

Abstract
Fossil evidence indicates that the globally distributed brown rat (Rattus norvegicus) originated in northern China and Mongolia. Historical records report the human-mediated invasion of rats into Europe in the 1500s, followed by global spread because of European imperialist activity during the 1600s–1800s. We analyzed 14 genomes representing seven previously identified evolutionary clusters, and tested alternative demographic models to infer patterns of range expansion, divergence times, and changes in effective population (Ne) size for this globally important pest species. We observed three range expansions from the ancestral population that produced the Pacific (diverged ∼16.1 kya), eastern China (∼17.5 kya), and Southeast (SE) Asia (∼0.86 kya) lineages. Our model shows a rapid range expansion from SE Asia into the Middle East and then continued expansion into central Europe 788 yr ago (1227 AD). We observed declining Ne within all brown rat lineages from 150–1 kya, reflecting population contractions during glacial cycles. Ne increased since 1 kya in Asian and European, but not in Pacific, evolutionary clusters. Our results support the hypothesis that northern Asia was the ancestral range for brown rats. We suggest that southward human migration across China between the 800s–1550s AD resulted in the introduction of rats to SE Asia, from which they rapidly expanded via existing maritime trade routes. Finally, we discovered that North America was colonized separately on both the Atlantic and Pacific seaboards, by evolutionary clusters of vastly different ages and genomic diversity levels. Our results should stimulate discussions among historians and zooarcheologists regarding the relationship between humans and rats.
The genus Rattus originated and diversified in eastern and central Asia, and fossil evidence (Smith and Xie 2008) suggests northern China and Mongolia as the likely ancestral range of the cold-hardy brown rat (Rattus norvegicus), yet their contemporary distribution includes every continent except Antarctica. As a human commensal, brown rats occupy urban and agricultural areas using food, water, and shelter provided by humans. Rats are one of the most destructive invasive mammals as they spread zoonotic diseases to humans (Himsworth et al. 2013), damage food supplies and infrastructure (Pimentel et al. 2000), and contribute to the extinction of native wildlife (Harper and Bunbury 2015). As an invasive species, brown rats outcompete native species for resources and are a primary target of eradication efforts (Jones et al. 2016). Brown rats have been domesticated as models for biomedical research with inbreeding, leading to disease phenotypes similar to humans (Atanur et al. 2013). Finally, they are a nascent model to study evolution within urban landscapes, as they likely experience multiple selection pressures given their global distribution across a range of habitats and climates (Johnson and Munshi-South 2017).
The historical record indicates that rats colonized Europe in the early 1500s, eastern North America by the 1750s, and the Aleutian Archipelago by the 1780s (Black 1983; Armitage 1993). These historic records provide independent estimates for assessing inferences from demographic models using genomic data. Few other species have archeological or written human records that can be used to corroborate genomic inferences, although the house mouse, domestic dogs, and livestock are notable exceptions. Thus, we paired these data sources to test how well demographic models of a rapid and recent global expansion match historic records on rat invasions.
Research into the global expansion of brown rats has focused on both the routes and timings of different invasions; questions of specific interest include the location of the ancestral range and when rats arrived in Europe. Black rats (R. rattus) reached southern Europe by 6 kya (Ervynck 2002) and Great Britain by the 300s AD (Yalden 2003), yet brown rats were not recorded in Europe until the 1500s AD. These dates imply vastly different phylogeographic histories for these two commensal rats, which are likely related to where they speciated within Asia: black rats on the Indian subcontinent and brown rats in the northern steppe. Previous phylogeographic studies of brown rats using mitochondrial DNA identified China as the ancestral range based on private haplotypes and ancestral state reconstructions, with multiple expansions into Southeast (SE) Asia, Europe, and North America (Lack et al. 2013; Song et al. 2014; Puckett et al. 2018). Inference from mitochondria has been limited because of the high haplotype diversity observed from locally intense but globally diffuse sampling strategies. Thus, key geographic regions especially around the Indian Ocean basin and the Middle East are unrepresented in current data sets; sampling these areas would allow us to distinguish clinal versus long-distance expansions, where multiple introductions occurred, and mito-nuclear discordance. A phylogeographic analysis using nuclear SNPs inferred hierarchical clustering along five range expansion routes (Puckett et al. 2016). From the putative ancestral range, brown rats expanded southward into SE Asia and eastward into China and Russia (Puckett et al. 2016). The eastward expansion extended to North America with two independent colonizations of the Aleutian Archipelago and sites along the Pacific coast of western North America. From SE Asia, rats expanded into Europe (Puckett et al. 2016) via the Middle East (Zeng et al. 2018), where the likely route was aboard ships conducting maritime trade across the Indian Ocean into the Red Sea and Persian Gulf before moving goods onto land. Although these trade routes were established by the 200s BC, they intensified in the 1400s–1500s AD (Tucker 2015). The fifth range expansion moved rats to eastern North America, the Caribbean, South America, western Africa, and Australasia during the age of European imperialism of the 1600s–1800s (Puckett et al. 2016) with the result that genetic diversity is similar across the Western hemisphere and in western Europe. Ultimately, our previous work inferred the following seven genomic clusters: Eastern China, SE Asia, Aleutian, Western North America, Northern Europe, Western Europe, and (Western Europe) Expansion. However, these range expansions were inferred from patterns of population clustering and not specific models that estimate the population tree topology or demographic parameters of the evolutionary lineages. Thus, we generated 10 whole-genome sequences (WGSs) to represent the previously identified clusters to infer the demographic history of brown rats. We pay particular attention to both divergence times and changes in effective population sizes (Ne) in relation to climatic changes and human history that may have influenced natural and human-mediated range expansions for this species.
