

Palangat et al. uncovered a noncanonical function of U2AF1, showing that it directly binds mature mRNA in the cytoplasm and negatively regulates mRNA translation.
Keywords: IL8, U2AF1, myeloid leukemia, splicing factor mutations, translation regulator
Abstract
Somatic mutations in the genes encoding components of the spliceosome occur frequently in human neoplasms, including myeloid dysplasias and leukemias, and less often in solid tumors. One of the affected factors, U2AF1, is involved in splice site selection, and the most common change, S34F, alters a conserved nucleic acid-binding domain, recognition of the 3′ splice site, and alternative splicing of many mRNAs. However, the role that this mutation plays in oncogenesis is still unknown. Here, we uncovered a noncanonical function of U2AF1, showing that it directly binds mature mRNA in the cytoplasm and negatively regulates mRNA translation. This splicing-independent role of U2AF1 is altered by the S34F mutation, and polysome profiling indicates that the mutation affects translation of hundreds of mRNA. One functional consequence is increased synthesis of the secreted chemokine interleukin 8, which contributes to metastasis, inflammation, and cancer progression in mice and humans.
Somatic mutations affecting the genes that encode core components of the splicing machinery, first discovered in blood malignancies (Yoshida et al. 2011), have now been observed in nearly every cancer type (Graubert et al. 2011; Dvinge et al. 2016; Seiler et al. 2018). These somatic mutations are largely present in genes, including SF3B1, SRSF2, ZRSR2, and U2AF1, which code for factors involved in selection of branchpoint, exonic sequences, and 3′ splice sites. Spliceosome mutations result in hundreds of changes in alternative splicing, which vary among tissues and patients (Inoue et al. 2016). The spectrum of mutations, the stoichiometry in tumors, and the clinical outcomes point toward splicing-factor mutations as oncogenic events (Seiler et al. 2018). However, there is little consensus on the molecular mechanism of action in human carcinogenesis.
The splicing factor U2AF, a heterodimer of U2AF1 and U2AF2, is responsible for the recognition and binding to the 3′splice site (U2AF1 to AG nucleotides and U2AF2 to the polypyrimidine tract), a critical initial step in the assembly of the spliceosome (Merendino et al. 1999; Wu et al. 1999; Zorio and Blumenthal 1999). A recurrent hotspot mutation in U2AF1 (S34F) in its first Zn-finger domain (Yoshida et al. 2011; Imielinski et al. 2012; Brooks et al. 2014) critical for RNA binding activity (Yoshida et al. 2015), alters RNA-binding specificity (Okeyo-Owuor et al. 2015; Fei et al. 2016; Jenkins and Kielkopf 2017; Kielkopf 2017) and splicing kinetics (Coulon et al. 2014), resulting in a wide variety of splicing outcomes, most frequently cassette exon inclusion (Graubert et al. 2011; Yoshida et al. 2011; Imielinski et al. 2012; Furney et al. 2013; Przychodzen et al. 2013; Ilagan et al. 2015; Okeyo-Owuor et al. 2015). In all cases, the tumor cells retain a wild-type allele of U2AF1 (Graubert et al. 2011). Most of the splicing changes observed in human tissue can be recapitulated in a human bronchial epithelial cell (HBEC) line carrying the heterozygous U2AF1-S34F mutation (Fei et al. 2016). However, mutant HBECs do not exhibit any visible morphological or altered growth phenotypes (Fei et al. 2016), indicating that the mutation is not a strong driver mutation. Although the S34F mutation mediates subtle splicing changes, these isoform shifts have not been definitively linked to a phenotype, leading some investigators to propose nuclear RNA processing defects such as alterations in 3′ UTRs (Park et al. 2016) or an increase in R-loops (Chen et al. 2018; Nguyen et al. 2018).
Here, we show that U2AF1 in association with its binding partner U2AF2, binds mature RNA in the cytoplasm and functions as a translational repressor. U2AF1 directly interacts with hundreds of spliced, polyadenylated mRNA in the cytoplasm, many of which are mTOR regulated and code for proteins involved in translation initiation and growth. Using PAR-CLIP, we show that U2AF heterodimer binding is often in the 5′ UTR near the start codon of these messages, suggesting an interplay between binding and translation initiation. Through RIP-seq and polysome profiling, we show that the cancer-associated S34F mutation correlates with loss of binding and translational derepression. We further provide evidence in support of a functional role for this noncanonical pathway through the translational regulation of the chemokine IL8. IL8 is translationally up-regulated in the mutant background, increases epithelial-to-mesenchymal transition (EMT) in culture, enhances the inflammatory response in mice, and blocking IL8 with neutralizing antibody reduces tumor burden. Moreover, elevated IL8 levels in human bone marrow correlate with relapsed/refractory acute myeloid leukemia. Thus, U2AF1 S34F has the potential to play both cell-autonomous and nonautonomous roles in cancer progression, and the mechanism described here suggests a possible therapeutic intervention.
Results
U2AF1-S34F mutation confers altered viability and sustained inflammatory cytokine secretion after DNA damage
To investigate the mechanism of the S34F mutation in oncogenesis, we used four isogenic HBEC lines created with gene editing (Fei et al. 2016): (1) wt/wt, the parent cell line; (2) wt/S34F, the heterozygous condition similar to that observed in patients (homozygous S34F mutant is lethal); (3) wt/−, a line with one allele carrying a frameshift mutation; and (4) wt/S34F−, a line derived from the wt/S34F line where the mutant allele is frameshifted, resulting in removal of the mutant U2AF1-S34F protein. Using RNA-seq, we observed modest changes (1.5-fold to 2.0-fold increase or decrease) in gene expression in the wt/S34F mutant cells and altered splicing outcomes (Supplemental Fig. S1A,B). Genes displaying changes in RNA levels were enriched for those associated with DNA metabolism, (Supplemental Table S1), suggesting either changes in cell cycle progression or DNA damage.
Although we did not observe change in growth rates (Fei et al. 2016), we did find that the S34F mutation conferred altered outcomes upon DNA damage. When irradiated with a high dose of X-rays (20 Gy), the S34F mutation conferred substantial growth advantage over a 6-d period in a cell proliferation assay (Fig. 1A), and this phenotype could be successfully rescued with the frame-shift mutation, indicating that the S34F mutation resulted in gain of function. A fraction of wt/S34F cells survived irradiation and stained positive for the senescence marker β-galactosidase and remained in a senescent state in culture for >30 d, whereas the wt/wt and wt/− mutant cells died 10–12 d after irradiation (Supplemental Fig. S1C). Thus, U2AF1 S34F provides resistance to clinically relevant doses of X-rays.
Figure 1.

U2AF1 S34F mutant cells show altered DNA damage response and inflammatory cytokine secretion. (A) Altered growth in wt/wt, wt/S34F, wt/−, and wt/S34F− mutant cells after irradiation with x-rays (20 Gy) measured using a cell proliferation assay at indicated times (average of three independent experiments ±SD). (B) Cytokine levels measured by ELISA in media collected at indicated times from wt/wt, wt/S34F, wt/−, and wt/S34F− mutant cells after irradiation with x-rays (20 Gy). IL8 secreted into media is presented as pg/mL (average of three independent experiments ±SD). (C) ELISA of steady state secretion of IL8 into media collected after 6 d in culture of HBEC (wt/wt, wt/S34F, wt/−, and wt/S34F−, (wt/wt vs. wt/S34F P = 0.0003; wt/wt vs. wt/S34F− P = 0.8533), human lung tumor cells (H441 and A549, H441S34F vs. H441S34F− P = 0.0614; A549S34F vs. A549S34F− P = 0.0281) and human myeloid leukemia cell line (NKM1, NKM1S34F vs. NKM1S34F− P = 0.0251) and with their respective S34F mutant allele knockout cell lines (average of three biological replicates ±SD). (D) ELISA of steady state secretion of IL1-α into media collected after 6 d in culture of wt/wt, wt/S34F, wt/−, and wt/S34F− mutant cells (average of three biological replicates ±SD wt/wt vs. wt/S34F P = 0.3446; wt/wt vs. wt/S34F− P = 0.6601). P values determined by two-sided t-test.
These results bear a resemblance to the phenomenon of oncogene-induced senescence, where nonproliferating cells contribute to oncogenesis through secretion of factors into the surrounding tissue (Krtolica et al. 2001; Bavik et al. 2006; Liu and Hornsby 2007; Coppé et al. 2008). We investigated whether these cells secreted analytes characteristic of senescent cells, and tested by ELISA for a panel of 10 cytokines or chemokines (IL1α, IL1β, IL2, IL4, IL6, IL8, IL10, IFNγ, and TNFα) in media collected postirradiation over a 30-d period. Two cytokines—IL8 and IL1α—showed altered secretion patterns. Strikingly, the senescent wt/S34F mutant cells exhibited sustained secretion of IL8 for over 30 d, and this phenotype could be rescued by frame-shifting the mutant allele (Fig. 1B). This sustained secretion over ∼1 mo may be due to the increased viability of wt/S34F cells in response to irradiation. However, even at steady state (no irradiation), the wt/S34F mutant cells secreted elevated levels of IL8 compared with wt/wt cells (17-fold increase), and this phenomenon was rescued in the wt/S34F− line (Fig. 1C). We confirmed this observation in human lung cancer cell lines A549 and H441 and the myeloid leukemia cell line NKM1, which were either gene edited to create the S34F mutation or correct a naturally occurring U2AF1-S34F mutation: Each cell line secreted elevated levels of IL8 in a mutant-dependent manner (Fig. 1C). In contrast, the wt/S34F HBECs showed decreased secretion of IL1α relative to wt/wt cells before and after irradiation, an effect which was also rescued in wt/S34F− cells (Fig. 1C; Supplemental Fig. S1D). In summary, U2AF1-S34F cells show radiation resistance and altered secretion of inflammatory cytokines even before treatment with X-rays. Because of the role of inflammatory cytokines in cancer (Grivennikov et al. 2010; Taniguchi and Karin 2018), we chose to study this phenotype further.
U2AF1 is present in the cytoplasm and binds mature mRNA
Surprisingly, we observed no changes in IL8 splicing by RNA-seq (Fei et al. 2016). Moreover, we did not observe any significant change in IL8 expression between wt/wt and wt/S34F mutant cells as measured by single-molecule RNA-FISH and gene expression (RT-qPCR) analyses (Fig. 2A; Supplemental Fig. S2A). Previous studies have shown IL8 expression is regulated both at the level of mRNA stability and translational control (Fan et al. 2011; Herranz et al. 2015; Laberge et al. 2015). Since there was no change in IL8 mRNA levels, the elevated secretion of IL8 in wt/S34F cells is controlled either at the level of translation or trafficking/secretion. We therefore sought to determine whether U2AF1 could be playing a role in regulation of this message independent of splicing.
Figure 2.

