

Abstract
Background:
The phosphodiesterase 3A (PDE3A) gene encodes a PDE that regulates cardiac myocyte cAMP levels and myocardial contractile function. PDE3 inhibitors are used for short term treatment of refractory heart failure (HF), but do not produce uniform long-term benefit.
Objectives:
We tested the hypothesis that drug target genetic variation could explain clinical response heterogeneity to PDE3 inhibitors in HF.
Methods:
PDE3A promoter studies were performed in a cloned luciferase construct. In human left ventricular (LV) preparations mRNA expression was measured by RT-PCR, and PDE3 enzyme activity by cAMP-hydrolysis.
Results:
We identified a 29-nt indel (insertion (INS)/deletion (DEL) polymorphism) in the human PDE3A gene promoter beginning 2214 nt upstream from the PDE3A1 translation start site. Transcription factor ATF3 binds to the INS and represses cAMP-dependent promoter activity. In explanted failing LVs who were homozygous for PDE3A DEL and had been treated with PDE3 inhibitors pre-cardiac transplant, PDE3A1 mRNA abundance and microsomal PDE3 enzyme activity were increased by 1.7 to 1.8-fold (p <0.05) compared to DEL homozygotes not receiving PDE3 inhibitors. The basis for the selective up-regulation in PDE3A gene expression in DEL homozygotes treated with PDE3 inhibitors was a cAMP response element (CRE) enhancer 61 nt downstream from the INS, which was repressed by INS. The DEL homozygous genotype frequency was also enriched in patients with HF.
Conclusion:
A 29-nt INS/DEL polymorphism in the PDE3A promoter regulates cAMP-induced PDE3A gene expression in patients treated with PDE3 inhibitors. This molecular mechanism may explain response heterogeneity to this drug class, and may inform a pharmacogenetic strategy for a more effective use of PDE3 inhibitors in HF.
Keywords: PDE3A, polymorphism, transcription regulation, ATF3, heart failure, β-adrenergic receptors
Condensed Abstract:
The phosphodiesterase 3A (PDE3A) gene encodes a PDE that regulates cardiomyocyte cAMP levels and contractile function. PDE3 inhibitors (PDE3i) are used for short term treatment of refractory heart failure (HF), but do not produce uniform long-term benefit. We identified a 29-nt insertion (INS)/deletion(DEL) polymorphism in the human PDE3A gene promoter that regulates PDE3A gene expression. In failing PDE3i-treated LVs homozygous for PDE3A DEL, PDE3A1 mRNA abundance and enzyme activity were increased compared to PDE3i-untreated DEL homozygotes and non-failing controls. This mechanism may explain response heterogeneity to PDEi and inform a pharmacogenetic strategy for an effective use of PDE3i in HF.
Introduction
Activation of β-adrenergic receptors (β-ARs) increases adenylate cyclase (AC) activity, resulting in cAMP syntheis, activation of protein kinase A (PKA) and phosphorylation of various downstream effectors. The cardiac β-AR/AC/cAMP/PKA axis has an important role in regulating heart rate and myocardial contraction and relaxation. In the failing human heart β-AR signal transduction is desensitized, and cAMP levels are decreased despite increased adrenergic activity (1). These changes may compromise cardiac function as well as redirect signaling to more biologically adverse “PKA-independent” pathways, such as Ca2+/calmodulin-dependent protein kinase II (CaMKII) (1).
Cyclic nucleotide phosphodiesterases (PDEs) hydrolyze the phosphodiester bond in cyclic adenosine or guanosine monophosphate (cAMP or cGMP) for conversion to their respective 5’ monophosphates (2). In the failing heart an increase in cAMP to more normal levels could potentially be achieved by inhibition of PDEs (3). However, several clinical trials have shown increased mortality (4,5) or lack of efficacy (6) in advanced heart failure with reduced ejection fraction (HFrEF) adult patients treated with PDE3 inhibitors. The increase in mortality appears to be due to pro-arrhythmic effects interacting with other clinical variables to produce an increase in sudden death (7,8), while the lack of effectiveness when a PDE3 inhibitor was used with a β-blocker appeared to be due in part to response heterogeneity (6) or an attenuation of initial effectiveness (9,14).
Effects of cAMP in the heart are classically attributed to the phosphorylation of cardiac myocyte proteins that affect excitation/contraction coupling, including L-Type Ca2+ channels (LTCCs), the sarcoplasmic reticulum ATPase 2 (SERCA2) regulatory protein phospholamban (PLN), ryanodine receptor 2 (RyR2), phosphatase 1 inhibitor and various contractile proteins (10). In order to regulate localized cAMP-mediated signaling PDEs are compartmentalized intracellularly (15), and PDE3A protein is present in a microdomain that includes SERCA2, PLN, PKA-RII and AKAP7/18 (16). Three isoforms of PDE3A have been identified in human myocardium, resulting from alternative transcriptional or translational start sites (17). PDE3A1, the longest isoform, is localized to microsomal fractions, while two shorter isoforms, PDE3A2 and PDE3A3, are present in microsomal and/or cytosolic fractions (17). Therefore, by virtue of its SR compartmentation, in the human heart PDE3A1 is a major regulator of PLN and SERCA2 activity.
Here we show that a 29-nucleotide insertion (INS) or deletion (DEL) “indel” polymorphism in the PDE3A promoter regulates transcription, via a downstream cAMP response element (CRE). In HFrEF patients, treatment with PDE3 inhibitors results in increased myocardial PDE3A1 mRNA expression and PDE3A catalytic activity in DEL homozygotes but not in INS homozygotes. The INS site binds the transcription factor ATF3, which produces a repressive effect on the increase in PDE3A transcriptional activity mediated by increased cAMP levels. These results suggest that PDE3 inhibitor treatment may be more efficacious in INS homozygote patients, where tolerance resulting from up-regulation of PDE3A gene expression would not be present and efficacy would be sustained.
