

Abstract
Eukaryotic genome replication depends on thousands of DNA replication origins (ORIs). A major challenge is to learn ORI biology in multicellular organisms in the context of growing organs to understand their developmental plasticity. We have identified a set of ORIs of Arabidopsis thaliana and their chromatin landscape at two stages of post-embryonic development. ORIs associate with multiple chromatin signatures including transcription start sites (TSS) but also proximal and distal regulatory regions and heterochromatin, where ORIs colocalize with retrotransposons. In addition, quantitative analysis of ORI activity led us to conclude that strong ORIs have high GC content and clusters of GGN trinucleotides. Development primarily influences ORI firing strength rather than ORI location. ORIs that preferentially fire at early developmental stages colocalize with GC-rich heterochromatin, but at later stages with transcribed genes, perhaps as a consequence of changes in chromatin features associated with developmental processes. Our study provides the set of ORIs active in an organism at the post-embryo stage that should allow us to study ORI biology in response to development, environment, and mutations with a quantitative approach. In a wider scope, the computational strategies developed here can be transferred to other eukaryotic systems.
Replication of the large and complex genomes of multicellular organisms occurs during S-phase, once per cell cycle. Full genome replication depends on the coordinated function of thousands of DNA replication origins (ORIs) scattered across the genome (Méchali 2010; Sanchez et al. 2012; Méchali et al. 2013). The association of pre-replication complexes (pre-RCs) with DNA specifies the genomic sites that can potentially act as ORIs. Some clues on the contribution of DNA sequence, chromatin marks, transcription factor binding sites, and GC content to ORI activity have been obtained (Méchali 2010; Costas et al. 2011; Leonard and Méchali 2013; Méchali et al. 2013; Gutierrez et al. 2016; Vergara and Gutierrez 2017). In spite of extensive efforts, the molecular determinants of ORIs in eukaryotes are still largely unknown.
Genomic approaches carried out in mammalian, insect, and plant cultured cells have helped to gain a picture supporting that ORIs frequently colocalize with chromatin marks associated with active chromatin (Sequeira-Mendes et al. 2009; Karnani et al. 2010; MacAlpine et al. 2010; Cayrou et al. 2011, 2015; Costas et al. 2011; Comoglio et al. 2015). ORIs in the euchromatin of multicellular organisms also associate with GC stretches, which may form G quadruplexes (G4) (Cayrou et al. 2012; Castillo Bosch et al. 2014; Valton et al. 2014). However large genomic regions lacking marks of active chromatin must be replicated, posing the question of whether there are multiple chromatin signatures that define ORI location.
The use of cultured cells, with which most studies have been done so far, to identify the determinants of ORIs in multicellular organism has some limitations (Mesner et al. 2013). Unlike in vitro culture conditions, cells within the body are subject to hormonal and developmental signals that influence cell proliferation and differentiation. These intra- and extracellular factors, lost in the in vitro cell culture studies, can be crucial for the integration of genome replication with cell proliferation and development. Therefore, one major challenge is to identify ORIs in the cells of a whole organism to assess potential effects of cell fate and developmental cues on ORI specification.
Several studies have shown that in eukaryotes only a subset of ORIs is activated at each replication round. Besides ORI specification, the factors that influence variability of ORI activity need to be identified. This has been approached in Caenorhabditis elegans using embryos of different ages (Pourkarimi et al. 2016; Rodríguez-Martínez et al. 2017) and Drosophila melanogaster salivary glands (Sher et al. 2012). Here, we have taken the challenge of identifying the active ORIs in a living organism analyzing their localization and plasticity during post-embryonic development. We used the plant Arabidopsis thaliana at two stages of vegetative development: 4-day-old seedlings (shortly after germination, when the hormonal and developmental signals necessary for vegetative growth have been established) and 10-day-old seedlings (before the transition to reproductive development). Since these seedlings contain a mixed population of dividing, endoreplicating, differentiating, embryo-derived, and stem cells (Fig. 1A; Gutierrez 2005), our approach should provide a collection of ORIs active in a wide variety of cell types. With this strategy we sought to obtain an understanding of (1) the molecular determinants of ORI specification and function, and (2) their relationships with cell proliferation and gene expression programs.
Figure 1.

DNA replication origin (ORI) identification in whole Arabidopsis seedlings and evaluation of reproducibility and quality of sequencing data sets. (A) Summary of basic steps for purification of nascent strands (NS) from seedlings at two developmental stages of Arabidopsis vegetative growth. Seedlings contain cells undergoing cell proliferation and the endocycle in different locations. In 4-d-old seedlings, the shoot and the root apical meristems contain dividing cells, whereas the cotyledons and the transition domain above the root apical meristem contain endocycling cells. The rest of the root is made up of different cell types, some of them dividing and some differentiated. In addition to all these organs and cell types, 10-d-old seedlings contain growing leaf primordia and lateral root primordia, with proliferating cells, as well as a longer root with more differentiated cells. (B) Flowchart summary of the assignment of the nascent strand score (NSS) and the ZPeaks algorithm used to identify ORIs. (C) Quality controls of the NS purification and dsDNA conversion step. The horizontal axis represents the following data sets: (class 1) biased peaks that do not overlap with the ORI set; (class 2) all peaks with dsDNA conversion bias; (class 3) biased peaks overlapping with ORIs; (class 4) all ORIs; and (class 5) ORIs that do not overlap with the biased set. (D) Representative genome browser view of a ∼35-kb region of Chromosome 1 to illustrate ORI identification in various sucrose gradient fractions of the three independent experiments in 4- and 10-d-old seedlings.
Results
Identification of ORIs and their replicative strength
Active ORIs are characterized by the presence of newly synthesized single-stranded DNA (ssDNA) molecules, also known as nascent strands (NS). NS purification from whole seedlings is challenging because of the limited amount of NS, even in highly proliferating cultured cells. Here, we have (1) implemented procedures to obtain sufficient amounts of a clean NS sample from whole plant seedlings, (2) designed protocols to reduce possible biases associated with NS preparation and dsDNA conversion, and (3) developed computational tools to analyze ORIs in a quantitative manner.
NS were isolated from Arabidopsis seedlings in two stages of vegetative development: 4-d-old seedlings soon after germination, and 10-d-old seedlings before the transition to reproductive development, which in both cases contain proliferating and endoreplicating cells of various differentiation stages and types. Our enhanced procedure yielded sufficient amounts of clean NS samples (Methods; Fig. 1A; Supplemental Fig. S1). Briefly, after nuclei purification, NS were purified from DNA replication bubbles by sucrose gradient centrifugation to isolate DNA fragments of appropriate size in several gradient fractions (300 bp < nascent strands < 2 kb, longer than Okazaki fragments but without compromising resolution). Any contaminating DNA fragmentation products were removed by λ-exonuclease (λ-exo) treatment because they are not protected by an RNA primer, as it occurs with bona fide NS. It has been reported that the λ-exo treatment produces a bias toward GC-rich DNA sequences (Foulk et al. 2015). However, this is significantly reduced provided that the treatment is carried out at least twice and under optimal conditions of substrate and λ-exo concentrations (Cayrou et al. 2011, 2015; Picard et al. 2014; Comoglio et al. 2015; Lombraña et al. 2016).
Purified NS were processed to generate libraries and submitted to sequencing. Nonredundant sequence reads were aligned to the Arabidopsis genome (TAIR10), and those uniquely mapping were kept for further analysis. To allow a stringent identification of ORIs, we carried out three independent experiments for each developmental stage and processed 2–3 consecutive sucrose gradient fractions in each case. In most cases, it was possible to detect the increase in the region covered by reads in fractions containing increased NS size, indicative of fork progression. Note that contaminating molecules derived from λ-exo-resistant non-nascent strands should have a similar region covered by reads. We reasoned that ORIs are in principle active in all the experiments, but with different probabilities of firing in the cell population. We identified a set of ORIs detected in at least two of the three gradient fractions analyzed and then in, at least, two of three biological replicates (experiments) and quantified their strength in each experiment through the excess of NS reads with respect to the genomic control by calculating their NSS (nascent strand score) (Methods; Fig. 1B). In this way, the NSS values fully characterize all samples, whereas ORI locations do not vary because they are obtained by combining all samples. We computed weighted averages over the set of ORIs using the NSS as weights, so that ORIs that are weak in all samples contribute little to the weighted averages, thus reducing the effect of possible false positives. Conversely, the strong correlation that we found between NSS in different samples makes it unlikely that we missed any strong ORI, as we verified by visual inspection in the genome browser. Genomic sites with a high NSS value could result from increased frequency of ORI usage in the cell population or from failures of replication fork progression. This can be discarded because replication fork arrest due to termination signals would occur in most cases in large bubbles and, consequently, nascent strands would appear in the sucrose gradient in fractions greater than those sequenced here. We identified ORIs using our own peak-calling ZPeaks algorithm because (1) it provides a well-defined profile of the NSS over all of the genome, crucial for our analysis, and (2) it localizes an ORI at the local maximum of the NSS over the ORI box called, which is needed for carefully centering the metaplots (Fig. 1B).
