

Abstract
Here, we describe single-tube long fragment read (stLFR), a technology that enables sequencing of data from long DNA molecules using economical second-generation sequencing technology. It is based on adding the same barcode sequence to subfragments of the original long DNA molecule (DNA cobarcoding). To achieve this efficiently, stLFR uses the surface of microbeads to create millions of miniaturized barcoding reactions in a single tube. Using a combinatorial process, up to 3.6 billion unique barcode sequences were generated on beads, enabling practically nonredundant cobarcoding with 50 million barcodes per sample. Using stLFR, we demonstrate efficient unique cobarcoding of more than 8 million 20- to 300-kb genomic DNA fragments. Analysis of the human genome NA12878 with stLFR demonstrated high-quality variant calling and phase block lengths up to N50 34 Mb. We also demonstrate detection of complex structural variants and complete diploid de novo assembly of NA12878. These analyses were all performed using single stLFR libraries, and their construction did not significantly add to the time or cost of whole-genome sequencing (WGS) library preparation. stLFR represents an easily automatable solution that enables high-quality sequencing, phasing, SV detection, scaffolding, cost-effective diploid de novo genome assembly, and other long DNA sequencing applications.
To date, the vast majority of individual higher organism whole-genome sequences lack information regarding the order of single- to multibase variants transmitted as contiguous blocks on homologous chromosomes, typically referred to as haplotypes. In addition, most sequenced genomes leave unresolved novel sequence not found in reference genomes, large structural variations, and other regions that are difficult to analyze with current technologies. For many early genome studies, this information was not critical and was overlooked. However, as we move toward a more complete understanding of how an individual's genome contributes to the myriad phenotypes they exhibit, this missing information will become necessary.
Numerous technologies, including direct single-molecule sequencing (Levene et al. 2003; Zhang et al. 2006; Ma et al. 2010; Olasagasti et al. 2010; Fan et al. 2011; Kitzman et al. 2011; Suk et al. 2011; Duitama et al. 2012; Peters et al. 2012; Selvaraj et al. 2013; Amini et al. 2014; Kuleshov et al. 2014; Zheng et al. 2016), have recently been developed to generate at least some of this information. Most are based on the process of cobarcoding (Peters et al. 2014), that is, the addition of the same barcode to the subfragments of single long genomic DNA molecules. After sequencing, the barcode information can be used to determine which reads are derived from the original long DNA molecule. This process was first described by Drmanac (2006) and implemented as a 384-well plate assay by Peters et al. (2012). These approaches have been technically challenging to implement, are expensive, have lower data quality, do not analyze individual DNA molecules separately (i.e., do not provide unique cobarcoding), or some combination of all four. In practice, most require a separate whole-genome sequence to be generated by standard methods to improve variant calling. Here, we describe the implementation of stLFR technology (Drmanac et al. 2014), an efficient approach for DNA cobarcoding with millions of barcodes enabled in a single tube. This is achieved by using the surface of a microbead as a replacement for a compartment (e.g., the well of a 384-well plate). Each bead carries many copies of a unique barcode sequence that is transferred to the subfragments of each long DNA molecule. These cobarcoded subfragments are then analyzed on common second-generation sequencing devices such as the BGISEQ-500, MGISEQ-2000, or equivalent.
Results
stLFR library process
In our implementation of this approach, we used a ligation-based combinatorial barcode generation strategy to create more than 3.6 billion different barcodes in three ligation steps. For a single sample, we used ∼10–50 million of these barcoded beads to capture ∼10–100 million long DNA molecules in a single tube. It is infrequent that two beads will share the same barcode because we sample 10–50 million beads from such a large library of total barcodes. Furthermore, in the case of using 50 million beads and 10 million long genomic DNA fragments, the vast majority of subfragments from each long DNA fragment are cobarcoded by a unique barcode. This makes stLFR more similar to long read single-molecule sequencing (e.g., Pacific Biosciences [PacBio] SMRT and Oxford Nanopore Technologies [Nanopore] sequencing) than other cobarcoding strategies like Chromium (10x Genomics) that cobarcode multiple long DNA fragments with the same barcode. A similar but informatically limited and less efficient approach using only ∼150,000 barcodes was recently described by Zhang et al. (2017). A detailed comparison of the differences between stLFR and similar technologies can be found in Supplemental Table S1.
stLFR is simple to perform and can be implemented with a relatively small investment in oligonucleotides to generate barcoded beads. Further, stLFR uses standard equipment found in most molecular biology laboratories and is sequencing technology agnostic. Finally, stLFR replaces standard second-generation sequencing library preparation methods, requires only 1 ng DNA, and does not add significantly to the cost of whole-genome or whole-exome library preparation with a total cost per sample of less than 30 dollars (Table 1).
Table 1.
stLFR equipment and reagent cost
The first step in stLFR is the insertion of a hybridization sequence at regular intervals along genomic DNA fragments. This is achieved through the incorporation of DNA sequences, by the Tn5 transposase, containing a single-stranded region for hybridization and a double-stranded sequence that is recognized by the enzyme and enables the transposition reaction (Fig. 1A). This step is done in solution, as opposed to having the insertion sequence linked directly to the bead (Zhang et al. 2017). This enables a very efficient incorporation of the hybridization sequence along the genomic DNA molecules. As previously observed (Amini et al. 2014), the transposase enzyme has the property of remaining bound to genomic DNA after the transposition event, effectively leaving the transposon-integrated long genomic DNA molecule intact. After the DNA has been treated with Tn5, it is diluted in hybridization buffer and added to 50 million ∼2.8 µm clonally barcoded beads in hybridization buffer. Each bead contains approximately 400,000 capture adapters, each containing the same barcode sequence. A portion of the capture adapter contains uracil nucleotides to enable destruction of unused adapters in a later step. The mix is incubated under optimized temperature and buffer conditions, during which time the transposon-inserted DNA is captured to beads via the hybridization sequence. It has been suggested that genomic DNA in solution forms balls with both tails sticking out (Jo et al. 2009). This may enable the capture of long DNA fragments toward one end of the molecule followed by a rolling motion that wraps the genomic DNA molecule around the bead. Approximately every 7.8 nm on the surface of each bead, there is a capture oligo. This enables a very uniform and high rate of subfragment capture. A 100-kb genomic fragment would wrap around a 2.8-µm bead approximately three times. In our data, 300 kb is the longest fragment size captured, suggesting larger beads may be necessary to capture longer DNA molecules. Beads are next collected, and individual barcode sequences are transferred to each subfragment through ligation of the nick between the hybridization sequence and the capture adapter (Fig. 1A). At this point the DNA/transposase complexes are disrupted, producing subfragments <1 kb in size. Owing to the large number of beads and high density of capture oligos per bead, the amount of excess adapter is four orders of magnitude greater than the amount of product. This huge unused adapter can overwhelm the following steps. In order to avoid this, we designed beads with capture oligos connected by the 5′ terminus. This enabled an exonuclease strategy to be developed that specifically degraded excess unused capture adapter.
