

Graphical Abstract
To the Editor:
Genetic hearing impairment is mostly nonsyndromic (80%), and >6,000 causal variants in >100 genes have been identified. Generally in hearing-impaired patients of Asian descent, GJB2 variants are most common (36%), followed by variants in SLC26A4 (MIM 605646), MYO15A (MIM 602666) and CDH23 (MIM 605516).1 Here we report seven novel variants in Filipino cochlear implantees, suggesting that the allelic spectrum for non-/syndromic hearing impairment in Filipinos is unique.
The UP Manila Research Ethics Board approved the study. Adult subjects and parents of pediatric patients provided informed consent. DNA samples and clinical data were obtained from 30 cochlear implantees with bilateral, severe-to-profound, congenital, non-progressive hearing loss as previously described.2–3 After excluding one patient with GJB2 c.[35delG];[235delC],2 twenty-nine DNA samples were submitted for exome sequencing, of which four are homozygous for SLC26A4 c.706C>G (p.(Leu236Val)).3 For the twenty-five GJB2-/SLC26A4-negative patients, homozygous/heterozygous coding variants within 132 non-/syndromic hearing impairment genes were selected if with MAF<0.005 in any gnomAD population. Rare variants were selected further if considered damaging by MutationTaster and/or >2 dbNSFP tools. Eleven Filipino-descent US families with no history of hearing impairment were ascertained for MAF estimation for speech delay (SDFIL) and were Sanger-sequenced for selected exome variants. Variants with zero MAF in the SDFIL cohort were then screened using samples from >88 unrelated Filipinos from the Cebu Longitudinal Health and Nutrition Survey, a cohort examined for various health outcomes. Screened exome variants were excluded due to increased MAF, lack of a second rare variant in an autosomal recessive gene and/or poor clinical correlation.
Of 30 Filipino cochlear implantees, we identified a genetic cause in sixteen (53%). Seven novel hearing loss variants were discovered (Table 1): CHD7 (MIM 608892) c.7312C>G (p.(Gln2438Glu)); COL4A3 (MIM 120070) c.764C>T (p.(Thr255Met)); DFNA5 (MIM 600994) c.1277_1279delATG (p.(Asp426del)); EYA4 (MIM 603550) c.1109G>A (p.(Arg370His)); MYH14 (MIM 608568) c.2971G>A (p.(Glu991Lys)); MYO15A c.263C>T (p.(Thr88Met)); and OTOA (MIM 607038) c.2301+1G>T. Patient #28 with the CHD7 variant has microcephaly and seizures, both known features of CHARGE syndrome (MIM 214800). Additionally he has left superior semicircular canal dehiscence (SSCD)3 but no vertigo or dizziness. Both DFNA5 c.1277_1279delATG and EYA4 c.1109G>A were previously identified through systematic clinical genetic screening and are annotated in ClinVar as variants of unknown significance. Patient #4 with the EYA4 variant complains of dizziness and balance problems; temporal bone findings include right enlarged vestibular aqueduct and a left jugular bulb diverticulum that impinges onto the ipsilateral vestibular aqueduct.3 COL4A3 c.764C>T was previously reported for familial kidney disease4 but not hearing impairment. MYH14 c.2971G>A is cited in LOVD as likely benign but was not clarified for nonsyndromic DFNA4A (MIM 600652) or peripheral neuropathy, myopathy, hoarseness and hearing loss (PNMHH; MIM 614369). This MYH14 variant is the only rare, damaging variant identified in patient #26, who has developmental delay and left foot inversion.
Table 1.
