

Abstract
Trehalose is used as an additive in thousands of foods, cosmetics, and pharmaceutical products, and it is being investigated as a therapeutic for multiple human diseases. However, its ability to be used as a carbon source by microbes is a concern, as highlighted by the recent finding that trehalose can be metabolized by and potentially enhance the virulence of epidemic Clostridioides difficile. Here, we show that trehalose analogues designed to resist enzymatic degradation are incapable of being used as carbon sources by C. difficile. Furthermore, we demonstrate that trehalose analogues, but not the known trehalase inhibitor validamycin A, inhibit native trehalose utilization by hypervirulent C. difficile. Thus, degradation-resistant trehalose analogues are valuable as trehalase inhibitors and as surrogates for or co-additives with trehalose in applications where enzymatic breakdown is a concern.
Trehalose (1), a non-mammalian disaccharide comprising two glucopyranosyl units connected via a 1,1-α,α-glycosidic bond, has unique stabilizing, sweetening, and texturizing properties that have driven its widespread use as an additive in the food industry.1 In addition, the ability of trehalose to preserve a wide range of biological materials, including proteins, DNA, cell lines, and tissues, has led to applications in the biotechnology and pharmaceutical industries.1 Trehalose is also gaining attention as a possible human therapeutic because it mitigates disease burden in models of neurodegenerative and cardiometabolic diseases.2 The remarkable increase in application of trehalose in these areas over the past two decades has been spurred by the introduction of an efficient biocatalytic process for its production in 1995.3 However, an emerging concern in this field is that many organisms possess trehalase or phosphotrehalase enzymes that hydrolyze trehalose or trehalose-6-phosphate (T6P), respectively (Scheme 1).4,5 With respect to trehalose therapeutic applications, the disaccharide’s bioavailability and thus efficacy could be reduced by the presence of trehalase in human intestines and kidneys.6,7 Another issue is that many microorganisms enlist trehalases to permit the use of trehalose as a carbon source. This causes concern not only due to the potential for microbial contamination of food and pharmaceutical products that contain trehalose as an additive, but also because the ingestion of such products by humans could potentially promote the growth of pathogens.
Scheme 1.
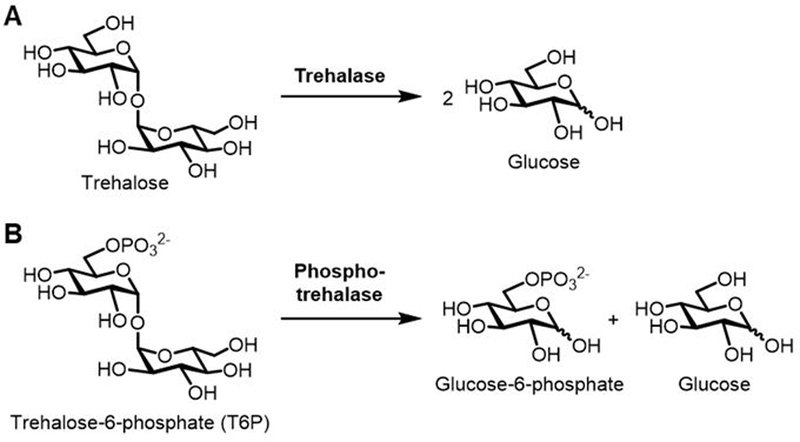
(A) Trehalase- and (B) phosphotrehalase-catalyzed hydrolysis of trehalose and trehalose-6-phosphate (T6P), respectively.
In 2018, it was shown that hypervirulent strains of Clostridioides difficile (formerly Clostridium difficile), a Gram-positive bacterium which causes an emerging and increasingly fatal intestinal infection, have acquired mechanisms for trehalose utilization that may enhance virulence.8,9 Specifically, epidemic C. difficile ribotypes RT027 and RT078 have evolved efficient trehalose uptake and phosphotrehalase (TreA)-mediated breakdown pathways that enable growth on low concentrations of trehalose.8 The expansion of these strains in humans is proposed to have been driven in part by trehalose consumption, which has escalated dramatically since trehalose received “generally regarded as safe” (GRAS) status by the FDA in 2000 and became a ubiquitous food additive.8 An important implication of this finding is that dietary additives such as trehalose may have unanticipated deleterious effects on the gut microbiota and human health, prompting further investigation and possibly strategies for mitigation.