Results
We sequenced two genomes each from SE Asia, Northern Europe, Western Europe, and the Western Europe-Expansion (hereafter, Expansion) evolutionary clusters, as well as one genome each from the Aleutian and Western North America clusters (NCBI BioProject accession number PRJNA344413) (Supplemental Table S1). Average sequencing depth was 28.2× (range, 24–38×). We estimated heterozygosity for each individual on the 20 autosomes separately. Samples from Eastern China had the highest average chromosomal heterozygosity (0.244), whereas the Aleutians and Western North America had the lowest heterozygosity (0.143 and 0.148, respectively) (Supplemental Fig. S1).
Geographic origins of range expansions
We estimated the directionality index (ψ) (Peter and Slatkin 2013), which measures asymmetries between pairwise site frequency spectra (SFS) to identify the geographic origins and directionality of range expansions. As input we combined a ddRADseq data set of global brown rat diversity (Puckett et al. 2016) with low-coverage WGS samples from Asia and Iran (Zeng et al. 2018), representing 45 global sampling sites and limited to the 32,127 SNPs genotyped in the ddRADseq data (Supplemental Table S2). We first tested the expansion across Asia and observed that northern sites served as source populations for southward range expansions across the continent (median absolute Z-score = 49.2) (Fig. 1A; Supplemental Table S3). When we compared SE Asia and the Middle East, we observed that both regions served as source and sink populations; median absolute Z-scores from SE Asia to Iran were 17.8, whereas the median was 50.2 in the opposite direction (Fig. 1B; Supplemental Table S3). Given the potential connectivity between central Asia and the Middle East, this region requires better sampling to fully describe the regional relationships. The Middle East showed a strong signal of serving as a source of brown rats moving into central Europe and then dispersing across the continent into the Iberian Peninsula, Fennoscandia, and Great Britain (median absolute Z-score = 62.8) (Fig. 1C; Supplemental Table S3). Because our previous work suggested two expansions into North America, we analyzed the eastern and western seaboards separately. Eastern North America showed a strong signature of expansion from Western Europe (median absolute Z-score = 25.6) (Fig. 1D; Supplemental Table S3) as expected based on patterns of genomic clustering. The eastern North America to western North America signatures from genomic clustering analyses (Puckett et al. 2016) were not observed in the directionality index data. Finally, we observed expansion from Russia (i.e., eastern Asia) to both the Aleutian Archipelago and San Diego, United States (Western North America cluster; median absolute Z-score = 30.4) (Fig. 1E; Supplemental Table S3).
Figure 1.

Estimates of regional brown rat range expansions based on pairwise ψ statistics: (A) eastern Asia; (B) SE Asia and the Middle East; (C) Europe; (D) western Europe and the Expansion range; and (E) Russia, the Aleutian Archipelago, and Western North America. Lines show directionality from inferred source (pink) to sink (yellow) populations, whereas thickness was scaled to the Z-score when the absolute value was greater than five.
Effective population size through time
We inferred the change in Ne over time using the multiple sequentially Markovian coalescent (MSMC) model (Schiffels and Durbin 2014) and scaled the estimates to years and Ne using the estimated mutation rate (µ) from the coalescent modeling analysis (see below) of 9.29 × 10−8 and three generations per year. (As Deinum et al. [2015] estimated µ of 2.96 × 10−9 and the precise generation time for rats is unknown, we present alternative estimates of the MSMC model in Supplemental Fig. S2.) We observed two distinct patterns in the MSMC results related to the Pacific (Aleutian and Western North America) and all other clusters. The Pacific clusters declined sharply in Ne beginning ∼50 kya (Fig. 2). MSMC is not accurate in its last two time periods (Schiffels and Durbin 2014); therefore, we present Ne of the third time lag, which was ∼200 yr ago and estimated at 1460 and 1550 effective individuals, respectively, in the Aleutian and Western North America clusters (Fig. 2).
Figure 2.
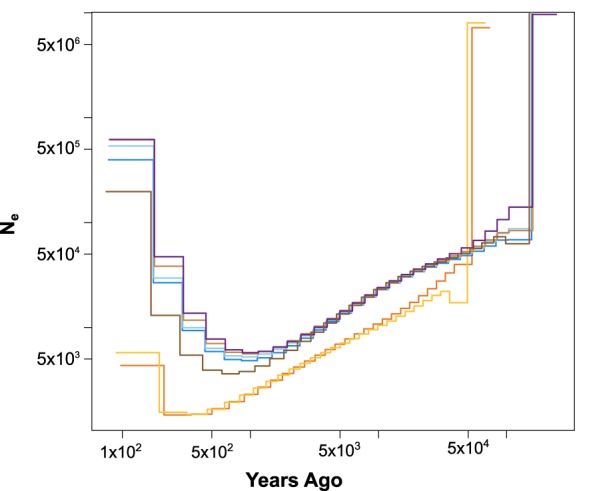
Plot of change in effective population size (Ne) over time using MSMC2, where the x-axis is years before the present. Each evolutionary cluster was represented by a different color: eastern China, dark brown; SE Asia, light brown; Aleutian, orange; Western North America, yellow; Northern Europe, purple; Western Europe, light blue; and Expansion, medium blue.
The second pattern was concordant between the Eastern China, SE Asia, Northern Europe, Western Europe, and Expansion clusters. Ne steadily declined from ∼150–1 kya before increasing in the most recent time periods (Fig. 2). Approximately 200 yr ago (the first reliable time step), Ne was 13,000 in Eastern China, 38,000 in SE Asia, 47,000 in Northern Europe, 29,000 in Western Europe, and 26,000 in Expansion (Fig. 2).