We also carried out experiments to evaluate a possible bias in peak detection introduced during the dsDNA conversion step prior to library preparation. The routine protocol for dsDNA conversion uses random primers mixed with heat-denatured genomic DNA and fast cooling to 37°C. We found that this protocol has a bias to enrich for specific genomic regions when compared with the untreated genomic DNA and determined 6479 dsDNA-converted biased regions. Therefore, we searched for conditions that reduced the dsDNA conversion bias and found that carrying the primer-annealing step slowly (Methods) halved the number of biased regions to 3223. Therefore, we used this procedure with our NS samples, which we believe is advisable in all ORI mapping approaches that require a dsDNA conversion step. To remove any residual bias, we used as background controls genomic DNA sheared, denatured, and converted into dsDNA as for the NS samples. The stringent strategy used to control for the dsDNA conversion bias has been shown to be effective (Fig. 1C) since (1) ∼80% of ORIs do not overlap with biased regions (class 5 in Fig. 1C) and possess high NSS and CDC6 (data from Costas et al. 2011) values, as expected for bona fide ORIs; (2) likewise, ORIs that overlap with biased regions present even higher CDC6 binding and higher NSS, despite the dsDNA conversion bias contributing negatively to the NSS, and (3) ∼84% of biased regions do not overlap with ORIs (class 1 in Fig. 1C) and show a vanishing NSS score as expected for random genomic regions.
The ORI midpoint was identified with a resolution of 25 bp, the bin size used in the ZPeaks algorithm. We found that when an ORI was identified in different samples, the midpoint varied ∼120 bp on average, which estimates the precision of our measurements. The ORI midpoint was then computed as the weighted average of the midpoints of the individual samples. The distribution of inter-ORI distance is highly skewed toward small values with a median of 27.6 kb, much smaller than the mean of 43.4 kb. We tested ZPeaks both by visual inspection of the overlap between sequencing reads and candidate ORIs (Fig. 1D) and by statistical tests. Together our strategy allowed us to identify a robust set of 2374 bona fide ORIs (Supplemental Table 1) that can be confidently used to analyze the determinants of ORI specification.
We found that the NSS values are broadly distributed, spanning several orders of magnitude. The NSS of different experiments correlated well with each other, with correlation coefficients ranging from 0.52 to 0.71, except between experiments 2 and 3 in 4-d-old seedlings (Supplemental Fig. S2). This result lends support to our approach and suggests that some intrinsic ORI properties influence their firing rates in all the experiments, although with important variations. Despite that the firing probability should saturate at 100% probability, the NSS did not show any sign of saturation, suggesting that they are far from 100% activity. To facilitate the comparison between the two developmental stages, we obtained scores that averaged the three independent experiments performed for each stage.
Validation of ORI activity
We validated ORI activity in an independently prepared NS sample by measuring the relative abundance of NS by qPCR across genomic regions that contained ORIs identified by SNS-seq. This approach can be used for a small number of ORIs but has the advantage of using a sample that consists of ssDNA, treated with λ-exo but not converted into dsDNA. To assess ORI activity in a stringent way, we selected ORIs belonging to either the class of ORIs with or without overlap with dsDNA-converted biased regions. As a control, we chose a region lacking any significant amount of reads. NS were prepared independently of those used for the SNS-seq experiments from 4- and 10-day-old seedlings and used as substrate in qPCR amplification reactions using primer pairs spanning ∼10–15 kb around the ORI sites (Supplemental Fig. S3; Supplemental Table 2). We detected clear peaks corresponding to active ORIs, including ORIs that overlapped with dsDNA-converted biased regions. These data demonstrate that the data set generated under our stringent conditions constitutes a bona fide genome-wide set of ORIs active in Arabidopsis seedlings.
Genomic landscape of ORI locations
To determine the preferences of ORI localization, we examined the association of ORIs with various genomic elements. Most ORIs (>78%) are associated with genic regions, including 1 kb upstream regions, much more than expected by chance. Within genes, ORIs locate more frequently in exons (Fig. 2A). Intergenic regions and TEs comprise ∼5% and ∼13% of ORIs, respectively, whereas these genomic regions represent a much larger fraction of the genome (∼15% and ∼21%, respectively). We recently discovered that ∼5% of ORIs active in cultured Arabidopsis cells colocalize with TEs, although in the gene-poor pericentromeric heterochromatin, the frequency of colocalization with TEs increases to ∼34%. These ORIs are much more frequently located within retrotransposons of the Gypsy and LINE families than in DNA transposons (Vergara et al. 2017). We found that in pericentromeric regions in seedlings, the frequency of ORIs located within TEs increases significantly compared to the overall genome (Fig. 2B). These ORI-TEs colocalize preferentially with retrotransposons, as in cultured cells, although in seedlings the fraction of ORIs located in DNA transposons is higher, in particular for the MuDR family (Fig. 2C). We found that TEs containing ORIs possess a higher GC content (43.6%) than those lacking ORIs (33.3%), in agreement with previous findings in Arabidopsis cultured cells (Vergara et al. 2017). We also found that ORIs have a preference to localize ∼0.5 kb downstream from the transcription start site (TSS) and tend to avoid transcription termination sites (TTS) at both developmental stages (Fig. 2D).
Figure 2.

Association of ORIs with genomic elements and TE families. (A) Relationship between ORI location and genomic elements. The overlap (in base pairs) between the indicated genomic elements and each ORI was computed and expressed as a percentage. A region of 1 kb upstream of the coding sequence was considered as the promoter. Note that TEs are large genomic elements that may have one or more TE genes associated with them. Here, the class TE refers to genomic regions that contain TEs but do not overlap with TE genes. (B) Frequency distribution of ORIs colocalizing with genes (green), TEs (blue), and nonannotated regions (gray) compared with the respective nucleotide coverage. (C) Frequency distribution of ORI-TEs (blue bars) in TE families in all the Arabidopsis genome, the nonpericentromeric regions and the pericentromeric regions compared with the respective TE family nucleotide coverage of total TE nucleotides (black bars). On the x-axis, retrotransposon families (red) and DNA transposon families (black). (D) Metaplots of the combined NSS of the three independent experiments of 4-d-old and 10-d-old seedlings with respect to the transcription start sites (TSS; left) or the transcription termination site (TTS; right), oriented in both cases with the transcribed RNAs.
Local properties of ORI locations
To study the properties of Arabidopsis ORIs in seedlings, we assessed the average local neighborhood of all ORIs by computing metaplots centered at the ORI midpoint with each ORI being weighted with its own NSS. To account for the asymmetry between the G and C and between the A and T bases, a feature of ORI, they were oriented along the 5′–3′ direction of the strand with positive GC skew. For this analysis each variable was transformed into Z-scores with respect to the set of control genomic regions, so that a positive value indicates that the trait in that state is larger than the average score over the whole genome. This allows comparing the strength of different genomic and epigenomic variables.
First we confirmed that the NSS of all experiments had a very prominent peak at the ORI midpoint (Fig. 3A,C; Supplemental Fig. S4). We examined the position of the pre-RC protein CDC6 (Costas et al. 2011) and found that it has a high peak centered at the ORI midpoint with a width of ±1 kb (Fig. 3A,C; Supplemental Fig. S4). This provides strong independent support to the peak-calling procedure used here to define ORI location based on nascent-strand mapping. We also found that ORI location coincides with a peak of nucleosome occupancy (Fig. 3A; Supplemental Fig. S4), as described for cultured cells in Arabidopsis and mammals (Stroud et al. 2012; Lombraña et al. 2013). Furthermore, regions around ORIs are more frequently transcribed than the genome average, with a broad peak of ±1 kb centered at the ORI midpoint (Fig. 3A; Supplemental Fig. S4).
Figure 3.

Features of the local neighborhood of ORIs in whole Arabidopsis seedlings. (A) Metaplots of NSS, CDC6, transcript content (RNA), and nucleosome (nucl.) content weighted with the combined Z-score of the three independent experiments of 4-d-old (top) and 10-d-old (bottom) seedlings. The metaplots for individual scores of each experiment are shown in Supplemental Figure S3. (B) Metaplots of GC, GC skew, and GC split weighted with the combined Z-score of the three independent experiments of 4-d-old (top) and 10-d-old (bottom) seedlings. The metaplots for individual scores of each experiment are shown in Supplemental Figure S4. (C) Heatmaps of the signals (square root) of several key features around ±2 kb of the ORI midpoint (0). ORIs have been ranked according to the first principal component of the complete set of features, computed over all 2374 ORIs. The color scale applies to all panels. Note that heatmaps of other features are shown in Figures 5 and 6 (see below).
The Arabidopsis genome is rich in A + T (63.8%). ORIs identified in cultured cells preferentially colocalize with short G + C-rich stretches (Costas et al. 2011). ORIs in seedlings also colocalize with G + C-rich regions showing a peak of ∼0.8 kb in width, centered at the ORI midpoint (Fig. 3B,C; Supplemental Fig. S4). The asymmetry between the G and C nucleotide, called GC skew, is a signature of ORIs both in prokaryotes and metazoa (MacAlpine et al. 2010; Cayrou et al. 2011; Arakawa and Tomita 2012; Xia 2012; Comoglio et al. 2015). The GC skew presents a strong peak centered at the ORI midpoint but asymmetrically distributed (−0.5 kb through 1.0 kb from ORI midpoint) (Fig. 3B,C; Supplemental Fig. S4).
The GC skew associated with DNA replication is attributed to the asymmetry of mutation processes on the leading and lagging strand. To test this we defined the GC split as the difference of GC skew downstream from and upstream of a genomic point. Under the hypothesis that the GC skew is produced by the different mutation processes in the leading and lagging strands, we expect that the GC split has a maximum coinciding with the ORI midpoint. However, we observed that ORI midpoints typically lay between a strong maximum of the GC split upstream of the ORI midpoint at approximately −0.3 kb, and a slightly less strong minimum at ∼0.4 kb (Fig. 3B; Supplemental Fig. S4). This result does not support the hypothesis that the different mutation processes at the leading and lagging strand are the sole cause of the GC skew.