We envision that trehalose analogues designed to retain the beneficial properties of trehalose for specific biotechnological or biomedical applications, but that are resistant to trehalase activity, will be valuable for addressing problems such as those described above. As a proof of concept, here we report the development of a group of trehalase-resistant trehalose analogues and their evaluation in C. difficile clinical isolates. Three analogues were investigated in this study (Figure 1A), including two epimers of trehalose bearing inverted hydroxyl groups at the 2- and 4-positions, named mannotrehalose (2) and lactotrehalose (3), respectively, which were previously reported to be trehalase-resistant.4,5,10,11 We also investigated 5-thiotrehalose (4) as a novel trehalase-resistant analogue, based on observations that other 5-deoxy-5-thio-modified glycosides are resistant to glycosidase activity.12 Gram-scale quantities of 3 were prepared through chemical synthesis according to a reported procedure.13 Semi-preparative-scale quantities (10–100 mg) of 2 and 4 were prepared using chemoenzymatic synthesis as we previously reported,14,15 which delivered the compounds from commercially available glucose derivatives in high yield in a single step.
Figure 1.
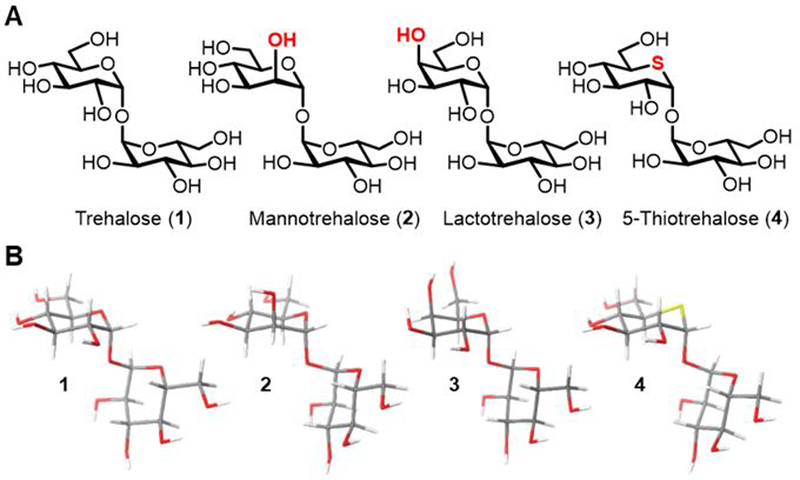
(A) Structures of trehalose (1) and synthetic trehalose analogues (2–4) used in this study. (B) Lowest energy conformers of 1–4 determined by conformational searches using MM3* with 2D NMR-determined glycosidic bond angles as constraints. See ESI for additional conformational analysis data.
To provide insight into the structural consequences of modifying the trehalose core, the conformations of 1–4 were determined using NMR spectroscopy and molecular modeling, as we previously reported for deoxyfluorinated trehalose analogues.16 The hexopyranosyl rings of all compounds adopted a classical 4C1 chair conformation as determined by vicinal 3JHH coupling constants. The dihedral angles across the glycosidic bonds (φH and ψH), which were determined using 3JCOCH coupling constants obtained via 2D excitation-sculptured indirect-detection NMR experiments (EXSIDE) (Figure S1), showed little perturbation of disaccharide structure arising from modification (Figure 1B and Table S1). These data suggest that analogues 2–4 generally mimic the solution conformation of native trehalose, and are therefore likely to retain biological activity of the native disaccharide. Consistent with this notion, recent data demonstrate that lactotrehalose (3) induces canonical fasting signaling in isolated murine hepatocytes, similar to the effects of native trehalose on hepatocyte metabolism.17,18
To evaluate whether 2–4 were resistant to enzymatic degradation by trehalase, we tested them using a commercially available model trehalase from porcine kidney. 10 mM of the analogues or native trehalose were incubated in the presence of trehalase or buffer alone, then the reactions were quenched and analyzed by thin-layer chromatography (TLC). Whereas trehalose was partially degraded to glucose after 1 h and completely degraded after 2 h, the three trehalose analogues were virtually unaffected by trehalase (Figure 2A), as only faint spots representing hydrolysis products of mannotrehalose (2) and 5-thiotrehalose (4) were observed at the 4 and 8 h time points. No degradation of lactotrehalose (3) was detected. These results demonstrate high hydrolytic stability of all three analogues 2–4 in the presence of trehalase.