U2AF1 binds RNA in the cytoplasm. (A) Single molecule FISH analysis of IL8 mRNA expression in wt/wt, wt/S34F mutant cells plotted as frequency histogram of IL8 mRNA per cell. (Inset) Single molecule RNAFISH image (gray) showing heterogeneity in IL8 RNA expression and nuclei counter stained with DAPI (blue) in wt/wt cells. (B) Quantitative immunofluorescence detection of cytoplasmic U2AF1 (gray) in PFA fixed cells showing heterogeneity in U2AF1 distribution in the cytoplasm and nuclei counterstained with DAPI (blue). (C) Box plot (min, max, first and third quartiles, and median) showing the fraction of U2AF1 in the nucleus and cytoplasm of individual wt/wt and wt/S34F cells. (D) Western blot analysis showing presence of U2AF1 in the cytosolic fraction. RBM10 and RPA32 (increasing amounts) served as controls for any nuclear protein contamination in the cytoplasmic fraction. (E) U2AF1 inteacts with mature IL8 mRNA in the cytoplasm. RNA isolated from immunoprecipitates of cytoplasmic U2AF1 (B) was analyzed by RT-qPCR for association of IL8, IL1α, DNA polH, and RPB3 mRNA. Fraction of IL1α, IL8, DNA polH, and RPB3 mRNA bound to U2AF1 (top) and their relative enrichment to U2AF1 in mutant cells (bottom). (F) Transcriptome-wide analysis of WT U2AF1 bound cytoplasmic mature polyadenylated mRNA by RIP-seq. Scatter plot of enrichment of U2AF1 bound RNA (gray), intron-less Histone RNA (red) and mTOR regulated RNA (blue). (G) Box plot analysis of mRNA enriched in RIP over input or control IgG in the top decile of RIP, mTOR sensitive mRNA, EIF4A sensitive mRNA, or background mRNA. (H) 3′ splice site-like “AG” sequence and polypyrimidine-rich TOP motifs in 5′-UTRs of mTOR-regulated mRNAs bound by cytoplasmic U2AF1. Gene ontology terms of wild-type U2AF1-enriched cytoplasmic mRNA.
We first asked whether U2AF1 interacts with IL8 mRNA. Fluorescently labeled U2AF1 has been shown to shuttle between the nucleus and cytoplasm in plant cells (Park et al. 2017) and using a heterokaryon assay in human cells (Gama-Carvalho et al. 2001). Also, splicing factors have been implicated in translational control (Sanford et al. 2004; Maslon et al. 2014; Aviner et al. 2017), leading us to ask whether there exists a cytoplasmic population of U2AF1 in human cells. We observed localization of U2AF1 in the cytoplasm of both wt/wt cells and S34F cells by immunostaining with ∼30% localized in the cytoplasm (Fig. 2B,C), and we could immunoprecipitate U2AF1 and U2AF2 from the cytoplasmic fraction with minimal nuclear contamination (Fig. 2D; Supplemental Fig. S2B). We note that wt/S34F cells are heterozygous and the antibody recognizes both wt and S34F forms of U2AF1. We next tested for the presence of mature spliced, polyadenylated mRNA in the cytoplasmic U2AF1-immunoprecipitates and detected the presence of mRNA for IL1α, IL8, and DNA pol H in substantial quantities, but very little of RPB3 mRNA (Fig. 2E; Supplemental Fig. S2C), suggesting preferential binding of U2AF1 to certain mRNAs and not others. In the wt/S34F mutant cells, binding of U2AF1 to IL8 mRNA was significantly lower (Fig. 2E; Supplemental Fig. S2D).
We next sought to generate a comprehensive transcriptome-wide view of U2AF1 interactions with RNA in the cytoplasm. RNA-IP under native conditions was performed on cytosolic extracts with anti-U2AF1 followed by sequencing (RIP-seq) of associated RNAs. Strikingly, we observed that mRNAs whose translation is regulated by mTOR are highly enriched in the U2AF1 RIP in wt/wt cells (Fig. 2F, blue circles; Supplemental Table S2). In fact, in a rank-ordered list of RIP/input for all messages exceeding a threshold for expression (1117 transcripts, RPKM > 1 in all fractions and samples; Supplemental Table S3) some of the top hits are mTOR-regulated mRNA (RPS6, IPO7, NPM1, RPS19). Moreover, mTOR-regulated mRNAs as a class (Thoreen et al. 2012) show greater enrichment in the U2AF1 IP than either the total pool of transcripts or another class of translationally regulated transcripts such as EIF4A-senstive mRNA (Fig. 2G; Rubio et al. 2014; Wolfe et al. 2014). IL8 did not meet our RIP-seq threshold criteria due to low expression levels but is reported to be sensitive to mTOR inhibitors (Herranz et al. 2015; Laberge et al. 2015). Finally, we took the top quintile of hits from lists ranked either by RIP/input or RIP/IgG and analyzed the GO terms. In both rankings the enriched transcripts are those involved in translational initiation and peptide biosynthesis (Fig. 2H). Thus, wt U2AF1 specifically binds mRNA in the cytosol, and many of these messages are mTOR-regulated and/or code for proteins involved in translation.
A noncanonical function for U2AF1 as a translational repressor
The decreased binding of U2AF1 to IL8 mRNA in wt/S34F cells, coupled with increased levels of IL8 in the media, led us to consider that U2AF1 might function as a translational repressor. To test this hypothesis we performed polysome profiling. We isolated polysomes from wt/wt, wt/S34F, and wt/S34F− mutant cells (Fig. 3A,B; Supplemental Fig. S3A) and determined the fractional amount of mRNA relative to monosomes. In addition to IL8 and IL1α, we tested the mRNAs of NPM1 and PABPC1, two mTOR-regulated RNAs that were either enriched or not enriched as measured by U2AF1 RIP-seq (Supplemental Table S3). NPM1 and IL8 show loss of binding in the wt/S34F cells and were enriched in the heavy polysome fractions (Fig. 3B; Supplemental Fig. S3B). PABPC1 mRNA shows no change (Fig 3B). The enhanced translational efficiency observed for NPM1 and IL8 mRNA by polysome analysis in mutant cells was also reflected in the steady state protein levels (Fig. 3C). We also tested steady state protein levels for several other genes: IPO7 (reduced binding in wt/S34F) and TSC2 (mTOR-sensitive but not detected above threshold in RIP) showed higher levels of protein; IL1α, RUNX1 and TRIM28 (no change or small gain in binding) showed no change or decreased translation, respectively (Fig. 3C).
Figure 3.

U2AF1 functions as a translational repressor in the cytoplasm. (A) 10%–50% linear sucrose gradient profile of polysomes isolated from wt/wt, wt/S34F, and wt/S34F− mutant cells. The positions of the small (S) and large (L) ribosomal subunits, monosomes, and polysomes are indicated. (B) Monosome and polysome fractions (three biological replicates) were subjected to semiquantitative RT-PCR for indicated genes, products fractionated on a 1.5% agarose gel and quantified. The mRNA in individual fractions normalized to the total in all fractions and the ratio of mRNA to monosome in the individual fractions plotted (average of three independent experiments ±SE). (C) Box plot analysis of normalized integrated fluorescence intensity (min, max, first and third quartiles, and median) after immunostaining PFA fixed wt/wt (black) and wt/S34F (green) cells for IL8, NPM1, IPO7, TSC2, RUNX1, and TRIM28. P values determined by two sided t-test.
We then tested for a direct repressive role of U2AF1 in translation that might be altered by the S34F mutation by sequencing the mRNA isolated from polysome fractions (Supplemental Table S4). Individual polysome profiles are shown in Figure 4A. NPM1, IL8, and IPO7 translational changes in wt/S34F cells were recapitulated in the genome-wide assay, while PABPC1 showed no change in translational efficiency with mutation status. The cytoplasmic binding targets of U2AF1 from RIP-seq were then rank-ordered by loss of binding which occurred in wt/S34F cells and divided into deciles (n ∼ 112 transcripts/decile, Fig. 4B). The average polysome profiles from the top and bottom deciles demonstrate that the transcripts that showed the greatest change in binding (decile 1) also showed a polysome profile that is characteristic of more efficient translation (Fig. 4C). In fact, when looking at a single ratio of polysome to monosomes over all deciles, we observe a systematic trend where loss of binding as measured by RIP correlates with an increase in polysome/monosme ratio, with the top two deciles showing the most pronounced effect (Fig. 4D, green). This effect is not observed in wt/wt cells (gray) and is rescued in wt/S34F− cells (red) (Fig. 4D). Supporting this result, when transcripts are rank ordered by enrichment in the U2AF1 RIP in wt/wt cells, there is a systematic trend toward less binding associated with greater translation (Fig. 4E,F). Thus, in independent measurements of RIP-seq and POLY-seq, U2AF1-bound transcripts show lower translation.
Figure 4.

U2AF1 directly represses translation of hundreds of genes. (A) Polysome profiles of indicated genes showing their enhanced distribution in heavy polysomes from wt/S34F cells. Normalization to unit area for each sample. (B) Rank order of S34F mutant-associated change in U2AF1 binding in RIP samples and binned into deciles (∼112 transcripts/decile). Bootstrap error bars. (C) Altered translation (sequence counts) of all mRNAs in the top (left plot) and bottom (right plot) deciles in B. (D) Change in translation (polysomes to monosome ratio) of all mRNAs in each decile (B) from wt/S34F (green), wt/wt (gray), and wt.S34F− (red) cells. (E) Rank order of U2AF1-associated mRNA from wt/wt cells relative to input and binned into deciles as in B. (F) Polysome to monosome ratio of mRNA in all deciles from E showing reduced binding by U2AF1 to mRNA in wt/wt cells does correlate with enhanced translation. (G) Polysome to monosome ratio for several different groups of transcripts. Each group contains three boxes reflecting the distribution of polysome/monosome ratios (from left to right): wt/wt, wt/S34F, wt/S34F−. The transcript groups, from left to right, are decile 1 of changes in RIP/input from wt/wt to wt/S34F, decile 10, mTOR-sensitive transcripts, and EIF4A-sensitive transcripts. (**) P < 0.001; (*)P < 0.05.
Loss of U2AF1 binding in wt/S34F cells is correlated with gain in translation efficiency on hundreds of messages, suggesting a functional association between U2AF1 and translational repression which is altered by the S34F mutation. The targets of this translational repression pathway in wt/wt cells are enriched in messages which themselves code for translation machinery (Fig. 2H), potentially resulting in direct and indirect changes in translation. U2AF1-bound messages are enriched for GO categories associated with multiple aspects of translation, but the messages that show the largest change in binding and translation with the S34F mutation are in a subset of these categories. For example, several translation initiation factors (EIF2S2, EIF5, and EIF4A2) show change in binding accompanied by translational up-regulation, along with proteins involved in cotranslational protein targeting (SRP72 and SSR3) and intracellular transport (IPO5, IPO7, XPOT, and NPM1) (Supplemental Tables S3, S4; Supplemental Fig. S4). Based on this observation, we checked other groups of messages known to be translationally regulated (Fig. 4G) and observed that EIF4A-regulated messages show increased translation in wt/S34F cells, despite not being direct binding targets (Fig. 2G) or showing changes in binding in the presence of S34F (Supplemental Fig. S4F). This result, however, is consistent with the change in EIF4A2 translation. Thus, there is the potential for widespread changes in the proteome due to the S34F mutation. Moreover, these data suggest a direction of causality between binding and translation: Loss of binding is predictive of increases in translation, but increases in translation are not predictive of loss of binding.