Methods
Tissue Procurement
Human subjects with end-stage heart failure were males and females of all ages, races and ethnic backgrounds who gave written consent to donate their hearts to the institutional review board-approved cardiac transplant tissue bank at the University of Colorado. Non-failing hearts that could not be used for transplant due to ABO or body size mismatch and had echocardiographic shortening fractions of ≥25% were obtained from organ donors, with consent for research use given by family members. Further details are described in the Online Appendix.
DNA extraction and genotyping
Genomic DNA was isolated using a modification of Chomczynski’s method (Supplement reference (SR) SR1). Briefly, frozen tissue was digested with proteinase K followed by phenol:chloroform extraction and isopropanol precipitation. PCR was performed using the BioReady Taq DNA polymerase (Bulldog). The resulting PCR product was separated in a 3% agarose gel. The following primers were used for genotype purposes: PDE3A F: 5’ CCACTGCCATTGACTAGCTG
PDE3A R: 5’ GCCAAAAGGAGATCCTTGAGAT
mRNA extraction and RT-PCR
RNA was extracted from left ventricular mid free wall 1 g aliquots of 44 NF and 98 nonischemic (NICM) or ischemic cardiomyopathy (ISCM) failing adult hearts. Tissue aliquots from ISCM hearts used for RNA extraction or biochemical measurements were from infarct-free areas. Tissue mRNA extraction was performed using the mirVana™ kit (Ambion) according to manufacturer’s recommendation. NRVMs were extracted using TRIzol (Thermo Fisher Scientific) and RT-PCR was performed as previously described (SR2) using PDE3A1, PLN or ATF3 specific primers:
Human PDE3A1 F: 5’ GCTCCGGAGCTCTCGGAAA
Human PDE3A1 R: 5’ CCAGCAGCGCCAGCAGAAA
Rat ATF3 F: 5’ CTGCCAAGTGTCGAAACAAG
Rat ATF3 R: 5’ GCAGGTTGAGCATGTAAATCAG
Human PLN F: 5’ CCAATACCTCACTCGCTCAG
Human PLN R: 5’ GATTCTGTAGCTTTTGACGTGC
β-adrenergic receptor density
Total β (β1, β2 and β3)-adrenergic receptor (AR) density and β1, β2 were measured in crude LV membranes by 125[I]CYP binding and displacement of the β1-AR fraction by CGP 20172a, by either of previously described methods (SR3,SR4).
NRVM preparation
Neonatal rat ventricular myocytes (NRVMs) were prepared from the hearts of 2-day-old rat pups by enzymatic digestion as previously described (SR2).
Construct design and cloning
Cloning of the PDE3A promoter region was performed using PCR primers that amplified the −1434/+440 region of the human promoter that contains the INS/DEL genotype. The resulting fragment was cloned in the Promega pGL3-basic vector using MluI and BglII sites as previously described (SR5). Cloning of the construct that lacks the cAMP response element (CRE) was done by amplifying the −1066/+440 region of the PDE3A promoter downstream from the CRE site. Mutations were generated in the 29 nucleotide INS region by PCR amplification of overlapping fragments, randomly designed as:
| Wild Type | 5’TTCTCATATCTACTTATGTCATAATATTA |
| Mutant | 5’CCTCTGCGCTCGTCCGCACTGCGGCGCCG |
Transfection and luciferase activity assays:
Firefly luciferase plasmid DNA was transfected using Lipofectamine 3000 (Thermo Fisher Scientific). Briefly, 1.5 μl of Lipofectamine 3000 and 2 μl of p300 reagents were transfected with 1 μg of plasmid DNA/160,000 cells. Twenty-four hours after transfection cells were treated with 300 μmol/L N6-Benzoyladenosine-3’,5’-Cyclic monophosphate, sodium salt or 5 µmol/L enoximone for 48 hours. Luciferase activity was determined as previously described (S5). siRNA transfection was performed using Lipofectamine RNAiMAX (Thermo Fisher Scientific) with minor modifications. Briefly, 1.5 μl of Lipofectamine RNAiMax and 24 μM scrambled (Dharmacon GE Healthcare Catalog # D-001810–10-05 ON-TARGET plus Non-targeting Pool) or ATF3 (Dharmacon GE Healthcare catalog # L-080117–02-0005 ON-TARGET plus Rat Atf3 (25389) siRNA SMART pool) siRNA were transfected in a 12-well dish (160,000 cells/well) 18 hours after cells were plated. cDNA transfection was performed 24 hours after siRNA transfection, and treated with 300 μmol/L N6-Benzoyladenosine-3’,5’-Cyclic monophosphate, sodium salt 4 hours after transfection. Cells were harvested 48 hours after treatment.
Electrophoretic Mobility Shift Assay (EMSA)
EMSA was performed as previously described (SR5). Briefly, 29 base pairs of the double stranded oligonucleotides of the wild type INS or mutant were labeled by T4 polynucleotide kinase. The reaction was performed using 100,000 cpm of wild type or mutant probe in a 15 μl binding reaction containing HeLa or human heart nuclear extracts.
Nuclear extract preparation
Human heart nuclear extracts were prepared as previously described (SR5). HeLa cell nuclear extracts were purchased from Active Motif. YY1 and ATF3 antibodies were purchased from Santa Cruz Biotechnology.
Preparation of subcellular fractions from human left ventricular myocardium
Approximately 150 mg of −80° C frozen left ventricular mid free wall myocardium immediately frozen upon collection was homogenized and separated into nuclear, cytosolic, and sarcoplasmic reticulum enriched microsomal fractions by differential sedimentation by a protocol adapted from previously published methods (SR6). Further details can be found in the Online Appendix.
PDE3 activity assay
cAMP-hydrolytic activity was quantified according to the method of Kincaid and Manganiello (SR7) as described in detail previously (SR8).
Statistical analysis
Statistical analyses were performed using R (SR9) or GraphPad Prism software. For detailed statistical analysis please see the Online Appendix.
