no. R901)
Urea (Fisher, cat. no. BP169)
Imidazole (Sigma-Aldrich, cat. no. 56750) ! CAUTION Imidazole is harmful; handle it with extra care.
RapiGest SF surfactant (Waters, cat. no. 186001186)
EDTA disodium salt dihydrate (Fisher, cat. no. BP120)
Glycerol (Fisher, cat. no. BP229)
SDS (Fisher, cat. no. BP166) ! CAUTION SDS is harmful if it is inhaled in powder form; wear a mask and handle it with extra care.
2-Mercaptoethanol (Sigma-Aldrich, cat. no. M6250) ! CAUTION 2-mercaptoethanol is toxic; avoid exposure and handle it in a fume hood.
Pierce bicinchoninic acid (BCA) protein assay kit (Thermo Scientific, cat. no. 23225) ! CAUTION BCA protein assay reagent B is very toxic; handle it with extra care.
Pierce silver stain kit (Thermo Scientific, cat. no. 24612)
Mouse monoclonal anti-6xHis, albumin free (Clontech, cat. no. 631212)
Mouse monoclonal anti-Flag M2 (Sigma-Aldrich, cat. no. F1804)
Mouse monoclonal anti-ubiquitin P4D1 (available from Cell Signaling Technology, cat. no. 3936)
Mouse monoclonal anti-Cul1, clone 2H4C9 (Invitrogen, cat. no. 32–2400)
EQUIPMENT
Incubator shaker, 30 °C
Growing flasks for yeast
Centrifuge (Avanti J-20 XP) with JLA-9.1 and JA-25.5 rotors
Nalgene Oak Ridge high-speed centrifuge tubes (Thermo Scientific, cat. no. 05–562-16A)
Magnetic stir bar
Stirring hot plate
Polycarbonate bottle assemblies, 1 liter (Beckman Coulter, cat. no. A98812)
Luer-Lok tip syringe, 10 ml (BD Biosciences, cat. no. 309604)
Luer-Lok tip syringe, 60 ml (BD Biosciences, cat. no. 309653)
Nalgene syringe filter, 0.2 μm (Thermo Scientific, cat. no. 190–2520)
Nalgene Rapid-Flow sterile disposable bottle top filter, 0.2 μm (Thermo Scientific, cat. no. 291–3320)
Microcentrifuges (Eppendorf 5430 R and Eppendorf 5415 D)
Heat block
Microcentrifuge tubes, 2.0 ml (Axygen, cat. no. 311–10-051)
Low-adhesion microcentrifuge tubes, 1.5 ml (USA Scientific, cat. no. 1415–2600)
Low-adhesion microcentrifuge tubes, 0.5 ml (USA Scientific, cat. no. 1405–2600)
Polypropylene conical centrifuge tube, 15 ml (Falcon, cat. no. 352096)
Polypropylene conical centrifuge tube, 50 ml (Falcon, cat. no. 352070)
Screw-cap tubes (Axygen, cat. no. SCT-150-W)
UV-visible photometer
Rocking platform
Vortex mixer with multiple sample head
Scissors
Spatula
Clean razor blades
Shaker rotisserie
Ball mill (Retsch M301) with CryoKit and 25-mm-diameter steel grinding ball
Six-tube magnetic stand (Ambion, cat. no. AM10055)
PrecisionGlide needle, 25G ×1 ½ inch (BD Biosciences, cat. no. 305127)
GeLoader pipette tips (Eppendorf, cat. no. 022351656)
Criterion Tris-HCl gels, 4–20% (wt/vol), 26 well, 15 μl (Bio-Rad, cat. no. 345–0034)
REAGENT SETUP
100× adenine Mix 2 g of adenine powder with 5 ml of 4 M NaOH in 1 liter of water. Autoclave the solution for 30 min. This solution can be made ahead of time and stored at room temperature (~23 °C) for several months.
100× uracil Mix 2 g of uracil powder with 5 ml of 4 M NaOH in 1 liter of water. Autoclave the solution for 30 min. This solution can be made ahead of time and stored at room temperature for several months.
YM-1 medium Resuspend 5.5 g of yeast extract, 11 g of peptone, 5.5 g of ammonium sulfate, 11 g of succinic acid, 1.61 g of yeast nitrogen base without amino acids or ammonium sulfate, 5.72 g of NaOH, 20 ml of 100× adenine and 20 ml of 100× uracil in 1 liter of water. Titrate the mixture to pH 5.8 with NaOH. Autoclave the solution for 60 min. This solution can be made ahead of time and stored at room temperature for several months.
20% (wt/vol) raffinose Dissolve 200 g of raffinose in 1 liter of water. Filter the solution with a bottle-top filter and autoclave it for 30 min. This solution can be made ahead of time and stored at room temperature for several months.
20% (wt/vol) galactose Dissolve 200 g of galactose in 1 liter of water. Filter the solution with a bottle-top filter and autoclave it for 30 min. This solution can be made ahead of time and stored at room temperature for several months.
DNase I Resuspend DNase I in lysis buffer to a final concentration of 10,000 U/ml. Aliquots can be stored at −20 °C for up to 2 years. Do not re-freeze the aliquots.
MG132 Resuspend MG132 in DMSO to a final concentration of 50 mg/ml. Aliquots can be stored at –20 °C for several months.
10×FLAG peptide Resuspend 3×FLAG peptide to a final concentration of 5 mg/ml in PBS. Aliquots of 100 μl can be made ahead of time and stored at –80 °C.
10% (wt/vol) RapiGest Dissolve one vial (1 mg) of RapiGest in 100 μl of water. Freshly prepare the solution and keep it at room temperature.