U2AF1-U2AF2 heterodimer directly binds mRNA in the cytoplasm
To investigate whether U2AF1 directly binds mRNA in the cytoplasm, we turned to photocativatable ribonucleoside enhanced cross-linking and immunoprecipitation (PAR-CLIP) analysis that allows for transcriptome-wide identification of protein–RNA interactions at nucleotide resolution (Hafner et al. 2010, 2012; Benhalevy et al. 2017). PAR-CLIP relies on the incorporation of the photocross linkable nucleoside analog 4-thiouridine (4-SU) into nascent RNA, and the specific site of protein-nucleic acid cross-linking is revealed by a characteristic T-to-C mutation in the sequenced cDNA. The frequency and density of T-to-C mutations is further used to computationally filter binding sites from the background of copurified fragments of abundant cellular RNAs (Corcoran et al. 2011).
We created stable HEK 293 (HEK T-REx Flp-In-293 cells) cell lines that conditionally expresses N-terminally Flag-HA tagged WT (FH-U2AF1-WT) or S34F mutant U2AF1 (FH-U2AF1-S34F) and used them in several variations of PAR-CLIP. Using biochemical fractionation, we isolated chromatin-associated, nuclear, and cytosolic cellular-cross-linked U2AF1-RNPs (Fig. 5A). Autoradiography of the immunoprecipitated and radiolabeled complexes fractionated by SDS-PAGE revealed in both cytoplasmic and nuclear fractions two bands of similar intensity migrating at ∼35 and ∼70 kDa, corresponding to RNPs containing U2AF1 and most likely its interaction partner U2AF2 (Fig. 5B). This data suggests that in the cytoplasm U2AF1 bound RNA in a heterodimeric complex with U2AF2, which is consistent with our observation that U2AF2 is present in the cytoplasm (Fig. 2D). Interestingly, immunoprecipitation of cross-linked U2AF1 RNPs from chromatin did not result in coprecipitation of the U2AF2 RNP; it remains unclear whether this difference was biological or merely reflects that the U2AF heterodimer dissociated under harsher experimental conditions for chromatin isolation. Ligation of 29-nt long 3′ adapters to the 3′ end of the protected RNA fragments of these RNPs resulted in further retention of the U2AF complexes on the SDS-PAGE (Supplemental Fig. S5A), further demonstrating that both U2AF1 and U2AF2 were cross-linked to RNA.
Figure 5.
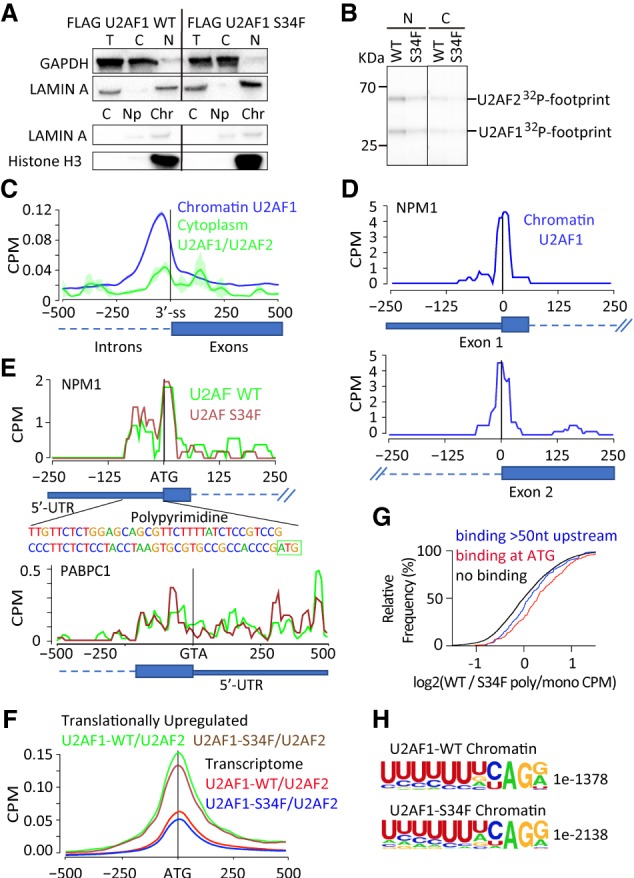
Direct association of U2AF1-U2AF2 heterodimer at start codons of translationally regulated mRNAs. (A) Western blot confirming fractionation of UV cross-linked 4SU treated HEK-293 cells expressing either WT- or S34F−U2AF1 to obtain cytoplasmic, total nuclear, or chromatin-associated RBPs [(T) total, (C) cytoplasmic, (N) nuclear, (NP) nucleoplasm, (Chr) chromatin]. (B) SDS-PAGE of 32P-labeled RBP-cross-linked RNA from the cytoplasmic fraction showing association of U2AF2 with U2AF1 in the cytoplasm after direct release from anti-Flag antibody beads. [(N) nuclear, (C) cytoplasmic, (Chr) chromatin]. (C) Enrichment of U2AF1 to 3′ splice sites of pre-mRNA in the chromatin fraction from the nucleus and lack thereof in the mature RNA in the cytoplasmic fraction. (D) Sequence coverage map with the characteristic “T to C” mutation upon binding of U2AF1 to the first exon and 3′ splice sites of NPM1 pre-mRNA in the chromatin fraction. (E) PAR-CLIP sequence coverage map with the characteristic “T to C” mutation showing binding of U2AF1-WT/U2AF2 and U2AF1-S34F/U2AF2 at the start codon and the 5′-UTR of NPM1 mRNA in the cytoplasm, and the lack thereof on the first exon of PABPC1 mRNA. The polypyrimidine tract in the 5′-UTR and the ATG start codon are indicated on the sequence. (F) PAR-CLIP sequence coverage showing altered binding by the S34F mutant at or near the start codon of translationally up-regulated genes (WT, Green; S34F, Brown), and the entire transcriptome (WT, Red; S34F, Blue). (G) Cumulative distribution showing that genes bound by WT-U2AF1 at or near the ATG codon is a predictor of translation up-regulation in wt/S34F cells (no binding at start codon, Black, n = 6211; binding at start codon, Red, n = 343, median = 0.29, P-value = 2.6 × 10−11; binding >50 nt upstream, Blue, n = 134, median = 0.11, P-value = 0.083). (H) MEME motif analysis of binding sites in 5′-UTRs showing the presence of a polypyrimidine tract, the binding site for U2AF2, and the characteristic CAG binding motif of U2AF1.
Since we did not detect any contaminating copurifying RNPs in our PAR-CLIP experiments, we directly isolated RNA from the immunoprecipitated U2AF complexes (U2AF1/2) without further purification and converted them into small RNA cDNA libraries for deep sequencing. Genome-aligned reads were grouped into clusters by PARalyzer (Corcoran et al. 2011) to identify those enriched for cross-link-induced T-to-C mutations. Altogether, we identified ∼96,000 and 26,000 clusters for U2AF1 WT and ∼45,000 and 28,000 clusters for U2AF1 S34F mapping to mRNA from complexes recovered from the chromatin and cytoplasm, respectively. As expected, we observed robust enrichment of 3′ splice sites in U2AF1 PAR-CLIP clusters in the chromatin fraction (Fig. 5C). In contrast, in the cytoplasm, U2AF binding sites distributed across the mRNA without enrichment at 3′ splice sites (Fig. 5C), consistent with the lack of intron/exon boundaries in cytosolic mRNA.
Since HEK 293 cells do not express IL8, we chose to investigate whether NPM1 was also bound by U2AF1 in PAR-CLIP experiments. NPM1 showed substantially reduced binding to U2AF1 in RIP-seq experiments and increased association with polysomes in the wt/S34F cell line. Indeed, chromatin-associated U2AF1 bound NPM1 mRNA at multiple sites, including at the 3′-ss located at the boundary between intron 1 and exon 2 (Fig. 5D; Supplemental Fig. S5B). Surprisingly, we also observed U2AF1/2 binding to the start codon and immediately upstream in the 5′-UTR in exon 1 of NPM1. U2AF1/2 association to the NPM1 start codon was preserved in the cytoplasm (Fig. 5E; Supplemental Fig. S5B), and further analysis of the NPM1 5'-UTR sequence context revealed a polypyrimidine tract followed by an “AG” dinucleotide located slightly upstream of the start codon, consistent with the primary sequence requirements for U2AF1/2 binding at 3′ splice sites. On the contrary, PABPC1, an mTOR-regulated RNA that was neither enriched in our RIP assay nor found to be translationally regulated by expression of U2AF1 S34F in our polysome profiling or sequencing assay showed no binding of the U2AF complex at or close to the start codon, indicative of the specificity of the interaction. The presence of U2AF at start codons for select messages suggests a possible regulatory role for this complex during translation initiation, which we interrogate in greater detail below.
Direct binding of U2AF at or near start codons of select mRNAs results in their translational repression
To determine whether there was any correlation between association of U2AF complex with 5′-UTRs and translational repression, we compiled all the genes that show increased translation in wt/S34F cells (Figs. 3, 4; Supplemental Figs. S3, S4) and aligned them centered at the start codon. Gene targets that were translationally up-regulated in wt/S34F cells compared with wt/wt HBECs showed an approximately threefold increase in binding by WT U2AF in the PAR-CLIP assay relative to other U2AF target mRNAs (Fig. 5F). Likewise, S34F U2AF still bound these translationally up-regulated messages compared with other target mRNAs, but there was a slight reduction compared with WT U2AF1/2. Strikingly, binding of WT U2AF1/2 at the start codon alone could independently predict whether a message would show translational up-regulation in the wt/S34F cells (Fig. 5G). The cumulative distribution indicates that when WT U2AF1/2 is not bound at the start codon (black line), there is roughly a 50/50 chance of seeing an increase in the polysome/monosome ratio in the wt/S34F cells. In contrast, when WT U2AF1/2 is bound at the start codon, one is much more likely to see those messages show increases in translation in a cell line that contains the S34F mutation, as evidenced by the rightward shift in the red curve. Even in comparison to a control analysis where messages are stratified by WT U2AF1/2 binding slightly upstream in the 5′-UTR (>50 nt, blue curve), the functional importance of U2AF binding to the ATG start codon is apparent. Taken together, our results suggest that binding of the U2AF complex around the start codon is functional for translational repression.
Lastly, MEME motif analysis of 5′-UTRs from translationally regulated target genes returned a polypyrimidine tract followed by a “AG” dinucleotide upstream of or close to the ATG codon (Fig. 5H), both bona fide binding motifs of U2AF2 and U2AF1, respectively, at 3′ splice sites. We note that this analysis integrated results from different high-throughput experiments (PAR-CLIP, RIP-seq, and polysome sequencing) performed across different cell lines and thus attests to the generality of our conclusions. Taken together, these PAR-CLIP studies identify a class of mRNA which show specific binding of U2AF1/2 near the ATG start codon, and this binding—independently of any other experimental measure—can predict the translational response in another cell line containing the S34F mutation.