Figure 1.

Overview of stLFR. (A) The first step of stLFR involves inserting a hybridization sequence approximately every 200–1000 bp on long genomic DNA molecules. This is achieved using transposons. The transposon-integrated DNA is then mixed with beads that each contain ∼400,000 copies of an adapter sequence that contains a unique barcode shared by all adapters on the bead, a common PCR primer site, and a common capture sequence that is complementary to the sequence on the integrated transposons. After the genomic DNA is captured to the beads, the transposons are ligated to the barcode adapters. There are a few additional library processing steps and then the cobarcoded subfragments are sequenced on a BGISEQ-500 or equivalent sequencer. (B) Mapping read data by barcode results in clustering of reads within 10- to 350-kb regions of the genome. Total coverage and barcode coverage from four barcodes are shown for the 1-ng stLFR-1 library across a small region on Chromosome 11. Most barcodes are associated with only one read cluster in the genome. (C) The number of original long DNA fragments per barcode are plotted for the 1-ng libraries stLFR-1 (blue) and stLFR-2 (orange) and the 10-ng stLFR libraries stLFR-3 (yellow) and stLFR-4 (gray). More than 80% of the fragments from the 1-ng stLFR libraries are cobarcoded by a single unique barcode. (D) The fraction of nonoverlapping sequence reads (blue) and captured subfragments (orange) covering each original long DNA fragment are plotted for the 1-ng stLFR-1 library.
In one approach to stLFR, two different transposons are used in the initial insertion step, allowing PCR to be performed after exonuclease treatment. However, this approach results in ∼50% less coverage per long DNA molecule because it requires that two different transposons were inserted next to each other to generate a proper PCR product. To achieve the highest coverage per genomic DNA fragment, we used a single transposon in the initial insertion step and added an additional adapter through ligation. This noncanonical ligation, termed 3′ branch ligation, involves the covalent joining of the 5′ phosphate from the blunt-end adapter to the recessed 3′ hydroxyl of the genomic DNA (Fig. 1A). A detailed explanation of this process was previously described (Wang et al. 2019). Using this method, it is theoretically possible to amplify and sequence all subfragments of a captured genomic molecule. In addition, this ligation step enables a sample barcode to be placed adjacent to the genomic sequence for sample multiplexing. This is useful because it does not require an additional sequencing primer to read this barcode. After the ligation step, PCR is performed, and the library is ready to enter any standard second-generation sequencing workflow. In the case of BGISEQ-500, the library is circularized as previously described (Drmanac et al. 2010). From single-stranded circles, DNA nanoballs are made and loaded onto patterned nanoarrays (Drmanac et al. 2010). These nanoarrays are then subjected to combinatorial probe-anchor synthesis (cPAS)–based sequencing on the BGISEQ-500 (Fehlmann et al. 2016; Huang et al. 2017; Mak et al. 2017). After sequencing, barcode sequences are extracted using a custom program (Methods). Mapping the read data by unique barcode shows that most reads with the same barcode are clustered in a region of the genome corresponding to the length of DNA used during library preparation (Fig. 1B). A protocol of this process and the process used to make clonally barcoded beads has been described by Cheng et al. (2018).
stLFR read coverage and variant calling
To demonstrate stLFR phasing and variant calling, we generated four libraries using 1 ng (stLFR-1 and stLFR-2) and 10 ng (stLFR-3 and stLFR-4) of DNA isolated from cell line GM12878. The number of beads added ranged from 10 million (stLFR-4), 30 million (stLFR-3), and 50 million (stLFR-1 and stLFR-2). Finally, the 3′ branch ligation method was used for libraries stLFR-1–3, and the two-transposon method was used for stLFR-4. Both stLFR-1 and stLFR-2 were sequenced to exhaustion with 336 and 660 Gb of total base coverage, respectively, using paired-end 100-base reads on a BGISEQ-500 instrument (for additional sequencing metrics, see Supplemental Table S2). We also down-sampled these libraries to enable comparisons at roughly similar coverages. stLFR-3 and stLFR-4 were also sequenced with paired-end 100-base reads, but to more modest levels of 117 and 126 Gb, respectively. Cobarcoded reads were mapped to build 37 of the human reference genomes using BWA-MEM (Li and Durbin 2009). The nonduplicate coverage ranged from 34× to 58×, and the number of long DNA molecules per barcode ranged from 1.2 to 6.8 (Table 2; Fig. 1C). As expected, the stLFR libraries made from 50 million beads and 1 ng genomic DNA (stLFR-1 and stLFR-2) had the highest single unique barcode cobarcoding rates of up to 85% (Fig. 1C). These libraries also observed the highest average nonoverlapping read coverage per long DNA molecule of 10.7%–12.1% and the highest average nonoverlapping base coverage of captured subfragments per long DNA molecule of 17.9%–18.4% (Fig. 1D). This coverage is ∼10× higher than previously demonstrated using 3 ng DNA and transposons attached to beads (Zhang et al. 2017). This suggests our solution-based transposition process is threefold more efficient at subfragment capture, that is, 40.7–47.4 subfragments per genomic fragment in 1 ng genomic DNA versus five subfragments captured in 3 ng at similar read coverage as reported by Zhang et al. (2017).
Table 2.