Doxycycline Doxycyline stocks are made up at 10 mg/ml in DMSO. Stocks can be stored at –20 °C for several months without loss of activity.
1 M HEPES (pH 8.0) Prepare 1 M HEPES solution in dH2O, adjust the pH to 8.0 and autoclave the solution. Stock solutions can be made ahead of time and stored at room temperature for several months.
1 M potassium acetate Prepare 1 M potassium acetate solution in dH2O and autoclave the solution. Stock solutions can be made ahead of time and stored at room temperature for several months.
1 M magnesium chloride Prepare 1 M magnesium chloride solution in dH2O and autoclave it. Stock solutions can be made ahead of time and stored at room temperature for several months.
1 M calcium chloride Prepare 1 M calcium chloride solution in dH2O and autoclave it. Stock solutions can be made ahead of time and stored at room temperature for several months.
0.5 M EDTA Prepare 0.5 M EDTA solution in dH2O and autoclave it. Stock solutions can be made ahead of time and stored at room temperature for several months.
1 M Tris (pH 7.5) Prepare 1 M Tris solution in dH2O, adjust the pH to 7.5 and autoclave it. Stock solutions can be made ahead of time and stored at room temperature for several months.
20% (wt/vol) SDS Prepare 20% (wt/vol) SDS solution in dH2O. Stock solutions can be made ahead of time and stored at room temperature for several months.
1 M imidazole Prepare 1 M imidazole solution in dH2O and autoclave it. Stock solutions can be made ahead of time and stored at room temperature for several months.
100 mM PMSF Prepare 100 mM PMSF solution in ethanol. Aliquots can be stored at –20 °C for several months.
500 mM sodium fluoride Prepare 500 mM sodium fluoride in H2O. Aliquots can be stored at –20 °C for several months.
8 M β-glycerophosphate Prepare 8 M β-glycerophosphate in H2O. Aliquots can be stored at –20 °C for several months.
100 mM sodium orthovanadate Prepare 100 mM Sodium orthovanadate solution in H2O. Aliquots can be stored at –20 °C for several months.
100 μg/ml leupeptin Prepare 100 μg/ml leupeptin in H2O. Aliquots can be stored at –20 °C for several months.
100 μg/ml bestatin Prepare 100 μg/ml bestatin in H2O. Aliquots can be stored at –20 °C for several months.
1 M benzamidine HCl Prepare 1 M benzamidine HCl in H2O. Aliquots can be stored at –20 °C for several months.
100 μg/ml pepstatin A Prepare 100 μg/ml pepstatin A in H2O. Aliquots can be stored at –20 °C for several months.
Lysis buffer Prepare lysis buffer containing 25 mM HEPES (pH 8.0), 150 mM potassium acetate, 10 mM magnesium chloride, 5 mM calcium chloride, 1 mM PMSF, 5 mM sodium fluoride, 80 mM β-glycerophosphate, 1 mM sodium orthovanadate, 1 μg/ml leupeptin, 1 μg/ml bestatin, 1 benzamidine HCl, 1 μg/ml pepstatin A, protease inhibitor cocktail PhosSTOP tablets (2 per 100 ml), protease inhibitor cocktail tablets (EDTA-free, 2 per 100 ml) and 15 μg/ml MG132. For mammalian cell protocol only, add iodoacetamide to a final concentration of 20 mM. Filter it using a 0.2-μm syringe filter before use. Freshly prepare the solution and keep it at 4 °C.
PBS Prepare 2× PBS solution containing 270 mM sodium chloride, 5 mM potassium chloride, 17 mM sodium phosphate and 2 mM potassium phosphate. Titrate the solution to pH 7.4–7.5 with 8 M NaOH. This solution can be made ahead of time and stored at room temperature for several months.
FLAG elution buffer Prepare FLAG elution buffer by diluting 10× FLAG peptide to 0.5 mg/ml in 1× PBS with 0.08% (vol/vol) Nonidet P-40. Aliquots can be stored at –20 °C for up to 2 years. Do not re-freeze the aliquots.
Buffer B (9.4 M urea lysis buffer) Prepare a 1.5× solution of buffer B containing 118 mM sodium phosphate, 12 mM Tris base and 9 M urea. Stir the solution overnight and titrate it to pH 8.0 using 8 M NaOH. Filter the solution before use (using 0.2 μm syringe filter). Freshly prepare the solution and keep it at room temperature.
Ni-NTA final wash buffer Prepare a solution of Ni-NTA final wash buffer containing 100 mM sodium phosphate, 11 mM Tris base and 1 M urea. Adjust the pH to 8.0 using 8 M NaOH. Filter the solution before use (using a 0.2-μm syringe filter). Freshly prepare the solution and keep it at room temperature.
Ni-NTA elution buffer Prepare a solution of 0.5× Ni-NTA final wash buffer containing 300 mM imidazole and 0.1% (wt/vol) RapiGest. Freshly prepare the solution and keep it at room temperature.
Sina’s sample buffer Prepare a 2× solution of Sina’s sample buffer containing 0.1 M Tris (pH 7.5), 10 mM EDTA, 10% (vol/vol) SDS, 20% (vol/vol) glycerol, 1% (vol/vol) 2-mercaptoethanol and a sprinkle of bromophenol blue (for color). This buffer can be made ahead of time and stored at room temperature for several months. ! CAUTION 2-mercaptoethanol is toxic; avoid exposure and handle it in a fume hood.
PROCEDURE
1| This protocol can be performed using yeast cells (option A) or mammalian cells (option B).
(A) Procedure for yeast cells ● TIMING 2.5 d
-
(i)
Section 1: cell collection and lysis (6–7 h). From an overnight culture grown in YM-1 with 2% (vol/vol) raffinose, inoculate 4 liters of the same medium, and incubate it with vigorous shaking at 30 °C for 1 h to achieve an optical density at 600 nm (OD600) of 0.3.
