

Abstract
The first asymmetric cooperative Lewis base/palladium catalyzed benzylic alkylation of acyclic esters is reported. This reaction proceeds via stereodefined C1-ammonium enolate nucleophiles. Critical to its success was the identification of benzylic phosphate electrophiles, which were uniquely reactive. Alkylated products were obtained with very high levels of enantioselectivity, and this method has been applied toward the synthesis of the thrombin inhibitor DX-9065a.
Keywords: benzylation, cooperative catalysis, enantioselectivity, Lewis bases, palladium
Enantioselective palladium-catalyzed allylic alkylation reactions are amongst the most versatile and robust methods for the construction of C(sp3)–C(sp3) bonds.[1] Under the action of a suitable Pd catalyst, carbogenic nucleophiles react efficiently with allylic electrophiles. These reactions typically proceed via cationic π-(allyl)PdIILn species, the reactivity and stereocontrol elements of which can be readily tuned by the supporting ligands. Despite possessing isostructural and isoelectronic characteristics, asymmetric Pd-catalyzed benzylic alkylation reactions using enolate nucleophiles are far less common.[2] This is attributable, in part, to the difficulty in forming Pd0/arene complexes and the relatively high energy required for dearomatizing ionization/oxidative addition (Figure 1 a).
Figure 1.
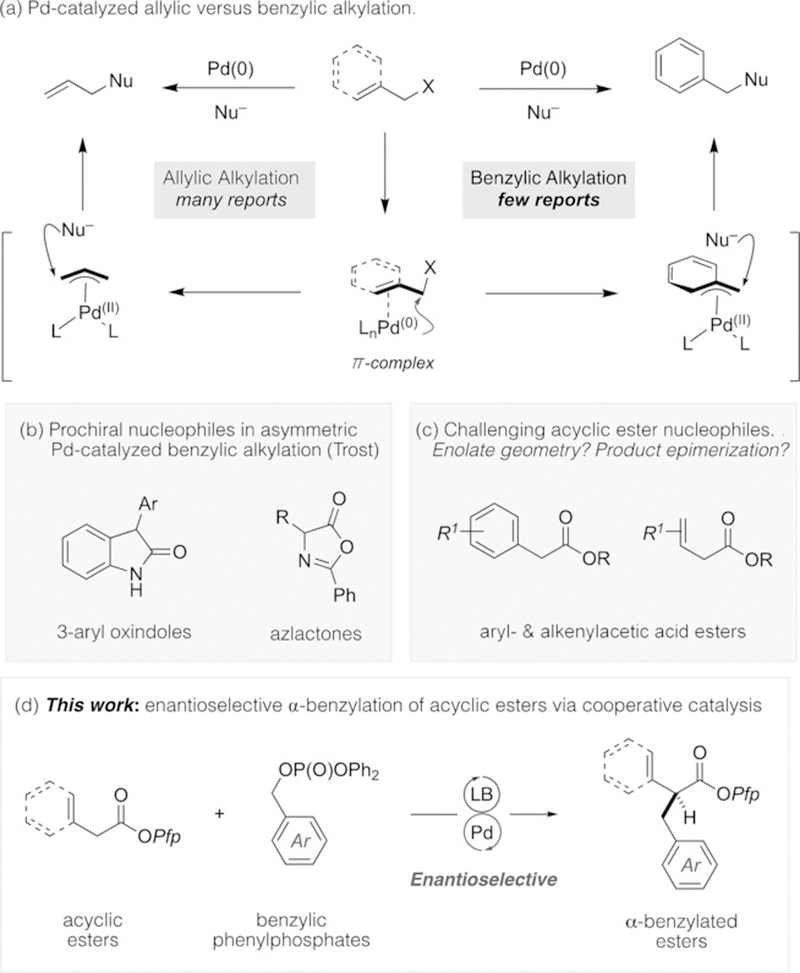
a) Palladium-catalyzed allylic versus benzylic alkylation. b) Competent prochiral nucleophiles in asymmetric palladium-catalyzed benzylation. c) Challenging acyclic ester nucleophiles. d) This work: Direct enantioselective α-benzylation.