U2AF1 regulates translation of IL8 via its 5′-UTR
Because we identified a role for U2AF1 in translation repression and discovered binding sites in the 5′-UTR and also near the start codon, we analyzed 5′-UTRs for the enrichment of the U2AF1 motif and found it to be pervasive, with 4608 out of 6441 experimentally determined 5′ UTRs containing the motif (FIMO, P < 0.01). We also identified a number of 3′-ss-like sequences in the IL8 5′ UTR. Since IL8 did not appear in most of our genome-wide analyses due to low expression levels, we sought to experimentally test whether these motifs could be functioning as cis-acting U2AF1 binding sites. We made several IL8-T2A-GFP reporters (Fig. 6A; Supplemental Fig. S6) with either the wild-type 5'-UTR of IL8 or mutations in 3′-ss-like sequences. This reporter contains no introns, and the GFP is not secreted, thereby testing the translational control mechanism in a splicing- and secretion-independent manner. We found that a point mutant in the canonical 3′-ss nearest the 5′ end (changing the trinucleotide sequence from “CAG” to “CAT”) showed translation derepression. wt/wt and wt/S34F mutant cells carrying either the wild-type or mutant IL8 reporter had similar levels of RNA expression (Fig 6B), and FACS analysis revealed that the wt/S34F mutant cells showed an increase (∼1.4-fold) in the fraction of the cells expressing GFP (Fig. 6C–E; Supplemental Fig. S6), consistent with our earlier findings (Fig. 3C; Supplemental Fig. S3C). Strikingly, we observed a fivefold or 10-fold increase for the 5′ UTR mutant IL8 reporter in cells that were either wt/wt or wt/S34F for U2AF1, respectively (Fig. 6C–E), indicating that a point mutation in the U2AF1 motif in the IL8 5'-UTR was sufficient to overcome translational repression. In addition, altering the 5′-UTR with multiple mutations either completely abolished translation (Supplemental Fig. S6B; 5′-UTR mt1) or enhanced it (Supplemental Fig. S6B; 5′-UTR mt2 and mt3), suggesting a significant regulatory role of the 5′-UTR in IL8 translation. The mutated 5′-UTR significantly compromised U2AF1 binding in both wt/wt and wt/S34F cells (Fig. 6F). However, there does not appear to be a quantitative relationship between the extent of U2AF1 binding loss (less than twofold) and the increase in translation of the reporter (approximately sixfold to 10-fold), suggesting possible cooperative effects on translational regulation.
Figure 6.
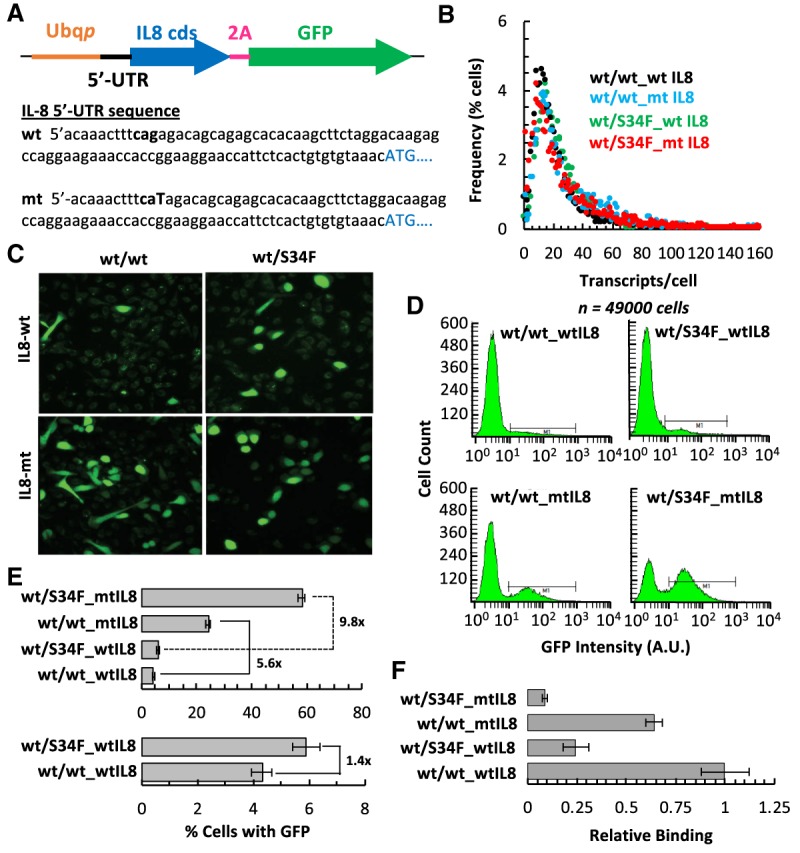
U2AF1 controls translation of IL8 via its 5′-UTR. (A) Schematic representation of the IL8-2A-GFP reporter construct. The wild-type IL8-5′-UTR sequence with the putative 3′ splice site-like sequence depicted in bold. The mutated base within the 3′ splice site in the mutant sequence is capitalized. (B) Quantitative smFISh analysis of reporter gene expression using probes specific to GFP and represented as a frequency histogram of the number of transcripts per cell. (C) Confocal live cell image of GFP expression from the wt- or mt-IL8-2A-GFP reporter integrated into wt/wt or wt/S34F mutant cells. (D) FACS of wt/wt or wt/S34F cells expressing wt- or mt-5′-UTR IL8-2A-GFP reporter gated above auto fluorescence from wt/wt or wt/S34F mutant control cells, respectively. (E) Percent cells expressing GFP from the wt- or mt-5′-UTR IL8-2A-GFP reporter as a function of wild-type or S34F U2AF1 status (mean of three biological replicates ±SD). (F) Association of wt- or mt-IL8-2A-GFP mRNA to U2AF1 in vivo as a function of wild-type or S34F U2AF1 status (mean of three biological replicates ±SD).
Overall, our data indicate that U2AF1 represses translation in the cytosol through a direct interaction with mRNA, that this repression is relieved in the presence of the S34F mutation on hundreds of messages, and that repression is mediated for IL8 by a cis-acting sequence in the 5′ UTR.
Elevated IL8 is associated with multiple measures of cancer progression
We next asked whether this change in IL8 translation was sufficient to mediate phenotypic consequences. We carried out experiments on EMT, inflammatory response, and tumor burden during cancer progression. EMT is a critical step in metastasis (Kalluri 2009; Thiery et al. 2009; Fernando et al. 2011; Shen et al. 2014; Ortiz-Montero et al. 2017), known to be induced by IL8 (Fernando et al. 2011; Shen et al. 2014; Ortiz-Montero et al. 2017), and is characterized by the induction of fibronectin and repression of E-Cadherin expression in epithelial cells (Kalluri 2009; Thiery et al. 2009). Purified recombinant IL8 by itself elicited EMT in MCF-7 cells (Fig. 7A,B), as determined from the emergence of a fibronectin-high and E-cadherin-low population in tissue culture, as observed previously. Conditioned media from the wt/S34F mutant cells also induced the expression of fibronectin and repression of E-cadherin, but the conditioned media from wt/wt cells did not (Fig. 7A,B). These data indicate that the media from wt/S34F cells was biologically active and was sufficient to induce EMT-related gene expression changes.
Figure 7.

IL8 can induce EMT, inflammation and tumor progression. (A) MCF-7 cells were cultured in either control, control + IL8 (1 nM), or conditioned media (EMT assay) from WT or S34F mutant cells. After 10 d in culture, cells were immunostained for E-cadherin (green) and Fibronectin (red). (B) Scatter plot showing EMT-associated changes in E-cadherin and Fibronectin expression observed by quantitative immunofluorescence detection of E-cadherin and Fibronectin in PFA fixed individual cells from A. (C) Representative H&E stained images (top panel) showing exent of peri- or intratumoral inflammation response in xenograft tumors and its quantitation (bottom panel). Scale bar, 20 µm. The number of independent tumors: H441 (wt/S34F), n = 16; H441 (wt/S34F−), n = 20; HCC78 (wt/S34F), n = 14; HCC78 (wt/S34F−), n = 9. P-values were determined by χ2 test. (D) Serum IL8 levels (gray bar) and the number of macroscopic liver nodules (black circles) from NOG mice injected (tail vein) with H441 isogenic cells, and treated with either isotype control or anti-IL8 antibodies for 10 wk (n = 3 for each treatment group). Two naïve NOG mice (the two samples on the left) were used as negative controls. (E) Principal component analysis of 12 cytokine levels in bone marrow from healthy and relapsed or refractory acute myeloid leukemia donors (RR-AML). (F) Heatmap showing the clustering of cytokine measurements. Rows are centered with unit variance scaling applied to rows. Both rows and columns are clustered using Euclidean distance and average linkage. The tree ordering for rows and columns places the higher median first. Patient and spliceosome mutational status are not part of the clustering and are indicated on the right. (G) IL8/VEGF ratio and IL8 levels from healthy and AML individuals. VEGF and IL8 account for 96% of the variation within the samples.
We then examined the role of the U2AF1-S34F mutation using xenografts. We used two different human lung cancer lines (HCC78 and H441) with naturally occurring S34F mutations isolated from patients and generated derived lines with frame-shifted mutant alleles as previously reported (Fei et al. 2016). H441 cells with or without S34F mutation were able to form subcutaneous xenograft tumors in athymic nude mice with no apparent differences in tumor sizes, while HCC78-derived cells were unable to establish tumor growth over a period of 150 d (Fei et al. 2016). Presentation of inflammatory cells at the periphery or inside the xenograft tumors (from H441) cells or residue cells (from HCC78 cells) were scored. In both cases, the occurrence of the tumor-induced inflammation was reduced by removing the S34F mutant copy from the human cell lines (Fig. 7C).
Next, we sought to assess the specific role of IL8 in mediating the ability of H441 cancer cells to form tumors in mice. One caveat in these studies is that IL8 is not present in rodents, but homologs of the IL8 receptors (CXCR1 and CXCR2) are present. NOG mice were injected with H441 cells via tail vein and were treated with neutralizing IL8 antibody or with an isotype-matched IgG three times a week for 10 wk. Serum IL8 levels and tumor burden were then assessed after the mice were sacrificed 3 d after the last antibody injection. Half of the mice (n = 3) were given intraperitoneal injections of the anti-IL8 neutralizing antibody, and the other half were injected with the control IgG. Tumors were observed in the liver, but predominantly in the lung (Fig. 7D; Supplemental Fig. S7). Although lung-tumor burden did not change (Supplemental Fig. S7), the tumor burden in the liver showed a significant decrease with a corresponding decrease in serum levels of IL8 (Fig. 7D). Taken together, studies in tissue culture and xenograft models demonstrate that EMT, immune infiltration, and tumor burden are sensitive to the U2AF1 mutational status, and these effects are consistent with and/or mediated by changes in IL8.