▲ CRITICAL STEP This experimental protocol uses a 4-liter yeast culture that can be scaled down proportionally based on need. For example, a 1-liter culture may suffice for MS analysis of ligase traps that are strongly expressed. For validation experiments, we recommend using extracts from 350 ml of yeast culture. Further details for validation experiments are described in Box 1.
-
(ii)
Add 440 ml of 20% (wt/vol) galactose and continue incubation with shaking until an OD600 of 1.0 is obtained (~5–6 h).
-
(iii)
Centrifuge the cells at 17,000g for 10 min at 4 °C. Discard the supernatant and wash the pellet by vortexing it in 200 ml of water. Resuspend the pellet by vortexing it in 3.5 ml of lysis buffer.
▲ CRITICAL STEP Multiple samples can be normalized to each other by OD600.
■ PAUSE POINT Pellets can be stored at –80 °C until ready to use.
-
(iv)
Use scissors or a razor blade to make several nonoverlapping 1-cm slits in the cap of a 50-ml Falcon tube. Remove the cap and fill the tube with ~50 ml of liquid nitrogen. Use a pipette to add the yeast sample, drop by drop, into the liquid nitrogen-containing tube, while refilling with liquid nitrogen to maintain at least a 30-ml volume. This creates frozen droplet-sized sample ‘beads’. Place the screw cap back on and discard the liquid nitrogen through the slits. Alternatively, if you do not have access to a ball mill, we have had success lysing cells in a bead beater with glass beads. Divide the cell lysate into 1.5-ml screw-cap tubes.
▲ CRITICAL STEP Drop the sample into liquid nitrogen slowly to avoid clumps. Be careful when performing this procedure, as liquid nitrogen can cause severe burns. The use of a face shield is highly recommended.
? TROUBLESHOOTING
■ PAUSE POINT Sample ‘beads’ can be stored at –80 °C until required.
-
(v)
Transfer the sample ‘beads’ to a precooled steel ball mill chamber. Grind the sample with the ball mill using five cycles of 2 min at 27 Hz. Cool the chambers intermittently by submerging them in liquid nitrogen for 2 min between cycles. Alternatively, if you do not have access to a ball mill, add glass beads and agitate in a bead-beater six times for 1.5 min, resting 2 min on ice in between rounds.
▲ CRITICAL STEP Cool the chambers in liquid nitrogen before adding samples. Do not close the metal chambers too tightly; bubbles should appear when they are submerged. Apply safety precautions while handling liquid nitrogen.
? TROUBLESHOOTING
-
(vi)
Transfer the powder with a spatula that has been precooled in liquid nitrogen to a 50-ml Falcon tube and resuspend it in 10 ml of lysis buffer on a rocking platform at 4 °C. Make sure that the sample is thoroughly suspended (~2 h). Transfer the sample to a 15-ml Falcon tube. Add 500 μl of DNase I (5,000 U) and incubate at 4 °C on a rocking platform for 30 min. Alternatively, if you have lysed the cells by bead beating, pool the lysate in a 15-ml Falcon tube and proceed with adding DNase I.
▲ CRITICAL STEP Precool the spatula in liquid nitrogen before use to prevent the sample from melting.
-
(vii)
Preclear the lysate by centrifuging it at 6,000g for 10 min at 4 °C using an Oak Ridge centrifuge tube. Transfer the supernatant to a new Oak Ridge tube and centrifuge it at 58,500g for 1 h at 4 °C.
? TROUBLESHOOTING
-
(viii)
Section 2: purification—stage I, native FLAG (12–16 h). In a 15-ml Falcon tube, use the magnet of a six-tube magnetic stand to wash 380 μl of anti-FLAG magnetic bead slurry three times with 4 ml of lysis buffer containing 0.08% (vol/vol) Nonidet P-40.
▲ CRITICAL STEP Use wide-bore tips to avoid damaging the beads during pipetting. Mix the beads well before removal, as they settle quickly.
-
(ix)
(Optional) Take 5 μl of lysate and check the protein concentration with a BCA kit according to the manufacturer’s instructions.
-
(x)
Save 30 μl of lysate (0.2%) for quality-control analysis (Step 1A(xxi)). Add the lysate to the beads and incubate the mixture on a rotating platform overnight at 4 °C. Expect a total volume of ~15 ml.
-
(xi)
Prepare buffer B for the Ni-NTA purification step. Keep the beads stirring overnight at room temperature.
-
(xii)
Section 3: purification—stage II, denaturing Ni-NTA (5–6 h). Adjust the pH of buffer B to 8.0 with 8 M NaOH and filter-sterilize it.
-
(xiii)
Wash the beads three times with 2 ml of 1× PBS containing 0.08% (vol/vol) Nonidet P-40. Save 4 μl of the FLAG flow-through (0.2%) for quality-control analysis (Step 1A(xxi)).
-
(xiv)
Transfer the sample to a 1.5-ml low-adhesion tube and elute it with 570 μl of FLAG elution buffer with gentle vortexing for 45 min at room temperature.
▲ CRITICAL STEP Use wide-bore pipette tips to avoid damaging the beads.
-
(xv)
Wash 60 μl of Ni-NTA agarose slurry three times with 600 μl of buffer B containing 10 mM imidazole, by spinning down the slurry each time at 800g for 2 min at room temperature in a microcentrifuge. Resuspend the Ni-NTA agarose beads in 60 μl of buffer B with 10 mM imidazole, and place the suspension into a 2.0-ml tube.
▲ CRITICAL STEP Use wide-bore pipette tips to avoid damaging the beads. Use a 25G × 1 ½ needle to aspirate the supernatant while avoiding beads.