Results
Identification of an INS/DEL polymorphism in the promoter region of the PDE3A gene
In order to determine if there are polymorphisms in the PDE3A promoter region that could affect mRNA expression and response to PDE3 inhibitor treatment, PDE3A1 genomic DNA region including the upstream 10 Kb promoter region was sequenced in 60 samples of failing left ventricular myocardium from the explanted hearts of transplant recipients that were outliers in a PDE3A1 mRNA expression screen. As shown in Figure 1, this resulted in the identification of a 29-nucleotide deletion (DEL)/insertion (INS) polymorphism beginning at c.−2214(c.−2214_−2215,INS TTCTCATATCTACTTATGTCATAATATTA; rs145697127). Polymorphism distribution in the study populations and the enrichment of the DEL homozygous genotype failing vs. non-failing LVs is presented and discussed in the Online Appendix.
Figure 1. Identification of a polymorphic region on the PDE3A promoter.

(A) An insertion of 29 nucleotides (nt) was identified 1130 bp upstream from the PDE3A1 first transcription and 2214 bp upstream from the first translation start sites. The 29 nt sequence is shown in the Figure and is recognized by the ATF3 transcription factor. A putative cAMP response element (CRE) is present 1069 bp upstream from the PDE3A1 transcription start site. Altered position of the INS polymorphism is shown in the Figure. (B) Representative agarose gel of PCR products from DEL homozygotes (DEL/DEL), heterozygotes (DEL/INS) and INS homozygote (INS/INS) patients.
Polymorphism effects on PDE3A promoter activity
To determine the effect of the INS/DEL polymorphism on PDE3A expression in response to agents that act through cAMP, the promoter region containing the indel polymorphism was cloned in a PGL3 basic vector and transfected into neonatal rat ventricular myocytes (NRVMs) treated with 6-Benz-cAMP or the PDE3 inhibitor enoximone. As shown in Figure 2A, treatment of this cAMP biosensor with 6-Benz-cAMP or enoximone resulted in increased luciferase activity of the DEL promoter construct, whereas the INS construct exhibited no evidence of up-regulation of PDE3A expression. Higher luciferase activity of the DEL promoter construct in response to increased cAMP levels suggested that the PDE3A promoter contains a cAMP-response element (CRE) that activates PDE3A expression in the presence of cAMP. Further analysis of the PDE3A promoter region revealed the presence of a CRE (– 5’TAAGTCATCT) 51 bp downstream from the INS region. The PDE3A DEL construct containing the CRE resulted in increased luciferase activity in response to cAMP or enoximone, while deletion of the CRE sequence attenuated cAMP-induced increases in promoter activity (Figure 2B). The increased cAMP-mediated luciferase activity in CRE DEL construct may be due to other putative binding sites downstream from the indel site (data not shown).
Figure 2. Deletion polymorphism results in increased cAMP-dependent promoter activity.
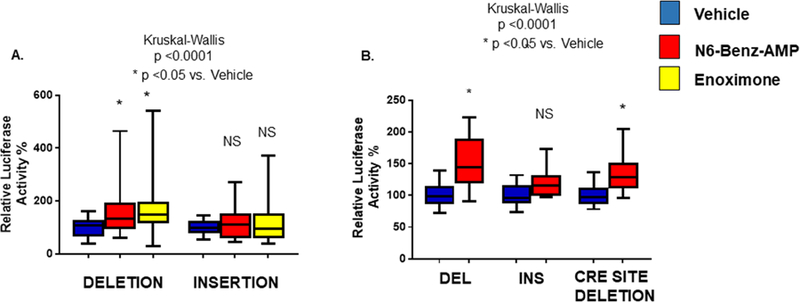
(A) NRVMs were transfected with DEL or INS promoter constructs. 24 hours after transfection cells were treated with 1 µ mol/L 6-Benz-cAMP or 5 µmol/L enoximone for 48 hours. Higher cAMP levels specifically increased activity of the DEL promoter construct. (B) NRVMs were transfected with the DEL promoter construct +/− a CRE binding site. 24 hours after transfection cells were treated with cAMP for 48 hours. Deletion of the CRE site attenuated the response to cAMP. ANOVA/ Kruskal-Wallis’ test significance is shown in the Figure (*). N= 8 different transfections, 4–6 wells/transfection.
The transcription factor ATF3 binds to the 29 nucleotide INS sequence and regulates cAMP-dependent PDE3A promoter activity
To determine if a specific transcription factor interacted with the INS sequence, an electrophoretic mobility shift assay (EMSA) was performed using HeLa cell nuclear extracts and the 29 nt INS sequence as a probe. As shown in Figure 3A, a prominent band (arrow) was observed in the EMSA using the wild type INS probe and HeLa nuclear extracts. Analysis of the INS region using Transfac showed potential binding sites for the transcription factors ATF3, GATA4 and EVI-1, but electrophoretic mobility assays using a GATA4 antibody in NRVM nuclear extracts or EVI-1 in vitro translated protein failed to show interaction of these transcription factors with the INS region (data not shown). However, as shown in Figure 3A, addition of an ATF3 antibody reduced binding to the wild type probe. The ATF3 antibody binds to the c-terminus of the protein where the DNA binding domain is located, and the addition of the antibody likely prevented ATF3 binding to the DNA. In addition, a YY1 antibody was used as negative control, and no decrease in binding was observed. To determine specificity of binding, a cold competitor (wild-type unlabeled probe) was added to the reaction and completely abolished binding. Furthermore, binding was not observed when a mutant probe was used (Figure 3A). Binding was also observed when human heart nuclear extract was used, and the addition of the ATF3 antibody abolished binding (Figure 3B).
Figure 3: ATF3 binds to the INS polymorphism and regulates PDE3A promoter activity.