Demographic model
Based on previous work on the hierarchical genetic clustering of brown rats (Puckett et al. 2016) and the range expansion results, we split the range into Asian- and European-derived clusters and inferred that SE Asia linked the two regions. Thus, we built our full demographic model by conducting model selection in two stages, in which we first identified the models that best represented divergence patterns in Asia (Supplemental Fig. S3) and Europe (Supplemental Fig. S4) separately and then combined those tree topologies into a global model for parameter estimation. Specifically, we tested 10 and four alternative topologies with the Asian and European clusters, respectively. For each model, we ran 50 replicates of fastsimcoal2 (Excoffier et al. 2013) and retained the run with the highest log likelihood; we then compared these log likelihoods between topology models and retained the model with the highest overall log likelihood. The best Asian model had an ancestral unsampled population with independent divergence events for Eastern China, SE Asia, and the “Pacific” cluster that diverged into the Aleutians and Western North America (Supplemental Fig. S3). For the European model, the best-supported model used SE Asia as the ancestral population and then inferred a series of divergences first into the Middle East and then Western Europe, followed by independent divergences of Northern Europe and the Expansion from Western Europe (Supplemental Fig. S4).
By using WGS data from 14 genomes, we modeled the nine-population topology (Fig. 3) inferred from combining the submodels (Supplemental Figs. S3, S4). We compared five models that varied the population growth rate parameters on tip and edge branches of the population tree based on patterns within our MSMC analysis (Fig. 2). We observed that the best-supported model included decreasing population size on ancestral branches and increasing size since the start of the range expansions (Supplemental Methods). We estimated that Eastern China diverged from the ancestral population 17.5 kya (90% highest density probably [HPD]: 14.6–36.2 kya) (Table 1). The Pacific cluster diverged from the ancestral population 16.1 kya (HPD: 0.64–13.6 kya), and then the Aleutians and Western North America diverged 9.5 kya (HPD: 0.15–6.84 kya). The divergence that led to the global expansion of rats occurred rapidly, when rats first expanded into SE Asia 865 yr ago (1150 AD; HPD: 361 BC–1677 AD). Our model estimated rats entered the Middle East 792 yr ago (HPD: 586–1781 AD). We estimated rapid divergence of rats into Europe, including the Western Europe divergence 788 yr ago (1227 AD; HPD: 589–1781 AD) and the Northern Europe divergence 547 yr ago (1468 AD; HPD: 952–1857 AD). Finally, we estimated the Expansion cluster diverged 463 yr ago (1552 AD; HPD: 1491–1845 AD).
Figure 3.

Inferred population tree topology and divergence times for the global expansion of brown rats. (A) The best-supported demographic model contained nine evolutionary clusters inclusive of two unsampled populations. The divergence times in generations and Ne are listed in Table 1. (B) Map of global sampling locations for the WGS demographic model in which evolutionary clusters were represented by different colors: Eastern China, dark brown; SE Asia, light brown; Aleutian, orange; Western North America, yellow; Middle East, gray-blue; Northern Europe, purple; Western Europe, light blue; and Expansion, medium blue.
Table 1.
Parameter estimates from the best-supported model of global brown rat demography for eight evolutionary clusters and an unsampled ancestral population using 14 whole genomes

We ran the cross-coalescence analysis within MSMC2 to estimate the rate of divergence between the seven clusters with high depth of coverage (i.e., excluding Iran). We observed that divergence was complete between both the Aleutians or Western North America and all other populations (Supplemental Fig. S5). The European clusters showed similar patterns of divergence with Eastern China with ∼60% divergence complete (Supplemental Fig. S5A). Cross-coalescence between the Aleutians and Western North America increased approximately 200 generations ago before decreasing to 50% (Supplemental Fig. S5C). The four clusters making up the most recent expansions (SE Asia, Northern Europe, Western Europe, and Expansion) had signatures of increasing cross-coalescence over the past 1000 generations (Supplemental Fig. S5B,D–F).
ddRAD demographic models
Our WGS data came from a limited number of geographic sites, and previous work identified population structure at the spatial scale of cities (Puckett et al. 2016); therefore, we used a ddRADseq data set (Supplemental Table S2) to further investigate regional population tree topologies to better understand patterns of global range expansion as more populations were represented than in our WGS data. Below we detail the motivation and results for each analysis. We estimated divergence time of the two Pacific clusters at 9.5 kya (Table 1), which was earlier than historic records of rats being introduced to the Aleutian Archipelago in 1780 AD. Thus, we estimated the population tree topology between eastern Asia and western North America (Supplemental Fig. S6). We observed that a model in which eastern China and Russia were sister populations with an admixture pulse from Russia into Adak Island (Aleutian cluster) was the best-supported model.
Our previous clustering results suggested that brown rats in the Philippines were diverged from other SE Asia countries and that there may be gene flow between Thailand and Cambodia (Puckett et al. 2016); therefore, we modeled the population tree within SE Asia. We observed that the Philippines were well diverged from mainland populations and that gene flow from Thailand into Cambodia was present (Supplemental Fig. S7). The population tree topology supported the geography in which Cambodia and Vietnam were sister populations that shared an ancestor with Thailand (Supplemental Fig. S7).
We split European populations between the Western and Northern evolutionary clusters and observed patterns concordant with geography; specifically, Norway and Sweden were sister populations and shared a common ancestor with the Netherlands on continental Europe (Supplemental Fig. S8). Similarly, France and Spain on the Iberian Peninsula shared a common ancestor with Great Britain, an island nation (Supplemental Fig. S9).