Figure 2.
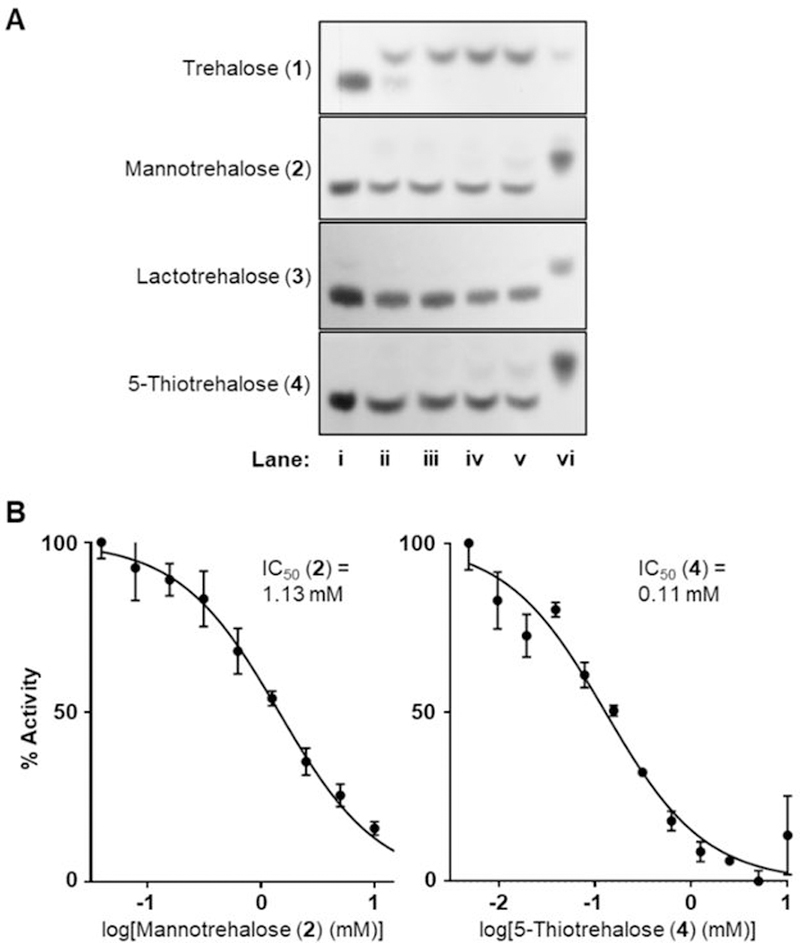
(A) Trehalose is hydrolyzed by porcine trehalase but compounds 2–4 are not. Compounds were incubated with trehalase for 1–8 hours, then reactions were analyzed by TLC. Lanes: (i) trehalose or analogue incubated 8 h without trehalase; (ii–v) trehalose or analogue incubated 1, 2, 4, or 8 h in the presence of trehalase; (vi) glucose (for 1) or corresponding glucose analogue (for 2–4) standards. (B) Compounds 2 and 4 inhibit porcine trehalase. Varying concentrations of analogues 2–4 were incubated in the presence of trehalose and trehalase, then released glucose was measured using a spectrophotometric assay and IC50 values determined. No inhibition was observed for compound 3 using this assay. Error bars represent the standard deviation from three replicates.