Pathogenic/likely pathogenic variants in Filipino cochlear implantees identified in this study†
| ID | Geno- type |
Gene | Expected MOI |
RefSeq NM_ |
cDNA Variant |
Amino Acid Variant |
Damaging Prediction |
CADD | gnomAD MAF Overall |
gnomAD MAF NFE |
gnomAD MAF EAS |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2‡ | Het | DFNA5 | DFNA5 | 004403 | c.1277_1279delATG | p.(Asp426del) | MT | -- | 0.0006 | 0 | 0.0005 |
| 4§ | Het | EYA4 | DFNA10 | 004100 | c.1109G>A | p.(Arg370His) | Fa,LRT,mLR, mSVM,MA,MT, PP2_HD/HV, PR,SI |
35 | 0.00004 | 0.00006 | 0 |
| 10‡ | Het | KCNQ4 | DFNA2A | 004700 | c.546C>G | p.(Phe182Leu) | MT | 22 | 0.0003 | 0 | 0.004 |
| 14¶ | Hom | OTOA | DFNB22 | 144672 | c.2301+1G>T | NA | MT | 27.1 | 0 | 0 | 0 |
| 15‡ | Cpd het | CDH23 | DFNB12; AR USH1D |
022124 | c.68–3C>T | NA | MT | -- | 0.0005 | 0.000008 | 0.008 |
| 15‡ | Cpd het | CDH23 | DFNB12; AR USH1D |
022124 | c.4762C>T | p.(Arg1588Trp) | LRT,MA,MT, PP2_HD/HV, PR,SI | 23.3 | 0.0002 | 0.00007 | 0.001 |
| 16 | Het | WFS1 | DFNA6/14/38; AD/AR Wolfram | 006005 | c.708C>G | p.(Ser236Arg) | Fa,LRT,mLR,MT | 12.9 | 0 | 0 | 0 |
| 17‡ | Hom | MYO15A | DFNB3 | 016239 | c.263C>T | p.(Thr88Met) | Fa, PP2_HD | 18.6 | 0.00007 | 0 | 0 |
| 21 | Het | COL4A3 | AD/AR Alport | 000091 |
c.764C >T |
p.(Thr2 55Met) |
Fa,mLR,mSVM, MT,PP2_HD/HV,SI | 33 | 0.00009 | 0.000009 | 0.00006 |
| 26 | Het | MYH14 | DFNA4A; AD PNMHH |
001145809 |
c.2971G >A |
p.(Glu991Lys) | Fa,mLR,mSVM,MA,MT,PP2_HD/HV,PR,SI | 28.9 | 0.00001 | 0 | 0.00007 |
| 28 | Het | CHD7 | AD CHARGE | 017780 |
c.731 2C> G |
p.(Gln24 38Glu) |
LRT,MT, PP2_HD/HV |
25.3 | 0 | 0 | 0 |
All variants listed were not found in Filipino controls from SDFIL and CLHNS. Novel variants are in bold font.
Confirmed to have nonsyndromic hearing impairment (if including GJB2-/SLC26A4-positive patients, total 9 of 15 or 60% of those with genetic variants).
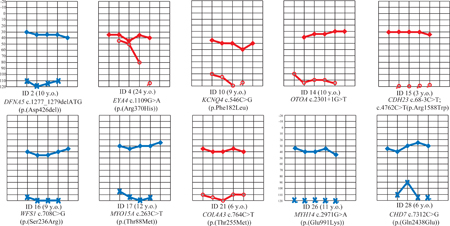
Prior to surgery, patient #4 had a steeply sloping audiogram with 45–50 dB hearing at 500–1000 kHz and profound loss at 4000 Hz. All other patients listed in Table 1 had flat audiograms with severe-to-profound hearing loss. Additional clinical information on these patients were previously provided in reference #3.
Patient #14 has global developmental delay and a history of maternal rubella, low birth weight, exchange transfusion for jaundice, antibiotic treatment and mechanical ventilation for neonatal pneumonia, and intraventricular hemorrhage.