North America presents the most complex scenario as invasion occurred on both the east and west coasts and shows patterns of cross-continent range expansions in both directions (Fig. 1D,E; Puckett et al. 2016). We modeled the population tree of North America for Vancouver, Canada (Expansion), and Berkeley, United States (admixed between Expansion and Western North America) separately. We fixed the global topology using four clusters (Udon Thani, Thailand, for SE Asia; Nottingham, Great Britain, for Western Europe; New York City [NYC], United States, for Expansion; and San Diego for Western North America) and then added in either Vancouver or Berkeley to understand variation along the Pacific seaboard. Our previous work (Puckett et al. 2016) identified that brown rats in Vancouver had high proportions of European ancestry with some Asian ancestry; we interpreted this result as original invasion by the Expansion cluster with gene flow from neighboring Pacific coast populations that contained Aleutian or Western North America ancestry. Our best-supported model showed admixture between the Expansion and Western North America clusters (Supplemental Fig. S10); the proportion from the Expansion cluster was 44%, which was low compared with our previous result of ∼90% European ancestry based off of clustering analyses. The pattern in Berkeley differed from that in Vancouver, whereas a model of population divergence between San Diego (Western North America) and Berkeley was observed before an admixture pulse from NYC (Expansion) (Supplemental Fig. S10). This admixture pulse was estimated as 3% of the total Berkeley ancestry, lower than previous estimates of high proportions of European ancestry.
Discussion
Our demographic modeling inferred that brown rats expanded from an ancestral range in northern Asia into eastern China, western North America, and SE Asia (Figs. 1, 2; Supplemental Fig. S1). We included an unsampled ghost population in our model to represent this ancestral range in northern Asia. Brown rat fossils have been described from northern China and Mongolia (Smith and Xie 2008), and our range expansion results (Fig. 1A) suggest eastern Russia as a possible part of the ancestral range. The Pacific cluster diverged from the ancestral population 16.1 kya, and divergence of the Aleutian and Western North America clusters occurred 9.5 kya (Table 1; Fig. 3). Our cross-coalescence analysis (Supplemental Fig. S5C) suggested that divergence between these clusters may be as recent as 100 generations ago, which would explain why the patterns of change in Ne were similar over time. These results also suggest an explanation for the wide HPD estimates for these clusters in our demographic model (Table 1). We emphasize that the divergence of these clusters does not identify the timing of the introduction to the Aleutian Archipelago or the Pacific coast of North America where samples were collected. The historic record indicates rats were moved to the Aleutian Archipelago by Russian fur traders in the 1780s (Black 1983). Our regional population model suggested a scenario with gene flow from a population in eastern Russia into Adak Island (Supplemental Fig. S6), thereby suggesting two introductions of rats to the Aleutians. We acknowledge that our demographic model was complex as it contained nine evolutionary clusters and a modest number of genomes; thus, more data will improve parameter estimates. Specifically, additional spatial sampling in eastern Russia and the Aleutians and ancient samples would better estimate divergence times to differentiate between models of contemporary or historic movement of rats. This question is particularly interesting because human migration into the region occurred ∼36 kya (Moreno-Mayar et al. 2018). Thus, when and how rats first moved across Beringia remain open questions.
We estimated that the expansion across Asia varied spatially. Our modeling supported an independent expansion into Eastern China from the ancestral range 17.5 kya (Table 1; Fig. 3A). Given the eastern location of Harbin, China (where this lineage was sampled), we felt that it was reasonable to assume this was an independent expansion instead of part of the broader southern expansion into SE Asia (Figs. 1A, 3A). The Harbin population contains high mitochondrial diversity, with the most divergent clades estimated to 96 kya (HPD: 70–128 kya) (Puckett et al. 2018). This high mitochondrial diversity may reflect movement from multiple ancestral populations into the eastern portion of the range before recombination, creating a unique nuclear genomic signature for the lineage.
We estimated that the SE Asia cluster diverged from the ancestral population 0.865 kya (1150 AD) (Table 1; Fig. 3). The timing of this divergence immediately raises the question of why rats did not expand sooner, as overland trade between China and SE Asia was established by the 500s AD (Lieberman 2009); maritime trade between these regions and the Indian Ocean basin was established before the 900s AD (Heng 2009). A partial explanation may be because of the intersection of climate and human demography across eastern Asia. The Medieval Climate Anomaly (850–1250 AD) aided agricultural expansion and human demographic growth in China, specifically prompting urban centers to expand outward at a time of human movement from northern arid lands to more agriculturally productive lands in the south (see references within Lieberman 2009). However, the end of this climatic period resulted in drought, famine, political instability, and ultimately human demographic contractions in both China and SE Asia; fortunes reversed in the late 1400s to mid-1500s as the climate improved and populations expanded again (Lieberman 2009). We hypothesize that this southward human demographic expansion facilitated the range expansion of brown rats, which explains the clinal pattern of ancestry from northern China across southern China into SE Asia that Zeng et al. (2018) observed. Thus, the founding of new agrarian communities and increasing inter-connectedness with urban centers would serve as stepping stones for rats to move from northern China to SE Asia during the two periods of human demographic expansion.
Our results regarding an ancestral range in the north with southward expansion into SE Asia stand in marked contrast to a different study that identified brown rats in SE Asia as the ancestral population with a northward expansion (Zeng et al. 2018). Both analyses used coalescent modeling approaches but with four primary differences: independent data sets, mutation rates, generation time, and tree topology. To address variation in the mutation rate, we ran our global model with the mutation rate fixed to 1.103 × 10−9 as estimated by Zeng et al. (2018) and observed a decreased model fit compared with when we allowed the mutation rate to be estimated as part of the model (Supplemental Table S4); therefore, we reported the model using the estimated rate of 9.29 × 10−8. Regarding the generation time, we converted generations to years using the estimate of three generations per year, whereas Zeng et al. (2018) used two generations per year. Thus, if both papers estimated a divergence given the same number of generations, the estimate of three generations per year would make those estimates more recent in time and two generations per year would estimate the event further back in time. Without direct field observations of rat fecundity and how it may vary with resources or climate, we are unable to identify the exact dates and thus acknowledge that discrepancy between the papers.