ORIs associate with multiple chromatin signatures
The mechanisms responsible for ORI specification in multicellular eukaryotes remain unknown. Studies in animals and plants have revealed a preferential association of ORIs with activating chromatin marks (Sequeira-Mendes et al. 2009; Cayrou et al. 2011, 2015; Costas et al. 2011; Picard et al. 2014; Comoglio et al. 2015; Pourkarimi et al. 2016; Rodríguez-Martínez et al. 2017). A simplistic interpretation of these observations may suggest that some combination of chromatin features may be sufficient for ORI specification. However, simple inspection of genomic data clearly shows that there is not a single epigenetic mark or combination of them common to all ORIs. Recent studies demonstrate the existence of three major classes of ORIs with different organization, chromatin environment, and sequence motifs (Cayrou et al. 2015), suggesting that ORIs are associated with different signatures.
To investigate the preferences of ORIs to occur in particular chromatin settings, we assigned each ORI midpoint to one of the high-resolution chromatin states defined for the Arabidopsis genome (Sequeira-Mendes et al. 2014). These states simplify the combinatorial complexity of DNA and histone marks across the Arabidopsis genome into nine chromatin states characterized by unique signatures, as reported for Drosophila and human cells (Ernst et al. 2011; Kharchenko et al. 2011). The Arabidopsis chromatin states show a preferred linear sequence defining proximal promoters (state 2), TSS (state 1), 5′ end of genes (state 3), long genes (state 7), 3′ end of genes (state 6), followed by repressed states containing Polycomb marks (states 5 and 4), and two types of heterochromatin (states 8 and 9) (Sequeira-Mendes and Gutierrez 2016).
The cumulative weight of ORIs in the chromatin states is higher for ORIs colocalizing with state 1 (TSS), which are the most numerous (Fig. 4A; Supplemental Fig. S5A). State 2 (proximal promoters and 5′ UTRs) and state 3 (5′ end of genes) ORIs also have a relatively high weight (Fig. 4A; Supplemental Fig. S5A). On the contrary, state 5 (PcG-repressed regions) and states 8 and 9 (the two heterochromatin types), state 7 (long coding genes), state 6 (3′ end of genes), and in particular state 4 (distal regulatory intergenic regions) contain more moderate amounts of ORIs.
Figure 4.

Association of ORIs with chromatin states. (A) Normalized weight of ORIs belonging to the nine chromatin states in the combined experiments of 4-d-old (transparent color) and 10-d-old (solid color) seedlings. The results of the three independent experiments are shown in Supplemental Figure S5A. Black circles indicate the fractions of ORIs in each chromatin state, and broken lines indicate the genome coverage of each chromatin states (both using the same scale of the y-axis). (B) Same as A, showing the propensity (instead of the cumulative weight) for ORIs in the nine chromatin states depicted. This reveals ORI types according to the associated chromatin states that have larger NSS than expected by chance based on the fraction of genome that they represent. The results of the three independent experiments are shown in Supplemental Figure S5B.
Because not all states have the same frequency in the genome, we transformed the normalized weight into propensities dividing by the genome fraction covering the same state, so that positive values identify states with a NSS weight larger than expected based on its genome coverage. We confirmed that ORIs close to TSS (state 1) show the highest values, followed by ORIs in proximal promoters (state 2) (Fig. 4B; Supplemental Fig. S5B). Among the rest, ORIs in distal regulatory intergenic regions (state 4), long genes (state 7), and 3′ end of genes (state 6) showed a negative propensity (Fig. 4B; Supplemental Fig. S5B), whereas ORIs in heterochromatin (states 8 and 9) showed a negative propensity in 10-d-old but not in 4-d-old seedlings.
Factors associated with ORI specification in different chromatin landscapes
The presence of ORIs across all chromatin states demonstrates that ORI activity is not associated with a single chromatin signature. To characterize ORI features in a quantitative manner, we calculated five traits (NSS, CDC6, GC content, GC skew, and the number of GGN [N = A,T,C,G] trinucleotides) over a region of 300 bp around the ORI midpoint for each state and experimental data set. All of these traits correlate positively with the NSS, suggesting that they contribute to ORI strength. We then computed weighted averages of these traits (using the NSS as weight) over the ORI of a given state and transformed them into Z-scores, so that a positive value indicates that the trait in that state is larger than the average score over the whole genome.
We found that the GGN score profile across states is very similar to that of CDC6 and, to a lower extent, to those of GC and GC skew. Moreover, these profiles were consistently similar in all experimental situations tested (Fig. 5A; Supplemental Fig. S6). This is relevant because it might be argued that λ-exo has a lowered activity on G-rich secondary structures (Foulk et al. 2015). As discussed below, both the CDC6 binding data, obtained in ChIP experiments, and ORIs mapped in Arabidopsis cultured cells, also locally enriched in GC, were obtained using independent procedures that do not rely on the use of λ-exo (Costas et al. 2011). We can see that ORIs in different chromatin states have different average NSS values (Fig. 5A). Thus, the average NSS of ORIs in active chromatin states 1 and 3 (TSS and 3′ end of genes) is significantly lower than for ORIs in repressed heterochromatin (states 8 and 9) and Polycomb regions (state 5; the two-tailed t-test P < 0.0001) in all cases except for state 3 and state 9 (P < 0.0046 and P < 0.0002 in 10- and 4-d-old seedlings, respectively). ORIs in Polycomb chromatin were consistently high in all other traits, which can explain their high NSS. In contrast, ORIs in heterochromatin are not particularly high in the other traits. The GC content is lowest for ORIs in the distal regulatory intergenic regions (state 4), which is the most AT-rich. Nevertheless, the GGN score of ORIs in state 4 is comparable to that of other states. The GGN score is the feature that correlates with the NSS, together with the CDC6 score (correlation coefficient 0.50 and 0.61 for the NSS of 4- and 10-d-old samples, respectively) (Fig. 5B,C; Supplemental Table 3). Consistent with this, the number of GGN motifs within ±150 nt of ORI midpoint is, with some exceptions, an indication of ORI strength. Furthermore, GGN trinucleotides in ORIs tend to occur in clusters larger than randomly in the average genome (Fig. 5D; Supplemental Fig. S6).
Figure 5.

Relevance of several genomic variables for ORI specification. (A) The weighted averages of the Z-scores for several variables (NSS, CDC6, GC content, GC skew, and GGN trinucleotide) are shown for each chromatin state, normalized with the average property across the entire genome and weighted with the combined NSS. The results of the three independent experiments are shown in Supplemental Figure S6. NSS values were statistically significant when comparing states 1 and 3 with states 5, 8, and 9 (two-tailed t-test P < 0.0001) in all cases except for state 3 and state 9 (P < 0.0046 and P < 0.0002 in 10- and 4-d-old seedlings, respectively). (B) Heatmaps of the GGN signal (square root) within ±2 kb of the ORI midpoint (0). ORIs have been ranked according to the first principal component of the complete set of features, computed over all 2374 ORIs. (C) Examples of the DNA sequences within ±150 nt of the ORI midpoint highlighting the GGN motifs (orange). In these two ORIs, 76 and 10 GGN motifs were present. The complete list of ORI sequences is provided in Supplemental Table 3. (D) Distribution of ORIs (n = 2374) with different number of GGN motifs (orange; Mean = 33.8, SD = 17.9) compared with the same number of randomly chosen genomic regions (gray; Mean = 20.6, SD = 7.4). Two-tailed t-test, P > 0.0001.
Together our results support the following conclusions regarding the definition of different classes of ORIs depending on the chromatin states typical of their neighborhood:
ORIs located in genic regions (states 1, 3, 6, and 7), associated with active transcription and more open chromatin, possess low or intermediate NSS values, despite their large cumulative weight, suggesting a more variable usage of ORI sites. This is in part explained by the relatively lower values of CDC6 and GGN, and in part suggests possible interference between replication and transcription.
The contrary holds for ORIs in heterochromatin, with low accessibility for replication proteins, which tend to have a high NSS, despite their low cumulative weight. This suggests that once a region is specified as a potential ORI in a disfavored chromatin landscape, it is used more frequently in all cells of the population. This also applies to other poorly transcribed regions, for example, as Polycomb chromatin. It can be also that ORIs in heterochromatin are more consistent in the different cell types leading to a stronger signal. On the other hand, active chromatin regions likely vary across cell types and ORIs associated with them may be more variable, thus reducing the signal.
Differences in the average NSS of ORIs of different states may either stem from a global effect that affects all ORIs in the same way or indicate strong variability of the NSS. To investigate how the chromatin states influence the variability of ORIs, we measured the correlation coefficients of the NSS over the ORI sets of a given state (Supplemental Fig. S7). They were close to 1 in most states, except state 1 (TSS), state 8 (AT-rich heterochromatin), and state 4 (intergenic, Polycomb, and AT-rich), which are most variable.
Interplay between DNA replication origins and transcriptional programs
The relationship between ORI activity and transcriptional programs during development has been demonstrated (Nordman et al. 2011; Lubelsky et al. 2014; Comoglio et al. 2015; Müller and Nieduszynski 2017; Siefert et al. 2017). We observed that this is highly dependent on the ORI type according to the chromatin state where it is located, as visualized by plotting the quantity of transcripts in the different chromatin states identified by RNA-seq of 4- and 10-d-old seedlings. The first observation is that ORI locations in all states are more transcribed than genomic regions of the same chromatin state at both developmental stages, except GC-rich heterochromatin (state 9), supporting a strong relationship between DNA replication and transcription. Next, we compared the transcription scores of the two developmental stages and found that repressed or less transcribed regions (states 5, 4, 8, and 9) showed more transcripts through ORI sites in 4-d-old than in 10-d-old seedlings (Fig. 6A,B; Supplemental Fig. S8). This is particularly striking for ORIs in Polycomb chromatin (state 5) and to a lesser extent in the AT-rich heterochromatin (state 8), where a strong enhancement of transcription in 4-d-old seedlings was observed. The opposite happens for the active regions of long genes (state 7), which are more transcribed in 10-d-old seedlings. In other words, repressed regions in 4-d-old seedlings are more actively transcribed in 10-d-old seedlings, suggesting that there are differences in the accessibility of the different chromatin states, which may affect ORI activity.