Variant calling statistics

For each library, variants were called using Sentieon's DNA-seq (Freed et al. 2017) using default settings. Comparing SNP and indel calls to Genome in a Bottle (GIAB) (Zook et al. 2014) allowed for the determination of true positive (TP), false positive (FP), and false negative (FN) rates (Table 2). In addition, we performed variant calling using the same settings on standard non-stLFR libraries made from GIAB reference material (NIST RM 8398) DNA and the same genomic DNA used to make stLFR libraries. We also compared precision and sensitivity rates to a Chromium library made by 10x Genomics (Zheng et al. 2016) and to those reported in the bead haplotyping library study by Zhang et al. (2017). Although direct comparisons can be difficult owing to differences in coverage, for most metrics of variant calling stLFR libraries performed similar to or better than the published results of the bead haplotyping library (Zhang et al. 2017) or Chromium libraries, especially when nonoptimized mapping and variant calling processes were used (Table 2, no filter). To further improve the variant calling performance in stLFR libraries, we used a machine learning algorithm trained against additional stLFR libraries made from GIAB samples GM12878, GM24385, GM24149, GM24143, and GM24631 (Supplemental Table S3; Methods). This led to the discovery of a few selection criteria that lowered the FP rate by ∼40%. This was achieved while increasing the FN rate by <10% in most stLFR libraries. Taking into account these variants, and the reduced number of FP variants after filtering, results in a similar FP rate and a two- to threefold higher FN rate than the filtered STD library for SNP calling (Table 2).
One potential issue with using GIAB data to measure the FP rate is that we were unable to use the GIAB reference material (NIST RM 8398) because of the rather small fragment size of the isolated DNA. For this reason, we used the GM12878 cell line and isolated DNA using a dialysis-based method capable of yielding very high molecular weight DNA (Methods). However, it is possible that our isolate of the GM12878 cell line could have a number of unique somatic mutations compared to the GIAB reference material and thus cause the number of FPs to be inflated in our stLFR libraries. To examine this further, single-nucleotide FP variants were compared across all of the NA12878 libraries (Supplemental Fig. S1A). One thousand seven hundred forty FP variants were shared between stLFR libraries 1–4 and both standard libraries made from GM12878 cell line DNA, but not shared with the standard library made from GIAB reference material. We also compared cell line DNA FPs with the Chromium library sequenced using Illumina technology and found that 1268 of these shared FPs were also present in the Chromium library (Supplemental Fig. S1B). Examination of the distribution of these shared FP variants across the genome versus randomly selected true positive variants (Supplemental Fig. S2) showed similar patterns with the vast majority of shared FP variants having distributions similar to randomly selected variants. Sixty shared FP variants were found within 100 bases of each other and could be the result of incorrect mapping owing to short insert sizes. This suggests that the stLFR process introduces very few FP errors, likely because of the low (∼1000×) amount of amplification used to make these libraries.
stLFR phasing performance
We developed a custom software program called LongHap (Methods) to make full use of the unique characteristics of stLFR data. Filtered variants called by each respective library were used for phasing within that library. In general, phasing performance was very high with >99% of all heterozygous SNPs in most samples placed into phase blocks with N50s ranging from 1.2–34.0 Mb, depending on the library type and the amount of sequence data (Supplemental Table S4). Comparison to GIAB data showed that short and long switch error rates were low (Supplemental Table S4) and comparable to previous studies (Mao et al. 2016; Zheng et al. 2016; Zhang et al. 2017). The stLFR-1 library with 336 Gb of total read coverage (44× unique genome coverage) achieved the highest phasing performance with a phase block N50 of 34.0 Mb (Fig. 2). Indeed, the entire length of most chromosomes was covered by a few phase blocks (Fig. 2). Even with only 100 Gb of data (24× unique genome coverage), the phase block N50 was still 14.4 Mb (Fig. 2). N50 length appeared to be mostly affected by length and coverage of long genomic fragments. This can be seen in the decreased N50 of stLFR-2 because the DNA used for this sample was slightly older and more fragmented than the material used for stLFR-1 (average fragment length of 52.5 kb versus 62.2 kb) and the ∼10-fold shorter N50s of stLFR-3 and stLFR-4 made from 10 ng of DNA (Supplemental Table S4).
Figure 2.

stLFR-1 phasing performance. The 221 phased blocks from the stLFR-1 library are depicted on chromosomes as alternating colors of gray and purple. Unphased regions are depicted in white. The inset table shows the performance of phasing with different sequence read coverage levels.
In an effort to make a fair comparison of stLFR phasing performance to a Chromium library—the Zhang et al. (2017) bead haplotyping method did not have read data available, making direct comparisons impossible—we attempted to phase only the high-confidence variant call set from GIAB. This removed any influence of the variant calling performance within a library from the phasing performance of that library. We also used HapCUT2 (Edge et al. 2017), a freely available software package designed for phasing cobarcoded and Hi-C sequencing data. Overall, the phasing performance of stLFR was similar to the Chromium library. The number of SNPs phased and the long and short switch error rates were essentially identical (Supplemental Table S4). Similar to the results from LongHap, stLFR-1 generated the longest phase block N50 of 15.1 Mb, which was similar to the N50 achieved by the Chromium library. In all cases, conclusions of performance differences are difficult owing to differences in input DNA length, total read coverage, and sequencing platforms used.
Structural variation detection
Previous studies have shown that long fragment information can improve the detection of structural variations (SVs) and described large deletions (4–155 kb) in NA12878 (Zheng et al. 2016; Zhang et al. 2017). To demonstrate the ability of stLFR to detect SVs, we examined barcode overlap data, as previously described (Zhang et al. 2017), for stLFR-1 and stLFR-4 libraries in these regions. In every case the deletion was observed in the stLFR-1 data, even at lower coverage (Fig. 3A; Supplemental Fig. S3). Closer examination of the cobarcoded sequence reads covering a ∼150-kb deletion in Chromosome 8 demonstrated that the deletion was heterozygous and found in a single haplotype (Fig. 3B,C). The 10-ng stLFR-4 library also detected most of the deletions, but the three smallest were difficult to identify because of the lower coverage per fragment (and thus less barcode overlap) of this library.
Figure 3.