Finally, we asked whether IL8 is elevated in relapsed/refractory acute myeloid leukemia (RR-AML), where spliceosome mutations are common (Papaemmanuil et al. 2016). We isolated serum from 24 RR-AML patients and 20 age-matched healthy donors. A third of the AML patients had known spliceosome mutations (U2AF1, n = 3; SRSF2, n = 3; and SF3B1, n = 2). Levels of 12 cytokines/growth factors were quantified using a Luminex assay (Supplemental Table S5). Principal component analysis (Metsalu and Vilo 2015) revealed that 96% of the variation in samples could be accounted for by two factors: VEGF and IL8 (Fig. 7E). Healthy individuals showed high VEGF and low IL8 levels, while the RR-AML patients showed low VEGF and high IL8 levels (Fig. 7F). IL8 alone is significantly elevated in RR-AML patients compared with healthy donors (P = 0.017, Fig. 7G), and the ratio of IL8/VEGF allows superior discrimination (P = 0.0007). On this “axis” of IL8/VEGF, U2AF1 mutations cluster at an intermediate point, where VEGF is decreasing and IL8 is increasing (Fig. 7F). Thus, U2AF1 S34F is not associated with the highest levels of IL8 in human AML but may function in a general pathway whereby IL8 begins to condition the niche which supports proliferation of leukemic blasts.
Discussion
Here we show that U2AF1 can function in a noncanonical role as a translational repressor in the cytosol. This pathway was revealed by the somatic S34F mutation which occurs in cancer, resulting in translational derepression of hundreds of mRNA, one of which (IL8) directly contributes to the oncogenic phenotype. Importantly, the phenotypes and the translational changes we report are rescued by frameshifting the mutant allele, arguing for a gain-of-function dominant-negative effect rather than simple loss-of-function or lower binding. Critically, we demonstrate a direct interaction between the U2AF1/2 heterodimer and cytosolic mRNA. We discovered a population of messages which show binding in the 5′-UTR, often proximal to the start codon, and these messages are also the ones that show translational changes in the heterozygous mutant cells.
The precise mechanism of action remains to be determined. The transcripts which are translationally up-regulated in wt/S34F cells share two common features, determined from independent assays: (1) direct interaction of U2AF1/2 with RNA near the start codon by PAR-CLIP and (2) decrease in binding by native RIP. Moreover, for IL8 we showed that mutation of an AG dinucleotide in the 5′-UTR resulted in loss of binding of U2AF1—both WT and S34F—and translational up-regulation. Yet, the position and amplitude difference between WT and S34F binding by PAR-CLIP are modest (i.e., Fig 5E,F). Also, the binding motifs in the chromatin-bound fraction do not show profound differences. It may be that there are subtle differences in the exact location of binding which might effect the efficiency of ribosome scanning or translation initiation. In addition, how U2AF1 finds its targets or shuttles between the nucleus and cytoplasm is unknown, but shuttling of U2AF1 and other splicing factors has been reported previously (Gama-Carvalho et al. 2001, 2006; Park et al. 2017). Likewise, there are reports of a role for splicing factors in translation mediated by direct interaction between SR proteins and the ribosome (Sanford et al. 2004; Maslon et al. 2014).
U2AF1 selectively targets transcripts that code for the translation machinery, and the transcripts that show the greatest derepression in the presence of the S34F mutation are involved in protein transport. Thus, there is the potential for both direct and indirect changes to the proteome. In fact, the substantial change we see in extracellular IL8 may be due both to direct changes due to decreased U2AF1 binding to the IL8 5′-UTR and also changes in translation, processing, and trafficking of mature IL8, which are indirect effects. IL8 is correlated with worse clinical outcomes in lung cancer (Ryan et al. 2014) and AML (Schinke et al. 2015) and our data provide a direct link between U2AF1 mutations and these clinical phenotypes. Since IL8 also contributes to recruitment of myeloid cells that suppress an adaptive immune response (Alfaro et al. 2016; Dominguez et al. 2017), one prediction is that cells with U2AF1 mutations or perhaps splicing factor mutations in general will be refractory to immunotherapy unless treated with neutralizing IL8 antibody. Importantly, the S34F mutation in U2AF1 does not lead to a general induction of senescence or the senescence-associated secretory phenotype. We detect neither a growth arrest (Fei et al. 2016) nor β-galactosidase staining in HBECs under untreated conditions. Although we discovered this pathway through an altered DNA damage response, the change in secretion is present before DNA damage and, in fact, the opposing changes in IL8 and IL1α are fundamentally different from the normal SASP response (Lasry and Ben-Neriah 2015). Nevertheless, the sustained viability after genotoxic stress leads to a population of cells which secrete IL8 for >30 d.
Translation initiation is often misregulated in cancer (Mamane et al. 2006) and the translation machinery is tightly controlled at the posttranscriptional level through the mTOR pathway, which is responsive to an array of environmental conditions (Laplante and Sabatini 2012). Here, we focus primarily on the cell nonautonomous effects of translational misregulation such as enhanced EMT and immune infiltration mediated by elevated IL8 levels even in the absence of stress. However, we speculate that the U2AF1 S34F mutation could also function in a cell-autonomous, progrowth manner in tissue through this mechanism of translational misregulation. For example, although we did not observe a growth advantage for WT/S34F under normal cell culture conditions, nutrient-limited or hypoxic conditions in tissue might provide a clonal growth advantage to cells with the S34F mutation and the subsequent disruption of translational regulation. The fact that this mutation is often found with a variant allele frequency near 0.5 suggests either an initiating event or early progression event resulting in clonal expansion of the mutant cells (Abelson et al. 2018).
Changes in the translational machinery might be particularly relevant for myelodysplasia, where splicing factor mutations are common (Haferlach et al. 2014). Myelodysplasia is a disease of differentiation. Acquired somatic mutations in hematopoietic stem cells result in a hypercellular marrow but ineffective hematopoiesis and cytopenia. One form of MDS (5q-MDS) is thought to be causally related to a deletion of the RPS14 gene, involved in ribosome biogenesis (Ebert et al. 2008). However, there are a number of related inherited bone-marrow failure syndromes such as Diamond-Blackfan Anemia, Shwachman-Diamond Syndrome, X-linked dyskeratosis congenita, and cartilage hair hypoplasia that also involve alterations in the ribosome machinery and are collectively referred to as “ribosomopathies” (Narla and Ebert 2010). It is unknown why germ-line alterations in the translational machinery result in specific hematopoietic defects, but one hypothesis is that rapidly proliferating hematopoietic progenitors might be particularly reliant on translational regulation and/or capacity. We speculate that U2AF1 mutations and the associated translational misregulation might operate in a mechanistically similar way.
Materials and methods
Plasmids, cell lines, and cell cultures
The plasmid expressing the IL8-2A-GFP reporters were constructed by inserting a synthetic DNA fragment coding for wt or mutant IL8-2A-GFP between NotI and SphI sites of a lentiviral packaging vector. Transcription from this reporter is driven off an Ubiquitin promoter. Construction of WT, S34F, and frameshift HBEC lines has been described previously (Fei et al. 2016). To generate reporter cell lines, WT or S34F mutant HBEC lines (Fei et al. 2016) were transduced with different titers of lentivirus packaged with either WT or mt 5′-UTR IL8-2A-GFP reporter constructs. Reporter RNA expression was measured by smFISH using fluorescently labeled probes (Biosearch Technologies) against GFP and cell populations with similar RNA levels were selected and expanded for further use. All HBEC lines were cultured in keratinocyte serum-free medium supplemented with bovine pituitary extract and EGF as prescribed by the manufacturer (Invitrogen) at 37°C and 5% CO2.
Irradiation and WST-1 assay
WT or S34F mutant cells (1 × 104/well) were plated in 96-well plates and grown for 24 h before irradiation with x-rays (20Gy) (X-Rad 320, Precision X-Ray) and cultured for 6 d after irradiation. Cell viability was measured at indicated times using the WST-1 (Roche/Sigma) assay measuring the conversion of tetrazolium salts to formazan by cellular tetrazolium reductase as per manufacturer's instructions for 1 h at 37°C and 5% CO2.
ELISA
HBECs were plated (1 × 106/well) in a six-well plate and irradiated with 20 Gy of X-rays. At indicated times, medium was collected, filtered for cell debris using a 0.22-mm filter, and snap-frozen until ELISA was performed. ELISA was performed in duplicate for each sample using Quansys Human Cytokine-Inflammation 9-plex kit (Quansys Biosciences) containing IL1α, IL1β, IL2, IL4, IL6, IL8, IL10, IFNγ, and TNFα. The cytokine and chemokine levels were determined from the standard curve developed with each experiment using standards provided by the manufacturer. The anylate levels (picograms per milliliter) are the mean of three independent experiments with standard deviation.
Immunofluorescence assay, image acquisition and analysis
Cells were seeded at ∼30% confluency on 1.5-mm thick round cover glass in 12-well plates and cultured under normal growth conditions (5% CO2, 37°C) up to ∼80% confluency. Cells were washed once with PBS and fixed in 4% PFA (made in PBS) for 15 min at 22°C. Cells were then washed thrice with PBS for 10 min each at 22°C and permeabilized in 0.1% Triton X-100 (in PBS) for 15 min at 22°C. Cells were washed thrice in PBS, blocked with 5% normal calf serum (in PBS) for 2 h at 22°C and incubated in primary antibody diluted in 5% normal calf serum at 4°C overnight. Cells were washed thrice in PBS and incubated with fluorescently labeled secondary antibodies diluted in 5% normal calf serum for 1 h at 22°C, washed thrice in PBS, then cover-glass mounted on a glass slide using ProLong Gold with DAPI (Invitrogen) and cured overnight at 22°C. The slides were imaged on a custom-built epifluorescence Rapid Automated Modular Mounting (RAMM Scope, ASI) base mounted with a Plan-Apochromatic 40X (NA 1.4) objective (Zeiss), illuminated with a LED light source (Model Spectra-6LCR-SA, Lumencor) and the emitted fluorescence collected with a CMOS camera (Hamamatsu ORCA-Flash 4.0). Z-stacks (0.5 µM) were acquired to cover the entire thickness of the cells and the stacks were sum projected. Cells or nuclei were segmented and the mean fluorescence intensity calculated using the open source software, CellProfiler 2.1.1 (http://cellprofiler.org). Source and dilutions of primary and secondary antibodies used for immunofluorescence: IL8 (1:1000; eBioscience, BMS136), mH2A1.1 (1:1500; Cell Signaling Technologies, D5F6N), mH2A1.2 (1:1000; EMD Millipore, MABE61), NPM1 (1:1000; Abcam, AB10530), IPO7 (1:1000; Invitrogen, PAS21764), U2AF1 (1:1500; Abcam, AB172614), U2AF2 (1:1000; Santa Cruz Biotechnology, SC53942), TSC2 (1:1000; ThermoFisher, 37-0500), RUNX1 (1:1000; ThermoFisher, MA5-15814), TRIM28 (1:1000; ThermoFisher, MA1-2023), fibronectin (1:1500; Abcam, AB23750), E-cadherin (1:1000; Life Technologies, 131700), antimouse IgG-Alexa 570 (1:2500; Life Technologies, A11004), and antirabbit IgG-Alexa 670 (1:2500; Life Technologies, A21244).