Although the factors above likely contributed to small differences between our results and that of Zeng et al. (2018), we think that the more substantial discrepancy results from the population tree topology. Specifically, Zeng et al. (2018) included admixed individuals containing SE Asian ancestry (that were geographically located in southern China) within the northern China cluster. The inclusion of these individuals may have biased model support between the alternative topologies, specifically supporting the model in which SE Asia was ancestral given that a small proportion of SE Asian alleles were within the northern Asia cluster. We believe their result was not owing to the true history of the populations but can instead be attributed to sample clustering. Finally, we observed that inclusion of an unsampled ancestral population improved our model fit. Unsampled populations can influence parameter estimates of Ne and migration rates and have been shown to improve or at least not harm parameter estimation within the full model (Beerli 2004). Adding an unsampled population to our model was important given the limited number of chromosomes genotyped, as large sample sizes decrease the effect of unsampled populations on parameter estimates (Slatkin 2005).
We estimated a rapid range expansion from SE Asia into Europe via the Middle East (Table 1). There was concordance between the phylogeographic patterns in our results and those of Zeng et al. (2018); however, we estimated the divergence time into the Middle East 792 yr ago (Table 1); they, 3100 yr ago (2066 yr when using three generations per year). These discrepancies were likely owing to how population size and divergence time interact in coalescent models; specifically, we observed greatly improved model fit when including an ancestral population rate change parameter (RAncestral) (Supplemental Methods) that decreased Ne before recent expansions as observed in our MSMC analysis (Fig. 2). Our model also estimated a significantly smaller lineage-specific Ne. Finally, we estimated that the Expansion cluster diverged from Western Europe around 1552 AD (Table 1). Both the divergence estimates into Europe and North America were older than historic records (Armitage 1993), and either indicates limits to parameter estimation for recent divergence events or an area for improvement within our model. For example, we estimated Ne in NYC at 1004 individuals (HPD: 300–1341) (Table 1), whereas an independent analysis estimated 260 individuals (Combs et al. 2018), indicating an area in which the model may have overestimated the parameters of interest.
Ancestral population size
We observed that Ne steadily declined in both the Pacific and Ancestral range populations ∼150 and 50 kya, respectively (Fig. 2). These declines began before the Last Glacial Maximum (22–18 kya), a climatic period when populations of many species declined because of range contractions and/or shifts. The more recent increasing population size appears related to the demographic and geographic range expansion mediated by rats’ commensal relationship with humans instead of climatic events alone.
Range expansion via human-mediated movements
Our results identified that the global range expansion of rats began in the early 1200s and then proceeded rapidly from SE Asia into Europe via the Middle East and was likely linked by maritime trade between those regions. This stands in marked contrast to previous assumptions that brown rats were transported westward along the Silk Road through central Asia into Europe. This is counterintuitive as overland trade routes from central China to Persia were established 2.1 kya (105 BC), and goods reached Rome by 46 BC (Tucker 2015). The Silk Road passed through part of the native range of brown rats, unlike black rats, which originated on the Indian subcontinent (Aplin et al. 2011). Assuming that rats evolved their commensal relationship with humans before their global range expansion, as observed with the house mouse (Suzuki et al. 2013), the availability of cities, road networks, and a flow of merchants naturally suggests a way to expand westward. The Silk Road may not have been the route for expansion because of the limited distance that merchants traveled along the route, as goods went further than the caravans containing the resources rats would need for survival (Tucker 2015). Further, high aridity and a lack of water sources may have limited rat movement via the Silk Road. Yet this does not preclude the idea that brown rats may have expanded westward via Silk Road cities and were then extirpated because of the collapse of those cities during changing geo-politics and shifts toward maritime trade (Tucker 2015). We instead suggest that pulses of southward human demographic expansion from northern China during favorable climatic conditions enabled the expansion of rats into SE Asia, from which they expanded westward. This hypothesis was supported by our range expansion models (Fig. 1) showing westward movement from the Middle East into central Europe and then expansion in all directions across Europe. We present this historical narrative as a hypothesis supported by our demographic model, and as a stimulus for interest in further study by historians and zooarchaeologists to examine the historical expansion of this globally important invader.
Methods
Whole-genome sequencing and data sets
We selected 10 individuals for whole-genome sequencing: two each representing evolutionary clusters within SE Asia (Philippines and Cambodia), Northern Europe (Sweden and Netherlands), Western Europe (England and France), and Expansion (New York, USA), as well as one sample each from the Aleutian Islands and Western North America (Fig. 3B; Supplemental Table S1). We generated paired-end reads for each sample (4 ng RNase A–treated genomic DNA) by sequencing on an Illumina HiSeq 2500 at the New York Genome Center. Initial bioinformatics were completed by the New York Genome Center, where genomes were mapped to the Rnor_5.0.75 reference (Gibbs et al. 2004) using BWA-MEM v0.7.8 (Li and Durbin 2010). Then duplicates were marked using Picard Tools v1.122, and indels were realigned with the GATK v3.4.0 IndelRealigner (McKenna et al. 2010). We sorted and indexed BAM files using SAMtools v1.3.1 (Li et al. 2009). Data for these 10 genomes are available on the NCBI SRA BioProject PRJNA344413 (Puckett et al. 2018).
We combined these 10 new WGSs with three existing data sets depending on the analysis. Specifically, we downloaded whole genomes from 11 brown rats and one black rat (Rattus rattus) collected in Harbin, China (ENA ERP001276), although to not bias estimates with unequal sample sizes, we ran analyses using only Rnor13 and Rnor14 (Deinum et al. 2015), which were randomly selected. We downloaded 54 low-depth WGS brown rats collected in cities across Russia, China, and Iran (Beijing Institute of Genomics BioProject CRA000345; accessions: CRR021172–CRR021339) (Zeng et al. 2018). Two of these samples (Iran5 and Iran9) were used in WGS analyses, whereby we mapped the raw reads to the Rnor_5 reference with Bowtie 2 (Langmead and Salzberg 2012) using the default parameters and then sorted and indexed in SAMtools. All 54 genomes were also mapped to the Rnor_6 reference with Bowtie 2, sorted, and indexed and then had a set of 32,127 SNPs extracted using a position list and the mpileup function in SAMtools to make the data comparable to genotypes from 326 brown rats collected from around the globe (Puckett et al. 2018). By using these data sources, we created four data sets that varied in input samples and processing depending on the resultant analysis; we describe the input data and analyses in detail below.