Outstanding studies by the groups of Fiaud, Kwano, and Tunge provided some resolution to this restriction;[3] however, the generation of enantioenriched products remains a major challenge. Focusing on the generation of stereochemistry at the electrophilic carbon atom, noteworthy contributions from Fiaud and Hirano/Murai have described the use of secondary benzyl electrophiles in reactions that proceed by partial kinetic resolution or by a dynamic kinetic asymmetric transformation (DYKAT), respectively.[4,5] To address stereochemical induction at the nucleophilic carbon atom, Trost and Czabaniuk reported the highly enantioselective benzylation of cyclic prochiral azlactone and 3-aryl oxindole nucleophiles using primary benzylic carbonates and phosphates (Figure 1 b).[6] Although few in number, these remain the most effective Pd-catalyzed benzylic alkylation methods available.[7] Thus far, the use of acyclic ester nucleophiles has not been described despite the clear utility of the products (Figure 1 c).
In response to long-standing challenges associated with the use of acyclic prochiral nucleophiles in asymmetric transition-metal-catalyzed transformations, our laboratory has embraced cooperative Lewis base/transition-metal catalysis as a general design principle.[8] Proceeding via C1-ammonium enolate nucleophiles, this construct results in a general reaction template that accommodates a variety of transition-metal-catalyzed processes.[9] Herein, we further advance our cooperative framework by demonstrating, for the first time, that C1-ammonium enolates effectively react with putative cationic π-(benzyl)PdII electrophiles, enabling the highly enantioselective benzylic alkylation of aryl- and alkenylacetic acid esters with π-extended benzylic phosphate electrophiles (Figure 1 d).
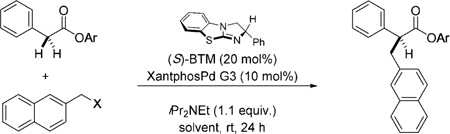
During their seminal benzylation studies, Trost and Czabaniuk described the critical role of the nucleofuge in determining both the facility of Pd0 oxidative addition and the obtained enantioselectivity.[6] Within our cooperative catalysis framework we have also observed the drastic effect that the nucleofuge plays in enantioselection.[8] Mindful of these observations, we surveyed a range of activated 2-naphthyl alcohol derivatives (Table 1).
Table 1:
Optimization studies.
 | |||||
|---|---|---|---|---|---|
| Entry[a] | X | Ar | Solvent | Yield [%][b] | er[c] |
| 1 | OTs | 1 | THF | 0 | – |
| 2 | OAc | 1 | THF | 0 | – |
| 3 | OCO2tBu | 1 | THF | 0 | – |
| 4 | OP(O)(OEt)2 | 1 | THF | 5 | – |
| 5 | OP(O)(OPh)2 | 1 | THF | 68 | 97:3 |
| 6 | OP(O)(OPh)2 | 1 | 1,4-dioxane | 60 | 93:7 |
| 7 | OP(O)(OPh)2 | 1 | CH2Cl2 | 40 | 89:11 |
| 8 | OP(O)(OPh)2 | 1 | toluene | 85 (81) | 99:1 |
| 9 | OP(O)(OPh)2 | 2 | toluene | 82 | 94:6 |
| 10 | OP(O)(OPh)2 | 3 | toluene | 21 | – |
| 11 | OP(O)(OPh)2 | 4 | toluene | 0 | – |
| 12 | OP(O)(OPh)2 | 5 | toluene | 75 | 99:1 |
| 13 | OP(O)(OPh)2 | 6 | toluene | 0 | – |
Reactions performed on 0.1 mmol scale.
Yields determined by 1H NMR analysis using 1,2,4,5-tetramethylbenzene as an internal standard. Yields of isolated products given in parentheses.
Determined by HPLC analysis on a chiral stationary phase.
Ms= methane-sulfonyl, Pfp = pentafluorophenyl.