Single molecule RNA fluorescence in situ hybridization (smRNA FISH)
Cells were plated at ∼30% confluency on 1.5-mm thick round cover glass in 12-well plates and cultured under normal growth conditions (5% CO2, 37°C) up to ∼80% confluency. Cells were washed once with PBS, fixed in 4% PFA (made in PBS) for 15 min at 22°C, and smRNA FISH performed using fluorescent probes directed against IL8 mRNA or GFP mRNA as described previously (Palangat and Larson 2016). Cells and nuclei were segmented using CellProfiler 2.1.1, single molecule RNA localization in cells and transcripts/cell calculated using software custom written in IDL (http://www.harrisgeospatial.com/SoftwareTechnology/IDL) and available at http://www.larsonlab.net.
RNA immunoprecipitation
Cells were grown to ∼90% confluency, and 5 × 107 cells were used for isolating cytoplasmic fraction as described previously (Gagnon et al. 2014). Briefly, the cells were washed in ice-cold PBS, suspended (1 mL of buffer/50 mg of cells) in hypotonic lysis buffer (10 mM Tris-HCl at pH 7.5, 10 mM NaCl, 3 mM MgCl2, 0.1% NP-40, 10% glycerol) supplemented with protease inhibitors (Roche-Sigma) and RNASin (Promega) as suggested by the manufacturer, and lysed for 10 min on ice. The cell suspension was centrifuged at 800g for 8 min at 4°C, and the supernatant was collected as cytoplasmic fraction. The cytoplasmic fraction was adjusted to 150 mM NaCl by addition of 5 M NaCl and centrifuged at 18000g for 15 min at 4°C. The supernatant was first precleared with antirabbit IgG-conjugated Dynabeads (Invitrogen) for 2 h at 4°C, and U2AF1-associated RNA was then immunoprecipitated from the cytoplasm (overnight at 4°C) with anti-U2AF1 antibodies (Abcam, AB172614). The immunoprecipitate was then adsorbed on antirabbit IgG-conjugated Dynabeads at twofold excess of secondary antibody bound beads relative to primary antibodies for 2 h at 4°C. The beads were washed four times with high-salt buffer (50 mM Tris-HCl at pH 7.5, 500 mM NaCl, 4 mM MgCl2, 0.05% NP-40) followed by four washes with low-salt buffer (50 mM Tris-HCl at pH 7.5, 150 mM NaCl, 4 mM MgCl2, 0.05% NP-40). The samples were suspended in low-salt buffer and used for RNA preparation or fractionation on polyacrylamide gels for Western blot analysis and probed with indicated antibodies.
Isolation of polysomes
Polysomes were isolated from cells cultured in one 10-cm dish at ∼80% confluency as described in Panda et al. (2017). The cells were incubated in medium containing 10 µg/mL cycloheximide for 10 min under normal culture conditions, washed with PBS, trypsinized, washed with ice-cold PBS containing 10 µg/mL cycloheximide, and lysed in lysis buffer (20 mM Tris at pH 7.2, 130 mM KCl, 15 mM MgCl2, 0.5% [v/v] NP-40, 0.2 mg/mL heparin, protease inhibitors (Sigma), RNASin (Promega), 2.5 mM DTT, 0.5% deoxycholic acid, and 10 µg/mL cycloheximide. The extract was clarified by centrifugation (8000g for 10 min at 4°C). The clarified lysate was loaded onto two 10%–50% linear sucrose gradient (10 mM Tris at pH 7.2, 60 mM KCl, 15 mM MgCl2, 1 mM DTT, 0.5% [v/v] NP-40, 0.1 mg/mL heparin) and centrifuged (40,000 rpm for 2 h at 4°C) in an SW41 Ti rotor (Beckman). UV profiling (254 nm) of ribosomes and fractionation of polysomes was done using a fractionator (Biocomp Instruments).
RNA isolation and RT-PCR
RNA was isolated from immune complexes using Qiagen RNA purification kit (Qiagen) as per the manufacturer's instructions, and from polysome fractions using phenol:chloroform extraction followed by ethanol precipitation using standard protocols. One-hundred nanograms of RNA was reverse transcribed using oligo-dT primer15 and Protoscript II (New England Biolabs) as per the manufacturer's instructions. qPCR of RIP samples was performed using iQ SYBR Green Supermix (Bio-Rad) using 1 µL of RT reaction product. Semi-quantitative PCR was performed using 1 µL of RT reaction product from polysome fractions for 18 cycles. The reaction products were separated on a 1.5% agarose gel and quantified using open source software ImageJ (https://imagej.nih.gov/ij).
EMT assay
MCF-7 cells were cultured in MEM supplemented with 5% FBS. For the EMT assay, MEM medium was replaced with filtered conditioned KSFM media collected from 3-d cultures of either WT or S34F mutant HBECs and cells cultured for another 6–8 d in conditioned medium, fixed and stained with antibodies against Fibronectin or E-Cadherin as described above.
FACS
Flow cytometric analysis of GFP expression in HBECs was done using BD FACSCalibur (BDBiosciences). GFP expression from wt/wt or wt/S34F cells expressing wt- or mt-5′-UTR IL8-2A-GFP reporter were analyzed using FACS. Cells were trypsinized, neutralized with Trypsin Neutralizing Reagent, and washed with PBS before FACS analysis. wt/wt cells served as a control for autofluorescence and the reporter cell lines were gated above the autofluorescence of wt/wt cells (Supplemental Fig. S6). Each experiment counted ∼50,000 events for each cell line, and the average of three independent experiments with SD is presented.
RIP- and polysome-seq and analysis
For each of the three cell lines used for RIP (wt/wt, wt/S34F, and wt/S34F−), three samples were collected and sequenced: input, U2AF1 RIP, and IgG nonspecific IP. RNA was extracted using TRIzol according to manufacturer's instructions. rRNA was removed using the NEBNext rRNA Depletion Kit rRNA Removal Kit (New England Biolabs) followed by cDNA library preparation (NEBNext Ultra RNA Library Prep kit for Illumina). The samples were sequenced on the illumina HiSeq 3000 platform. Sequences were aligned to the human genome version hg19 using TopHat (Trapnell et al. 2012). Abundances of transcript-mapped RNA sequences were estimated using rnacounter python program. RNA isolated from individual sucrose gradient fractions were used for polysome sequencing as described above.
The following steps are used to generate a list of transcripts for analysis:
Only transcripts that show abundance of RPKM > 1 in all samples (except the IgG nonspecific binding control, where the RPKM cutoff is 0.05) and both biological replicates are used for analysis.
RPKM values from the two biological replicates are averaged to generate the data in Extended Data Table 3.
For each cell line (wt/wt, wt/s34f, wt/−, and wt/S34F−) the log2 ratio of the IP/input is tabulated, and for the wt/wt cell line the log2 ratio of IP/IgG is also included.
For decile analysis, transcripts are ranked either by the difference in log2 ratios between wt/wt and wt/s34f cells or the log2 ratio in wt/wt cells.
The following steps are used to compute individual and composite polysome profiles from the sequencing read count data:
For each sample (wt/wt, wt/s34f, and wt/S34F−), 12 fractions were collected from the sucrose gradient, and the bottom/heaviest 10 fractions (Fig. 3A, fractions 3–12) were sequenced.
Fractions 10–12 were pooled to increase coverage. Fraction 5 and 6 correspond to the monosome fractions.
Reads mapping to particular message are normalized by the total reads in that sample and multiplied by 1 × 106, generating counts per million (CPM) for each transcript in each fraction.
- The CPM in each fraction constitutes the polysome profile (poly(x), where “x” designates the sucrose fraction) that is now normalized for read depth across fractions/samples. This profile can be further normalized by
A single “polysome/monosome ratio” is then the ratio of the heaviest fraction to the monosome fractions (i.e. Fig. 4D,F) {poly(10–12)/[poly(5)+poly(6)]}.
Splice-site finding in 5′ UTRs
Experimental 5′ UTRs reported in ref 40 were analyzed with FIMO (http://meme-suite.org/tools/fimo). Settings: motif width = 6; input sequences = 6441 sequences between 8 and 3067 bp in length. The search motif position weight matrix was [0.054 0.655 0.002 0.289], [1.000 0.000 0.000 0.000 0.000 0.000 1.000 0.000], [0.253 0.141 0.493 0.113], [0.241 0.189 0.199 0.371], [0.259 0.236 0.236 0.270].
Stable cell lines expressing wild-type and S34F mutant U2AF1 and PAR-CLIP
To determine the binding of U2AF1 to its specific mRNA targets in vivo, we created two stable HEK 293 (HEK T-REx Flp-In-293, Clontech) cell lines that conditionally (doxycycline inducible) express either wild-type or S34F mutant U2AF1 with an N-terminal Flag-HA tag. Briefly, pFRT-TO-Flag-HA-U2AF1 plasmid carrying the cDNA for wild-type U2AF1 was constructed using the gateway technology (Invitrogen) as recommended by the manufacturer. The S34F mutant U2AF1 was constructed from the wild-type plasmid by site-directed Quick Change II mutagenesis kit, Agilent Technologies). Plasmids were transfected into Flp-In T-REx HEK293 cells (Invitrogen) together with pOG44 plasmid expressing Flp-recombinase (Invitrogen) using Lipofectamine 2000 transfection reagent (Invitrogen). Stable cell lines were selected with 10 µg/mL blasticidin (Gibco) and 100 µg/mL hygromycin B. HEK293 cells stably expressing either the wild-type or S34F mutant U2AF1 (10 × 15 cm plates at 70% confluency) were induced with doxycycline for (24 h) followed by incubation with 4S-uridine (100 µM for 16 h at 37°C; to isolate chromatin-associated RBPs, the 4S-uridine incubation was at 200 µM for 8 h) and UV cross-linked (365 nm 400 mJ/cm2). The cells were fractionated into nuclear and cytoplasmic fractions as described previously (Gagnon et al. 2014) with a few modifications. Cells were lysed by gentle resuspension in a modified HLB buffer (10 mM Tris at pH 7.5, 50 mM NaCl, 3 mM MgCl2, 0.1% NP-40, 10% glycerol) for 2 min. To isolate chromatin-associated RNA-binding proteins, the nuclear pellet was washed twice with the same buffer and extracted using MWS buffer (10 mM Tris-HCl at pH 7.0, 4 mM EDTA, 0.3 M NaCl, 1 M urea, and 1% [v/v] NP-40). The chromatin pellet was washed twice in MWS buffer and resuspended in RIPA buffer, followed by sonication (3 × 10 sec; 60% amplitude on a VCX130 Vibra-Cell Ultrasonic Liquid Processor). To isolate the total nuclear fraction, nuclei were directly lysed in NP-40 buffer, followed by sonication. Proteins were Immunoprecipitated using the anti-Flag M2 magnetic beads (Sigma).