Figure 6.

ORIs and transcriptional activity. (A) The average Z-score of the transcription score with respect to the average transcription score of the entire genome is shown for ORIs (left) and genomic locations (right) belonging to all chromatin states. (B) Heatmaps of the RNA score (square root) within ±2 kb of the ORI midpoint (0) computed over the set of all 2374 ORIs in 4-d-old and 10-d-old seedlings. ORIs have been ranked according to the first principal component of the complete set of features, computed over all 2374 ORIs.
ORI specification and usage during vegetative development
The systematic differences in transcriptional activity and chromatin organization observed between early (4-d-old) and late (10-d-old) vegetative stages support the idea that chromatin organization, ORI specification, and the transcriptional program change during vegetative development. To further investigate these differences, we identified ORIs that have a higher NSS value in 4-d-old seedlings than in 10-d-old seedlings and vice versa. We first analyzed the variation of ORI frequency in different chromatin states as a function of the threshold used to define the preferred ORIs in each developmental stage (Methods). To simplify the analysis, we grouped ORIs in three classes: genic chromatin (states 2, 1, 3, 7, and 6), Polycomb chromatin (states 5 and 4), and heterochromatin (states 8 and 9). We found that ORIs preferentially used in 4-d-old seedlings have a strong preference for being located in heterochromatin, whereas ORIs preferentially used in 10-d-old seedlings are located in genic states (Fig. 7A; Supplemental Fig. S9). Using the same threshold for both developmental times, led us to identify 71 ORIs preferentially used in 4-d-old seedlings and 18 ORIs preferentially used in 10-d-old seedlings (Supplemental Table 4).
Figure 7.
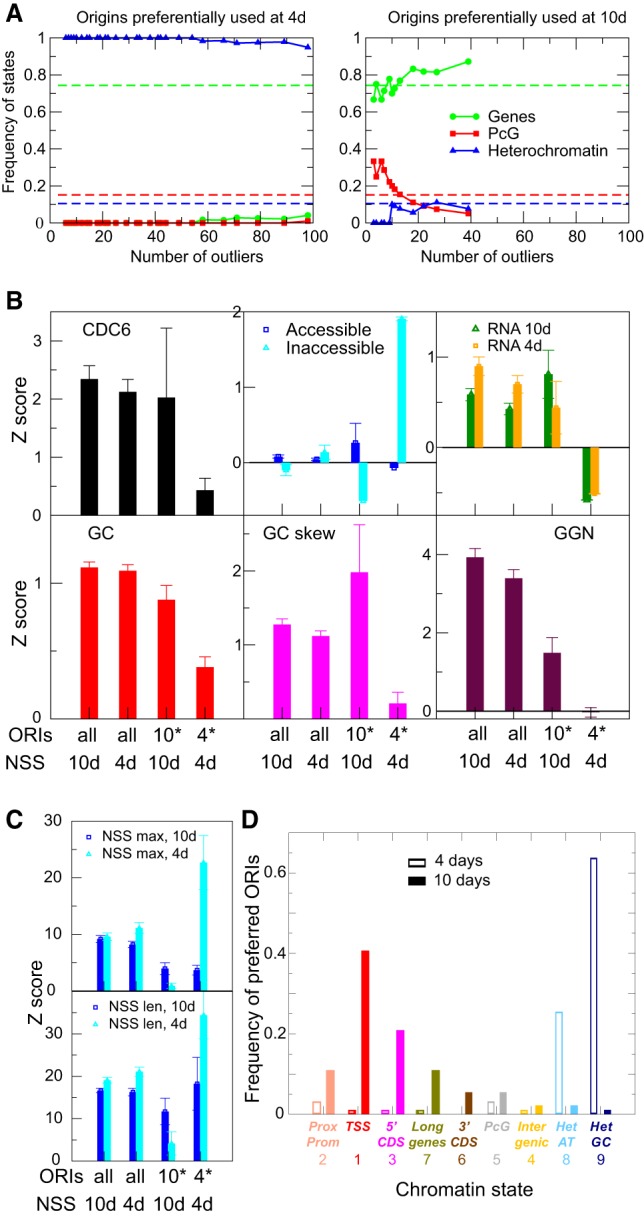
Properties of differentially activated ORIs during vegetative development. (A) Distribution into chromatin states of ORIs stronger in 4-d-old seedlings (left) or in 10-d-old seedlings (right), as a function of the number of ORIs obtained by varying the threshold. To simplify the analysis, ORIs have been grouped into those associated with genes (states 2, 1, 3, 7, and 6), with Polycomb chromatin (states 4 and 5), and with heterochromatin (states 8 and 9). Dotted lines represent the average values for the entire genome. (B) Weighted average of several genomic and epigenomic properties for the set of all ORIs and the ORIs preferred at 4- or 10-d-old seedlings, as indicated. For the set of all ORIs, we have used as weight the NSS of the two developmental stages. For the sets of differentially expressed ORIs, we have only used the stronger weight. Measurements were transformed to Z-score with respect to the whole genome, which produces positive values when they are higher than a generic genomic region. (C) Weighted average of the scores NSSmax (upper) and NSSlen (lower) measured at 10 d (dark blue) and 4 d (light blue) for the set of all ORIs weighted with the NSS of the two developmental stages and the set of differentially expressed ORIs weighted with the stronger NSS. Note that although ORIs stronger at 10 d have weak NSS score both at 4 and 10 d, ORIs stronger at 4 d have a score weak at 10 d but strong at 4 d. (D) Distribution into chromatin states at the reference threshold t = 0.9 of ORIs preferentially used at two developmental stages defined as in A colocalizing with different chromatin states in 4-d-old (empty bars) and 10-d-old seedlings (solid bars).
ORIs more active in 4-d-old seedlings possess lower CDC6, GC, GC skew, and GGN scores than average (Fig. 7B). The most distinctive feature of these ORIs is that they are located in nontranscribed regions, inaccessible to DNase I (Fig. 7B), consistent with their heterochromatic nature. ORIs more active in 10-d-old seedlings are slightly more accessible and transcribed than generic ORIs (Fig. 7B). These ORIs appear to be weaker than average, based on their GGN score (Fig. 7B). These features are visually compared for ORIs preferentially used in 4- and 10-d-old seedlings in the heatmaps shown in Supplemental Figure S11. We also show the heatmaps of the different scores at the ORI midpoint in the two sets of ORIs with indication of their genomic location in Supplemental Figure S12.
Based on ORI identification in animal cells in culture, developmentally regulated ORIs are not very efficient (Besnard et al. 2012). The quantitative parameter of ORI activity (NSS) showed that every genomic location associated with an ORI possesses a certain firing efficiency. In agreement with studies in animal cells in culture (Besnard et al. 2012; Comoglio et al. 2015), it seems that modulation of ORI activity rather than selection of different genomic locations determines ORI usage at different developmental stages and perhaps in different cell types. The differences between developmental stages may arise from origin activation or differential failures of replication forks to continue. To distinguish between these possibilities, we defined two NSS scores: the NSSmax score, which measures the maximum number of nascent DNA over all the 25-bp windows that cover the ORI, and the NSSlen score, which measures the number of windows for which the Z-score of the number of NS reads is larger than 1. The NSSmax score assesses the frequency of origins activation, and the NSSlen score assesses the resistance of ORI to failures. These scores are correlated (their correlation coefficient is r = 0.67 at 10 d and r = 0.72 at 4 d). In particular, although NSSmax is well conserved between 4 d and 10 d (r = 0.82), NSSlen is more variable between developmental stages (r = 0.66).
The NSS value of ORIs preferred at 10 d is lower than the average both at 10 and 4 d (Fig. 7C). In contrast, ORIs more active at 4 d have NSS values smaller than average at 10 d but much larger than average at 4 d (Fig. 7C). We also analyzed the correlations between developmental stages restricted to ORIs of a given chromatin state. We found that the NSSlen score is particularly variable for chromatin states 1 and 3, close to the TSS, suggesting that conflicts with transcription may increase the variability of the NS length. In contrast, the NSSmax value is more variable than the NSSlen for heterochromatin states 8 and 9 (Supplemental Fig. S10), suggesting that differences between developmental stages for these ORIs are mainly driven by changes in initiation events. ORIs preferentially activated in 4-d-old plants occur much more frequently in the two types of heterochromatin (Fig. 7D; Supplemental Table 4), mainly in pericentromeric regions and, among them, more skewed toward Gypsy elements (66.7%), in line with data obtained in Arabidopsis cultured cells (Vergara et al. 2017). In contrast, ORIs preferentially activated in 10-d-old plants colocalize more frequently with genic regions, typically in the 5′ end of genes, TSS, and proximal promoters (Fig. 7D). Because ORI activation depends on chromatin accessibility, our data suggest that ORI usage changes significantly in a locus-specific manner during post-embryonic development, likely according to changes in chromatin organization.
Discussion
Genomic features of Arabidopsis DNA replication origins
In this study, we generated a whole-body ORI map of A. thaliana plants at two different developmental stages. ORI activity was assessed quantitatively from the sequencing data by determining a nascent strand score (NSS) that measures the propensity of a certain genomic location to behave as an ORI. We identified 2374 ORIs genome-wide and showed that Arabidopsis ORIs are organized in discrete sites rather than in large initiation zones, in agreement with ORI mapping in cells with similar genome size (Comoglio et al. 2015; Lombraña et al. 2016; Pourkarimi et al. 2016; Rodríguez-Martínez et al. 2017). A comparison of ORI locations in cultured cells and seedlings revealed a coincidence of ∼14%–25%, depending on the threshold tolerance. This amount of ORIs common to both sources reveals the existence of technical and biological variables in ORI usage, for example, the presence of many different cell types in the seedling compared to the cell culture, that need to be identified in the future.