SV detection. (A) Previously reported deletions in NA12878 were also found using stLFR data. Heat maps of barcode sharing for each deletion can be found in Supplemental Figure S3. (B) A heat map of barcode sharing within windows of 2 kb for a region with a ∼150 kb heterozygous deletion on Chromosome 8 was plotted using a Jaccard index as previously described (Zhang et al. 2017). Regions of high overlap are depicted in dark red. Those with no overlap in beige. Arrows demonstrate how regions that are spatially distant from each other on Chromosome 8 have increased overlap marking the locations of the deletion. (C) Cobarcoded reads are separated by haplotype and plotted by unique barcode on the y-axis and Chromosome 8 position on the x-axis. The heterozygous deletion is found in a single haplotype. Heat maps were also plotted for overlapping barcodes between Chromosomes 5 and 12 for a patient cell line with a known translocation (Dong et al. 2016) (D) and GM20759, a cell line with a known transversion in Chromosome 2 (Dong et al. 2017) (E).
To evaluate stLFR performance for detecting other types of SVs, we made libraries from a cell line from a patient with a known translocation between Chromosomes 5 and 12 (Dong et al. 2016) and GM20759, a cell line with a known inversion on Chromosome 2 (Dong et al. 2017). stLFR libraries were able to identify the inversion and the translocation in the respective cell lines (Fig. 3D,E). Down-sampling the amount of reads per library showed that a strong signal of the translocations was detected even with as little as 5 Gb of read data (∼1.7× total coverage) (Supplemental Fig. S4A–H). Finally, examination of both SVs in the stLFR-1 library resulted in no obvious pattern (Supplemental Fig. S4I–L).
De novo assembly with stLFR
For 1-ng input libraries, up to 85% of fragments were cobarcoded by a single unique barcode. This means the majority of barcodes should be associated with reads derived from very small regions of the genome (<300 kb). We believe this type of data should help simplify and improve de novo assembly. To test if stLFR can be used for de novo assembly, we used stLFR-1 and stLFR-2 libraries and the software package Supernova 2.1.1 (10x Genomics). This software was not designed for stLFR and as a result does not allow for data with more than 4.7 million barcodes to be used. Because of this limitation, we had to reduce the total number of barcodes for each stLFR library by combining more than 10 million barcodes into a total of 4.7 million barcodes for this analysis. This is not ideal because it reduces the amount of information, but we were still able to use stLFR data with this software package. Contig and scaffold N50s of ∼100 kb and ∼30 Mb, respectively, were achieved for both libraries (Table 3). Plotting the assembled contigs against chromosome sequences from Genome Reference Consortium Human Build 38 (GRCh38) showed high concordance (Fig. 4). Analysis of the resulting assemblies with the Quality Assessment Tool for Genome Assemblies (QUAST) (Gurevich et al. 2013) and comparison to other assemblies of NA12878 using Chromium (Zheng et al. 2016) or Nanopore (Jain et al. 2018) technologies suggested that the stLFR-derived assemblies were very complete and harbored few misassembled regions (Supplemental Table S5). These assemblies and those made using Nanopore or other long read technologies are still very far from perfect, and variant calling made on these assemblies typically underperforms that of variant calling after mapping to the reference genome.
Table 3.
NA12878 de novo assembly statistics
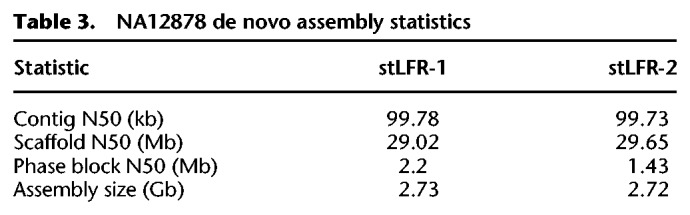
Figure 4.

Dot plots of de novo–assembled NA12878. The scaffolds from the de novo assemblies of stLFR-1 (A) and stLFR-2 (B) were compared against chromosomes from GRCh38 using dot plots.
Discussion
Here, we describe an efficient whole-genome sequencing (WGS) library preparation technology, stLFR, that enables the cobarcoding of subfragments of long genomic DNA molecules with a single unique clonal barcode in a single-tube process. Using microbeads as miniaturized virtual compartments allows a practically unlimited number of clonal barcodes to be used per sample at a negligible cost. Our optimized hybridization-based capture of transposon-inserted DNA on beads, combined with 3′-branch ligation and exonuclease degradation of the excess capture adapters, successfully barcodes up to ∼20% of subfragments in DNA molecules as long as 300 kb in length. This is achieved without DNA amplification of initial long DNA fragments and the representation bias that comes with it. In this way, stLFR solves the cost and limited cobarcoding capacity of emulsion-based methods.
The quality of variant calls using stLFR is very high and possibly, with further optimization, will approach that of standard WGS methods, but with the added benefit that cobarcoding enables advanced informatics applications. Using stLFR, we demonstrated high-quality, near complete phasing of the genome into long phase blocks with extremely low error rates, detection of SVs, and de novo assembly of a human genome. All of this was achieved from a single library that did not require special equipment or additional library preparation costs.
As a result of efficient barcoding, we successfully used as little as 1 ng of human DNA (600× genome coverage counting top and bottom strands of each DNA molecule) to make stLFR libraries and achieved high-quality WGS with most subfragments uniquely cobarcoded. Less DNA can be used, but stLFR does not use DNA amplification during cobarcoding and thus does not create overlapping subfragments from each individual long DNA molecule. For this reason, overall genomic coverage suffers as the amount of DNA is lowered. In addition, stLFR is currently capable of capturing and retaining about 10%–20% of each original long DNA molecule followed by PCR amplification. Because of random sampling, if 10%–20% sequence read coverage of each original DNA fragment is desired, this requires a relatively high sequencing read duplication rate to achieve. One potential solution is to remove the PCR step. This would eliminate duplicate reads and potentially reduce the false positive and false negative error rates. In addition, improvements such as optimizing the distance of insertion between transposons and increasing the length of sequencing reads to paired-end 200 bases are relatively easy to enable and would increase the coverage and overall quality. For some applications, such as structural variation detection, using less DNA and less coverage may be desirable. As we demonstrate in this paper, as little as 5 Gb of sequence coverage can faithfully detect inter- and intrachromosomal translocations; in these cases, the duplication rate is negligible. Indeed, stLFR may represent a simple and cost-effective replacement for long-mate-pair libraries in a clinical setting. Another potential application may be a phased high-quality genome in which really long phasing block N50s are not necessary. In this case, adding more DNA to stLFR libraries (such as 10 ng in stLFR-3) can enable this with a low duplication rate.