Patterns of range expansion
We explored the geographic patterns of the global range expansion using the directionality index (Peter and Slatkin 2013) calculated from the SFS. The directionality index identifies the expected geographic location that acted as the center of a range expansion event. This analysis used the combined ddRADseq genotypes from Puckett et al. (2018) and WGS data from Zeng et al. (2018) at 32,127 SNPs made using SAMtools. We removed sampling sites represented by a single individual for a final data set containing 276 individuals from 45 locations. The VCF was converted into PLINK format and then imported into the rangeExpansion package for R (Peter and Slatkin 2013). We calculated the directionality index, ψ, for all population pairs using the get.all.psi function. To determine significance, we calculated the standard error of the upper triangle of the pairwise ψ matrix excluding the diagonal, thereby allowing us to calculate the Z-score for each population pair. For each region of interest, we plotted data for each pair of populations in which the absolute Z-score was greater than five and visually assessed the geographic patterns of source and sink populations.
Estimates of Ne through time
We estimated the change in effective population size over time in each evolutionary cluster using MSMC2 (Schiffels and Durbin 2014). To call variants, we used SAMtools mpileup across all samples (10 WGS genomes sequenced here and two Chinese genomes) with a minimum mapping quality of 18 and the coefficient to downgrade mapping qualities for excessive mismatches at 50. We then used the variant calling in BCFtools v1.3 with the consensus caller and excluded indels that limited the data set to biallelic SNPs, before pipping the output to the bamCaller.py script that produced per chromosome masks and VCF files for each individual. As there was not a brown rat reference panel, we phased the 12 individuals plus two inbred lines (SS/Jr and WKY/NHsd; NCBI SRA accessions ERR224465 and ERR224470, respectively) (Atanur et al. 2013) for each of the 20 autosomes using fastPHASE v1.4.8 (Scheet and Stephens 2006). We generated genome-wide masks for each chromosome using SNPable (http://lh3lh3.users.sourceforge.net/snpable.shtml) and then converted to a BED file with the makeMappabilityMask.py script. Finally, we used the generate_multihetsep.py script to create the MSMC2 input files before running the program within and between population clusters. Specifically, we estimated change in Ne over time for each of the seven evolutionary clusters using two haplotypes for the Aleutian and Western North American clusters and four haplotypes for each other cluster. We also estimated the proportion of population divergence over time using the cross-population analysis and combined results from individual populations with the cross-population analysis using the combineCrossCoal.py script provided.
WGS demographic modeling
We inferred the demographic history of rats by modeling alternative scenarios that compared the observed and expected SFSs for each evolutionary cluster. We combined the 10 genomes sequenced in this study, two genomes from Harbin, China, and two genomes from Mahmudabad, Iran (Supplemental Table S1). We limited SNP calling to sites observed in 10 of 12 genomes (-minInd; excluding those from Iran, which had lower depth of coverage), to the 20 autosomes, and to bases that had a minimum mapping quality (-minmapq) of 30 and minimum Q score (-minQ) of 20 using ANGSD v0.915 (Korneliussen et al. 2014). We estimated genotype likelihoods using the function implemented in SAMtools (-GL 1) (Li et al. 2009). This resulted in 2.18 billion sites across the autosomes. We reran the genotype likelihood function (-GL 1) for each evolutionary cluster with the same minimum mapping qualities and Q-scores as above, applied the sites flag with the results from the analysis above, and included the R. rattus individual from China as the outgroup allowing for identification of ancestral and derived alleles. These genotype likelihoods were the input into the realSFS function in ANGSD to generate the pairwise SFS for each chromosome; the genotype likelihood for each chromosome was scaled by the number of sites genotyped and then summed across all chromosomes.
Given the large number of evolutionary clusters to model, we first modeled the population tree topology relationships between the Asian and European clusters separately. For the Asian cluster, we modeled the relationships between Eastern China, SE Asia, Aleutian, and Western North America by comparing five four-population models and five five-population models that included an unsampled population (Supplemental Fig. S3; Supplemental Code). For each scenario, we ran 50 replicates in fastsimcoal2 v2.6.0.3 (Excoffier et al. 2013), where each replicate had the following parameters: 1 × 105 simulations (-n 100000 -N 100000), stopping criteria of 0.001 (-M 0.001), and minimum and maximum ECM loops of 10 and 50, respectively (-l 10 -L 50). For these initial topology models, we did not allow population size to change through time, and we set the mutation rate at 2.5 × 10−8 mutations per generation. We identified the highest log likelihood for each model from the 50 replicates and then identified the highest log likelihood between the alternative topology models and retained that model for downstream analyses. We did not use AIC for model selection because we generated pairwise SFS for all population pairs (Excoffier et al. 2013). The best-supported scenario (model 6 in Supplemental Fig. S3) had a topology that included an ancestral unsampled population with independent divergence of Eastern China, SE Asia, and the Pacific clusters. For the European clusters, we modeled four scenarios of a five-tree topology between the SE Asia, Middle East, Northern Europe, Western Europe, and the Expansion. Our previous work on brown rat phylogeography suggested that rats expanded into Europe from SE Asia (Puckett et al. 2016), and Zeng et al (2018) showed that the Middle East served as an intermediary point between SE Asia and Europe; thus, we tested the topology between the three European clusters (Supplemental Code) using the same approach and fastsimcoal2 parameters as described above. The best-supported scenario (model 2 in Supplemental Fig. S4) had an initial divergence of Western Europe from the Middle East, with Northern Europe and the Expansion diverging independently from Western Europe.