? TROUBLESHOOTING
-
(xvi)
Save 20 μl of FLAG eluate (3.5%) for quality-control analysis (Step 1A(xxi)). Transfer the remainder of the FLAG eluate to the tube containing the Ni-NTA agarose beads. Add 1.14 ml of 1.5× buffer B and 17 μl of 1 M imidazole. Incubate the mixture at room temperature for 3.5 h on a rotisserie.
▲ CRITICAL STEP This step and all subsequent steps are done at room temperature.
-
(xvii)
Centrifuge the tube at 800g for 3 min, and then carefully transfer the beads to a 1.5-ml low protein-binding tube. Wash the beads three times with 1 ml of buffer B containing 10 mM imidazole. Centrifuge the tube at 800g for 3 min. Save 35 μl (3.5%) of the Ni-NTA flow-through for quality-control analysis (Step 1A(xxi)).
▲ CRITICAL STEP For each wash, a new tube should be used to reduce background. Use wide-bore pipette tips to avoid damaging the beads. Use a 25G × 1 ½ needle to aspirate the supernatant in order to avoid picking up any beads.
? TROUBLESHOOTING
-
(xviii)
Wash the beads twice with 1 ml of Ni-NTA wash buffer containing 10 mM imidazole. Centrifuge at 800g for 3 min to collect the beads after each wash.
▲ CRITICAL STEP For each wash, a new tube should be used to reduce the background.
? TROUBLESHOOTING
-
(xix)
Elute the beads using 90 μl of Ni-NTA elution buffer. Vortex the tube at room temperature for 20 min. To collect the eluate, place an Eppendorf GeLoader tip into the bead bed and gently remove the liquid by pipetting. Save 10 μl of imidazole elution (11%) for quality-control analysis (Step 1A(xxi)), and then freeze the remainder in liquid nitrogen for MS analysis.
? TROUBLESHOOTING
-
(xx)
Send the samples for MS analysis. Although some laboratories are capable of performing MS studies, most will choose to collaborate with others for this analysis or use the service of a protein chemistry core facility.
-
(xxi)
Section 4: quality control after purification (1 d). To determine the recovery efficiency of each purification step, aliquots of saved samples are used for western blotting analysis, as well as for silver staining. To do this, samples are diluted 1:2 in 2× Sina’s sample buffer and boiled for 5 min before loading on 4–20% (wt/vol) Criterion Tris-HCl 26-well SDS-PAGE gel. We generally analyze 0.0008–0.008% of the total cell extract, 0.5% of the FLAG elution and 2% of the imidazole elution. For silver stains, we run 6% of the imidazole elution with 1–10 ng of BSA as a control.
? TROUBLESHOOTING
Box 1 |. Variations to the protocol for validation of substrates.
Transform yeast strains or transiently transfect stable mammalian cell lines expressing the cognate Ligase trap, or a negative control ligase trap, with a construct that expresses the candidate substrate fused to a repeating epitope tag for which a sensitive antibody exists, such as 13×Myc or 5×HA. Endogenous antibodies are often not sufficiently sensitive to detect the small pool of ubiquitylated substrate.
Treating mammalian cells with MG132 before collection can increase background binding. Therefore, it is usually preferable not to use MG132 for validation. However, it may be necessary for some very unstable substrates.
Use 350 OD600 yeast cell pellets or one or two 245 × 245 mm plates of mammalian cells for each sample.
Lyse each sample in 1.2 ml of lysis buffer (yeast) or 1 ml of lysis buffer per confluent 245 × 245 mm plate (mammalian cells).
For yeast validation, load 0.006% of the input, 5% of the FLAG elution and 17% of the Ni-NTA elution. For mammalian cell validation, load 0.08% of the input, 2.5% of the FLAG elution and 50% of the Ni-NTA elution.
(B) Procedure for mammalian cells ● TIMING 5 d
-
(i)
Section 1: cell collection and lysis (3 d). Warm seven 500-ml bottles of DMEM, and then to each add 55 ml of FBS and 55 μl of 10 mg/ml doxycycline. For each bottle, trypsinize three near-confluent T150 flasks of the appropriate stable cell line, and place them in the bottle. Then, mix by inversion and distribute 60 ml each to nine 245 × 245 mm dishes. Grow the cells at 37 °C/8% CO2 until they are barely subconfluent, or for ~3 d.
▲ CRITICAL STEP This experimental protocol uses 63 245 × 245 mm dishes, and this amount of material yields the best results. For ligases that are expressed at a relatively high level, 30 dishes may be sufficient. For validation experiments, we recommend using extracts from one or two dishes. Further details for validation experiments are described in Box 1.
-
(ii)
Four hours before collection, treat the cells with 5 μM MG132 to inhibit the proteasome.
-
(iii)
Decant the medium and add 50 ml of cold 1× PBS + 5 mM EDTA to each plate. Spray off the cells with a disposable 10-ml pipette, and collect the liquid containing the cells in a 50-ml conical tube. Centrifuge the cells at 500g for 5 min (4 °C). Discard the supernatant and wash the pellet by resuspending it in 1 ml of 1× PBS per tube, pooling this resuspension from 10 or 11 50-ml conical tubes into one 15-ml conical tube, spinning it at 500g for 5 min (4 °C) and removing the supernatant. Freeze the pellets in liquid nitrogen, or immediately resuspend the pellets by pipetting them in a total of 6 ml of lysis buffer for all 63 plates.
■ PAUSE POINT Pellets can be stored at –80 °C until ready to use.
-
(iv)
Pool the lysates and sonicate each sample three times for 5 s at 30% amplitude, resting them on ice between rounds.