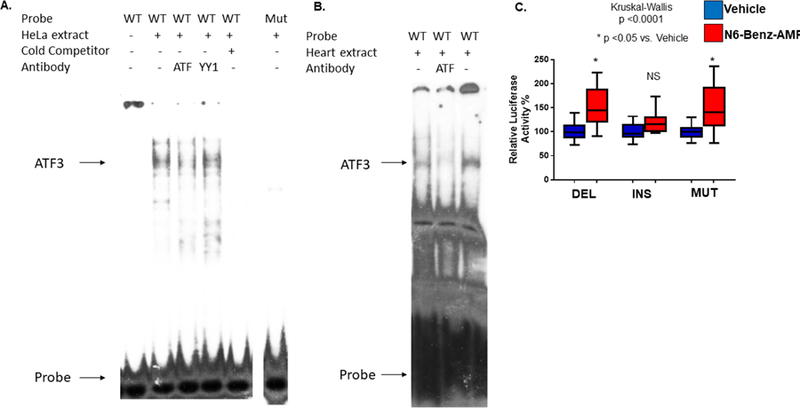
(A) EMSA of the 29 nucleotide probe and mutant. Lane 1: Wild type (WT) probe; 2: Wild type probe and HeLa nuclear extract; 3: Same as lane 2 plus ATF3 antibody; 4: Same as lane 2 plus YY1 antibody; 5: Same as lane 2 plus 100 ng of WT cold competitor; 6 Mutant (Mut) probe and HeLa nuclear extract. (B) EMSA of the 29 nucleotide probe and human left ventricle (LV) nuclear extracts. Lane 1: Wild type probe and human non-failing nuclear extract; 2: Same as Lane 1 plus ATF3 antibody; 3: Wild type probe and human non-failing nuclear extract; (C) NRVMs were transfected with luciferase constructs containing the INS, DEL, or mutant constructs. These results indicate ATF3 binds to the 29-nt INS.
To determine if abolishing binding had an effect on luciferase activity the mutant construct was transfected into NRVMs where treatment with 6-Benz-cAMP was associated with an increase in luciferase activity (Figure 3C).
Down-regulation of ATF3 prevents cAMP-mediated repression of the PDE3 promoter
We next determined if down-regulation of ATF3 had an effect on INS promoter activity. As shown in Figure 4A, NRVMs transfected with an ATF3 siRNA had a 5-fold down-regulation of ATF3 mRNA. Importantly, in NRVMs incubated with 6-Benz-cAMP, down-regulation of ATF3 converted the INS promoter construct to a luciferase activity level comparable to the DEL construct, where down-regulation of ATF3 had no effect (Figure 4B).
Figure 4: Down-regulation of ATF3 prevents cAMP-mediated inhibition of the PDE3A INS promoter.
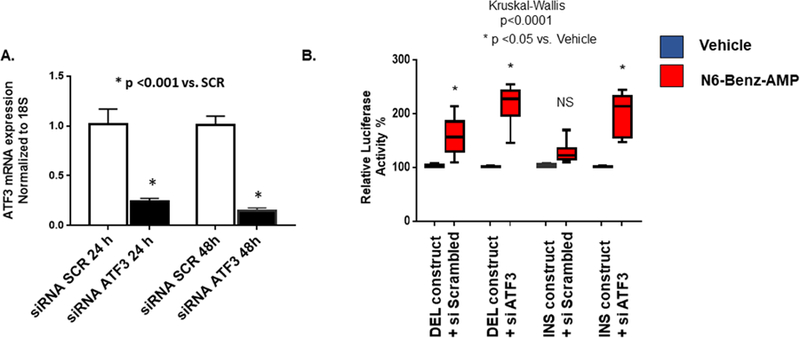
(A) NRVMs were transfected with siScrambled or siATF3. Cells were harvested 24 or 48 hours after transfection. ATF3 mRNA levels were normalized to 18S. A ~ 5 fold down-regulation of ATF3 was observed. (B) Down-regulation of ATF3 results in increased PDE3A INS polymorphism promoter activity in NRVMs treated with 300 μmol/L N6-Benzoyladenosine-3’,5’-Cyclic monophosphate, sodium salt. These results suggest ATF3 is necessary for regulation of promoter activity in response to higher cAMP levels. ANOVA/ Kruskal-Wallis’ test significance is shown in the Figure (* in comparison to untreated). N.S. – not significant. N= 6–10 different transfections, 3–6 wells/transfection.
Myocardial PDE3A1 mRNA abundance is increased in failing LVs, preferentially in DEL polymorphism genotypes
Demographic/other characteristics of hearts used in gene expression and biochemical experiments are given in Online Table 1. For data used in molecular/biochemistry assays there were no clinically meaningful differences in ages (range 47.6–51.5 years) among the non-failing, failing untreated and failing treated with PDE3 inhibitors clinical/treatment phenotypes. Since they originated from organ donors, non-failing hearts were nearly gender balanced (52.2% male) compared to failing LVs (79). Non-failing hearts (86.8% males in PDE3 inhibitor treated HF patients, 74.5% males in untreated patients. The LVEFs exhibited severe dysfunction in the failing groups (17% in PDE3 inhibitor treated, 18.7% in untreated) compared to non-failing (63.0%).
PDE3 inhibitor treatment is associated with increased PDE3A1 mRNA abundance in the failing hearts of patients with the DEL promoter polymorphism
As described in the Methods (Online Appendix) an initial 2-way ANOVA was performed on the PDE3A1 mRNA abundance data, which revealed evidence of an effect by clinical/treatment phenotype (non-failing, failing untreated with PDE3 inhibitors, failing treated with PDE3 inhibitors) as well as by genotype. Accordingly, in order to determine if treatment of HFrEF patients with PDE3 inhibition affects PDE3A1 mRNA levels, transcript abundance was analyzed in non-failing LVs (n = 41), and in failing hearts from patients who had (n = 30) or hadn’t (n = 66) been treated with the PDE3 inhibitors enoximone or milrinone. There were no differences in baseline characteristics including LVEF between PDE inhibitor treated and untreated patients (Online Table 1). In the failing-treated group DEL homozygotes and DEL carriers exhibited respective increases in PDE3A1 abundance (by 45% and 33%, p= 0.046 and 0.025) compared to failing untreated patients (Figure 5). In addition, failing LVs from DEL/DEL or DEL carrier patients treated with PDE3 inhibitors had higher mRNA abundance compared to non-failing LVs (by respective values of 65% (p = 0.031) and 71% (p = 0.0007) (Figure 5). In contrast, nosignificant differences in PDE3A1 mRNA abundance was observed across the 3 clinical/treatment phenotypes LVs that were INS homozygote (ANOVA p = 0.43) or heterozygotes (p = 0.052) (Figure 5).