Abbreviations: AD, autosomal dominant; AR, autosomal recessive; CADD, Combined Annotation-Dependent Depletion; Cpd het, compound heterozygous; DFNA#, nonsyndromic autosomal dominant hearing loss; DFNB#, nonsyndromic autosomal recessive hearing loss; EAS, East Asian; Fa, FATHMM; gnomAD, Genome Aggregation Database; HD, HumDiv; Het, heterozygous; Hom, homozygous; HV, HumVar; LRT, Likelihood Ratio Test; MA, MutationAssessor; MAF, minor allele frequency; mLR, MetaLR; MT, MutationTaster; mSVM, MetaSVM; NFE, non-Finnish European; PNMHH, peripheral neuropathy, myopathy, hoarseness and hearing loss; PP2, PolyPhen2; PR, PROVEAN; RefSeq, Reference Sequence; SI, SIFT; USH1D, Usher syndrome Type I
Variants CDH23 c.68–3C>T and c.4762C>T (p.(Arg1588Trp)), KCNQ4 (MIM 603537) c.546C>G (p.(Phe182Leu)), and WFS1 (MIM 606201) c.708C>G (p.(Ser236Arg)) have been previously reported for hearing impairment. Patient #10 with the KCNQ4 variant has SSCD on the left3 but no vestibular symptoms. WFS1 c.708C>G was reported in a patient with compound heterozygous WFS1 variants and autosomal recessive Wolfram syndrome (MIM 222300).5 Patient #16 has no second WFS1 coding variant but has birth history of cord coil, white matter disease and mild motor delay.
Clinical data helped identify the correct gene when multiple potentially causal variants were present. Patients with pathogenic variants had higher pre-surgical audiometric thresholds at >1kHz (Wilcoxon p<0.05). However there was no significant difference in post-surgical thresholds, suggesting that carriage of the genetic variants reported here does not determine the outcome of cochlear implantation, with average implant-aided hearing at 38dB across frequencies. Therefore for carriers of these variants, cochlear implantation remains an excellent option for rehabilitation.
Acknowledgments:
This work was funded by grants PCHRD-DOST FP150010 and UP Manila-NIH 2008–005 (to C.M.C.).
Footnotes
Conflict of Interest: None
Data Availability Statement: ClinVar accession numbers pending
Web URLs
Cebu Longitudinal Health and Nutrition Survey (CLHNS), www.cpc.unc.edu/projects/cebu
ClinVar, www.ncbi.nlm.nih.gov/clinvar/
dbNSFP, sites.google.com/site/jpopgen/dbNSFP
Genetic Variant Interpretation Tool, www.medschool.umaryland.edu/Genetic_Variant_Interpretation_Tool1.html/
Genome Aggregation Database, gnomad.broadinstitute.org
Hereditary Hearing Loss Homepage, hereditaryhearingloss.org
Integrative Genomics Viewer, software.broadinstitute.org/software/igv/
Leiden Open Variation Database, http://www.lovd.nl/3.0/home
MutationTaster, www.mutationtaster.org
Online Mendelian Inheritance of Man, www.omim.org
UCSC Genome Browser, genome.ucsc.edu
References
- 1.Sloan-Heggen CM, et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum Genet 2016;135:441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chiong CM, et al. GJB2 variants and auditory outcomes among Filipino cochlear implantees. Audiol Neurotol Extra 2013:3:1–8. [Google Scholar]
- 3.Chiong CM, et al. The SLC26A4 c.706C>G (p.Leu236Val) Variant is a Frequent Cause of Hearing Impairment in Filipino Cochlear Implantees. Otol Neurotol 2018;39:e726–e730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mallett AJ, et al. Massively parallel sequencing and targeted exomes in familial kidney disease can diagnose underlying genetic disorders. Kidney Int 2017;92:1493–1506. [DOI] [PubMed] [Google Scholar]
- 5.Zmyslowska A, et al. Serum Metabolic Fingerprinting Identified Putatively Annotated Sphinganine Isomer as a Biomarker of Wolfram Syndrome. J Proteome Res 2017;16:4000–4008. [DOI] [PubMed] [Google Scholar]