-
(v)
Add 100 μl of DNase I (1,000 U) and incubate the samples at 4 °C on a rocking platform for 30 min.
-
(vi)
Add Nonidet P-40 to 0.1% (vol/vol) and mix the tube by inverting. Divide the samples into 2-ml tubes, and preclear the lysate by centrifuging at 20,000g for 15 min at 4 °C. Transfer the supernatant to a new 2-ml tube, and centrifuge it at 20,000g for 15 min at 4 °C.
? TROUBLESHOOTING
-
(vii)
Section 2: purification—stage I, native FLAG (12–16 h). In a 15-ml Falcon tube, wash 100 μl of anti-FLAG magnetic bead 50% slurry three times with 1 ml of lysis buffer containing 0.1% (vol/vol) Nonidet P-40.
▲ CRITICAL STEP Use wide-bore tips to avoid damaging the beads during pipetting. Mix the beads well before removal, as they settle quickly.
-
(viii)
Take 5 μl of the lysate and check the protein concentration with a BCA kit according to the manufacturer’s instructions. We aim to use 100 mg of protein for each MS experiment.
-
(ix)
Save 15 μl of lysate (0.25%) for quality-control analysis (Step 1B(xx)). Add the lysate to the beads and incubate them on a rotating platform overnight at 4 °C.
-
(x)
Prepare buffer B for the Ni-NTA purification step. Keep the beads stirring overnight at room temperature.
-
(xi)
Section 3: purification-stage II, denaturing Ni-NTA (5–6 h). Adjust the pH of buffer B to 8.0 with 8 M NaOH and filter-sterilize the solution.
-
(xii)
Wash the beads five times with 2 ml of 1× PBS containing 0.1% (vol/vol) Nonidet P-40. Save 4 μl of FLAG flow-through (0.2%) for quality-control analysis (Step 1B(xx)).
-
(xiii)
Transfer the sample to a 1.5-ml low-adhesion tube and elute with 480 μl of FLAG elution buffer with gentle vortexing for 30 min at room temperature.
▲ CRITICAL STEP Use wide-bore pipette tips to avoid damaging the beads.
-
(xiv)
Wash 40 μl of Ni-NTA agarose slurry three times with 600 μl of buffer B containing 10 mM imidazole, spinning down the slurry each time at 800g for 3 min in a microcentrifuge. Resuspend the Ni-NTA agarose beads in 60 μl of buffer B with 10 mM imidazole and place them into a 2.0-ml tube.
▲ CRITICAL STEP Use wide-bore pipette tips to avoid damaging the beads. Use a 25G × 1 ½ needle to aspirate the supernatant while avoiding beads.
? TROUBLESHOOTING
-
(xv)
Save 20 μl of FLAG eluate (3.5%) for quality-control analysis (Step 1B(xx)). Transfer the remainder of the FLAG eluate to the tube containing the Ni-NTA agarose beads. Add 1.14 ml of 1.5× buffer B and 17 μl of 1 M imidazole. Incubate the mixture at room temperature for 3.5 h on a rotisserie.
▲ CRITICAL STEP This step and all subsequent steps are done at room temperature.
-
(xvi)
Centrifuge the mixture at 800g for 3 min, and then carefully transfer the beads to a 1.5-ml low protein-binding tube. Wash the beads three times with 1 ml of buffer B containing 10 mM imidazole. Centrifuge the beads at 800g for 3 min. Save 35 μl (3.5%) of the Ni-NTA flow-through for quality-control analysis (Step 1B(xx)).
▲ CRITICAL STEP For each wash, a new tube should be used to reduce the background. Use wide-bore pipette tips to avoid damaging the beads. Use a 25G × 1 ½ needle to aspirate the supernatant and avoid picking up any beads.
? TROUBLESHOOTING
-
(xvii)
Wash the beads twice with 1 ml of Ni-NTA wash buffer containing 10 mM imidazole. Centrifuge the tube at 800g for 3 min to collect the beads after each wash.
▲ CRITICAL STEP For each wash, a new tube should be used to reduce the background.
? TROUBLESHOOTING
-
(xviii)
Elute the beads using 90 μl of Ni-NTA elution buffer. Vortex at room temperature for 20 min. To collect the eluate, place an Eppendorf GeLoader tip into the bead bed and gently remove the liquid by pipetting. Save 10 μl of imidazole elution (11%) for quality-control analysis (Step 1B(xx)) and freeze the remainder in liquid nitrogen for MS analysis.
? TROUBLESHOOTING
-
(xix)
Send the samples for MS analysis. Although some laboratories are capable of performing MS studies, most will choose to collaborate with others for this analysis or use the service of a protein chemistry core facility.
-
(xx)
Section 4: quality control after purification (1 d). To determine the recovery efficiency of each purification step, aliquots of saved samples are used for western blotting analysis, as well as for silver staining. To do this, samples are diluted 1:2 in 2× Sina’s sample buffer and boiled for 5 min before loading on a 4–20% (wt/vol) Criterion Tris-HCl 26-well SDS-PAGE gel. We generally analyze 0.0005% of the total cell extract, 0.78% of the FLAG elution and 0.28–2.8% of the imidazole elution. For silver stains, we run 5% of the imidazole elution with 1–10 ng of BSA as a control.
? TROUBLESHOOTING
? TROUBLESHOOTING
Troubleshooting advice can be found in Table 1.
TABLE 1 |.
Troubleshooting table.