Figure 5. PDE3A1 mRNA abundance analyzed by RT-PCR in non-failing and failing LVs, by −2214 INS/DEL genotype.
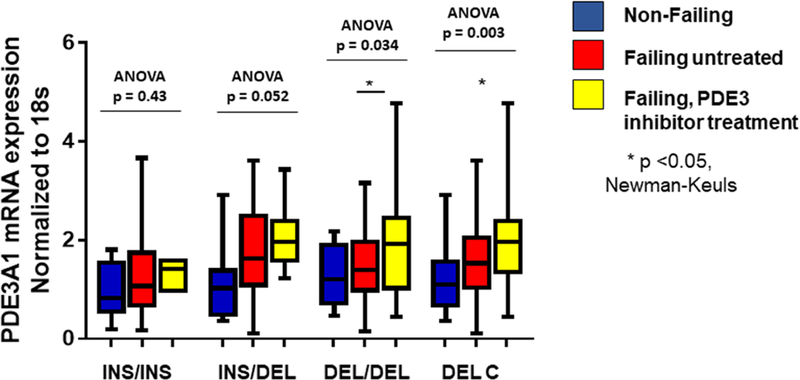
PDE3A1 mRNA abundance analyzed by RT-PCR in non-failing and failing LVs with and without PDE3 inhibitor treatment, by INS/DEL genotype reveals increased expression in DEL homozygous or DEL carriers failing LVs with previous PDE inhibitor (PDEi) treatment. Values are normalized to 18S rRNA and then to Non-failing Ins/Ins as 100%, represented as 1.0 on the y axis. ANOVA/Newman-Keuls test significance is shown in the Figure (* in comparison to untreated). N.S. – not significant.
PDE3 inhibitor treatment is associated with increased microsomal PDE3 enzyme activity in DEL carriers failing hearts
PDE3 enzyme activity was measured in microsomal fractions prepared from 20 non-failing and 47 failing hearts, 22 of which had been treated with PDE inhibitors (Figure 6). In the LVs of the 17 HFrEF patients treated with PDE3 inhibitors, enzyme activity was increased in DEL homozygotes when compared to patients not treated with PDE3 inhibitors (by 56%, p=0.020). DEL carrier failing LVs from patients treated with PDE3 inhibitors exhibited a trend for higher enzyme activity, by 32% (p = 0.056) vs. untreated patients. For comparisons of failing LVs from PDE3 inhibitor treated patients to non-failing LVs, DEL homozygotes had 27% (p = 0.092) and DEL carriers 31% (p = 0.026) higher enzyme activity. In contrast INS homozygotes (ANOVA p = 0.97) or heterozygotes (p = 0.68) did not exhibit differences in PDE3 enzyme activity among the 3 clinical/treatment phenotypes (Figure 6).
Figure 6. Microsomal PDE3 enzyme activity in non-failing and failing human LVs (LVs).
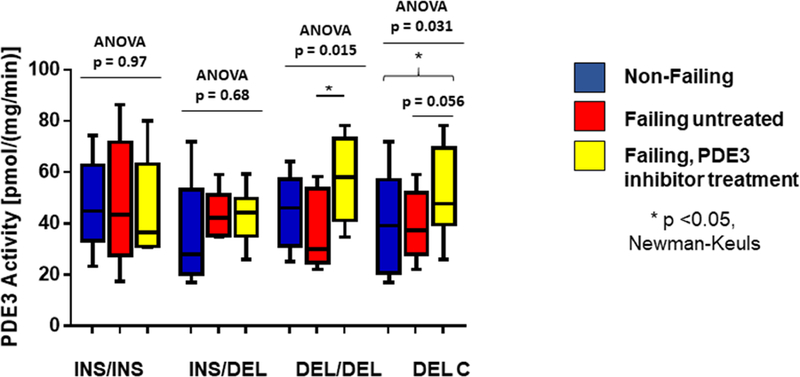
PDE3 activity by genotype in non-failing and failing hearts separated by presence or absence of PDE3 inhibitor treatment, showing increased activity in PDE inhibitor treated failing LVs in DEL homozygotes or DEL carriers. ANOVA/Newman-Keuls test significance is shown in the Figure (* in comparison to untreated). N.S. – not significant.
PLN mRNA expression is affected by PDE3A indel genotypes
The PDE3A1 mRNA data support a PDEA promoter indel effect on PDE3A gene transcription in response to localized cAMP levels that are regulated by the same gene’s protein product, giving rise to a cAMP-PDE3A transcriptional negative feedback loop in DEL carriers that is inhibited in INS homozygotes. This feedback loop is most apparent in PDE3 inhibitor-treated failing hearts, which in DEL carriers exhibit an increase in PDE3A1 mRNA abundance and SR-enriched microsomal PDE3 enzyme activity. This implies that a PDE3 inhibitor-induced increase in cAMP is sensed in the nucleus, leading to higher levels of PDE3A1 transcription in DEL and lower levels in INS alleles. We next investigated if the expression of another cAMP-responsive gene exhibiting co-regulation by ATF3 might be affected by cAMP levels modulated by PDE3 inhibitors and polymorphisms of PDE3A. We chose PLN, whose promoter activity is negatively regulated by ATF3 and positively influenced by a CRE (18). In NRVMs over-expression of adenylate cyclase 6 (ADCY6) was previously shown to repress PLN expression in an ATF3-dependent manner (18), presumably related to ADCY6 generated cAMP and PKA activation upregulating the protein expression of ATF3. Heterozygotes and DEL carriers treated with a PDE3 inhibitors displayed increased PLN mRNA abundance when compared to non-failing hearts (respective p values 0.0007 and 0.0008 for abundances 165% and 81% higher) but untreated failing hearts were not different from the non-failing group (respective p values 0.12 and 0.07) (Figure 7). In heterozygotes and DEL carriers PDE3 inhibitor treated failing hearts also exhibited greater PLN mRNA abundance than untreated failing hearts (by respective amounts of 51% (p= 0.035) and 40% (p=0.023), while a trend for increased PLN mRNA abundance was observed in DEL homozygotes (by 36%, p=0.15). PLN expression was not different in INS homozygotes (ANOVA p = 0.74).