Using nonoptimized software, we demonstrated that stLFR can enable de novo assembly of the human genome. To be clear, this de novo assembly was far from perfect and indeed, the majority of analyses in this paper utilized mapping to the reference human genome to achieve high-quality results. However, we believe that with improvements to the stLFR process and the algorithms that use this type of data, the “perfect genome” (Peters et al. 2014) may be within reach. stLFR has the advantage over direct single-molecule long read technologies in that it utilizes cost-effective low error rate second-generation sequencing. However, there are situations in the assembly process when longer contiguous sequence is important. There are several strategies that potentially could be used to achieve this. One strategy uses transposon insertion to create a 9-base sequence overlap between adjacent subfragments. Frequently, these neighboring subfragments are captured and sequenced. In these cases, it may be possible to synthetically double the read length (e.g., for 200-base reads, two neighboring captured subfragments would create two 200-base reads with a 9-base overlap, or 391 bases).
In this paper libraries were made with 50 million beads, however using more is possible. This will enable many types of cost-effective analyses in which hundreds of millions of barcodes would be useful. We envision this type of cheap massive barcoding can be useful for RNA analyses, such as full-length mRNA sequencing from thousands of cells by combination with single-cell technologies or deep population sequencing of 16S RNA in microbial samples. Phased chromatin mapping by the Assay for Transposase-Accessible Chromatin (ATAC-seq) (Buenrostro et al. 2013) or methylation studies are all also possible with stLFR. Finally, in an effort to share what we believe to be a very important technology, we made a detailed protocol freely available for academic use (Cheng et al. 2018).
Methods
High-molecular-weight DNA isolation
Long genomic DNA was isolated from cell lines following a modified version of the RecoverEase DNA isolation kit (Agilent Technologies) protocol (https://www.agilent.com/cs/library/usermanuals/public/200600.pdf). Briefly, approximately 1 million cells were pelleted and lysed with 500 µL of lysis buffer. After a 10-min incubation at 4°C, 20 µL RNace-It ribonuclease cocktail in 4 mL digestion buffer was added directly to the lysed cells and incubated on a 50°C heat block. After 5 min, 4.5 mL Proteinase K solution (∼1.1 mg/mL Proteinase K, 0.56% SDS, and 0.89× TE) was added and the mix was incubated at 50°C for an additional 2 h. The genomic DNA was then transferred to dialysis tubing with a 1000-kD molecular weight cutoff (Spectrum Laboratories) and dialyzed overnight at room temperature in 0.5× TE buffer. Dialyzed DNA was transferred to a microcentrifuge tube and quantified using a Quant-iT Broad-Range dsDNA Assay Kit (Thermo Fisher Scientific). DNA was used directly following quantification.
Barcoded bead construction
Barcoded beads are constructed through a split and pool ligation-based strategy using three sets of double-stranded barcode DNA molecules. A common adapter sequence was attached to Dynabeads M-280 Streptavidin (Thermo Fisher Scientific) magnetic beads with a 5′ dual-biotin linker. Three sets 1536 of barcode oligos containing regions of overlapping sequence were constructed by Integrated DNA Technologies. Ligations were performed in 384-well plates in a 15-µL reaction containing 50 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 1 mM ATP, 2.5% PEG-8000, 571 units T4 ligase, 580 pmol barcode oligo, and 65 million M-280 beads. Ligation reactions were incubated for 1 h at room temperature on a rotator. Between ligations, beads were pooled into a single vessel through centrifugation, collected to the side of the vessel using a magnet, and washed once with high-salt wash buffer (50 mM Tris-HCl [pH 7.5], 500 mM NaCl, 0.1 mM EDTA, and 0.05% Tween 20) and twice with low-salt wash buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, and 0.05% Tween 20). Beads were resuspended in 1× ligation buffer and distributed across 384-well plates, and the ligation steps were repeated.
stLFR using two transposons
Two picomoles Tn5 coupled transposons were inserted into 40 ng genomic DNA in a 60-µL reaction of 10 mM TAPS-NaOH (pH 8.5), 5 mM MgCl2, and 10% DMF for 10 min at 55°C. Next, 1.5 µL transposon-inserted DNA was transferred to 248.5 µL hybridization buffer consisting of 50 mM Tris-HCl (pH 7.5), 100 mM MgCl2, and 0.05% TWEEN 20. From 10 to 50 million barcoded beads were resuspended in the same hybridization buffer. The diluted DNA was added to the barcoded beads, and the mix was heated for 10 min at 60°C with occasional light mixing. The DNA-bead mix was transferred to a tube revolver in a laboratory oven and incubated for 50 min at 45°C, after which 500 µL ligation mix containing 50 mM Tris-HCl (pH 7.8), 10 mM DTT, 1 mM ATP, 2.5% PEG-8000, and 4000 units T4 ligase was added directly to the DNA-bead mix. The ligation reaction was incubated for 1 h at room temperature on a revolver, 110 µL 1% SDS was added, and the mix was incubated for 10 min at room temperature to remove the Tn5 enzyme. Beads were collected to the side of the tube with a magnet and washed once with low-salt wash buffer and once with NEB2 buffer (New England Biolabs). Excess barcode oligos were removed using 10 units UDG (New England Biolabs), 30 units APE1 (New England Biolabs), and 40 units Exonuclease 1 (New England Biolabs) in 100 µL 1× NEB2 buffer. This reaction was incubated for 30 min at 37°C. Beads were collected to the side of the tube and washed once with low-salt wash buffer and once with 1× PCR buffer (1× PfuCx buffer [Agilent Technologies], 5% DMSO, 1 M Betaine, 6 mM MgSO4, and 600 µM dNTPs). The PCR mix containing 1× PCR buffer, 400 pmol of each primer, and 6 µL PfuCx enzyme (Agilent Technologies) was heated for 3 min to 95°C then cooled to room temperature. This mix was used to resuspend beads, and the combined mixture was incubated for 10 min at 72°C followed by 12 cycles of 10 sec at 95°C, 30 sec at 58°C, and 2 min at 72°C.