Next, we tested the ability of analogues 2–4 to inhibit porcine trehalase-catalyzed hydrolysis of native trehalose using a commercially available UV spectrophotometric assay that couples trehalose hydrolysis to NADPH production (Figure 2B). Both mannotrehalose (2) and 5-thiotrehalose (4) exhibited trehalase inhibition, with IC50 values of 1.132 mM and 0.106 mM, respectively. Unexpectedly, we observed no inhibition for lactotrehalose (2), despite this compound being previously reported to inhibit porcine trehalase.11 Since the no-trehalose/no-analogue controls and experiments with 2 and 4 behaved as expected, and multiple independently synthesized batches of compound 3 yielded the same results, there is no indication the assay was functioning improperly. Regardless of this inconsistency with the literature regarding lactotrehalose (3), based on our data it is clear that at least compounds 2 and 4 inhibit trehalase, with the novel sulfur-modified analogue 4 exhibiting 10-fold higher potency than the trehalose epimer 2.
Having confirmed that compounds 2–4 were resistant to enzymatic breakdown, and that at least 2 and 4 were trehalase inhibitors, we evaluated the analogue panel in C. difficile clinical isolates. First, we tested whether the analogues could serve as the sole carbon source for the trehalose-utilizing epidemic C. difficile ribotypes RT027 and RT078, as well as two control strains RT003 and RT053, which cannot grow on low trehalose alone.8 The strains were evaluated for growth in defined minimal medium (DMM) containing either 20 mM glucose, 10 mM trehalose, or 10 mM analogues 2–4 (Figure 3A). As previously observed,8 all strains grew in 20 mM glucose, whereas only RT027 and RT078 grew on an equivalent concentration (10 mM) of trehalose. By contrast, no growth was observed for RT027 or RT078 (or the control strains RT053 and RT003) when either lactotrehalose (3) or 5-thiotrehalose (4) were used, demonstrating that—in contrast to native trehalose—neither of these trehalase-resistant compounds support the growth of C. difficile. On the other hand, mannotrehalose (2) was efficiently used as the sole carbon source by all strains, despite the inability of this compound to be hydrolyzed by porcine trehalase. Interestingly, deletion of the phosphotrehalase TreA did not affect growth of C. difficile on mannotrehalose in RT027 (Figure S2), suggesting the existence of an alternative pathway by which this unnatural disaccharide is metabolized.
Figure 3.
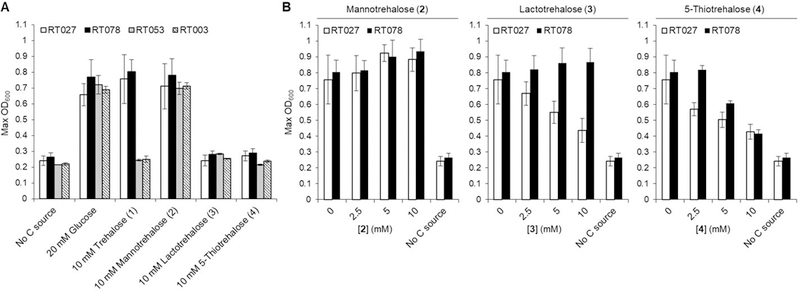
(A) C. difficile ribotypes RT027 and RT078 grow on trehalose and mannotrehalose, but not lactotrehalose or 5-thiotrehalose. RT053 and RT003 are control strains that do not grow on low trehalose. The indicated strains were grown in DMM containing the indicated carbon source(s) and growth was assessed by monitoring OD600 over 16 h. (B) Growth of RT027 and RT078 on low trehalose is blocked by 5-thiotrehalose (both strains) and lactotrehalose (RT027 only) in a dose-dependent manner. The indicated strains were grown in DMM containing 10 mM trehalose (or no carbon source if noted) in the presence of varying concentrations of trehalose analogues. Error bars represent the standard deviation from three biological replicates.