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 1A(iv) | Sample aggregates in large clumps | Sample was dropped too quickly in liquid nitrogen | Maintain maximum liquid nitrogen volume, and introduce sample drops more slowly |
| 1A(v) | Sample is stuck to the inside of the chamber after grinding | Unknown | Scrape off as much sample as possible with a cold spatula. It is not necessary to remove all of the sample |
| 1A(vii) 1B(vi) |
After the second centrifugation, the sample is cloudy | Unknown | Sample cloudiness does not necessarily compromise sample quality. Proceed with the protocol, and monitor sample quality and quantity |
| 1A(xv,xvii,xviii), 1B(xiv,xvi,xvii) |
Bead volume decreases during washing | Beads may be accidentally aspirated | Avoid the bead bed when aspirating. Keep the needle bore against the side of the tube. Tilt the tube if it helps prevent the removal of beads |
| 1A(xix), 1B(xviii) |
Protein did not elute from Ni-NTA beads | Incorrect pH of elution buffer | Ensure that the pH of the elution buffer is correct |
| 1A(xxi), 1B(xx) |
No visible bands on the western blot or silver stain | Insufficient material | Samples may need to be loaded at higher quantities, depending on the level of ligase expression. Consider overexpressing the ligase of interest, as this is often the limiting factor for purifying enough material |
● TIMING
Step 1A, yeast: 2.5 d
Section 1, cell collection and lysis: 6–7 h
Section 2, purification—stage I (native FLAG): 12–16 h
Section 3, purification—stage II (denaturing Ni-NTA): 5–6 h
Section 4, quality control post purification: 1 d
Step 1B, mammalian cells: 5 d
Section 1, cell collection and lysis: 3 d
Section 2, purification—stage I (native FLAG): 12–16 h
Section 3, purification—stage II (denaturing Ni-NTA): 5–6 h
Section 4, quality control post purification: 1 d
ANTICIPATED RESULTS
Figure 2 shows a representative western blot (Fig. 2a) of ligase trap purifications. Polyubiquitinated material exhibits retarded mobility in SDS-PAGE and runs as a ladder of bands or a smear. To validate the ubiquitination of candidate substrates in vivo, we generate yeast strains expressing 13×-Myc epitope-tagged candidate substrates and perform the two-step purification protocol on a smaller scale. Western blot analyses of these samples show that the putative Grr1 substrate, Sfg1, is specifically purified as a polyubiquitinated species with the Grr1 ligase trap, but not with two other control ligase traps (Mfb1 and Ufo1) expressed at similar levels (Fig. 2b).
Figure 2 |.
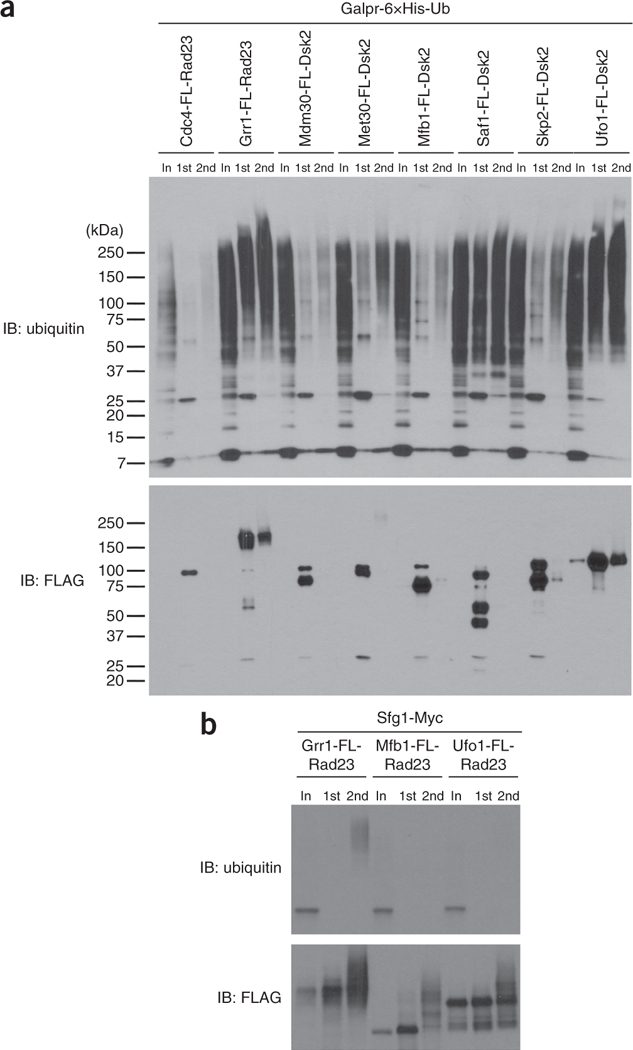
Quality-control post-purification results for yeast purification. (a) Western blot analysis of different ligase traps showing purification of polyubiquitinated species. Each ligase trap (F-box-FLAG-UBA fusion protein) is listed at the top. Western blots were probed with an anti-ubiquitin (P4D1) antibody (gift from E. Wayner) and an anti-FLAG antibody (Sigma-Aldrich). Loaded per lane, relative to input (In), is 500× of the FLAG IP (1st) and 2,000× of the Ni-NTA IP (2nd). FL, FLAG. This image is reproduced with permission from ref. 22. (b) Western blot analysis of the Grr1 substrate, Sfg1, expressed in cells containing the ligase trap of Grr1, or the negative controls Mfb1 and Ufo1. This image is adapted with permission from ref. 22.
By using the yeast protocol above, we identified 17 known substrates and 18 novel substrates of eight F-box proteins in budding yeast22. Our work also demonstrated that ligase traps with different UBAs, Rad23 or Dsk2, performed well in identifying target substrates. Furthermore, we showed that UBAs can be fused to either the N or the C terminus of the F-box protein with little difference in the ability to capture substrate.
