

Abstract
Summary
The rapid advances of omics technologies have generated abundant genomic data in public repositories and effective analytical approaches are critical to fully decipher biological knowledge inside these data. Meta-analysis combines multiple studies of a related hypothesis to improve statistical power, accuracy and reproducibility beyond individual study analysis. To date, many transcriptomic meta-analysis methods have been developed, yet few thoughtful guidelines exist. Here, we introduce a comprehensive analytical pipeline and browser-based software suite, called MetaOmics, to meta-analyze multiple transcriptomic studies for various biological purposes, including quality control, differential expression analysis, pathway enrichment analysis, differential co-expression network analysis, prediction, clustering and dimension reduction. The pipeline includes many public as well as >10 in-house transcriptomic meta-analytic methods with data-driven and biological-aim-driven strategies, hands-on protocols, an intuitive user interface and step-by-step instructions.
Availability and implementation
MetaOmics is freely available at https://github.com/metaOmics/metaOmics.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
With the rapid advances of high-throughput ‘-omics’ technologies in the past decades, production of various kinds of omics data has become affordable and prevalent. Large amounts of transcriptomic data have been generated using microarray or RNA sequencing platforms for different biological aims and have been stored in data repositories such as GEO, ArrayExpress and SRA. However, individual studies are often of small or moderate sample size, which yield limited statistical power and low reproducibility. The combination of multiple transcriptomic studies of a related hypothesis using meta-analysis has become an emerging and effective practice to improve statistical power, accuracy and generalizability in biological investigations. In existing transcriptomic meta-analysis publications, the project rationale, objectives and data inclusion/exclusion criteria are often vaguely reported, since the analyses are intended to be exploratory and assist further hypothesis generation. The data pre-processing procedures, such as gene matching, gene filtering and outlier detection/exclusion, are often ambiguous and ir-reproducible as well. For further information, see Tseng et al. (2012) for a detailed review. Supplementary Figure S1A shows the number of publications in PubMed related to ‘transcriptomic meta-analysis’ each year, demonstrating its rapid development and wide application. Despite this popularity, a thoughtful analysis pipeline with step-by-step instructions and an intuitive interface for biologists to conveniently conduct data-driven investigations is lacking. In addition, existing omics meta-analyses often only focus on the detection of differentially expressed (DE) genes, pathways and network analysis, leaving many powerful statistical learning tools unexplored. For example, Integrative Array Analyzer (iArray) (Pan et al., 2006) and NetworkAnalyst (Xia et al., 2015) provided tools for conventional DE gene and pathway detection and network visualization. In this paper, we introduce a comprehensive analytical pipeline and browser-based software suite, called MetaOmics, to meta-analyze multiple transcriptomic studies for various biological purposes, including seven modules for quality control (MetaQC), differential expression analysis (MetaDE), pathway enrichment analysis (MetaPath), differential co-expression network analysis (MetaNetwork), classification analysis (MetaPredict), clustering analysis (MetaClust) and dimension reduction (MetaPCA; Supplementary Fig. S1B and S1C). The pipeline includes a large number of public and >10 in-house transcriptomic meta-analysis methods with biology-driven strategies and hands-on protocols. The modularized software structure of MetaOmics will allow for its future extension as new methodologies become available.
2 Overview and workflow of MetaOmics
Figure 1 demonstrates a general workflow of implementing the eight modules (shaded in grey) in MetaOmics. After data input, genes are annotated, matched and properly filtered in the MetaPreprocess module. Inclusion of poor quality studies in the meta-analyses can weaken statistical power and distort the final conclusion. The next module ‘MetaQC’ incorporates biological pathway databases and gene co-expression information to provide objective and quantitative measures for quality control (QC) and help determine inclusion/exclusion of studies for meta-analysis. After QC, users can select any of the six analytical modules depending on their desired biological exploration. For users not familiar with the different types of omics data analyses, Supplementary Box S2 outlines the basics and rationale of these statistical learning approaches. The most common first choice is to identify DE candidate markers. The ‘MetaDE’ module allows the implementation of 12 meta-analysis methods. The pipeline and defaults follow our previously published statistical characterization and application guidelines to advise selection of the method and related parameters. Beyond DE gene identification, users may be interested in detecting differential expression profiles at the pathway level or co-expression network level under a meta-analytic framework. The ‘MetaPath’ module includes two advanced tools, Meta-analysis for Pathway Enrichment (MAPE) and comparative pathway integrator (CPI), for meta-analytic pathway analysis. The ‘MetaNetwork’ module integrates multiple transcriptomic studies to detect differential co-expression networks and to infer regulatory changes under different disease conditions. MetaOmics also contains three statistical learning tools that were developed in-house for transcriptomic meta-analysis. Prediction analysis (a.k.a. classification analysis or supervised machine learning) is a popular analysis to translate omics findings into clinical decisions. The ‘MetaPredict’ module implements the MetaKTSP algorithm, which combines multiple transcriptomic studies using a non-parametric top scoring pair approach for robust and accurate prediction across different experimental platforms. When class labels of patients are unknown, cluster analysis can identify novel disease subtypes, an important component of personalized medicine. The ‘MetaClust’ module contains an effective MetaSparseKmeans algorithm which performs simultaneous sample clustering and common intrinsic gene selection from multiple transcriptomic studies for this purpose. Finally, dimension reduction via methods such as principal component analysis (PCA) is a powerful exploratory tool to analyze high-dimensional omics data. The ‘MetaPCA’ module implements a meta-analytic approach of the PCA algorithm for simultaneous dimension reduction and feature selection in multiple transcriptomic studies. Each of these in-house methodologies have been thoroughly developed, rigorously evaluated in many applications and published in high-profile journals. Supplementary Table S1 outlines the advantages and additional features compared to existing tools.