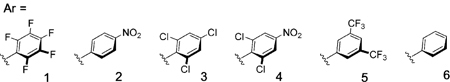
Employing benzotetramisole (BTM)[10,11] as the Lewis base catalyst and Buchwald’s XantphosPd G3 precatalyst[12] we quickly identified diphenylphosphate as the only competent nucleofuge (entries 1–5), which furnished the desired product in high yield and excellent enantioselectivity (68 %, er 97:3). Further assessment of the solvent identified toluene, which gave the product in an enhanced 85 % yield and er 99:1. Finally, evaluation of various electron-deficient aryl esters (2–5) (entries 9–12) offered no improvement, although ester 5 did function with notable efficiency (entry 12). Phenyl ester 6 was ineffective (Entry 13).[13]
With an optimized procedure in hand, we proceeded to evaluate the scope of arylacetic Pfp ester nucleophiles (Figure 2). As expected, a wide variety of arylacetic esters performed well and gave the corresponding products with excellent levels of enantioselectivity. Notable examples include the tolerance of o-bromophenyl moieties (13 and 14), the performance of 2- and 3-thiophene-derived esters (15 and 16), and the facility with which arene-rich systems can be constructed (20–23). Evaluation of the electrophile scope using the Pfp ester of 2-naphthylacetic acid (Figure 3) revealed the tolerance of acetylene units (27), pinacolboronic esters (28), nitriles (29), acrylates (30), and Lewis basic N-heterocycles (31). Extension to 1-naphthyl electrophiles (32–34) as well as regioisomeric benzothiophene (35) and benzofuran (36) heterobenzylic electrophiles was also possible.[14] Whereas π-extended electrophiles functioned effectively, simple monocyclic benzylic phosphates were unreactive, presumably owing to the aforementioned energy required for dearomatization.[2–6] Pfp esters derived from alkylacetic acids are also unreactive within this cooperative catalysis framework.
Figure 2.
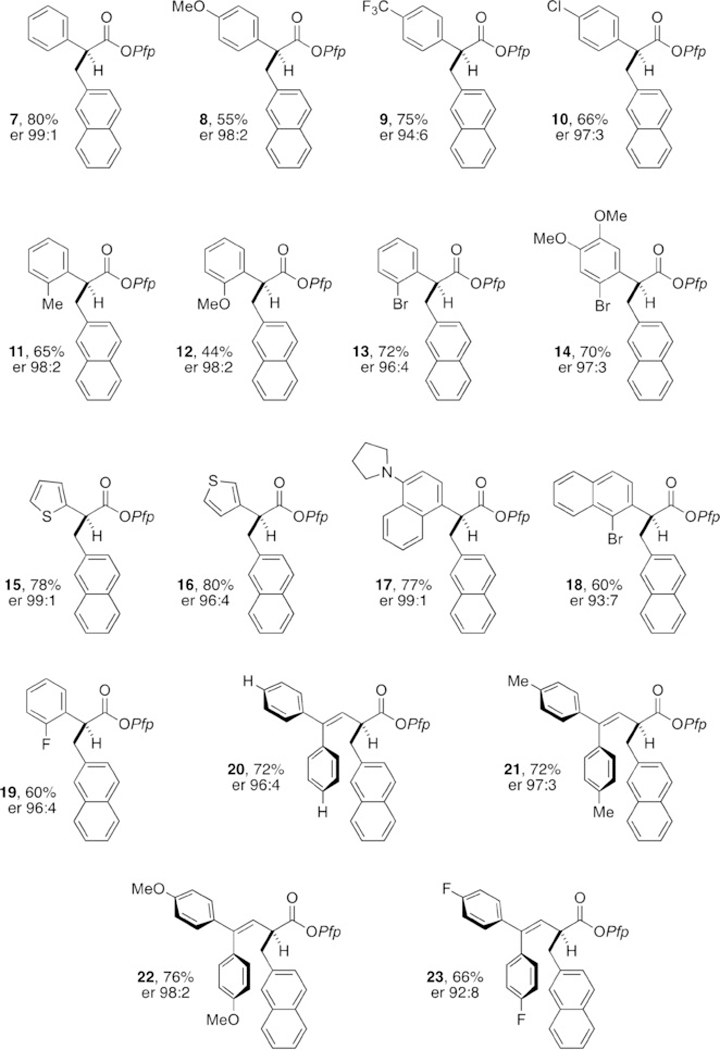
Nucleophile scope. Reactions performed on 0.1 mmol scale. Yields of isolated produts after purification by column chromatography are given. The er values were determined by HPLC analysis on a chiral stationary phase.