Next, we evaluated whether the trehalose analogues could prevent hypervirulent C. difficile ribotypes RT027 and RT078 from using native trehalose as a carbon source. These strains were tested for growth in DMM containing 10 mM trehalose and supplemented with 0–10 mM of trehalose analogues 2–4 (Figure 3B). Consistent with the sole carbon source assays, mannotrehalose (2) treatment did not impair growth of RT027 or RT078 on trehalose, and in fact appeared to enhance growth moderately. Lactotrehalose (3) activity was ribotype-dependent, as growth of RT027, but not RT078, was inhibited by increasing concentrations of 3. Most promisingly, 5-thiotrehalose (4) blocked the growth of both RT027 and RT078 in a dose-dependent manner, exhibiting an approximate two-thirds reduction in growth when administered at 10 mM. We also tested the effect of 5-thiotrehalose treatment when RT078 was grown on 20 mM glucose instead of 10 mM trehalose and found no growth defect under these conditions (Figure S3). This result confirmed that analogue activity was specific for trehalose metabolism. As well, 5-thiotrehalose was non-toxic to HEK 293 cells (Table S2), implying compatibility of this analogue with mammalian systems. Finally, we found that the known trehalase inhibitor validamycin A had no effect on trehalose utilization by hypervirulent C. difficile up to 1 mM (Figure S4), further underscoring the value of trehalose-based inhibitors in the context of C. difficile metabolism, and potentially other areas.
The precise mechanism by which trehalose analogues prevent utilization of exogenous trehalose by RT027 and RT078 is not yet known. C. difficile is proposed to transport exogenous trehalose into the cell via a phosphotransferase system (PTS), generating cytosolic T6P that can then undergo hydrolysis by the phosphotrehalase TreA to yield glucose and glucose-6-phosphate.8 One possibility is that analogues 3 and 4 are taken up through the PTS and converted into their corresponding T6P analogues, which subsequently inhibit intracellular TreA. Alternatively, the analogues may act directly on the PTS, blocking uptake and subsequent utilization of native trehalose. While we performed initial characterization of compounds 2–4 in this work using porcine trehalase as a model enzyme, there can be wide differences in trehalase and phosphotrehalase sequences between taxa. A more detailed investigation of these compounds’ mechanism(s) of action in C. difficile will be achievable with recombinant TreA phosphotrehalase and relevant T6P analogues, both of which we are currently working toward. The latter is a significant synthetic challenge, but our chemoenzymatic methods can potentially be adapted to improve the accessibility of T6P analogues. In the meantime, the data presented here demonstrate the value of trehalose analogues as tools to probe and inhibit trehalose metabolism in C. difficile. Of particular interest, 5-thiotrehalose (4) was shown for the first time to be (i) trehalase-resistant, (ii) an improved inhibitor of purified trehalase (in comparison to known trehalose analogues), (iii) an inhibitor of trehalose utilization by C. difficile strains RT027 and RT078, and (iv) non-toxic to mammalian cells. Interestingly, we observed significant differences in activity of the three analogues against the various C. difficile strains tested. Thus, in addition to probing the analogues’ mechanism of action in C. difficile, it will be of considerable interest to expand the compound class and the clinical strains being evaluated to obtain a more robust structure–activity profile. To conclude, this study represents the first reported strategy for addressing the possible link between dietary trehalose and C. difficile hypervirulence, which was first proposed in early 2018. More broadly, we expect that trehalase-resistant trehalose analogues will serve as trehalose surrogates or co-additives that retain or enhance the sugar’s beneficial characteristics for food additive and emerging therapeutic applications, but are non-caloric, have improved bioavailability, and do not contribute to microbial growth and pathogenesis.
This work was funded by a grant to B.M.S. and P.J.W. from the National Institutes of Health (R15 AI117670), as well as a Henry Dreyfus Teacher−Scholar Award to B.M.S from The Camille & Henry Dreyfus Foundation (TH-17–034). This work was also supported by grants to R.A.B. from the National Institute of Allergy and Infectious Disease at the NIH (R01AI123278 and U01AI124290). This work was also supported by grants to B.J.D. from the NIH (R56 DK115764), Children’s Discovery Institute (MI-FR-2014–426 and MI-II2017–593), AGA-Gilead Sciences Research Scholar Award in Liver Disease, The AGA-Allergan Pilot Research Award in Non-Alcoholic Fatty Liver Disease, and the Robert Wood Johnson Foundation. Dr. Daniel Holmes of the Michigan State University NMR Facility is thanked for assistance with EXSIDE experiments. Dr. Mallary Wacker is thanked for assistance with the mammalian cell toxicity experiments.