stLFR with 3′ branched adapter ligation
This method starts with the same hybridization insertion conditions but using only one transposon as opposed to two transposons. After capture and barcode ligation steps, as described above, beads were collected to the side of the tube and washed with low-salt wash buffer. An adapter digestion mix of 90 units Exonuclease I (New England Biolabs) and 100 units Exonuclease III (New England Biolabs) in 100 µL 1× TA Buffer (Teknova) was added to the beads and incubated for 10 min at 37°C. The reaction is stopped and the Tn5 enzyme is removed by adding 11 µL 1% SDS. Beads were collected to the side of the tube and washed once with low-salt wash buffer and once with 1× NEB2 buffer (New England Biolabs). Excess capture oligo was removed by adding 10 units UDG (New England Biolabs) and 30 units APE1 (New England Biolabs) in 100 µL 1× NEB2 buffer (New England Biolabs) and incubating for 30 min at 37°C. Beads were collected to the side of the tube and washed once with high-salt wash buffer and once with low-salt wash buffer, then 300 pmol of a second adapter was ligated to the bead-bound subfragments with 4000 units T4 ligase in 100 µL ligase buffer containing 50 mM Tris-HCl (pH 7.8), 10 mM MgCl2, 0.5 mM DTT, 1 mM ATP, and 10% PEG-8000 on a revolver at room temperature for 2 h. Beads were collected to the side of the tube and washed once in high-salt wash buffer and once in 1× PCR buffer. The PCR mix and conditions were the same as the two-transposon process described above.
Sequence mapping and variant calling
Raw read data were first demultiplexed by the associated barcode sequence using the barcode split tool (GitHub; https://github.com/stLFR/stLFR_read_demux) (Supplemental Code). Barcode assigned and clipped reads were mapped to the hs37d5 reference genome with BWA-MEM (Li and Durbin 2009). The resulting BAM file was then sorted by chromosomal coordinates with SAMtools (Li et al. 2009) and duplicates were marked with Picard MarkDuplicate function (http://broadinstitute.github.io/picard). Short variant (SNPs and indels) calling was performed using Sentieon's DNA-seq (Freed et al. 2017) optimization of the GATK's HaplotypeCaller (McKenna et al. 2010). To further improve the FP rate in stLFR libraries, we developed a binary classification model for variant filtering based on XGboost (Chen and Guestrin 2016). TPs and FPs from samples GM12878, GM24385, GM24149, GM24143, and GM24631 (Supplemental Table S3) were generated using VCFeval (Cleary et al. 2015) by comparing to Genome in a Bottle (GIAB) high-confidence variant lists (Zook et al. 2014) for each sample and labeled for model training. Using custom software (https://github.com/stLFR/extremevariantfilter) (Supplemental Code), mapping quality (MQ), MQ rank sum, strand odds ratio, Fisher strand bias, read position rank sum, quality by depth, reference allele depth, alternate allele depth, percentage of reads supporting the reference allele, the ratio of alternate depth to reference depth, and an encoding of genotype (homozygous or heterozygous) were extracted from the labeled VCFs and used as features for model training. Models were trained individually for SNPs and InDels, and generalizability of the models was tested by training on four of the five samples and testing on the fifth.
LongHap
We developed a novel phasing algorithm, LongHap (https://github.com/stLFR/stLFR_LongHap) (Supplemental Code), specifically designed for the uniqueness of stLFR's data. A seed-extension strategy was used in the phasing process. It initially starts from one pair of seeds, composed of the most upstream heterozygous variant in the chromosome. The seeds are extended by linking the other downstream candidate variants until no more variants can be added to the extending seeds (Supplemental Fig. S5). In this extending process, the candidate variants at different loci will not be equally treated (i.e., the upstream variant has higher priority compared with the downstream ones across the chromosome). Each two heterozygous loci have two possible combinations along the two different alleles. Taking variant T2/G2 and G3/C3, for example (Supplemental Fig. S5), one combination pattern is T2-G3 and G2-C3, whereas another one is T2-C3 and G2-G3. The score of each combination is calculated by the number of long DNA fragments spanning the two loci, which is equivalent to the number of unique barcodes with reads mapping to these two loci. As shown in Supplemental Figure S5, the final score of the former combination is 3, which is three times more than the latter. The variant T2/G2 is added to the extending seeds and the process repeats. If any barcode supports both of the alleles at one specific locus, it will be ignored when calculating the linkage score. This helps to decrease the switch error rate. When a conflict in linking downstream candidate variants occurs, as the variant A4/C4 in Supplemental Figure S5 shows, a simple decision will be made by comparing the linked loci number to allow further extending candidate variants. In this case, there are two linked loci in the left scenario, and there is only one in the right scenario. LongHap will choose the left combination pattern as the final phasing result.
Variant phasing with HapCUT2
SNPs were phased with HapCUT2 (https://github.com/vibansal/HapCUT2) (Edge et al. 2017) using its 10x Genomics data pipeline. The BAM file was first converted into a format that carries barcode information in a similar format as a 10x Genomics barcoded BAM. Specifically, a “BX” field was added to each line reflecting the barcode information of that read. GIAB variants or variants called by GATK for each library were used as the input for phasing, and the phasing result was summarized and compared against the GIAB phased VCF file (Zook et al. 2014) using the calculate_haplotype_statistics.py tool of HapCUT2.
SV detection
Structural variants were detected by calculating shared barcodes between regions of the genome as previously described (Zhang et al. 2017). Duplicate reads were first removed. The mapped cobarcoded reads were scanned using a sliding window (the default value is 2 kb) along the genome, every window recorded how many barcodes have been found within this 2-kb window, and a Jaccard index was calculated for the shared barcodes ratio between the window pairs. Structural variant events were identified by the Jaccard index sharing metric between window pairs.