Figure 3a shows a representative western blot of a β-TrCP ligase trap purification from mammalian cells. A silver stain of the Ni-NTA elution is used to assess purity and yield from each purification (Fig. 3b).
Figure 3 |.

Quality-control post-purification results for mammalian purification. (a) Western blot analysis of different ligase traps showing purification of polyubiquitinated species (as indicated). Western blots were probed with an anti-6×His antibody (Clontech), an anti-FLAG antibody (Sigma-Aldrich) and an anti-Cul1 antibody (Invitrogen). (b) Silver stain of 5% of the Ni-NTA elution.
Using the mammalian protocol, we identified 12 known substrates and 11 new substrates of the human F-box protein β-TrCP43. We showed that ligase trapping was an especially accurate method of ubiquitin ligase substrate identification: of the known and candidate substrates that we tested, 88% were either previously described or were validated by us.
ACKNOWLEDGMENTS
This work was supported by US National Institutes of Health (NIH) grants R01 GM059691 and GM070539 to D.P.T., and by a predoctoral fellowship 13PRE14190011 from the American Heart Association to T.B.L.
Footnotes
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
References
- 1.Breitschopf K, Bengal E, Ziv T, Admon A & Ciechanover A A novel site for ubiquitination: the N-terminal residue, and not internal lysines of MyoD, is essential for conjugation and degradation of the protein. EMBO J. 17, 5964–5973 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schulman BA Twists and turns in ubiquitin-like protein conjugation cascades. Protein Sci. 20, 1941–1954 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Finley D, Ulrich HD, Sommer T & Kaiser P The ubiquitin-proteasome system of Saccharomyces cerevisiae. Genetics 192, 319–360 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Komander D & Rape M The ubiquitin code. Annu. Rev. Biochem. 81, 203–229 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Benanti JA, Cheung SK, Brady MC & Toczyski DP A proteomic screen reveals SCFGrr1 targets that regulate the gLycoLytic-gLuconeogenic switch. Nat. Cell Biol. 9, 1184–1191 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Yen HC & Elledge SJ Identification of SCF ubiquitin ligase substrates by global protein stability profiling. Science 322, 923–929 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Yen HC, Xu Q, Chou DM, Zhao Z & EUedge SJ Global protein stability profiling in mammalian cells. Science 322, 918–923 (2008). [DOI] [PubMed] [Google Scholar]
- 8.Emanuele MJ et al. Global identification of modular cullin-RING ligase substrates. Cell 147, 459–474 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barral Y, Jentsch S & Mann C G1 cyclin turnover and nutrient uptake are controlled by a common pathway in yeast. Genes Dev. 9, 399–409 (1995). [DOI] [PubMed] [Google Scholar]
- 10.Koepp DM, Kile AC, Swaminathan S & Rodriguez-Rivera V The F-box protein Dia2 regulates DNA replication. Mol. Biol. Cell 17, 1540–1548 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Landry BD, Doyle JP, Toczyski DP & Benanti JA F-box protein specificity for G1 cyclins is dictated by subcellular localization. PLoS Genet. 8, e1002851 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pant V & Lozano G Limiting the power of p53 through the ubiquitin proteasome pathway. Genes Dev. 28, 1739–1751 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Galligan JT et al. Proteomic analysis and identification of cellular interactors of the giant ubiquitin ligase HERC2. J. Proteome Res. 14, 953–966 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martinez-Noel G et al. Identification and proteomic analysis of distinct UBE3A/E6AP protein complexes. Mol. Cell Biol. 32, 3095–3106 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davis MA et al. The SCF-Fbw7 ubiquitin ligase degrades MED13 and MED13L and regulates CDK8 module association with mediator. Genes Dev. 27, 151–156 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pierce NW, Kleiger G, Shan SO & Deshaies RJ Detection of sequential polyubiquitylation on a millisecond timescale. Nature 462, 615–619 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gardner RG, Shearer AG & Hampton RY In vivo action of the HRD ubiquitin ligase complex: mechanisms of endoplasmic reticulum quality control and sterol regulation. Mol. Cell Biol. 21, 4276–4291 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tagwerker C et al. A tandem affinity tag for two-step purification under fully denaturing conditions: application in ubiquitin profiling and protein complex identification combined with in vivo cross-linking. Mol. Cell Proteomics 5, 737–748 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Elsasser S & Finley D Delivery of ubiquitinated substrates to protein-unfolding machines. Nat. Cell Biol. 7, 742–749 (2005). [DOI] [PubMed] [Google Scholar]
- 20.Grabbe C & Dikic I Functional roles of ubiquitin-like domain (ULD) and ubiquitin-binding domain (UBD) containing proteins. Chem. Rev. 109, 1481–1494 (2009). [DOI] [PubMed] [Google Scholar]
- 21.Sims JJ, Haririnia A, Dickinson BC, Fushman D & Cohen RE Avid interactions underlie the Lys63-linked polyubiquitin binding specificities observed for UBA domains. Nat. Struct. Mol. Biol. 16, 883–889 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mark KG, Simonetta M, Maiolica A, Seller CA & Toczyski DP Ubiquitin ligase trapping identifies an SCFSaf1 pathway targeting unprocessed vacuolar/lysosomal proteins. Mol. Cell 53, 148–161 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bennett EJ, Rush J, Gygi SP & Harper JW Dynamics of cullin-RING ubiquitin ligase network revealed by systematic quantitative proteomics. Cell 143, 951–965 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuchay S et al. FBXL2- and PTPL1-mediated degradation of p110-free p85β regulatory subunit controls the PI(3)K signalling cascade. Nat. Cell Biol. 15, 472–480 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wimuttisuk W et al. Novel Cul3 binding proteins function to remodel E3 ligase complexes. BMC Cell Biol. 15, 28 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peng J et al. A proteomics approach to understanding protein ubiquitination. Nat. Biotechnol. 21, 921–926 (2003). [DOI] [PubMed] [Google Scholar]