Most Arabidopsis ORIs in seedlings (∼78%) associate with genic elements, in particular the 5′ end of genes, reinforcing the strong preference of ORIs for genic regions demonstrated in metazoan cultured cells and embryos (Sequeira-Mendes et al. 2009; MacAlpine et al. 2010; Cayrou et al. 2015; Comoglio et al. 2015; Rodríguez-Martínez et al. 2017). Similar to the situation in cultured cells (Costas et al. 2011), Arabidopsis ORIs colocalize with transposable elements (TEs) less frequently than expected at random. However, as reported in cultured cells (Vergara et al. 2017), we found that ORIs in the gene-poor pericentromeric regions increase their tendency to colocalize with TEs, in particular with retrotransposons of the Gypsy and LINE families. Thus, we conclude that Arabidopsis ORIs have a high preference to associate with genes in euchromatin and with both genes and transposons in pericentromeric heterochromatin.
GGN clusters are a strong determinant of ORI strength
The local GC content is much higher than genome average in ORIs, a common feature of ORIs in animal and plant cells (MacAlpine et al. 2010; Cayrou et al. 2011, 2015; Costas et al. 2011; Besnard et al. 2012). The number of GGN trinucleotides within ±150 nt around the ORI midpoint correlates with the NSS value, although it explains <30% of the variance (r2 = 0.28). From a structural point of view, four consecutive GGN motifs may form G-rich secondary structures, such as G4 with two tetrads (Sen and Gilbert 1988; Chen and Yang 2012). Bioinformatics programs look for G4 motifs with at least three consecutive G (Todd et al. 2005). Thus, the overlap between origins and G4 predicted with this parameter is not particularly strong; for instance, using quadparser (Huppert and Balasubramanian 2005) with the standard maximum loop length of 7 bases, we found 1232 predicted G4 and only 53 of them overlap with our set of origins (2% of ORIs and 4% of G4s). Results were similar using the more permissive loop length of 15 bases. G4 with just two tetrads have been experimentally demonstrated in the thrombin binding aptamer d(G2T2G2TGTG2T2G2) (Macaya et al. 1993), the Bombyx mori telomeric sequence d(AG2T2AG2T2AG2T2AG2) (Saccà et al. 2005), and in a large-scale study of the human genome that identified more than 70,000 G4 with two tetrads, amounting to ∼8%–10% of all observed G4 (Chambers et al. 2015). Moreover, the stability of G4 is largest for loops of length 1, such as those in consecutive GGN motifs, and it has been observed that GjNGj sequence motifs form a robust parallel stranded structure motif with 1-nt loop (Chen and Yang 2012). Stabilizing interactions between distinct G4 structures have been observed (Palumbo et al. 2009), suggesting that there could be synergy between the large number of GGN motifs observed in Arabidopsis ORIs. Only 35 (1.4%) ORIs contain fewer than four GGN motifs, while the maximum frequency is between 8 and 16 GGN, finding up to 77 GGN out of the theoretical maximum of 100 in the 300-nt windows. Our finding that GGN motifs are enriched in ORIs is consistent with reports that the presence of G4 influences ORI activity in animal cells (Besnard et al. 2012; Cayrou et al. 2012, 2015; Valton et al. 2014). Moreover, the GGN-rich regions could well be part OGRE-like motifs, which have been identified in metazoans (Cayrou et al. 2015).
The enrichment of ORIs in GGN motifs might be related to a reduced efficiency of λ-exo to digest G-mediated secondary structures. However, we carried out λ-exo treatments under optimal enzyme/substrate conditions. In addition, consider that (1) we observed a strong correlation between occurrence of GGN motifs at ORIs and the CDC6 binding score, and (2) Arabidopsis ORIs were found to be locally enriched in GC in cultured cells using procedures that do not rely on λ-exo-treated samples (Costas et al. 2011). The enrichment in GGN stems from two genomic properties of Arabidopsis ORIs, the high GC content and the GC skew, which concur to produce one G-rich strand. As mentioned above, the GC skew associated with replication has been associated with the mutational asymmetry between the leading and lagging strand (Lobry 1996). However, we found that the asymmetry between G and C starts approximately 300 nt before the ORI midpoint (the local NSS maximum). Thus, replication asymmetry cannot be the sole cause of the GC skew at Arabidopsis ORIs, which may be likely generated by positive selection of GGN clusters. Moreover, many GGN clusters are tandems of quasi-repeats, so that a likely mutational mechanism is through trinucleotides insertions produced by the slippage of the polymerase. The three hypothesized mechanisms (replication asymmetry, insertion, and selection) may cooperate in the formation and maintenance of GGN clusters.
Other structural features of Arabidopsis DNA replication origins
Besides GGN clusters, an important feature associated with ORIs is chromatin accessibility because more accessible, transcriptionally active states, are more prone to be ORI locations. The second driving factor is the propensity to bind the pre-RC protein CDC6, with an affinity significantly larger in the repressed than in the active chromatin states, probably to compensate their reduced accessibility. Finally, despite both transcription and replication are affected by chromatin accessibility, we observed an overall negative correlation between the ORI strength and transcription, in particular, ORIs colocalizing with TSS are among the weakest ones. There are reports of a preferential location of ORIs in actively transcribed genes (Aladjem 2004; MacAlpine et al. 2004; Saha et al. 2004; Goren et al. 2008). However, in these cases ORIs are not in close proximity to the TSS. We hypothesize that the negative correlation may originate from possible interference between replication and transcription (Aguilera and García-Muse 2013).
Our data demonstrate the existence of different types of ORIs according to their features, primarily their chromatin landscape. There are multiple signatures that can accommodate ORIs, in agreement with observations in animal cells (Cayrou et al. 2015), although they show an overall preference for localizing in the TSS (state 1) and adjacent states with open chromatin. Long genes (state 7), the 3′ end of coding sequences (state 6), and distal regulatory intergenic regions (state 4) are significantly depleted of ORIs. ORIs located within PcG chromatin (state 5) as well as within heterochromatin (states 8 and 9) tend to be stronger than average. Thus, it is conceivable that finding an appropriate local ORI landscape within repressed and compact chromatin may favor that it is used in more cells of the population, leading to a higher NSS value.
Developmental preferences of DNA replication origin activation and the chromatin landscape
We found that ORI activity undergoes systematic changes in the course of development. ORIs that are stronger in 10-d-old seedlings are particularly frequent in the TSS (state 1) and adjacent chromatin regions (states 2 and 3). In contrast, ORIs preferentially used in 4-d-old seedlings locate in heterochromatin (state 9). These trends are consistent with our transcriptional analysis that revealed lower repression of typically repressed chromatin states in 4-d-old seedlings compared to 10-d-old seedlings, suggesting that chromatin organization may be different at these two stages of vegetative development. The increased frequency of ORIs in TEs, in particular of the Gypsy family, in 4-d-old seedlings suggests that the role of these TEs as ORIs could be important at early developmental stages where the dynamics of cytosine methylation suggests differences in the repression level of heterochromatin (Bouyer et al. 2017). This is a major difference with recent studies in C. elegans, where early pregastrula embryos are depleted of ORIs in heterochromatin, whereas ORIs have a preference for noncoding regions and enhancers in post-gastrula embryos (Rodríguez-Martínez et al. 2017). These differences between animal and plants suggest different mechanisms of coupling ORI activity, developmental programs, and heterochromatin dynamics. It is also worth noting that the spatial organization of the Arabidopsis genome reveals that typical TADs and distal enhancers as in animals are lacking, or very infrequent (Wang et al. 2015; Liu et al. 2016; Vergara and Gutierrez 2017).
Polycomb complexes regulate gene expression associated with developmental phase transitions in Arabidopsis (Kuwabara and Gruissem 2014). ORIs associated with Polycomb chromatin (state 5) are underrepresented but they behave as strong ORIs, suggesting that once a genomic site is chosen as an ORI, this location tends to be used in many cells. This is in agreement with the finding that Polycomb factors associate with efficiently used ORIs in mammalian cells (Cayrou et al. 2011, 2015; Picard et al. 2014). Developmentally regulated genes in animal pluripotent stem cells share H3K4me3 and H3K27me3 marks, typical of bivalent chromatin (Bernstein et al. 2006). A possibility is that some of the H3K27me3 regions colocalizing with ORIs contain bivalent chromatin, although this has not been experimentally demonstrated. Bivalent chromatin in Arabidopsis seedlings has been identified in regulatory regions (states 2 and 4) using sequential re-ChIP experiments (Sequeira-Mendes et al. 2014). We found that H3K27me3 (and H3K4me3) is enriched in ORIs located in proximal promoters (state 2) but not in distal regulatory regions (state 4).
Our genome-wide results have defined the main DNA and chromatin properties associated with different ORI classes in a living organism and at two stages of post-embryonic growth. These properties show that ORI activity is compatible with a variety of signatures and demonstrate the existence of various classes of ORIs defined by their strength, DNA features, and chromatin landscape. The feasibility to study ORI activity in a developmentally and genetically tractable organism opens new avenues to determine how ORI activity is regulated in response to developmental cues, in association with transcriptional programs, in response to environmental challenges, and in a variety of mutant backgrounds.