Figure 3.
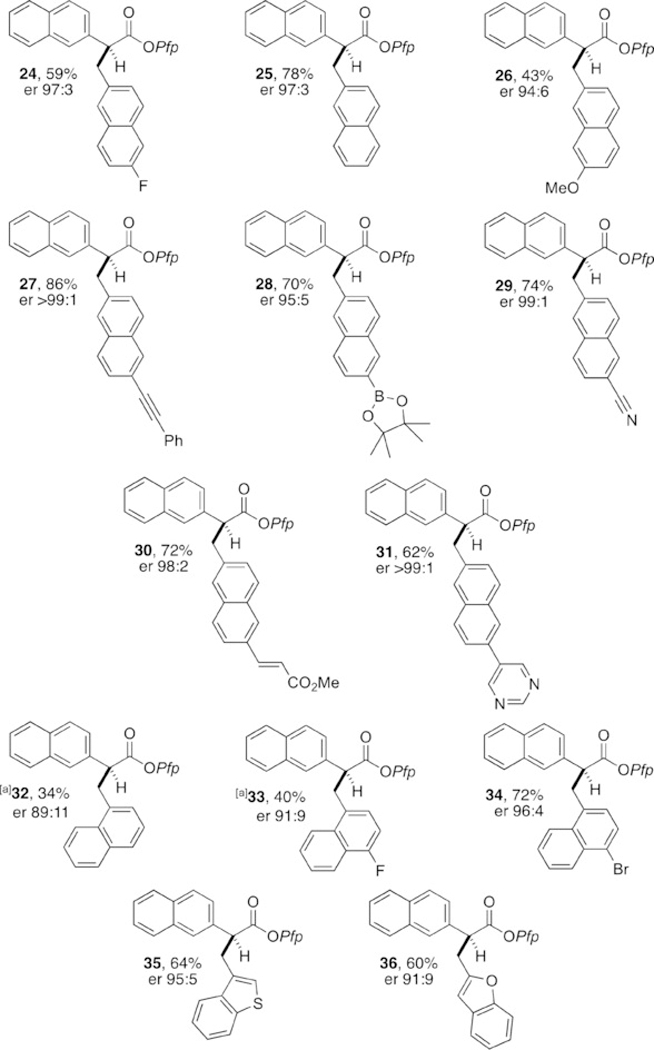
Electrophile scope. Yields of isolated produts after purification by column chromatography are given. The er values were determined by HPLC analysis on a chiral stationary phase. [a] Isolated as the corresponding benzylamide (see the Supporting Information for details).
Our interest in this process stems not only from the well-documented challenges associated with enantioselective catalysis via cationic π-(benzyl)Pd intermediates, but also from the potential of such reactions to address the synthesis of therapeutically relevant chiral molecules. Here, we demonstrate the utility of this method towards the synthesis of the thrombin inhibitor DX-9065A (41),[15] a selective inhibitor of the coagulant enzyme activated factor X (FXa).[16] Ethyl ester 40 is a key intermediate en route to 41 and was previously prepared as a 1:1 diastereomeric mixture at the ester-bearing stereocenter; crystallization provided 41 as a single diastereomer.[15] We envisioned the stereocontrolled preparation of 39 (and thence 40) using the method described here. In the event, direct alkylation of ester 37 with benzylic phosphate 38 gave 39 in 83 % isolated yield as a single diastereomer, demonstrating complete catalyst control over stereoinduction (Scheme 1). Thereafter, transesterification gave the key ethyl ester 40 in quantitative yield.
Scheme 1.