Supplementary Material
Footnotes
Conflicts of interest
There are no conflicts to declare.
Electronic Supplementary Information (ESI) available: Experimental methods and supporting data. See DOI: 10.1039/x0xx00000x
References
- 1.Ohtake S and Wang YJ, J. Pharm. Sci., 2011, 100, 2020–2053. [DOI] [PubMed] [Google Scholar]
- 2.Hosseinpour-Moghaddam K, Caraglia M and Sahebkar A, J. Cell Physiol., 2018, 233, 6524–6543. [DOI] [PubMed] [Google Scholar]
- 3.Maruta K, Nakada T, Kubota M, Chaen H, Sugimoto T, Kurimoto M and Tsujisaka Y, Biosci. Biotechnol. Biochem., 1995, 59, 1829–34. [DOI] [PubMed] [Google Scholar]
- 4.Walmagh M, Zhao R and Desmet T, Int. J. Mol. Sci., 2015, 16, 13729–13745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Neill MK, Piligian BF, Olson CD, Woodruff PJ and Swarts BM, Pure Appl. Chem, 2017, 89, 1223–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeBosch BJ, Heitmeier MR, Mayer AL, Higgins CB, Crowley JR, Kraft TE, Chi M, Newberry EP, Chen Z, Finck BN, Davidson NO, Yarasheski KE, Hruz PW and Moley KH, Sci. Signal., 2016, 9, ra21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sergin I, Evans TD, Zhang X, Bhattacharya S, Stokes CJ, Song E, Ali S, Dehestani B, Holloway KB, Micevych PS, Javaheri A, Crowley JR, Ballabio A, Schilling JD, Epelman S, Weihl CC, Diwan A, Fan D, Zayed MA and Razani B, Nat. Commun., 2017, 8, 15750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Collins J, Robinson C, Danhof H, Knetsch CW, van Leeuwen HC, Lawley TD, Auchtung JM and Britton RA, Nature, 2018, 553, 291–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Collins J, Danhof H and Britton RA, Gut Microbes, 2018, doi: 10.1080/19490976.2018.1491266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ryu S-I, Kim J-E, Huong NT, Woo E-J, Moon S-K and Lee S-B, Enzym. Microb. Technol., 2010, 47, 249–256. [Google Scholar]
- 11.Kim H-M, Chang Y-K, Ryu S-I, Moon S-G and Lee S-B, J. Mol. Catal. B Enzym., 2007, 49, 98–103. [Google Scholar]
- 12.Yuasa H, Izumi M and Hashimoto H, Curr. Top. Med. Chem., 2009, 9, 76–86. [DOI] [PubMed] [Google Scholar]
- 13.Bassily RW, El-Sokkary RI, Silwanis BA, Nematalla AS and Nashed MA, Carbohydr. Res., 1993, 239, 197–207. [DOI] [PubMed] [Google Scholar]
- 14.Urbanek BL, Wing DC, Haislop KS, Hamel CJ, Kalscheuer R, Woodruff PJ and Swarts BM, ChemBioChem, 2014, 15, 2066–2070. [DOI] [PubMed] [Google Scholar]
- 15.Meints LM, Poston AW, Piligian BF, Olson CD, Badger KS, Woodruff PJ and Swarts BM, J. Vis. Exp., 2017, e54485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rundell SR, Wagar ZL, Meints LM, Olson CD, O’Neill MK, Piligian BF, Poston AW, Hood RJ, Woodruff PJ and Swarts BM, Org. Biomol. Chem., 2016, 14, 8598–8609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Higgins CB, Zhang Y, Mayer AL, Fujiwara H, Stothard AI, Graham MJ, Swarts BM and DeBosch BJ, JCI Insight, 2018, 3, 120794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y, Higgins CB, Fortune HM, Chen P, Stothard AI, Mayer AL, Swarts BM and DeBosch BJ, Nat. Commun., 2019, 10, 1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.