Conclusions
Most general features of ORIs appear to be shared by multicellular organisms as evolutionary distant as plants, insects, worms, and mammals. These include their preferential association with genic regions, with open and transcribed chromatin, and local GC-richness. Our study also revealed that although mammalian ORIs colocalize with G-rich strands potentially able to form G4 structures (Cayrou et al. 2015), canonical G4-forming sequences do not occur at Arabidopsis (this work) or C. elegans ORIs (Rodríguez-Martínez et al. 2017). Instead, we found that Arabidopsis ORIs are enriched in GGN trinucleotides, which might also be a feature of many G-rich sequences of mammalian ORIs. Additionally, we identified ORIs along all chromatin states, indicating that ORI function is compatible with multiple chromatin signatures. Furthermore, beyond the DNA and chromatin features, Arabidopsis ORIs are located more frequently in heterochromatin at very early stages of vegetative development, perhaps as a consequence of not being fully assembled and related to the post-embryonic nature of plant organogenesis. This is in contrast with the largely invariant nature of ORIs through metazoan embryogenesis (Pourkarimi et al. 2016; Rodríguez-Martínez et al. 2017).
Methods
Plant growth
Arabidopsis thaliana seeds (Col-0 ecotype) were stratified for 48 h and grown in Murashige and Skoog (MS) medium supplemented with 1% (w/v) sucrose and 1% (w/v) agar in a 16-h:8-h light/dark regime at 22°C, for either 4 or 10 d.
Purification of short nascent strands (SNS)
Total genomic DNA and SNS preparations were obtained under RNase-free conditions, by an optimization of the protocol described (for full details, see Supplemental Methods; Sequeira-Mendes et al. 2009). Nuclei were isolated from 4 or 10 d post-sowing (dps) Arabidopsis seedlings as described (Chodavarapu et al. 2010) to reduce contamination with polyphenols and other secondary metabolites (for detailed protocol of SNS purification, see Supplemental Methods). Twelve grams of whole seedlings were collected, frozen, ground in liquid nitrogen, and resuspended in 10 mL per gram of Honda Buffer Modified for 30 min in a rotary shaker at 4°C (HBM; 2% [p/v] PVP10 [Sigma-Aldrich], 25 mM Tris-HCl, pH 7.6, 440 mM sucrose [Merck], 10 mM magnesium chloride, 0.1% Triton X-100, 10 mM β-mercaptoethanol) and the nuclei centrifuged 10 min at 3000g and 4°C. The nuclear pellet was resuspended in 5 mL per gram of Nuclei Isolation Buffer (NIB; 2% [p/v] PVP10 [Sigma-Aldrich], 20 mM Tris-HCl, pH 7.6, 250 mM sucrose [Merck], 5 mM magnesium chloride, 5 mM potassium chloride, 0.1% Triton X-100, 10 mM β-mercaptoethanol), loaded onto a 15/50% gradient of Percoll in NIB and centrifuged 20 min at 50g and 4°C with slow brake, centrifuged 5 min at 1100g and 4°C, washed twice with 10 mL of NIB and 4°C, and resuspended in 20 mL of lysis buffer per 12 g of starting material (0.5% [p/v] PVP10, 50 mM Tris-HCl, pH 8.0, 10 mM EDTA pH 8.0, 1% SDS, 10 mM β-mercaptoethanol) by agitation 15 min at 4°C. Proteins were digested with Proteinase K (100 µg/mL) and DNA extracted twice with phenol, pH 8.0, and with phenol:chloroform:IAA. DNA was precipitated, washed twice with 70% ethanol, air dried, and resuspended in 1 mL of TE (10 mM Tris-HCl, pH 8.0, 1 mM EDTA) containing 160 units of RNaseOUT (Invitrogen). DNA was incubated at 4°C overnight without pipetting or vortexing.
Purified DNA was denatured and size-fractionated in a neutral 5%–10% sucrose gradient. Fractions (1 mL) were collected from the top and the DNA was ethanol-precipitated. Normally, fractions 3 (∼100–600 nt), 4 (∼300–800 nt), and 5 + 6 + 7 (∼500–3000 nt) were processed further by treating with polynucleotide kinase (0.67 units/µL; PNK, Fermentas) to phosphorylate 5′-hydroxyl ends in the presence of 1.34 mM dATP for 30 min at 37°C and then digested with λ-exonuclease (Gerbi and Bielinsky 1997; Costas et al. 2011; Cayrou et al. 2015; Comoglio et al. 2015). The λ-exonuclease digestion was carried out with 5 units/µL of enzyme (Thermo Fisher Scientific) following the manufacturer's instructions at 37°C overnight. The efficiency of the digestion was monitored by adding 40 ng of phosphorylated linearized plasmid to an aliquot of each reaction tube. The phosphorylation and λ-exonuclease treatments were repeated at least twice. RNA was digested with 0.05 µg/mL RNase A (Roche) and 0.16 units/µL RNase I (Thermo Fisher Scientific) for 30 min at 37°C. RNases were digested with Proteinase K. The ssDNA of purified SNS was converted into dsDNA: First, SNS and 2 pmol random hexamer primers (Roche) were denatured together 5 min at 100°C, then a slow annealing was achieved by cooling down the samples from 80°C to room temperature; second, the dsDNA was synthesized by using 0.17 units/µL of Klenow fragment for 1 h at 37°C; third, the fragments were ligated with 2 units/µL of Taq DNA Ligase (New England BioLabs) for 45 min at 45°C; finally, dsDNA was extracted, precipitated, resuspended in Milli-Q water, and quantified before library preparation. The same method of dsDNA conversion was applied to sheared and denatured genomic DNA to be used as sequencing control.
RNA purification
Total RNA from 4 dps seedlings was isolated using TRIzol (Invitrogen) according to the manufacturer's instructions. Total RNA was treated with DNase I (Roche) before proceeding with library preparation.
Next-generation sequencing
DNA libraries of both SNS DNA (sucrose gradient fractions 3, 4, and 5 + 6 + 7 combined) and genomic DNAs were first sheared by an S2 focused-ultrasonicator (Covaris) for 2 min (Intensity 5, Duty Cycle 10%, Cycles per Burst 200), and then used as inputs to generate sequencing libraries by Ovation Ultralow V1 library prep kits (NuGen). The libraries were subjected to deep sequencing on HiSeq 2000 per manufacturer instructions (Illumina). In two of the three experiments, we used different amplification protocols for library generation with the purpose of estimating possible bias introduced by this crucial but unavoidable step. RNA-seq libraries were made by TruSeq Stranded mRNA library prep kit and NeoPrep (Illumina), and subjected to deep sequencing on HiSeq 2000 per manufacturer instructions (Illumina). Single-end sequenced reads (51 nt) were aligned to the reference Arabidopsis genome (TAIR10), using the Bowtie alignment tool (Langmead et al. 2009), allowing up to one mismatch and discarding multihit reads. PCR duplicate reads were removed using an in-house script (for full details, see Supplemental Methods).
Peak-calling
For each sample and each fraction, we called ORIs with our own peak-calling algorithm ZPeaks (U Bastolla, R Peiró, J Sequeira-Mendes, et al., in prep.) that can be accessed at https://github.com/ugobas/Zpeaks. ZPeaks (1) provides a well-defined, genome-wide profile of nascent strand score (NSS), instrumental for weighting candidate ORIs and generic genomic locations, and (2) localizes an ORI at the local maximum of the NSS over the ORI box called, needed for centering the metaplots. We tested ZPeaks by visual inspection of the overlap between experiment and control reads and candidate ORIs as well as by the statistical analysis of the ORIs properties. Furthermore, our procedure was robust with respect to false positive ORIs because (1) it requires that each ORI is detected in several independent experiments, and (2) it weights each ORI with its NSS, so that spurious ORIs have low NSS and contribute little to the average properties.
Thus, ZPeaks computes optimally smoothed profiles of the reads of the experiment and the control, obtains from them a normalized smoothed profile, calls peaks when the profile is above a user-specified threshold, and sets the ORI location at the maximum of the normalized profile (for a detailed description, see Supplemental Methods). More in detail, the algorithm works as follows, once the sequencing reads have been aligned to the reference Arabidopsis TAIR10 genome: (1) The WIG files (normalized read counts) are input to ZPeaks and the number of reads is rescaled so that its mean number over each chromosome is the same both for the experiment e and the control c; (2) the profiles of the rescaled experiment and control are smoothed; (3) differences between the smoothed experiment and control are obtained as a Z-score; (4) for a chosen threshold T, the program counts the number of bins with zi > T, N(T); (5) we then joined together consecutive bins with a nascent strand score NSSei > T separated by less than 200 nucleotides, obtaining boxes that represent candidate origins; and (6) finally, the putative ORI is set at the bin where zi is maximum within the box, and the limits of the box are reduced in such a way that the ORI is at the center and the new box is contained into the original one. One may expect that the threshold parameter T may be objectively determined by clustering all genomic bins in two clusters through some clustering algorithm such as k-means, Expectation Maximization (that assumes that the scores zi are distributed according to a Gaussian distribution), or Hidden Markov Models (that also exploits the positional order of the bins along the chromosome). We followed such strategies, but the thresholds that we obtained were low, a sizable fraction of the genome satisfied zi > T, and visual inspection showed that most candidate ORIs were not reliable. Thus, we had no better choice than selecting an arbitrary threshold T and determining bona fide ORIs by combining different experiments, as explained below.