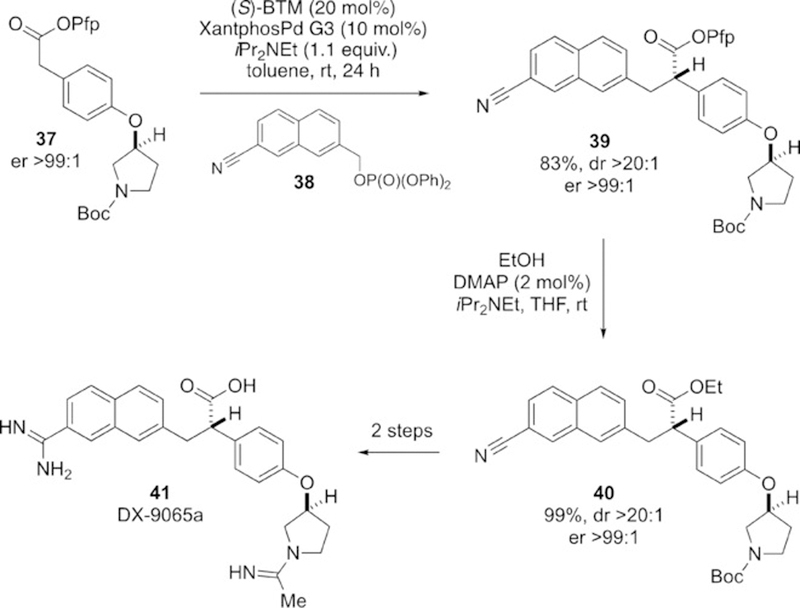
Synthesis of DX-9065a.
In conclusion, we have demonstrated the first enantioselective palladium-catalyzed benzylic alkylation of acyclic ester nucleophiles. Critical to the success of this reaction was 1) the identification of the uniquely effective phosphate nucleofuge and 2) the cooperative action of a Lewis base catalyst, which governs the in situ production of stereodefined C1-ammonium enolate nucleophiles as well as the enantioelectivity of the reaction. This is complementary to the ligand-centered enantiocontrol typical of palladium catalysis, and further demonstrates the potential of cooperative catalysis to address challenges in reactivity and stereocontrol that are beyond single catalysts. Our current efforts are directed towards the union of monocyclic benzyl electrophiles with C1-ammonium enolates and will be reported in due course.
Supplementary Material
Acknowledgements
We gratefully acknowledge Indiana University and the National Institutes of Health (R01GM121573) for generous financial support. We thank Dr. Maren Pink and Dr. Chun-Hsing Chen (IU) for X-ray crystallography. This project was partially supported by the IU Vice Provost for Research through the Research Equipment Fund.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- [1].a) For reviews, see:Tsuji J, Tetrahedron 2015, 71, 6330–6348 [Google Scholar]; b) Trost BM, Tetrahedron 2015, 71, 5708–5733 [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Weaver JD, Recio A III, Grenning AJ, Tunge JA, Chem. Rev 2011, 111, 1846–1913 [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Trost BM, Machacek MR, Aponick AP, Acc. Chem. Res 2006, 39, 747–760 [DOI] [PubMed] [Google Scholar]; e) Trost BM, Crawley ML, Chem. Rev 2003, 103, 2921–2944 [DOI] [PubMed] [Google Scholar]; f) Trost BM, Van Vranken DL, Chem. Rev 1996, 96, 395–422. [DOI] [PubMed] [Google Scholar]
- [2].a) For a review describing the reactivity of transition-metal η3-benzyl complexes, see: Trost BM, Czabaniuk LC, Angew. Chem. Int. Ed 2014, 53, 2826–2851; Angew. Chem. 2014, 126, 2868–2895 [DOI] [PubMed] [Google Scholar]; b) for a review on palladium-catalyzed benzylic alkylation using (hetero)benzylic electrophiles, see: Le Bras J, Muzart J, Eur. J. Org. Chem 2016, 2565–2593.