- 27.Gururaja T, Li W, Noble WS, Payan DG & Anderson DC Multiple functional categories of proteins identified in an in vitro cellular ubiquitin affinity extract using shotgun peptide sequencing. J. Proteome Res. 2, 394–404 (2003). [DOI] [PubMed] [Google Scholar]
- 28.Hitchcock AL, Auld K, Gygi SP & Silver PA A subset of membrane-associated proteins is ubiquitinated in response to mutations in the endoplasmic reticulum degradation machinery. Proc. Natl. Acad. Sci. USA 100, 12735–12740 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kirkpatrick DS, Weldon SF, Tsaprailis G, Liebler DC & Gandolfi AJ Proteomic identification of ubiquitinated proteins from human cells expressing His-tagged ubiquitin. Proteomics 5, 2104–2111 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Jeon HB et al. A proteomics approach to identify the ubiquitinated proteins in mouse heart. Biochem. Biophys. Res. Commun. 357, 731–736 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Meierhofer D, Wang X, Huang L & Kaiser P Quantitative analysis of global ubiquitination in HeLa cells by mass spectrometry. J. Proteome Res. 7, 4566–4576 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Franco M, Seyfried NT, Brand AH, Peng J & Mayor U A novel strategy to isolate ubiquitin conjugates reveals wide role for ubiquitination during neural development. Mol. Cell Proteomics 10.1074/mcp.M110.002188 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Danielsen JM et al. Mass spectrometric analysis of lysine ubiquitylation reveals promiscuity at site level. Mol. Cell Proteomics 10 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilson MD, Saponaro M, Leidl MA & Svejstrup JQ MultiDsk: a ubiquitin-specific affinity resin. PLoS One 7, e46398 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Layfield R et al. Purification of poly-ubiquitinated proteins by S5a-affinity chromatography. Proteomics 1, 773–777 (2001). [DOI] [PubMed] [Google Scholar]
- 36.Anindya R, Aygun O & Svejstrup JQ Damage-induced ubiquitylation of human RNA polymerase II by the ubiquitin ligase Nedd4, but not Cockayne syndrome proteins or BRCA1. Mol. Cell 28, 386–397 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Hjerpe R et al. Efficient protection and isolation of ubiquitylated proteins using tandem ubiquitin-binding entities. EMBO Rep. 10, 1250–1258 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shi Y et al. A data set of human endogenous protein ubiquitination sites. Mol. Cell Proteomics 10 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lopitz-Otsoa F et al. Integrative analysis of the ubiquitin proteome isolated using Tandem Ubiquitin Binding Entities (TUBEs). J. Proteomics 75, 2998–3014 (2012). [DOI] [PubMed] [Google Scholar]
- 40.Tan MK, Lim HJ, Bennett EJ, Shi Y & Harper JW Parallel SCF adaptor capture proteomics reveals a role for SCFFBXL17 in NRF2 activation via BACH1 repressor turnover. Mol. Cell 52, 9–24 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Behrends C, Sowa ME, Gygi SP & Harper JW Network organization of the human autophagy system. Nature 466, 68–76 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sowa ME, Bennett EJ, Gygi SP & Harper JW Defining the human deubiquitinating enzyme interaction landscape. Cell 138, 389–403 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Loveless TB et al. DNA damage regulates translation through β-TRCP targeting of CReP. PLoS Genet. 11, e1005292 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim TY et al. Substrate trapping proteomics reveals targets of the β-TrCP2/FBXW11 ubiquitin ligase. Mol. Cell Biol. 35, 167–181 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peng J & Gygi SP Proteomics: the move to mixtures. J. Mass Spectrom. 36, 1083–1091 (2001). [DOI] [PubMed] [Google Scholar]
- 46.Xu G, Paige JS & Jaffrey SR Global analysis of lysine ubiquitination by ubiquitin remnant immunoaffinity profiling. Nat. Biotechnol. 28, 868–873 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim W et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 44, 325–340 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Theurillat JP et al. Prostate cancer. Ubiquitylome analysis identifies dysregulation of effector substrates in SPOP-mutant prostate cancer. Science 346, 85–89 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kronke J et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 343, 301–305 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thompson JW et al. Quantitative Lys-e-Gly-Gly (diGly) proteomics coupled with inducible RNAi reveals ubiquitin-mediated proteolysis of DNA damage-inducible transcript 4 (DDIT4) by the E3 ligase HUWE1. J. Biol. Chem. 289, 28942–28955 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ordureau A, Munch C & Harper JW Quantifying ubiquitin signaling. Mol. Cell 58, 660–676 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sultan M et al. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science 321, 956–960 (2008). [DOI] [PubMed] [Google Scholar]
- 53.Kulathu Y, Akutsu M, Bremm A, Hofmann K & Komander D Two-sided ubiquitin binding explains specificity of the TAB2 NZF domain. Nat. Struct. Mol. Biol. 16, 1328–1330 (2009). [DOI] [PubMed] [Google Scholar]
- 54.Rahighi S et al. Specific recognition of linear ubiquitin chains by NEMO is important for NF-kB activation. Cell 136, 1098–1109 (2009). [DOI] [PubMed] [Google Scholar]
- 55.van Wijk SJ, Muller S & Dikic I Shared and unique properties of ubiquitin and SUMO interaction networks in DNA repair. Genes Dev. 25, 1763–1769 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McEwan DG & Dikic I The Three Musketeers of Autophagy: phosphorylation, ubiquitylation and acetylation. Trends Cell Biol. 21, 195–201 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]