Figure 7. PDE3 inhibitor treatment results in increased PLN in PDE3 inhibitor-treated DEL carriers.
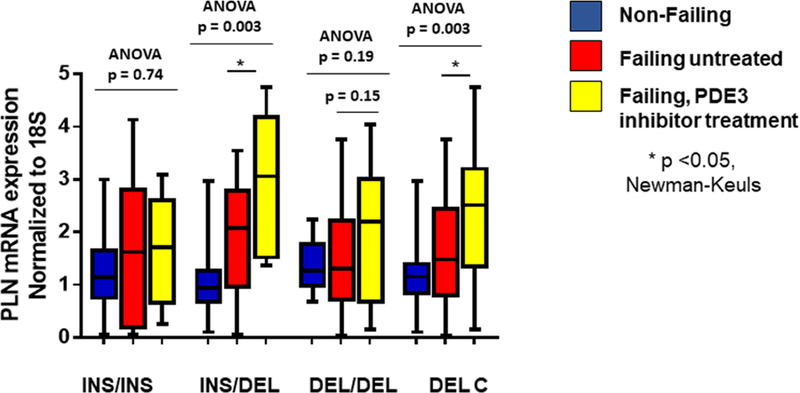
PLN mRNA abundance is increased in treated DEL/INS and DEL carriers when compared to untreated patients. mRNA levels were normalized to 18S and relative normalization is to all non-failings. ANOVA/Newman-Keuls test significance is shown in the Figure (* in comparison to untreated). N.S. – not significant.
Since PLN and PDE3A gene transcription are co-regulated by the ATF3 repressor and enhanced through a CRE, it would be expected that the steady state mRNA abundances of each would be related across PDE3A genotypes. Online Figure 1 demonstrates that is the case, with a significant positive correlation between PLN and PDE3A mRNA expression (r2 = 0.28, p<0.0001; Online Figure 1).
Discussion
We identified a 29-nucleotide indel (INS/DEL) polymorphism in the human PDE3A promoter region at c.−2214 that regulates gene transcription in response to cAMP (Central Illustration). In DEL promoter constructs, a cAMP generated molecularly positive but pharmacologically negative feedback loop is created by increasing PDE3A gene expression that would subsequently limit or reduce increases in cAMP in microdomains where the increased levels of PDE3A1 protein are localized by membrane attachment. In the absence of the INS the cAMP mediated effects on PDE3A promoter activity are mediated by a CRE present 51 bp downstream from where the INS begins. In human failing LVs PDE3A1 mRNA was increased in DEL genotypes compared to INS homozygotes, driven by effects in hearts that had been treated with PDE3 inhibitors. In failing LVs treated with PDE3 inhibitors microsomal PDE3 enzyme activity was also increased in DEL genotypes, proportional to gene dose. In HeLa cell or human LV nuclear extracts the INS binds to the transcription factor ATF3. In INS promoter constructs and in failing LVs explanted from INS homozygotes there was complete prevention of the negative feedback loop of increases in cAMP or PDE3 inhibition leading to up-regulation in PDE3A gene expression and microsomal PDE3 enzyme activity. Importantly, down-regulation of ATF3 prevented the inhibitory effect of the INS sequence in response to cAMP stimulation.
Central Illustration. A PDE3A Promoter Indel Polymorphism Regulates Transcription.
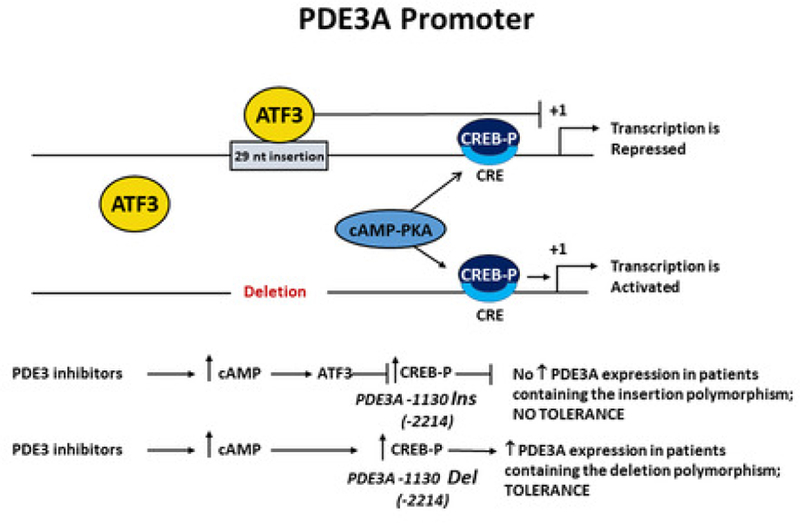
A 29-nucleotide insertion (INS) prevents up-regulation of PDE3A1 mRNA levels by blocking activation of the CRE site in response to increase cAMP levels. DEL of that region results in up-regulation of PDE3 mRNA through derepression of the CRE site. PDE3 inhibition results in increased cAMP levels and PDE3A1 mRNA abundance in the DEL polymorphism, but increased PDE3A mRNA abundance is not observed in the INS polymorphism.