- [3].a) Legros J-Y, Fiaud J-C, Tetrahedron Lett 1992, 33, 2509–2510 [Google Scholar]; b) Legros J-Y, Toffano M, Fiaud J-C, Tetrahedron 1995, 51, 3235–3246 [Google Scholar]; c) Legros J-Y, Toffano M, Fiaud J-C, Tetrahedron: Asymmetry 1995, 6, 1899–1902 [Google Scholar]; d) Toffano M, Legros J-Y, Fiaud J-C, Tetrahedron Lett 1997, 38, 77–80 [Google Scholar]; e) Legros J-Y, Primault GI, Toffano M, RiviHre M-A, Fiaud J-C, Org. Lett 2000, 2, 433–436 [DOI] [PubMed] [Google Scholar]; f) Kuwano R, Kondo Y, Matsuyama Y, J. Am. Chem. Soc 2003, 125, 12104–12105 [DOI] [PubMed] [Google Scholar]; g) Kuwano R, Kondo Y, Org. Lett 2004, 6, 3545–3547 [DOI] [PubMed] [Google Scholar]; h) Kuwano R, Kusano H, Chem. Lett 2007, 36, 528–529 [Google Scholar]; i) Li T-R, Maliiszewski ML, Xiao W-J, Tunge JA, Org. Lett 2018, 20, 1730–1734 [DOI] [PubMed] [Google Scholar]; j) Torregrosa RR, Ariyarathna Y, Chattopadhyay K, Tunge JA, J. Am. Chem. Soc 2010, 132, 9280–9282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Legros J-Y, Boutros A, Fiaud J-C, Toffano M, J. Mol. Catal. A 2003, 196, 21–25; see also Ref. [3b]. [Google Scholar]
- [5].Tabuchi S, Hirano K, Murai M, Angew. Chem. Int. Ed 2016, 55, 6973–6977; Angew. Chem. 2016, 128, 7087–7091. [DOI] [PubMed] [Google Scholar]
- [6].a) Trost BM, Czabaniuk LC, Chem. Eur. J 2013, 19, 15210–15218 [DOI] [PubMed] [Google Scholar]; b) Trost BM, Czabaniuk LC, J. Am. Chem. Soc 2012, 134, 5778–5781 [DOI] [PubMed] [Google Scholar]; c) Trost BM, Czabaniuk LC, J. Am. Chem. Soc 2010, 132, 15534–15536. [DOI] [PubMed] [Google Scholar]
- [7].a) Nickel-catalyzed methods permit asymmetric benzylic alkylations using organometallic nucleophiles. For selected examples, see: Taylor BLH, Swift EC, Waetzig JD, Jarvo ER, J. Am. Chem. Soc 2011, 133, 389–391 [DOI] [PubMed] [Google Scholar]; b) Taylor BLH, Harris MR, Jarvo ER, Angew. Chem. Int. Ed 2012, 51, 7790–7793; Angew. Chem. 2012, 124, 7910–7913 [DOI] [PubMed] [Google Scholar]; c) Greene MA, Yonova IM, Williams FJ, Jarvo ER, Org. Lett 2012, 14, 4293–4296 [DOI] [PubMed] [Google Scholar]; d) Harris MR, Hanna LE, Greene MA, Moore CE, Jarvo ER, J. Am. Chem. Soc 2013, 135, 3303–3306 [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Wisniewska HM, Swift EC, Jarvo ER, J. Am. Chem. Soc 2013, 135, 9083–9090 [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Yonova IM, Johnson AG, Osborne CA, Moore CE, Morrissette NS, Jarvo ER, Angew. Chem. Int. Ed 2014, 53, 2422–2427; Angew. Chem. 2014, 126, 2454–2459 [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Zhou Q, Srinivas HD, Dasgupta S, Watson MP, J. Am. Chem. Soc 2013, 135, 3307–3310 [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Arp FO, Fu GC, J. Am. Chem. Soc 2005, 127, 10482–10483 [DOI] [PubMed] [Google Scholar]; i) Binder JT, Cordier CJ, Fu GC, J. Am. Chem. Soc 2012, 134, 17003. [DOI] [PMC free article] [PubMed] [Google Scholar]; Do H-Q, Chandrashekar ERR, Fu GC, J. Am. Chem. Soc 2013, 135, 16288–16291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Schwarz KJ, Pearson CM, Cintro-Rosado G, Liu P, Snaddon TN, Angew. Chem. Int. Ed 2018, 57, 7800–7803; Angew. Chem. 2018, 130, 7926–7929 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Fyfe JWB, Kabia O, Pearson CM, Snaddon TN, Tetrahedron 2018, 10.1016/j.tet.2018.04.021 [DOI] [PMC free article] [PubMed]; c) Schwarz KJ, Amos JL, Klein JC, Do DT, Snaddon TN, J. Am. Chem. Soc 2016, 138, 5214–5217. [DOI] [PubMed] [Google Scholar]