Combining peaks into consensus boxes (potential ORIs)
Our strategy consisted of determining a robust set of ORIs detected in at least two independent experiments and two fractions for each experiment and weighting each candidate ORI with the NSS value of each experiment in such a way that the results are scarcely dependent on false positives with low score. We analyzed two developmental stages (4- and 10-d-old seedlings) and three experiments for each stage (exp1, exp2, exp3), obtaining six different samples. For each of them, either two (F3 and F4) or three (F3, F4, F5 + 6 + 7) consecutive fractions of the sucrose gradients for size selection of nascent strands were sequenced. We called candidate ORIs with a tolerant threshold (z > 1.8), and for each sample we selected candidate regions, or boxes, that were identified in at least two fractions of the same gradient. Boxes with size smaller than 200 bp were eliminated, and boxes closer than 200 bp were joined. In this way, we obtained six data sets of high quality ORIs, with the following numbers: 842 (4d_exp1), 1938 (4d_exp2), 3008 (4d_exp3), 3298 (10d_exp1), 1686 (10d_exp2), 3107 (10d_exp3).
To increase the reliability of candidate ORIs, we selected only those boxes that had been found in at least two of six independent samples, obtaining a total of 2374 highly reliable candidate ORIs. We matched the boxes with nonvanishing overlap, and if an ORI had multiple overlaps, we selected the largest overlap. The center of the combined box was computed as the weighted average of the location with maximum score present in the associated boxes, weighting more the boxes with high NSS and small size. When we matched different fractions, the fraction F5 + 6 + 7, which contains larger nascent strands, was used to confirm boxes but not to locate their center, in order to obtain better resolution. The limits of the combined box were set in such a way that all of the bins are above the threshold in all fractions.
Scoring ORIs in different samples
For each ORI, we obtained their score NSSek in the six samples, where e labels the experiment, k labels the ORI, and NSSek is the maximum value of the score over all bins included in the box that contains the ORI. For each sample, we used the corresponding scores as weights, and we obtained the average values and the metaplots of genomic and epigenetic marks as the weighted average over the set of ORIs. We also generated combined scores by averaging the scores of all 4-d-old and all 10-d-old seedling samples.
Quantitative real-time PCR (qPCR)
The qPCR analysis was performed using GoTaq Master Mix (Promega) according to the manufacturer's instructions in an ABI Prism 7900HT apparatus (Applied Biosystems). For each region, a subset of unique and specific primers (Supplemental Table 2) were designed. The quantification was determined using a standard curve (five serial fourfold dilutions of gDNA), and SNS enrichment was normalized against a region lacking ORIs (negative control).
Detection of preferentially activated ORIs
Despite that most strong ORIs in one sample are strong also in the others, we identified a reduced number of ORIs whose strength is significantly different from one sample to the other. For this purpose, we considered the combined NSS of the two developmental stages (4- and 10-d-old seedlings) and rescaled them in such a way that the average value over the set of origins was the same for both samples. For each ORI k, we computed the mean and standard deviation of the rescaled NSS ek across the two developmental stages. We assumed that mean and standard deviation are related through the power law relation (SD) = a|mean|α, and fitted the exponent α = 0.79. We then adopted the difference score Diff = (SD)/a|mean|α, that is, the standard deviation divided by its expected value based on the fit, and we considered ORIs to be differentially expressed if Diff is larger than a threshold. Finally, we studied the distribution of these outliers across chromatin states as a function of the threshold. The analysis presented in the main text was obtained by combining the NSS scores measured in the three independent experiments. To confirm that the same qualitative results are obtained in all experiments separately, we present the same analysis performed with the NSS scores of the three experiments treated separately in Supplemental Figure S9.
Analysis of ORIs in heterochromatin and TE families was carried out as described in Vergara et al. (2017).
Data access
The SNS-seq data sets generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE109668.
Supplementary Material
Acknowledgments
We thank E. Martinez-Salas and M. Gomez for discussions and comments during this project and the critical reading of the manuscript, C. Becker and N. Schandry for useful suggestions, members of our laboratories for continuous feedback, and V. Mora-Gil for technical assistance. We thank S. Jacobsen, C. Hale, S. Feng, and the BSCRC BioSequencing Core for Illumina DNA Sequencing and discussions. This research was supported by Secretaría de Estado de Investigación, Desarrollo e Innovación, grants BFU2012-34821, BFU2013-50098-EXP, and BFU2015-68396-R to C.G., BIO2016-79043-P to U.B. and AGL2013-43244-R to J.M.C., as well as by institutional grants from Fundacion Ramon Areces and Banco de Santander to the CBMSO.
Author contributions: The work was conceived by C.G., J.S.-M., and U.B. J.S.-M. and Z.V. implemented protocols for purification and analysis of nascent strands, with the help of C.C. and I.A. in the initial steps of the work. U.B. developed the computational and statistical analysis, with the initial help of R.M.-G. J.S.-M., Z.V., U.B., and R.P. generated and analyzed data. J.M. and J.M.C. analyzed ORIs in heterochromatin. C.G. and U.B. wrote the manuscript with the input of all authors.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.240986.118.
References
- Aguilera A, García-Muse T. 2013. Causes of genome instability. Annu Rev Genet 47: 1–32. 10.1146/annurev-genet-111212-133232 [DOI] [PubMed] [Google Scholar]
- Aladjem MI. 2004. The mammalian β globin origin of DNA replication. Front Biosci 9: 2540–2547. 10.2741/1415 [DOI] [PubMed] [Google Scholar]
- Arakawa K, Tomita M. 2012. Measures of compositional strand bias related to replication machinery and its applications. Curr Genomics 13: 4–15. 10.2174/138920212799034749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. 2006. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125: 315–326. 10.1016/j.cell.2006.02.041 [DOI] [PubMed] [Google Scholar]
- Besnard E, Babled A, Lapasset L, Milhavet O, Parrinello H, Dantec C, Marin JM, Lemaitre JM. 2012. Unraveling cell type–specific and reprogrammable human replication origin signatures associated with G-quadruplex consensus motifs. Nat Struct Mol Biol 19: 837–844. 10.1038/nsmb.2339 [DOI] [PubMed] [Google Scholar]
- Bouyer D, Kramdi A, Kassam M, Heese M, Schnittger A, Roudier F, Colot V. 2017. DNA methylation dynamics during early plant life. Genome Biol 18: 179 10.1186/s13059-017-1313-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo Bosch P, Segura-Bayona S, Koole W, van Heteren JT, Dewar JM, Tijsterman M, Knipscheer P. 2014. FANCJ promotes DNA synthesis through G-quadruplex structures. EMBO J 33: 2521–2533. 10.15252/embj.201488663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayrou C, Coulombe P, Vigneron A, Stanojcic S, Ganier O, Peiffer I, Rivals E, Puy A, Laurent-Chabalier S, Desprat R, et al. 2011. Genome-scale analysis of metazoan replication origins reveals their organization in specific but flexible sites defined by conserved features. Genome Res 21: 1438–1449. 10.1101/gr.121830.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayrou C, Coulombe P, Puy A, Rialle S, Kaplan N, Segal E, Méchali M. 2012. New insights into replication origin characteristics in metazoans. Cell Cycle 11: 658–667. 10.4161/cc.11.4.19097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayrou C, Ballester B, Peiffer I, Fenouil R, Coulombe P, Andrau JC, van Helden J, Méchali M. 2015. The chromatin environment shapes DNA replication origin organization and defines origin classes. Genome Res 25: 1873–1885. 10.1101/gr.192799.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers VS, Marsico G, Boutell JM, Di Antonio M, Smith GP, Balasubramanian S. 2015. High-throughput sequencing of DNA G-quadruplex structures in the human genome. Nat Biotechnol 33: 877–881 10.1038/nbt.3295 [DOI] [PubMed] [Google Scholar]
- Chen Y, Yang D. 2012. Sequence, stability, structure of G-quadruplexes and their drug interactions. Curr Protoc Nucleic Acid Chem Chapter 17: Unit17.5 10.1002/0471142700.nc1705s50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chodavarapu RK, Feng S, Bernatavichute YV, Chen PY, Stroud H, Yu Y, Hetzel JA, Kuo F, Kim J, Cokus SJ, et al. 2010. Relationship between nucleosome positioning and DNA methylation. Nature 466: 388–392. 10.1038/nature09147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comoglio F, Schlumpf T, Schmid V, Rohs R, Beisel C, Paro R. 2015. High-resolution profiling of Drosophila replication start sites reveals a DNA shape and chromatin signature of metazoan origins. Cell Rep 11: 821–834. 10.1016/j.celrep.2015.03.070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costas C, de la Paz Sanchez M, Stroud H, Yu Y, Oliveros JC, Feng S, Benguria A, López-Vidriero I, Zhang X, Solano R, et al. 2011. Genome-wide mapping of Arabidopsis thaliana origins of DNA replication and their associated epigenetic marks. Nat Struct Mol Biol 18: 395–400. 10.1038/nsmb.1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. 2011. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473: 43–49. 10.1038/nature09906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foulk MS, Urban JM, Casella C, Gerbi SA. 2015. Characterizing and controlling intrinsic biases of λ exonuclease in nascent strand sequencing reveals phasing between nucleosomes and G-quadruplex motifs around a subset of human replication origins. Genome Res 25: 725–735. 10.1101/gr.183848.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerbi SA, Bielinsky AK. 1997. Replication initiation point mapping. Methods 13: 271–280. 10.1006/meth.1997.0526 [DOI] [PubMed] [Google Scholar]