Fig. 1.
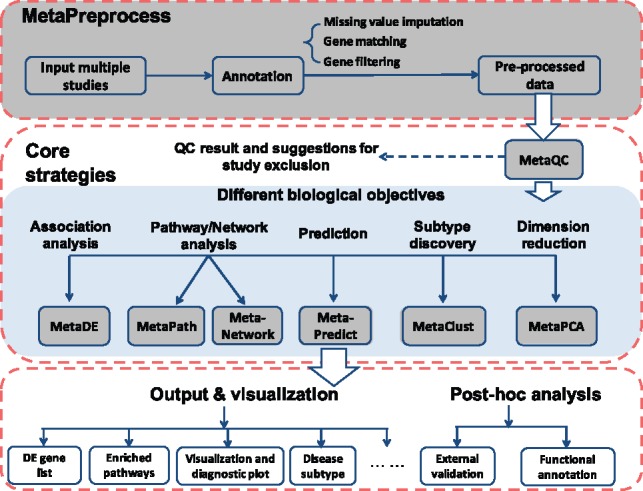
General workflow of data pre-processing and implementation of the seven analytical modules in MetaOmics. All modules are shaded in grey
Each of the seven analytical modules generates its own outputs, such as DE gene lists, pathway annotation, classification model or cluster assignment. MetaOmics also creates extensive visualization and diagnostic plots to assist users with selecting tuning parameters and/or interpreting the results. Post hoc analyses, such as external validation and functional annotation, are also included in the tool.The modules in MetaOmics can be creatively used in selected order to evaluate and understand the biological findings and generate further hypothesis. For example, MetaClust can first be used to cluster samples in multiple transcriptomic studies and identify disease subtypes. Based on the cluster assignment, disease subtypes of interest can be selected and MetaDE, MetaPath and MetaNetwork can then be used to investigate their DE genes, functional annotations and differential co-expression networks.
3 Case study and demonstration
In Supplementary Material, we present comprehensive applications of the seven modules to three real applications using breast cancer, prostate cancer and leukemia datasets. Due to space limit, here we only discuss three selected modules using the breast cancer example. This case study contained four breast cancer datasets, including one RNA-seq study from TCGA and three microarray studies from GEO (GSE7390, GSE2034 and GSE4922), for comparing estrogen receptor positive (ER+) and negative patients. The four datasets included 406 (319/87), 198 (134/64), 286 (209/77) and 245 (211/34) ER+/ER- samples and 10 330 genes.
MetaDE: The MetaDE module can conveniently integrate count data from RNA-seq and continuous measurements from microarray studies. We used ‘LIMMA’ for microarray and ‘edgeR’ for RNA-seq count data for individual study analysis, and chose the ‘AW-Fisher’ method to perform meta-analysis. Supplementary Figure S2A shows a heatmap of 731 significant DE genes at an FDR cutoff of 10−15 with samples ordered in columns by study and ER groups and genes displayed in rows by adaptive weight groups. The results showed that the majority of DE genes were common up-regulated or down-regulated genes (weight = 1, 1, 1, 1), indicating a generally homogeneous signal across the four studies. A follow-up pathway analysis in the module showed the most top enriched pathways to be cancer related, such as cell cycle and DNA replication.
MetaClust: For implementing MetaClust module, we ignored the ER status label and jointly clustered the samples of all four studies. Supplementary Figure S2B shows heatmaps corresponding to clustering of the four studies. In the gene list output, we found a large overlap with PAM50 gene list, a set of 50 intrinsic genes widely used for classifying breast cancer subtypes.
MetaNetwork: Supplementary Figure S2C shows one selected basic module with differentially co-expression network (DCN) that was highly connected in ‘ER+’ but lost connections in ‘ER-’ (left) as well as an example of reverse pattern (right). Enriched pathways of the top detected DCNs include smooth muscle contraction and extracellular matrix activity.
Detailed results of the three (leukemia, breast and prostate cancers) applications can be found in Supplementary Material.
Funding
Research reported in this publication was supported by NCI of the National Institutes of Health under award number R01CA190766 and NLM of the National Institute of Health under award number R21LM012752 to T.M., Z.H. and G.C.T. Wang was partly supported by National Nature Science Foundation of China (11701391).
Conflict of Interest: none declared.
Supplementary Material
References
- Pan F. et al. (2006) Integrative array analyzer: a software package for analysis of cross-platform and cross-species microarray data. Bioinformatics, 22, 1665–1667. [DOI] [PubMed] [Google Scholar]
- Tseng G.C. et al. (2012) Comprehensive literature review and statistical considerations for microarray meta-analysis. Nucleic Acids Res., 40, 3785–3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J.G. et al. (2015) NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data. Nat. Protoc., 10, 823–844. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.