- [9].a) For other examples of the use of C1-ammonium enolates in combination with transition-metal catalysis, see: Spoehrle SS, West TH, Taylor JE, Slawin AMZ, Smith AD, J. Am. Chem. Soc 2017, 139, 11895–11902 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jiang Z, Beiger JJ, Hartwig JF, J. Am. Chem. Soc 2017, 139, 87–90 [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lu X, Ge L, Cheng C, Chen J, Cao W, Wu X, Chem. Eur. J 2017, 23, 7689–7693 [DOI] [PubMed] [Google Scholar]; d) Song J, Zhang Z-J, Gong L-Z, Angew. Chem. Int. Ed 2017, 56, 5212–5216; Angew. Chem. 2017, 129, 5296–5300 [DOI] [PubMed] [Google Scholar]; e) Song J, Zhang Z-J, Chen S-S, Fan T, Gong L-Z, J. Am. Chem. Soc 2018, 140, 3177–3180. [DOI] [PubMed] [Google Scholar]
- [10].a) Liu P, Yang X, Birman VB, Houk KN, Org. Lett 2012, 14, 3288–3291 [DOI] [PubMed] [Google Scholar]; b) Bumbu VD, Birman VB, J. Am. Chem. Soc 2011, 133, 13902–13905 [DOI] [PubMed] [Google Scholar]; c) Yang X, Lu G, Birman VB, Org. Lett 2010, 12, 892–895 [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Birman VB, Li X, Org. Lett 2006, 8, 1351–1354 [DOI] [PubMed] [Google Scholar]; e) for a review describing the varied reactivity of isothioureas as enantioselective Lewis base catalysts, see: Merad J, Pons J-M, Chuzel O, Bressy C, Eur. J. Org. Chem 2016, 5589–5610.
- [11].a) For reviews discussing Lewis base catalysis and C1-ammonium enolates, see: Morrill LC, Smith AD, Chem. Soc. Rev 2014, 43, 6214–6226 [DOI] [PubMed] [Google Scholar]; b) Paull DH, Weatherwax A, Lectka T, Tetrahedron 2009, 65, 6771–6803 [DOI] [PMC free article] [PubMed] [Google Scholar]; c) France S, Guerin DJ, Miller SJ, Lectka T, Chem. Rev 2003, 103, 2985–3012. [DOI] [PubMed] [Google Scholar]
- [12].Bruno NC, Tudge MT, Buchwald SL, Chem. Sci 2013, 4, 916–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Electron-deficient aryl esters are electron-deficient enough to undergo displacement by isothiourea Lewis base catalysts. For a discussion of this process and the phenolate rebound/aryloxide turnover mechanism that underpins this chemistry, see: Hartley WC, O’Riordan TJC, Smith AD, Synthesis 2017, 49, 3303–3310. [Google Scholar]
- [14]. For examples of unreactive or unstable aryl phosphate electrophiles, see the Supporting Information.
- [15].Nagahara T, Yokoyama Y, Inamura K, Katakura S, Komoriya S, Yamaguchi H, Hara T, Iwamoto M, J. Med. Chem 1994, 37, 1200–1207. [DOI] [PubMed] [Google Scholar]
- [16].Pinto DP, Smallheer JM, Cheney DL, Knabb RM, Wexler RR, J. Med. Chem 2010, 53, 6243–6274. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.