We identified ATF3 as a transcription factor that binds to the PDE3A promoter 29nt insertion. ATF3 is a transcription factor involved in the stress response (19) that can activate transcription when co-expressed with its heterodimeric partners, whereas it represses transcription as a homodimer. In NRVMs Gao et al (18) showed that over-expression of adenylate cyclase 6 results in up-regulation of ATF3 and repression of PLN expression, presumably mediated through cAMP generation and PKA activation. Importantly, the putative ATF3 binding site in the PDE3A promoter 29nt INS sequence is similar to the one identified in the rat or human PLN promoter (INS – 5’ TATGACATAA 3’; rat PLN – 5’ TGACATCACAT 3’; human PLN TGATGTCACAT; complementary sequence depicted) (18). We showed that PLN mRNA expression is also affected by the PDE3A indel polymorphism. Decreased cAMP levels or PKA signaling in a nuclear microdomain in DEL carrier patients treated with a PDE3 inhibitor may be responsible for increased PLN mRNA abundance. Although ATF3 expression was unchanged in human failing hearts in response to enoximone or milrinone treatment, as previously shown by others (20) up-regulation of ATF3 occurs in an acute and transient manner suggesting that the increase in cAMP in response to PDE3A inhibition may result in a transient increase in ATF3 expression. In DEL carriers this would result in increased PDE3A gene transcription, and through decreased levels of cAMP, an up-regulation of PLN gene expression.
What could be the biologic signaling implications and consequences of the PDE3A promoter INS/DEL polymorphism? In the PDE3A-SERCA2-PLN microdomain SR attached PDE3A1 and to a lesser extent PDE3A2 (17) regulate cAMP level and therefore PKA activation and PLN phosphorylation (15). The SR PDE3A-SERCA2-PLN microdomain is the site of a fundamental defect in contractile function in the failing human heart (1,3,21,22), where the local concentration of cAMP and therefore PKA activity and PLN phosphorylation are regulated by SR membrane bound PDE3A isoforms (15–17). For these reasons we measured PDE3 activity in sarcoplasmic reticulum (SR)-enriched microsomes, as well as mRNA expression of the transcript encoding for the PDE3A1 isoform that is restricted to microsomal fractions (17,23). In LV free wall aliquots of failing human hearts, we found that across all genotypes microsomal fraction PDE3 enzyme activity and PDE3A1 mRNA were unchanged, but were increased in DEL homozygotes treated with PDE3 inhibitors. It would be predicted that in DEL homozygotes, increased diffusible cAMP such as from endogenous β-adrenergic stimulation or by therapeutic administration of β-agonists or PDE3 inhibitors would be met with activation of the CRE-mediated up-regulation in PDE3A gene transcription and an increase in PDE3A1 and possibly other PDE3A transcripts creating a pharmacologically negative feedback loop resulting in lack of PDE3 inhibitor effectiveness in DEL genotypes. Alternatively, in HF patients increased cAMP levels in INS homozygotes could result in increased SR-Ca2+ cycling and improved myocyte survival and cardiac function, as previously shown in models of PLN down-regulation (21).
What are the implications of the PDE3A promoter polymorphism findings for treatment with PDE3 inhibitors? Treatment with this drug class has been evaluated in multiple clinical trials in acute and chronic heart failure, and it is currently only used in patients with decompensated heart failure who are refractory to diuretics and vasodilators (24). PDE3 inhibitors have also been or are used as a bridge to transplant (25,26,SR3), and in weaning from cardiopulmonary by-pass. Although in adults with advanced heart failure an initial response is typically hemodynamically beneficial, long-term treatment with PDE3 inhibitors may lead to adverse effects that include arrhythmias and sudden cardiac death (4,5), attenuation of favorable hemodynamic or clinical effects and overt progression of heart failure (11–14). In adult heart failure patients, administration of PDE3 inhibitors can result in tachyphylaxis (9,10) that can be observed as early as 72 hours into continuous infusion (9). In contrast, in children with heart failure PDE3 inhibitors may have better safety and efficacy profiles (26) that could relate to differences in β-AR signaling (27,28) and/or PDE3A activity (28). Thus, there is ample evidence that treatment with PDE3 inhibitors may be accompanied by loss of effectiveness in some patients, which may result in a shift of β-adrenergic signaling to PKA-independent pathways that have greater adverse effects on myocyte biology (1) or allow the Ca2+ mobilizing or other pharmacologic properties of milrinone (10,29) or other compounds to predominate.
In addition, PDE inhibitor-induced up-regulation in PDE expression resulting in PDE inhibitor loss of effectiveness has been observed for other phosphodiesterases (30,31), and cAMP-induced up-regulation in PDE3A gene expression has been reported in porcine ovarian cumulus cells (32). However, this is the first report of a genetic polymorphism in a PDE gene where one variant can mediate cAMP up-regulation in transcriptional activity and the counterpart variant cannot.
In summary, the data from the current study suggest that patients who are INS homozygotes would have less loss of effectiveness to PDE3 inhibitor administration, due to a lack of up-regulation in PDE3A gene transcription and increased PDE3 activity. To be clinically relevant this possibility would need to be tested in a prospective clinical trial with results stratified by PDE3A genotype, to determine the pharmacogenetic consequences of the PDE3A promoter indel polymorphism. In view of the demonstration that in adult HF patients receiving a background of β-blocker therapy PDE3 inhibitors are safe and have evidence of an efficacy signal in subpopulations (6), a pharmacogenetic clinical trial of a PDE3 inhibitor would seem justified.
Limitations
The clinical component of the work was conducted entirely as a retrospective analysis in explanted human hearts, in tissue available from our human heart biobank. Although 166 end-stage LVs were available, fewer than half (N = 41) were from patients who had received PDE3 inhibitors. The sample size was limited to the number of PDE3 inhibitor exposed hearts in the biobank, and for some genetic subsets the N was relatively small (e.g., N = 5 for PDE3 inhibitor treated INS/INS). No prospective clinical study or trial data are included, and the findings in explanted hearts should be considered hypothesis generating for a subsequent trial or trials in advanced HFrEF patients stratified by INS/DEL genotype.