PAR-CLIP was performed on the subcellular fractions isolated above as described previously (Hafner et al. 2010; Benhalevy et al. 2017) with a few modifications. Briefly, after ribonuclease cleavage of the unprotected RNA, a 3′-adaptor was ligated with the complexes associated to beads and were then washed three times with high-salt wash buffer. To isolate the chromatin-associated RBPs, the ligated RBP-RNA was separated by SDS-PAGE and the U2AF1 cross-linked 3′-adaptor ligated footprints were eluted using proteinase K. To isolate U2AF1/U2AF2-associated RNAs from the cytoplasmic and nuclear fractions, the ligated footprints were retrieved directly from the bead-associated U2AF1-U2AF2 complex with proteinase K digestion. The recovered footprints were further size selected for 19- to 35-nt fragments after electrophoresis on urea (8 M) PAG (15%). A 5′-adaptor was then ligated to the size selected 3′-ligated footprints and reverse transcribed using SuperScript IV reverse transcriptiase (Invitrogen) as per the manufacture's instructions. The library was PCR amplified and sequenced on an Illumina HiSeq 3000 platform. Sequences were further processed through PARpipes (https://github.com/ohlerlab/PARpipe) implementing bowtie for alignment and paralyzer for cluster identification (Corcoran et al. 2011). Coverage plots were made using ngs.plot (Shen et al. 2014).
Mouse experiments
All procedures related to mouse experiments and husbandry were approved by the Institutional Animal Care and Use Committee at the National Human Genome Research Institute (NHGRI; protocol: G-10-6) and Weill Cornell Medicine (WCM; protocol: 2015-017). Mice (six mice treated with IL8 or control IgGs were female; one of the naive controls was female, and another was male); NOD background (Taconic #NOG)) were 7–8 wk at the beginning of the experiment. Xenograft tumors of H441- and HCC78-derived isogenic cells were described previously (Fei et al. 2016). Briefly, 1 million to 5 million cancer cells were mixed with matrigel (Corning) and implanted subcutaneously in 6- to 8-wk-old nude mice (nu/nu, JAX). Tumors were harvested once they reached 10% of the body weight or up to 150 d after tumor inoculation and were fixed in formalin. Tissue sectioning and H&E staining were done at a paid service at Histoserv. Peri- or intratumoral inflammation were quantified by a pathologist.
To evaluate xenograft tumor formation in mouse lung and liver, 1 million isogenic H441 cells (with or without S34F mutation) were mixed at equal cell number in 100 µL of PBS and were injected into NOG mice (Taconic) via tail vein. Anti-IL8 and isotype control IgG1 (R&D Systems) were given to the mice via intraperitoneal injection three times a week (Monday, Wednesday, and Friday) starting 1 d after the tail vein injection for 10 wk. Half of the mice (n = 3) were given the anti-IL8 neutralizing antibody, and the other half with the control IgG. Three days after the last antibody treatment, blood was collected from live mice by submandibular bleeding, and serum levels of human IL8 were measured by ELISA. Mice were then euthanized by CO2, and the macroscopic tumor nodules on the lung and liver were counted.
Human peripheral blood collection and serum isolation
Blood samples from relapsed or refractory acute myeloid leukemia patients and healthy donors were collected following informed consent on one of two IRB-approved NHLBI protocols (04-H-0012 [https://clinicaltrials.gov/ct2/show/NCT00071045] and 07-H-0113 [https://clinicaltrials.gov/ct2/show/NCT00442195]). RR-AML patient's average age was 55 (age range: 22–81; 14 males, 10 females) and healthy donor average age was 53 (age range: 24–84; nine males, 11 females). Serum was extracted from serum separator tubes (BD) per the manufacturer's instructions and stored in aliquots at −80°C.
Luminex assay
The Human XL Cytokine Discovery Premixed Luminex Performance Assay Kit (FCSTM18 from R&D Systems) was used to determine levels of 12 analytes (IL8/CXCL8, IFN-γ, IL-10, IL-2, IL-5, TNF-α, VEGF, IL-6, IL-4, IL-12 p70, IL-1β, and GM-CSF) in the 44 human serum samples. All serum samples were run as one batch with technical duplicates, blanks, standards, and high and low controls as per the manufacturer's instructions. All reported values for protein concentration in picograms per milliliter were used in the analysis, including those extrapolated beyond the range of the standards, unless the fluorescence intensity was below that of the blanks. Averaged values were used for samples run in duplicates.
Computer code and software availability
All open source software and custom written software used in this study has been referenced in the Materials and Methods with web links or provided upon request.
Data availability
PAR-CLIP sequencing, ribosome-depleted RNA-seq from polysome and monosome fractions, and RIP-seq data are available on the NCBI Short-Read Archive (SRA) under the Gene Expression Omnibus accession number GSE126912. Additional data are available in the Supplemental Material.
Supplementary Material
Acknowledgments
We thank Dr. Harold Varmus for support, making available cell lines, reagents, other resources, and for critical reading of the manuscript; Dr. Alan Hinnebusch, Dr. Sandra Wolin, Dr. Nicholas Guydosh, and Dr. Jeffrey Chao for critical reading of the manuscript and valuable suggestions. M.P. and D.R.L. acknowledge Dr. Heather Kalish from the Micro Analytical Immunochemistry Unit, National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health (NIH) for performing ELISA, and Katherine McKinnon and Sophia Brown from the Center for Cancer Research FACS Core Facility at NIH for support with FACS. D.L.F. acknowledges the support of Dr. Benjamin Durham, M.D., from Memorial Sloan Kettering Cancer Center for evaluation of tumor histology; Sukanya Goswami (Weill Cornell Medicine), Danielle Miller-O'Mard (National Human Genome Research Institute [NHGRI]), and members of the transgenic mouse core at NHGRI for technical assistance with the mouse experiments. This work was supported in part by the Intramural Research Programs of the National Cancer Institute and the National Heart, Lung, and Blood Institute of the National Institutes of Health.
Author contributions: M.P. and D.R.L. designed the study. M.P. performed all of the experiments except for the RIP-seq, polysome-seq, and PAR-CLIP experiments were performed by D.G.A., mouse experiments performed by D.L.F., and human patient serum collection and cytokine analysis performed by K.E.L. and C.S.H. M.P. and D.R.L. analyzed the data. M.P. and D.R.L. wrote the paper with input from all the authors.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.319590.118.
References
- Abelson S, Collord G, Ng SWK, Weissbrod O, Mendelson Cohen N, Niemeyer E, Barda N, Zuzarte PC, Heisler L, Sundaravadanam Y, et al. 2018. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature 559: 400–404. 10.1038/s41586-018-0317-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfaro C, Teijeira A, Oñate C, Pérez G, Sanmamed MF, Andueza MP, Alignani D, Labiano S, Azpilikueta A, Rodriguez-Paulete A, et al. 2016. Tumor-produced interleukin-8 attracts human myeloid-derived suppressor cells and elicits extrusion of neutrophil extracellular traps (NETs). Clin Cancer Res 22: 3924–3936. 10.1158/1078-0432.CCR-15-2463 [DOI] [PubMed] [Google Scholar]
- Aviner R, Hofmann S, Elman T, Shenoy A, Geiger T, Elkon R, Ehrlich M, Elroy-Stein O. 2017. Proteomic analysis of polyribosomes identifies splicing factors as potential regulators of translation during mitosis. Nucleic Acids Res 45: 5945–5957. 10.1093/nar/gkx326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bavik C, Coleman I, Dean JP, Knudsen B, Plymate S, Nelson PS. 2006. The gene expression program of prostate fibroblast senescence modulates neoplastic epithelial cell proliferation through paracrine mechanisms. Cancer Res 66: 794–802. 10.1158/0008-5472.CAN-05-1716 [DOI] [PubMed] [Google Scholar]
- Benhalevy D, McFarland H, Sarshad A, Hafner M. 2017. PAR-CLIP and streamlined small RNA cDNA library preparation protocol for the identification of RNA binding protein target sites. Methods 118-119: 41–49. 10.1016/j.ymeth.2016.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks AN, Choi PS, de Waal L, Sharifnia T, Imielinski M, Saksena G, Pedamallu CS, Sivachenko A, Rosenberg M, Chmielecki J, et al. 2014. A pan-cancer analysis of transcriptome changes associated with somatic mutations in U2AF1 reveals commonly altered splicing events. PLoS One 9: e87361 10.1371/journal.pone.0087361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Chen JY, Huang YJ, Gu Y, Qiu J, Qian H, Shao C, Zhang X, Hu J, Li H, et al. 2018. The augmented R-loop is a unifying mechanism for myelodysplastic syndromes induced by high-risk splicing factor mutations. Mol Cell 69: 412–425 e416. 10.1016/j.molcel.2017.12.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. 2008. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6: 2853–2868. 10.1371/journal.pbio.0060301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran DL, Georgiev S, Mukherjee N, Gottwein E, Skalsky RL, Keene JD, Ohler U. 2011. PARalyzer: definition of RNA binding sites from PAR-CLIP short-read sequence data. Genome Biol 12: R79 10.1186/gb-2011-12-8-r79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulon A, Ferguson ML, de Turris V, Palangat M, Chow CC, Larson DR. 2014. Kinetic competition during the transcription cycle results in stochastic RNA processing. Elife 3: e03939 10.7554/eLife.03939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez C, McCampbell KK, David JM, Palena C. 2017. Neutralization of IL-8 decreases tumor PMN-MDSCs and reduces mesenchymalization of claudin-low triple-negative breast cancer. JCI Insight 2: e94296 10.1172/jci.insight.94296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvinge H, Kim E, Abdel-Wahab O, Bradley RK. 2016. RNA splicing factors as oncoproteins and tumour suppressors. Nat Rev Cancer 16: 413–430. 10.1038/nrc.2016.51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert BL, Pretz J, Bosco J, Chang CY, Tamayo P, Galili N, Raza A, Root DE, Attar E, Ellis SR, et al. 2008. Identification of RPS14 as a 5q- syndrome gene by RNA interference screen. Nature 451: 335–339. 10.1038/nature06494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Ishmael FT, Fang X, Myers A, Cheadle C, Huang SK, Atasoy U, Gorospe M, Stellato C. 2011. Chemokine transcripts as targets of the RNA-binding protein HuR in human airway epithelium. J Immunol 186: 2482–2494. 10.4049/jimmunol.0903634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei DL, Motowski H, Chatrikhi R, Prasad S, Yu J, Gao S, Kielkopf CL, Bradley RK, Varmus H. 2016. Wild-type U2AF1 antagonizes the splicing program characteristic of U2AF1-mutant tumors and is required for cell survival. PLoS Genet 12: e1006384 10.1371/journal.pgen.1006384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernando RI, Castillo MD, Litzinger M, Hamilton DH, Palena C. 2011. IL-8 signaling plays a critical role in the epithelial–mesenchymal transition of human carcinoma cells. Cancer Res 71: 5296–5306. 10.1158/0008-5472.CAN-11-0156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furney SJ, Pedersen M, Gentien D, Dumont AG, Rapinat A, Desjardins L, Turajlic S, Piperno-Neumann S, de la Grange P, Roman-Roman S, et al. 2013. SF3B1 mutations are associated with alternative splicing in uveal melanoma. Cancer Discov 3: 1122–1129. 10.1158/2159-8290.CD-13-0330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon KT, Li L, Janowski BA, Corey DR. 2014. Analysis of nuclear RNA interference in human cells by subcellular fractionation and Argonaute loading. Nat Protoc 9: 2045–2060. 10.1038/nprot.2014.135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gama-Carvalho M, Carvalho MP, Kehlenbach A, Valcárcel J, Carmo-Fonseca M. 2001. Nucleocytoplasmic shuttling of heterodimeric splicing factor U2AF. J Biol Chem 276: 13104–13112. 10.1074/jbc.M008759200 [DOI] [PubMed] [Google Scholar]