The best-supported Asian and European models were concordant with the range expansion results; thus, we combined the topologies into a nine-population model. We were able to do this as each model contained the SE Asia cluster. By using this nine-population model, we tested five scenarios of lineage-specific population expansion and contraction (Supplemental Methods). We ran 50 replicates of each model using the fastsimcoal2 parameters as described above; however, we estimated the mutation rate parameter instead of fixing it. As with the submodels, we retained the replicate with the highest likelihood. The best-supported model allowed for independent growth rate parameters for the nine tip branches and two ancestral branches (Pacific and Ancestral Range). To explore the effect of jointly estimating the mutation rate (µ) with divergence times and Ne parameters, we reran the best topology fixing the mutation rate at either the fastsimcoal2 default value of 2.5 × 10−8 (Nachman and Crowell 2000) or the rate estimated from Zeng et al. (2018) of 1.103 × 10−9. We ran 50 iterations of each model with the same settings as described above and retained the iteration with the best likelihood from each model. The model that jointly estimated µ with the other parameters had the highest likelihood, whereas the default mutation rate had a higher likelihood than the estimated rate from a similar analysis (Supplemental Table S4).
By using the point estimates from the best model in which µ was jointly estimated, we generated 500 samples of pairwise SFS, each containing 100,000 sites that served as pseudo-observed data for estimating parameter ranges under the best-supported model. We calculated the 90% HPD from these 500 data sets using the HDInterval v0.1.3 package (https://cran.r-project.org/web/packages/HDInterval/index.html) in R (R Core Team 2013). We used three generations per year to convert parameter estimates; all time calculations were performed since 2015.
ddRADseq demographic modeling
Although our WGS had many more loci, there was limited geographic representation, as well as fewer individuals sampled; therefore, we built regional models from the ddRADseq data set to explore additional population tree topologies. We estimated the SFS of each population in ANGSD using the reference aligned Illumina reads instead of the previously called SNPs.
We built regional models within the evolutionary clusters for eastern Asia/Pacific, SE Asia, Northern Europe, and Western Europe. We used this reductive approach to limit the number of parameters being estimated. Within each region, we compared topologies between populations suggested by previous population structure analyses (Puckett et al. 2016). We used the same fastsimcoal2 run parameters as described above; however, we did not create pseudo-observed data sets for parameter estimation, unless noted, as our interest was in topology. A secondary reason we did not further explore population parameters within the regions was that we observed these data sets tended to overestimate divergence times, likely because of unsorted variation remaining within populations until coalescence with the unsampled ancestral population. Finally, we investigated population topology and admixture proportions in Vancouver and Berkeley because each site was identified as admixed in our previous analysis.
Chromosomal diversity
By using the genotypes from the WGS data created with ANGSD, we estimated heterozygosity on each chromosome for each individual. We exported the data into PLINK v1.9 (Purcell et al. 2007; Chang et al. 2015) and estimated heterozygosity (--het) on each chromosome.
Supplementary Material
Acknowledgments
We thank Joshua Schraiber and anonymous reviewers for comments that improved the manuscript. This work was funded by National Science Foundation grants DEB 1457523 and MRI 1531639 to J.M.-S. The mammal collections at the University of Alaska Museum of the North, University of California–Berkeley Museum of Vertebrate Zoology, the Burke Museum at the University of Washington, and the Museum of Texas Tech University also graciously provided tissue samples.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.235754.118.
References
- Aplin KP, Suzuki H, Chinen AA, Chesser RT, ten Have J, Donnellan SC, Austin J, Frost A, Gonzalez JP, Herbreteau V, et al. 2011. Multiple geographic origins of commensalism and complex dispersal history of black rats. PLoS One 6: e26357 10.1371/journal.pone.0026357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armitage P. 1993. Commensal rats in the New World, 1492–1992. Biologist 40: 174–178. [Google Scholar]
- Atanur SS, Diaz AG, Maratou K, Sarkis A, Rotival M, Game L, Tschannen MR, Kaisaki PJ, Otto GW, Ma MC, et al. 2013. Genome sequencing reveals loci under artificial selection that underlie disease phenotypes in the laboratory rat. Cell 154: 691–703. 10.1016/j.cell.2013.06.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerli P. 2004. Effect of unsampled populations on the estimation of population sizes and migration rates between sampled populations. Mol Ecol 13: 827–836. 10.1111/j.1365-294X.2004.02101.x [DOI] [PubMed] [Google Scholar]
- Black L. 1983. Record of maritime disasters in Russian America, part one: 1741–1799. In Proceedings of the Alaska Maritime Archaeology Workshop, May 17–19, Vol Alaska Sea Grant Report no. 83-9 University of Alaska- Fairbanks, Sitka, AK. [Google Scholar]
- Chang CC, Chow CC, Tellier LCAM, Vattikuti S, Purcell SM, Lee JJ. 2015. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4: 7 10.1186/s13742-015-0047-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combs M, Puckett EE, Richardson J, Mims D, Munshi-South J. 2018. Spatial population genomics of the brown rat (Rattus norvegicus) in New York City. Mol Ecol 27: 83–98. 10.1111/mec.14437 [DOI] [PubMed] [Google Scholar]
- Deinum EE, Halligan DL, Ness RW, Zhang Y-H, Cong L, Zhang J-X, Keightley PD. 2015. Recent evolution in Rattus norvegicus is shaped by declining effective population size. Mol Biol Evol 32: 2547–2558. 10.1093/molbev/msv126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ervynck A. 2002. Sedentism or urbanism? On the origin of the commensal black rat (Rattus rattus). In Bones and the man: studies in honour of Don Brothwell (ed. Dobney K, O'Connor T), pp. 95–109. Oxbow Books, Oxford. [Google Scholar]