- Goren A, Tabib A, Hecht M, Cedar H. 2008. DNA replication timing of the human β-globin domain is controlled by histone modification at the origin. Genes Dev 22: 1319–1324. 10.1101/gad.468308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez C. 2005. Coupling cell proliferation and development in plants. Nat Cell Biol 7: 535–541. 10.1038/ncb0605-535 [DOI] [PubMed] [Google Scholar]
- Gutierrez C, Desvoyes B, Vergara Z, Otero S, Sequeira-Mendes J. 2016. Links of genome replication, transcriptional silencing and chromatin dynamics. Curr Opin Plant Biol 34: 92–99. 10.1016/j.pbi.2016.10.005 [DOI] [PubMed] [Google Scholar]
- Huppert JL, Balasubramanian S. 2005. Prevalence of quadruplexes in the human genome. Nucleic Acids Res 33: 2908–2916. 10.1093/nar/gki609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnani N, Taylor CM, Malhotra A, Dutta A. 2010. Genomic study of replication initiation in human chromosomes reveals the influence of transcription regulation and chromatin structure on origin selection. Mol Biol Cell 21: 393–404. 10.1091/mbc.e09-08-0707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko PV, Alekseyenko AA, Schwartz YB, Minoda A, Riddle NC, Ernst J, Sabo PJ, Larschan E, Gorchakov AA, Gu T, et al. 2011. Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nature 471: 480–485. 10.1038/nature09725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwabara A, Gruissem W. 2014. Arabidopsis Retinoblastoma-related and Polycomb group proteins: cooperation during plant cell differentiation and development. J Exp Bot 65: 2667–2676. 10.1093/jxb/eru069 [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard AC, Méchali M. 2013. DNA replication origins. Cold Spring Harb Perspect Biol 5: a010116 10.1101/cshperspect.a010116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Wang C, Wang G, Becker C, Zaidem M, Weigel D. 2016. Genome-wide analysis of chromatin packing in Arabidopsis thaliana at single-gene resolution. Genome Res 26: 1057–1068. 10.1101/gr.204032.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobry JR. 1996. Asymmetric substitution patterns in the two DNA strands of bacteria. Mol Biol Evol 13: 660–665. 10.1093/oxfordjournals.molbev.a025626 [DOI] [PubMed] [Google Scholar]
- Lombraña R, Almeida R, Revuelta I, Madeira S, Herranz G, Saiz N, Bastolla U, Gómez M. 2013. High-resolution analysis of DNA synthesis start sites and nucleosome architecture at efficient mammalian replication origins. EMBO J 32: 2631–2644. 10.1038/emboj.2013.195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombraña R, Álvarez A, Fernández-Justel JM, Almeida R, Poza-Carrión C, Gomes F, Calzada A, Requena JM, Gómez M. 2016. Transcriptionally driven DNA replication program of the human parasite Leishmania major. Cell Rep 16: 1774–1786. 10.1016/j.celrep.2016.07.007 [DOI] [PubMed] [Google Scholar]
- Lubelsky Y, Prinz JA, DeNapoli L, Li Y, Belsky JA, MacAlpine DM. 2014. DNA replication and transcription programs respond to the same chromatin cues. Genome Res 24: 1102–1114. 10.1101/gr.160010.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAlpine DM, Rodriguez HK, Bell SP. 2004. Coordination of replication and transcription along a Drosophila chromosome. Genes Dev 18: 3094–3105. 10.1101/gad.1246404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAlpine HK, Gordan R, Powell SK, Hartemink AJ, MacAlpine DM. 2010. Drosophila ORC localizes to open chromatin and marks sites of cohesin complex loading. Genome Res 20: 201–211. 10.1101/gr.097873.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macaya RF, Schultze P, Smith FW, Roe JA, Feigon J. 1993. Thrombin-binding DNA aptamer forms a unimolecular quadruplex structure in solution. Proc Natl Acad Sci 90: 3745–3749. 10.1073/pnas.90.8.3745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méchali M. 2010. Eukaryotic DNA replication origins: many choices for appropriate answers. Nat Rev 11: 728–738. 10.1038/nrm2976 [DOI] [PubMed] [Google Scholar]
- Méchali M, Yoshida K, Coulombe P, Pasero P. 2013. Genetic and epigenetic determinants of DNA replication origins, position and activation. Curr Opin Genet Dev 23: 124–131. 10.1016/j.gde.2013.02.010 [DOI] [PubMed] [Google Scholar]
- Mesner LD, Valsakumar V, Cieślik M, Pickin R, Hamlin JL, Bekiranov S. 2013. Bubble-seq analysis of the human genome reveals distinct chromatin-mediated mechanisms for regulating early- and late-firing origins. Genome Res 23: 1774–1788. 10.1101/gr.155218.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller CA, Nieduszynski CA. 2017. DNA replication timing influences gene expression level. J Cell Biol 216: 1907–1914. 10.1083/jcb.201701061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordman J, Li S, Eng T, MacAlpine D, Orr-Weaver TL. 2011. Developmental control of the DNA replication and transcription programs. Genome Res 21: 175–181. 10.1101/gr.114611.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palumbo SL, Ebbinghaus SW, Hurley LH. 2009. Formation of a unique end-to-end stacked pair of G-quadruplexes in the hTERT core promoter with implications for inhibition of telomerase by G-quadruplex-interactive ligands. J Am Chem Soc 131: 10878–10891. 10.1021/ja902281d [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard F, Cadoret JC, Audit B, Arneodo A, Alberti A, Battail C, Duret L, Prioleau MN. 2014. The spatiotemporal program of DNA replication is associated with specific combinations of chromatin marks in human cells. PLoS Genet 10: e1004282 10.1371/journal.pgen.1004282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pourkarimi E, Bellush JM, Whitehouse I. 2016. Spatiotemporal coupling and decoupling of gene transcription with DNA replication origins during embryogenesis in C. elegans. eLife 5: e21728 10.7554/eLife.21728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Martínez M, Pinzón N, Ghommidh C, Beyne E, Seitz H, Cayrou C, Méchali M. 2017. The gastrula transition reorganizes replication-origin selection in Caenorhabditis elegans. Nat Struct Mol Biol 24: 290–299. 10.1038/nsmb.3363 [DOI] [PubMed] [Google Scholar]
- Saccà B, Lacroix L, Mergny JL. 2005. The effect of chemical modifications on the thermal stability of different G-quadruplex-forming oligonucleotides. Nucleic Acids Res 33: 1182–1192. 10.1093/nar/gki257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S, Shan Y, Mesner LD, Hamlin JL. 2004. The promoter of the Chinese hamster ovary dihydrofolate reductase gene regulates the activity of the local origin and helps define its boundaries. Genes Dev 18: 397–410. 10.1101/gad.1171404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez MP, Costas C, Sequeira-Mendes J, Gutierrez C. 2012. Regulating DNA replication in plants. Cold Spring Harb Perspect Biol 4: a010140 10.1101/cshperspect.a010140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen D, Gilbert W. 1988. Formation of parallel four-stranded complexes by guanine-rich motifs in DNA and its implications for meiosis. Nature 334: 364–366. 10.1038/334364a0 [DOI] [PubMed] [Google Scholar]
- Sequeira-Mendes J, Gutierrez C. 2016. Genome architecture: from linear organisation of chromatin to the 3D assembly in the nucleus. Chromosoma 125: 455–469. 10.1007/s00412-015-0538-5 [DOI] [PubMed] [Google Scholar]
- Sequeira-Mendes J, Díaz-Uriarte R, Apedaile A, Huntley D, Brockdorff N, Gómez M. 2009. Transcription initiation activity sets replication origin efficiency in mammalian cells. PLoS Genet 5: e1000446 10.1371/journal.pgen.1000446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sequeira-Mendes J, Araguez I, Peiro R, Mendez-Giraldez R, Zhang X, Jacobsen SE, Bastolla U, Gutierrez C. 2014. The functional topography of the Arabidopsis genome is organized in a reduced number of linear motifs of chromatin states. Plant Cell 26: 2351–2366. 10.1105/tpc.114.124578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sher N, Bell GW, Li S, Nordman J, Eng T, Eaton ML, MacAlpine DM, Orr-Weaver TL. 2012. Developmental control of gene copy number by repression of replication initiation and fork progression. Genome Res 22: 64–75. 10.1101/gr.126003.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siefert JC, Georgescu C, Wren JD, Koren A, Sansam CL. 2017. DNA replication timing during development anticipates transcriptional programs and parallels enhancer activation. Genome Res 27: 1406–1416. 10.1101/gr.218602.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroud H, Otero S, Desvoyes B, Ramirez-Parra E, Jacobsen SE, Gutierrez C. 2012. Genome-wide analysis of histone H3.1 and H3.3 variants in Arabidopsis thaliana. Proc Natl Acad Sci 109: 5370–5375. 10.1073/pnas.1203145109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd AK, Johnston M, Neidle S. 2005. Highly prevalent putative quadruplex sequence motifs in human DNA. Nucleic Acids Res 33: 2901–2907. 10.1093/nar/gki553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valton AL, Hassan-Zadeh V, Lema I, Boggetto N, Alberti P, Saintome C, Riou JF, Prioleau MN. 2014. G4 motifs affect origin positioning and efficiency in two vertebrate replicators. EMBO J 33: 732–746. 10.1002/embj.201387506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergara Z, Gutierrez C. 2017. Emerging roles of chromatin in the maintenance of genome organization and function in plants. Genome Biol 18: 96 10.1186/s13059-017-1236-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergara Z, Sequeira-Mendes J, Morata J, Peiró R, Henaff E, Costas C, Casacuberta JM, Gutierrez C. 2017. Retrotransposons are specified as DNA replication origins in the gene-poor regions of Arabidopsis heterochromatin. Nucleic Acids Res 45: 8358–8368. 10.1093/nar/gkx524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Liu C, Roqueiro D, Grimm D, Schwab R, Becker C, Lanz C, Weigel D. 2015. Genome-wide analysis of local chromatin packing in Arabidopsis thaliana. Genome Res 25: 246–256. 10.1101/gr.170332.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X. 2012. DNA replication and strand asymmetry in prokaryotic and mitochondrial genomes. Curr Genomics 13: 16–27. 10.2174/138920212799034776 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.