- Gama-Carvalho M, Barbosa-Morais NL, Brodsky AS, Silver PA, Carmo-Fonseca M. 2006. Genome-wide identification of functionally distinct subsets of cellular mRNAs associated with two nucleocytoplasmic-shuttling mammalian splicing factors. Genome Biol 7: R113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graubert TA, Shen D, Ding L, Okeyo-Owuor T, Lunn CL, Shao J, Krysiak K, Harris CC, Koboldt DC, Larson DE, et al. 2011. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat Genet 44: 53–57. 10.1038/ng.1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov SI, Greten FR, Karin M. 2010. Immunity, inflammation, and cancer. Cell 140: 883–899. 10.1016/j.cell.2010.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, Schnittger S, Sanada M, Kon A, Alpermann T, et al. 2014. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 28: 241–247. 10.1038/leu.2013.336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano MJ, Jungkamp AC, Munschauer M, et al. 2010. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141: 129–141. 10.1016/j.cell.2010.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Renwick N, Farazi TA, Mihailović A, Pena JT, Tuschl T. 2012. Barcoded cDNA library preparation for small RNA profiling by next-generation sequencing. Methods 58: 164–170. 10.1016/j.ymeth.2012.07.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herranz N, Gallage S, Mellone M, Wuestefeld T, Klotz S, Hanley CJ, Raguz S, Acosta JC, Innes AJ, Banito A, et al. 2015. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol 17: 1205–1217. 10.1038/ncb3225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilagan JO, Ramakrishnan A, Hayes B, Murphy ME, Zebari AS, Bradley P, Bradley RK. 2015. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res 25: 14–26. 10.1101/gr.181016.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, et al. 2012. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 150: 1107–1120. 10.1016/j.cell.2012.08.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue D, Bradley RK, Abdel-Wahab O. 2016. Spliceosomal gene mutations in myelodysplasia: molecular links to clonal abnormalities of hematopoiesis. Genes Dev 30: 989–1001. 10.1101/gad.278424.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins JL, Kielkopf CL. 2017. Splicing factor mutations in myelodysplasias: insights from spliceosome structures. Trends Genet 33: 336–348. 10.1016/j.tig.2017.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R. 2009. EMT: when epithelial cells decide to become mesenchymal-like cells. J Clin Invest 119: 1417–1419. 10.1172/JCI39675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielkopf CL. 2017. Insights from structures of cancer-relevant pre-mRNA splicing factors. Curr Opin Genet Dev 48: 57–66. 10.1016/j.gde.2017.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. 2001. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci 98: 12072–12077. 10.1073/pnas.211053698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laberge RM, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L, Curran SC, Davalos AR, Wilson-Edell KA, Liu S, et al. 2015. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol 17: 1049–1061. 10.1038/ncb3195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. 2012. mTOR signaling in growth control and disease. Cell 149: 274–293. 10.1016/j.cell.2012.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasry A, Ben-Neriah Y. 2015. Senescence-associated inflammatory responses: aging and cancer perspectives. Trends Immunol 36: 217–228. 10.1016/j.it.2015.02.009 [DOI] [PubMed] [Google Scholar]
- Liu D, Hornsby PJ. 2007. Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res 67: 3117–3126. 10.1158/0008-5472.CAN-06-3452 [DOI] [PubMed] [Google Scholar]
- Mamane Y, Petroulakis E, LeBacquer O, Sonenberg N. 2006. mTOR, translation initiation and cancer. Oncogene 25: 6416–6422. 10.1038/sj.onc.1209888 [DOI] [PubMed] [Google Scholar]
- Maslon MM, Heras SR, Bellora N, Eyras E, Cáceres JF. 2014. The translational landscape of the splicing factor SRSF1 and its role in mitosis. Elife 3: e02028 10.7554/eLife.02028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merendino L, Guth S, Bilbao D, Martínez C, Valcárcel J. 1999. Inhibition of msl-2 splicing by Sex-lethal reveals interaction between U2AF35 and the 3′ splice site AG. Nature 402: 838–841. 10.1038/45602 [DOI] [PubMed] [Google Scholar]
- Metsalu T, Vilo J. 2015. ClustVis: a web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res 43: W566–W570. 10.1093/nar/gkv468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narla A, Ebert BL. 2010. Ribosomopathies: human disorders of ribosome dysfunction. Blood 115: 3196–3205. 10.1182/blood-2009-10-178129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HD, Leong WY, Li W, Reddy PNG, Sullivan JD, Walter MJ, Zou L, Graubert TA. 2018. Spliceosome mutations induce R loop-associated sensitivity to ATR inhibition in myelodysplastic syndromes. Cancer Res 78: 5363–5374. 10.1158/0008-5472.CAN-17-3970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okeyo-Owuor T, White BS, Chatrikhi R, Mohan DR, Kim S, Griffith M, Ding L, Ketkar-Kulkarni S, Hundal J, Laird KM, et al. 2015. U2AF1 mutations alter sequence specificity of pre-mRNA binding and splicing. Leukemia 29: 909–917. 10.1038/leu.2014.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz-Montero P, Londoño-Vallejo A, Vernot JP. 2017. Senescence-associated IL-6 and IL-8 cytokines induce a self- and cross-reinforced senescence/inflammatory milieu strengthening tumorigenic capabilities in the MCF-7 breast cancer cell line. Cell Commun Signal 15: 17 10.1186/s12964-017-0172-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palangat M, Larson DR. 2016. Single-gene dual-color reporter cell line to analyze RNA synthesis in vivo. Methods 103: 77–85. 10.1016/j.ymeth.2016.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda AC, Martindale JL, Gorospe M. 2017. Polysome fractionation to analyze mRNA distribution profiles. Bio Protoc 7: e2126 10.21769/BioProtoc.2126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, Potter NE, Heuser M, Thol F, Bolli N, et al. 2016. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med 374: 2209–2221. 10.1056/NEJMoa1516192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SM, Ou J, Chamberlain L, Simone TM, Yang H, Virbasius CM, Ali AM, Zhu LJ, Mukherjee S, Raza A, et al. 2016. U2AF35(S34F) promotes transformation by directing aberrant ATG7 pre-mRNA 3′ end formation. Mol Cell 62: 479–490. 10.1016/j.molcel.2016.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HY, Lee KC, Jang YH, Kim SK, Thu MP, Lee JH, Kim JK. 2017. The Arabidopsis splicing factors, AtU2AF65, AtU2AF35, and AtSF1 shuttle between nuclei and cytoplasms. Plant Cell Rep 36: 1113–1123. 10.1007/s00299-017-2142-z [DOI] [PubMed] [Google Scholar]
- Przychodzen B, Jerez A, Guinta K, Sekeres MA, Padgett R, Maciejewski JP, Makishima H. 2013. Patterns of missplicing due to somatic U2AF1 mutations in myeloid neoplasms. Blood 122: 999–1006. 10.1182/blood-2013-01-480970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio CA, Weisburd B, Holderfield M, Arias C, Fang E, DeRisi JL, Fanidi A. 2014. Transcriptome-wide characterization of the eIF4A signature highlights plasticity in translation regulation. Genome Biol 15: 476 10.1186/s13059-014-0476-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan BM, Pine SR, Chaturvedi AK, Caporaso N, Harris CC. 2014. A combined prognostic serum interleukin-8 and interleukin-6 classifier for stage 1 lung cancer in the prostate, lung, colorectal, and ovarian cancer screening trial. J Thorac Oncol 9: 1494–1503. 10.1097/JTO.0000000000000278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanford JR, Gray NK, Beckmann K, Caceres JF. 2004. A novel role for shuttling SR proteins in mRNA translation. Genes Dev 18: 755–768. 10.1101/gad.286404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinke C, Giricz O, Li W, Shastri A, Gordon S, Barreyro L, Bhagat T, Bhattacharyya S, Ramachandra N, Bartenstein M, et al. 2015. IL8–CXCR2 pathway inhibition as a therapeutic strategy against MDS and AML stem cells. Blood 125: 3144–3152. 10.1182/blood-2015-01-621631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler M, Peng S, Agrawal AA, Palacino J, Teng T, Zhu P, Smith PG; Cancer Genome Atlas Research Network, Buonamici S, Yu L. 2018. Somatic mutational landscape of splicing factor genes and their functional consequences across 33 cancer types. Cell Rep 23: 282–296 e284. 10.1016/j.celrep.2018.01.088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen T, Yang Z, Cheng X, Xiao Y, Yu K, Cai X, Xia C, Li Y. 2014. CXCL8 induces epithelial–mesenchymal transition in colon cancer cells via the PI3K/Akt/NF-κB signaling pathway. Oncol Rep 37: 2095–2100. 10.3892/or.2017.5453 [DOI] [PubMed] [Google Scholar]
- Taniguchi K, Karin M. 2018. NF-κB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol 18: 309–324. 10.1038/nri.2017.142 [DOI] [PubMed] [Google Scholar]
- Thiery JP, Acloque H, Huang RY, Nieto MA. 2009. Epithelial-mesenchymal transitions in development and disease. Cell 139: 871–890. 10.1016/j.cell.2009.11.007 [DOI] [PubMed] [Google Scholar]
- Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM. 2012. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 485: 109–113. 10.1038/nature11083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7: 562–578. 10.1038/nprot.2012.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe AL, Singh K, Zhong Y, Drewe P, Rajasekhar VK, Sanghvi VR, Mavrakis KJ, Jiang M, Roderick JE, Van der Meulen J, et al. 2014. RNA G-quadruplexes cause eIF4A-dependent oncogene translation in cancer. Nature 513: 65–70. 10.1038/nature13485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Romfo CM, Nilsen TW, Green MR. 1999. Functional recognition of the 3′ splice site AG by the splicing factor U2AF35. Nature 402: 832–835. 10.1038/45590 [DOI] [PubMed] [Google Scholar]
- Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, et al. 2011. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478: 64–69. 10.1038/nature10496 [DOI] [PubMed] [Google Scholar]
- Yoshida H, Park SY, Oda T, Akiyoshi T, Sato M, Shirouzu M, Tsuda K, Kuwasako K, Unzai S, Muto Y, et al. 2015. A novel 3′ splice site recognition by the two zinc fingers in the U2AF small subunit. Genes Dev 29: 1649–1660. 10.1101/gad.267104.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorio DA, Blumenthal T. 1999. Both subunits of U2AF recognize the 3′ splice site in Caenorhabditis elegans. Nature 402: 835–838. 10.1038/45597 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
PAR-CLIP sequencing, ribosome-depleted RNA-seq from polysome and monosome fractions, and RIP-seq data are available on the NCBI Short-Read Archive (SRA) under the Gene Expression Omnibus accession number GSE126912. Additional data are available in the Supplemental Material.