- Excoffier L, Dupanloup I, Huerta-Sánchez E, Sousa VC, Foll M. 2013. Robust demographic inference from genomic and SNP data. PLoS Genet 9: e1003905 10.1371/journal.pgen.1003905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs RA, Weinstock GM, Metzker ML, Muzny DM, Sodergren EJ, Scherer S, Scott G, Steffen D, Worley KC, Burch PE, et al. 2004. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature 428: 493–521. 10.1038/nature02426 [DOI] [PubMed] [Google Scholar]
- Harper GA, Bunbury N. 2015. Invasive rats on tropical islands: their population biology and impacts on native species. Global Ecol Conserv 3: 607–627. 10.1016/j.gecco.2015.02.010 [DOI] [Google Scholar]
- Heng D. 2009. Sino-Malay trade and diplomacy from the tenth through the fourteenth century. Ohio University Press, Athens, OH. [Google Scholar]
- Himsworth CG, Parsons KL, Jardine C, Patrick DM. 2013. Rats, cities, people, and pathogens: a systematic review and narrative synthesis of literature regarding the ecology of rat-associated zoonoses in urban centers. Vector Borne Zoonotic Dis 13: 349–359. 10.1089/vbz.2012.1195 [DOI] [PubMed] [Google Scholar]
- Johnson MTJ, Munshi-South J. 2017. Evolution of life in urban environments. Science 358: eaam8327 10.1126/science.aam8327 [DOI] [PubMed] [Google Scholar]
- Jones HP, Holmes ND, Butchart SHM, Tershy BR, Kappes PJ, Corkery I, Aguirre-Muñoz A, Armstrong DP, Bonnaud E, Burbidge AA, et al. 2016. Invasive mammal eradication on islands results in substantial conservation gains. Proc Natl Acad Sci 113: 4033–4038. 10.1073/pnas.1521179113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korneliussen TS, Albrechtsen A, Nielsen R. 2014. ANGSD: analysis of next generation sequencing data. BMC Bioinformatics 15: 356 10.1186/s12859-014-0356-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lack J, Hamilton M, Braun J, Mares M, Van Den Bussche R. 2013. Comparative phylogeography of invasive Rattus rattus and Rattus norvegicus in the U.S. reveals distinct colonization histories and dispersal. Biol Invasions 15: 1067–1087. 10.1007/s10530-012-0351-5 [DOI] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2010. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26: 589–595. 10.1093/bioinformatics/btp698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman V. 2009. Strange parallels: Southeast Asia in global context, c. 800–1830. Cambridge University Press, Cambridge. [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Mayar JV, Potter BA, Vinner L, Steinrücken M, Rasmussen S, Terhorst J, Kamm JA, Albrechtsen A, Malaspinas A-S, Sikora M, et al. 2018. Terminal Pleistocene Alaskan genome reveals first founding population of Native Americans. Nature 553: 203 10.1038/nature25173 [DOI] [PubMed] [Google Scholar]
- Nachman MW, Crowell SL. 2000. Estimate of the mutation rate per nucleotide in humans. Genetics 156: 297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter BM, Slatkin M. 2013. Detecting range expansions from genetic data. Evolution 67: 3274–3289. 10.1111/evo.12202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimentel D, Lach L, Zuniga R, Morrison D. 2000. Environmental and economic costs of nonindigenous species in the United States. Bioscience 50: 53–65. 10.1641/0006-3568(2000)050[0053:EAECON]2.3.CO;2 [DOI] [Google Scholar]
- Puckett EE, Park J, Combs M, Blum MJ, Bryant JE, Caccone A, Costa F, Deinum EE, Esther A, Himsworth CG, et al. 2016. Global population divergence and admixture of the brown rat (Rattus norvegicus). Proc R Soc B Biol Sci 283: 20161762 10.1098/rspb.2016.1762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puckett EE, Micci-Smith O, Munshi-South J. 2018. Genomic analyses identify multiple Asian origins and deeply diverged mitochondrial clades in inbred brown rats (Rattus norvegicus). Evol Appl 11: 718–726. 10.1111/eva.12572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, et al. 2007. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81: 559–575. 10.1086/519795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2013. R : a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna: https://www.R-project.org/. [Google Scholar]
- Scheet P, Stephens M. 2006. A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase. Am J Hum Genet 78: 629–644. 10.1086/502802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffels S, Durbin R. 2014. Inferring human population size and separation history from multiple genome sequences. Nat Genet 46: 919–925. 10.1038/ng.3015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slatkin M. 2005. Seeing ghosts: the effect of unsampled populations on migration rates estimated for sampled populations. Mol Ecol 14: 67–73. 10.1111/j.1365-294X.2004.02393.x [DOI] [PubMed] [Google Scholar]
- Smith AT, Xie Y. 2008. A guide to the mammals of China. Princeton University Press, Princeton, NJ. [Google Scholar]
- Song Y, Lan Z, Kohn MH. 2014. Mitochondrial DNA phylogeography of the Norway rat. PLoS One 9: e88425 10.1371/journal.pone.0088425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Nunome M, Kinoshita G, Aplin KP, Vogel P, Kryukov AP, Jin ML, Han SH, Maryanto I, Tsuchiya K, et al. 2013. Evolutionary and dispersal history of Eurasian house mice Mus musculus clarified by more extensive geographic sampling of mitochondrial DNA. Heredity 111: 375–390. 10.1038/hdy.2013.60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker J. 2015. The Silk Road: China and the Karakorum Highway. I.B.Tauris & Company, New York. [Google Scholar]
- Yalden DW. 2003. Mammals in Britain: a historical perspective. British Wildlife 14: 243–251. [Google Scholar]
- Zeng L, Ming C, Li Y, Su L-Y, Su Y-H, Otecko NO, Dalecky A, Donnellan S, Aplin K, Liu X-H, et al. 2018. Out of Southern East Asia of the brown rat revealed by large-scale genome sequencing. Mol Biol Evol 35: 149–158. 10.1093/molbev/msx276. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.