For every window pair (X, Y) across the genome, the Jaccard index is calculated as follows:
De novo assembly
For each library, barcodes with a minimum of 10 reads were selected, and these barcodes were degenerated into the list of ∼4.7 million barcodes from the Long Ranger 2.2.2 software package (10x Genomics) barcode whitelist (located in unpacked software at /longranger-2.2.2/longranger-cs/2.2.2/tenkit/lib/python/tenkit/barcodes/4M-with-alts-february-2016.txt). stLFR FASTQ files were converted into a format that resembles Chromium FASTQ files and were then used as input for Supernova 2.0.1 (10x Genomics). Specifically, for Supernova runs, options maxreads = 2,100,000,000 and nopreflight were used. A pseudohap assembly output was generated with the mkoutput function of Supernova, and scaffolds with a minimum length of 10 kb were compared against the human reference GRCh38 with QUAST 5.0.0 (Gurevich et al. 2013). In addition, pseudohap2 assembly outputs were also generated from Supernova, and each haplotype was aligned to GRCh38 with function NUCmer of MUMmer 4.0.0 (Delcher et al. 1999; Kurtz et al. 2004), with options -c 1000 -l 100. The alignment delta file was filtered with the delta-filter function of MUMmer to filter for one-to-one alignments. Scaffolds of at least 500 kb and alignments of at least 50 kb from the resulting alignment list were plotted into dot plots.
Data access
All sequencing data reported in this study have been submitted to the China National GeneBank (CNGB) Nucleotide Sequence Archive (CNSA; https://db.cngb.org/cnsa/) under accession number CNP0000066 and to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject) under accession number PRJEB27414.
Competing interest statement
Employees of BGI and Complete Genomics have stock holdings in BGI.
Supplementary Material
Acknowledgments
We acknowledge the ongoing contributions and support of all Complete Genomics and BGI-Shenzhen employees, in particular, the many highly skilled individuals that work in the libraries, reagents, and sequencing groups that make it possible to generate high-quality whole-genome data. We thank Z. Dong, Z. Yang, and W. Xie for providing cell lines for the translocation analysis. This work was supported in part by the Shenzhen Peacock Plan (No. KQTD20150330171505310) and the National Key Research and Development Program of China (No. 2017YFC0906501). B.A.P. is a recipient of and this work was partially supported by the Research Fund for International Young Scientists, National Natural Science Foundation of China (31550110216).
Authors contributions: R.D. and B.A.P. conceived the study. O.W., R.C., X.C., M.K.Y.W., H.K.L., D.C., L.W., F.F., Y.Z., S.D., D.N., A.A., X.X., R.D., and B.A.P. developed the molecular biology process of stLFR. R.Y.Z., S.D., S.G., N.B., and A.C. performed the sequencing. Q.M., J.T., Y.S., Y.Z., E.A., Y.X., C.V., S.N., W.T., J.W., X.L., X.Q., H.W., and Y.D. developed algorithms for and performed analyses on stLFR data. O.W., C.X., J.S.L., W.Z., H.Y., J.W., K.K., X.X., R.D., and B.A.P. coordinated the study. O.W., R.D., and B.A.P. wrote the manuscript. All authors reviewed and edited the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.245126.118.
Freely available online through the Genome Research Open Access option.
References
- Amini S, Pushkarev D, Christiansen L, Kostem E, Royce T, Turk C, Pignatelli N, Adey A, Kitzman JO, Vijayan K, et al. 2014. Haplotype-resolved whole-genome sequencing by contiguity-preserving transposition and combinatorial indexing. Nat Genet 46: 1343–1349. 10.1038/ng.3119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. 2013. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10: 1213–1218. 10.1038/nmeth.2688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Guestrin C. 2016. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, pp. 785–794. ACM, San Francisco, CA. [Google Scholar]
- Cheng X, Wu M, Chin R, Lam H, Chen D, Wang L, Fan F, Zou Y, Chen A, Zhang W, et al. 2018. A simple bead-based method for generating cost-effective co-barcoded sequence reads. Protoc Exch 10.1038/protex.2018.116 [DOI] [Google Scholar]
- Cleary JG, Braithwaite R, Gaastra K, Hilbush BS, Inglis S, Irvine SA, Jackson A, Littin R, Rathod M, Ware D, et al. 2015. Comparing variant call files for performance benchmarking of next-generation sequencing variant calling pipelines. bioRxiv 10.1101/023754 [DOI]
- Delcher AL, Kasif S, Fleischmann RD, Peterson J, White O, Salzberg SL. 1999. Alignment of whole genomes. Nucleic Acids Res 27: 2369–2376. 10.1093/nar/27.11.2369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Z, Zhang J, Hu P, Chen H, Xu J, Tian Q, Meng L, Ye Y, Wang J, Zhang M, et al. 2016. Low-pass whole-genome sequencing in clinical cytogenetics: a validated approach. Genet Med 18: 940–948. 10.1038/gim.2015.199 [DOI] [PubMed] [Google Scholar]
- Dong Z, Wang H, Chen H, Jiang H, Yuan J, Yang Z, Wang WJ, Xu F, Guo X, Cao Y, et al. 2017. Identification of balanced chromosomal rearrangements previously unknown among participants in the 1000 Genomes Project: implications for interpretation of structural variation in genomes and the future of clinical cytogenetics. Genet Med 20: 697–707. 10.1038/gim.2017.170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drmanac R. 2006. Nucleic acid analysis by random mixtures of non-overlapping fragments. International patent application no. PCT/US2006/022950.
- Drmanac R, Sparks AB, Callow MJ, Halpern AL, Burns NL, Kermani BG, Carnevali P, Nazarenko I, Nilsen GB, Yeung G, et al. 2010. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science 327: 78–81. 10.1126/science.1181498 [DOI] [PubMed] [Google Scholar]
- Drmanac R, Peters BA, Alexeev A. 2014. Multiple tagging of long DNA fragments. International application no. PCT/US2014/030649.
- Duitama J, McEwen GK, Huebsch T, Palczewski S, Schulz S, Verstrepen K, Suk EK, Hoehe MR. 2012. Fosmid-based whole genome haplotyping of a HapMap trio child: evaluation of Single Individual Haplotyping techniques. Nucleic Acids Res 40: 2041–2053. 10.1093/nar/gkr1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edge P, Bafna V, Bansal V. 2017. HapCUT2: robust and accurate haplotype assembly for diverse sequencing technologies. Genome Res 27: 801–812. 10.1101/gr.213462.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan HC, Wang J, Potanina A, Quake SR. 2011. Whole-genome molecular haplotyping of single cells. Nat Biotechnol 29: 51–57. 10.1038/nbt.1739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehlmann T, Reinheimer S, Geng C, Su X, Drmanac S, Alexeev A, Zhang C, Backes C, Ludwig N, Hart M, et al. 2016. cPAS-based sequencing on the BGISEQ-500 to explore small non-coding RNAs. Clin Epigenetics 8: 123 10.1186/s13148-016-0287-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed DN, Aldana R, Weber JA, Edwards JS. 2017. The Sentieon Genomics Tools—a fast and accurate solution to variant calling from next-generation sequence data. bioRxiv 10.1101/115717 [DOI]
- Gurevich A, Saveliev V, Vyahhi N, Tesler G. 2013. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29: 1072–1075. 10.1093/bioinformatics/btt086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Liang X, Xuan Y, Geng C, Li Y, Lu H, Qu S, Mei X, Chen H, Yu T, et al. 2017. A reference human genome dataset of the BGISEQ-500 sequencer. Gigascience 6: 1–9. 10.1093/gigascience/gix024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M, Koren S, Miga KH, Quick J, Rand AC, Sasani TA, Tyson JR, Beggs AD, Dilthey AT, Fiddes IT, et al. 2018. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat Biotechnol 36: 338–345. 10.1038/nbt.4060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo K, Chen YL, de Pablo JJ, Schwartz DC. 2009. Elongation and migration of single DNA molecules in microchannels using oscillatory shear flows. Lab Chip 9: 2348–2355. 10.1039/b902292a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitzman JO, Mackenzie AP, Adey A, Hiatt JB, Patwardhan RP, Sudmant PH, Ng SB, Alkan C, Qiu R, Eichler EE, et al. 2011. Haplotype-resolved genome sequencing of a Gujarati Indian individual. Nat Biotechnol 29: 59–63. 10.1038/nbt.1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov V, Xie D, Chen R, Pushkarev D, Ma Z, Blauwkamp T, Kertesz M, Snyder M. 2014. Whole-genome haplotyping using long reads and statistical methods. Nat Biotechnol 32: 261–266. 10.1038/nbt.2833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. 2004. Versatile and open software for comparing large genomes. Genome Biol 5: R12 10.1186/gb-2004-5-2-r12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levene MJ, Korlach J, Turner SW, Foquet M, Craighead HG, Webb WW. 2003. Zero-mode waveguides for single-molecule analysis at high concentrations. Science 299: 682–686. 10.1126/science.1079700 [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Xiao Y, Huang H, Wang Q, Rao W, Feng Y, Zhang K, Song Q. 2010. Direct determination of molecular haplotypes by chromosome microdissection. Nat Methods 7: 299–301. 10.1038/nmeth.1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak SST, Gopalakrishnan S, Carøe C, Geng C, Liu S, Sinding MS, Kuderna LFK, Zhang W, Fu S, Vieira FG, et al. 2017. Comparative performance of the BGISEQ-500 vs Illumina HiSeq2500 sequencing platforms for palaeogenomic sequencing. Gigascience 6: 1–13. 10.1093/gigascience/gix049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Q, Ciotlos S, Zhang RY, Ball MP, Chin R, Carnevali P, Barua N, Nguyen S, Agarwal MR, Clegg T, et al. 2016. The whole genome sequences and experimentally phased haplotypes of over 100 personal genomes. Gigascience 5: 42 10.1186/s13742-016-0148-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olasagasti F, Lieberman KR, Benner S, Cherf GM, Dahl JM, Deamer DW, Akeson M. 2010. Replication of individual DNA molecules under electronic control using a protein nanopore. Nat Nanotechnol 5: 798–806. 10.1038/nnano.2010.177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters BA, Kermani BG, Sparks AB, Alferov O, Hong P, Alexeev A, Jiang Y, Dahl F, Tang YT, Haas J, et al. 2012. Accurate whole-genome sequencing and haplotyping from 10 to 20 human cells. Nature 487: 190–195. 10.1038/nature11236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters BA, Liu J, Drmanac R. 2014. Co-barcoded sequence reads from long DNA fragments: a cost-effective solution for “perfect genome” sequencing. Front Genet 5: 466 10.3389/fgene.2014.00466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaraj S, Dixon JR, Bansal V, Ren B. 2013. Whole-genome haplotype reconstruction using proximity-ligation and shotgun sequencing. Nat Biotechnol 31: 1111–1118. 10.1038/nbt.2728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suk EK, McEwen GK, Duitama J, Nowick K, Schulz S, Palczewski S, Schreiber S, Holloway DT, McLaughlin S, Peckham H, et al. 2011. A comprehensively molecular haplotype-resolved genome of a European individual. Genome Res 21: 1672–1685. 10.1101/gr.125047.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Xi Y, Zhang W, Wang W, Shen H, Wang X, Zhao X, Alexeev A, Peters B, Albert A, et al. 2019. 3′ Branch ligation: a novel method to ligate non-complementary DNA to recessed or internal 3′OH ends in DNA or RNA. DNA Res 26: 45–53. 10.1093/dnares/dsy037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Zhu J, Shendure J, Porreca GJ, Aach JD, Mitra RD, Church GM. 2006. Long-range polony haplotyping of individual human chromosome molecules. Nat Genet 38: 382–387. 10.1038/ng1741 [DOI] [PubMed] [Google Scholar]
- Zhang F, Christiansen L, Thomas J, Pokholok D, Jackson R, Morrell N, Zhao Y, Wiley M, Welch E, Jaeger E, et al. 2017. Haplotype phasing of whole human genomes using bead-based barcode partitioning in a single tube. Nat Biotechnol 35: 852–857. 10.1038/nbt.3897 [DOI] [PubMed] [Google Scholar]
- Zheng GX, Lau BT, Schnall-Levin M, Jarosz M, Bell JM, Hindson CM, Kyriazopoulou-Panagiotopoulou S, Masquelier DA, Merrill L, Terry JM, et al. 2016. Haplotyping germline and cancer genomes with high-throughput linked-read sequencing. Nat Biotechnol 34: 303–311. 10.1038/nbt.3432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zook JM, Chapman B, Wang J, Mittelman D, Hofmann O, Hide W, Salit M. 2014. Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nat Biotechnol 32: 246–251. 10.1038/nbt.2835 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.