Conclusions
In conclusion, this study is the first to identify a polymorphic region in the PDE3A promoter that regulates gene transcriptional activity in response to changes in cAMP concentration or with PDE inhibitor treatment. The DEL allele of this polymorphism may be a genetic biomarker for the development or progression of heart failure (see Supplement). In addition, on theoretical grounds, heart failure patients who are INS homozygotes may experience less tolerance to PDE3 inhibitors, and PDE3 inhibitors may exhibit greater effectiveness in this genetic subpopulation, a hypothesis that will need to be tested in a prospective clinical trial.
Supplementary Material
CLINICAL PERSPECTIVES.
Competency in Medical Knowledge:
Phosphodiesterase-3 (PDE3) inhibitors produce short term hemodynamic benefit in patients with heart failure, but long-term use has been ineffective. A common genetic variant in the promoter region of the PDE3A gene that regulates transcriptional activity may mediate tolerance to this line of therapy.
Translational Outlook:
Clinical studies are needed to test the hypothesis that patients with heart failure patients who are homozygous for the PDE3A INS promoter variant and possibly those who are heterozygous gain sustained hemodynamic benefit from PDE3 inhibitor drugs.
Acknowledgements:
The authors thank Laura Hofstatter and Rachel Rosenberg for assistance in manuscript preparation and submission.
Funding: This work was supported by Leducq Foundation Transatlantic Networks of Excellence grant FLQ-06 CVD 02 awarded to Drs. Bristow and Movsesian; NIH 2R01 HL48013 awarded to Dr. Bristow; R21 HL097123, AHA 11IRG5070006 and 13GRNT16950045 awarded to Dr. Sucharov; NIH T32 HL007822 and American Academy of Pediatric fellowship grant awarded to Dr. Nakano; the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development (Merit Review CARA-029–09F and CARA-027–12S awarded to Dr. Movsesian) and the American Heart Association (grant-in-aid to Dr. Movsesian); and ARCA biopharma.
Abbreviations:
- PDE
phosphodiesterase
- PDE3A
human phosphodiesterase 3A gene
- LV
left ventricle
- HF
heart failure
- HFrEF
Heart failure with reduced LV ejection fraction
- β−AR
β-adrenergic receptor
- INS
insertion
- DEL
deletion
- Indel
insertion/deletion polymorphism
- HET
heterozygote
Footnotes
Disclosures: Intellectual property relevant to this work has been licensed by the University of Colorado to ARCA biopharma, in which Dr. Bristow is an officer, director and equity holder. No other authors have any relevant disclosures.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bristow MR. Treatment of chronic heart failure with β-adrenergic receptor antagonists: A convergence of receptor pharmacology and clinical cardiology. Circ Res 2011;109:1176–94. [DOI] [PubMed] [Google Scholar]
- 2.Movsesian M, Stehlik J, Vandeput F, Bristow MR. Phosphodiesterase inhibition in heart failure. Heart Fail Rev 2009;14:255–63. [DOI] [PubMed] [Google Scholar]
- 3.Feldman MD, Copelas L, Gwathmey JK, et al. Deficient production of cyclic AMP: pharmacologic evidence of an important cause of contractile dysfunction in patients with end-stage heart failure. Circulation 1987;75:331–9. [DOI] [PubMed] [Google Scholar]
- 4.Packer M, Carver JR, Rodeheffer RJ, et al. Effect of oral milrinone on mortality in severe chronic heart failure. The PROMISE Study Research Group. N Engl J Med 1991;325:1468–75. [DOI] [PubMed] [Google Scholar]
- 5.Cohn JN, Goldstein SO, Greenberg BH, et al. A dose-dependent increase in mortality with vesnarinone among patients with severe heart failure. Vesnarinone Trial Investigators. N Engl J Med 1998;339:1810–6. [DOI] [PubMed] [Google Scholar]
- 6.Metra M, Eichhorn E, Abraham WT, et al. Effects of low-dose oral enoximone administration on mortality, morbidity and exercise capacity in patients with advanced heart failure. The randomised, double-blind, placebo-controlled, parallel group ESSENTIAL Trials. Eur Heart J 2009;30:3015–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holmes JR, Kubo SH, Cody RJ, Kligfield P. Milrinone in congestive heart failure: observations on ambulatory ventricular arrhythmias. Am Heart J 1985;110:800–6. [DOI] [PubMed] [Google Scholar]
- 8.Teerlink JR, Jalaluddin M, Anderson S, et al. Ambulatory ventricular arrhythmias in patients with heart failure do not specifically predict an increased risk of sudden death. PROMISE (Prospective Randomized Milrinone Survival Evaluation) Investigators. Circulation 2000;101:40–6. [DOI] [PubMed] [Google Scholar]
- 9.Maisel AS, Wright CM, Carter SM, Ziegler M, Motulsky HJ. Tachyphylaxis with amrinone therapy: association with sequestration and down-regulation of lymphocyte beta-adrenergic receptors. Ann Intern Med 1989;110:195–201. [DOI] [PubMed] [Google Scholar]
- 10.Farah AE, Frangakis CJ. Studies on the mechanism of action of the bipyridine milrinone on the heart. Basic Res Cardiol 1989;84 Suppl 1:85–103. [DOI] [PubMed] [Google Scholar]
- 11.Simonton CA, Chatterjee K, Cody RJ, et al. Milrinone in congestive heart failure: acute and chronic hemodynamic and clinical evaluation. J Am Coll Cardiol 1985;6:453–59. [DOI] [PubMed] [Google Scholar]
- 12.LeJemtel TH, Gumbardo D, Chadwick B, Rutman HI, Sonnenblick EH. Milrinone for long-term therapy of severe heart failure: clinical experience with special reference to maximal exercise tolerance. Circulation 1986;73(3 Pt 2):III213–8. [PubMed] [Google Scholar]
- 13.Narahara K Oral enoximone therapy in chronic heart failure: a placebo-controlled randomized trial. The Western Enoximone Study Group. Am Heart J 1991;121:1471–79. [DOI] [PubMed] [Google Scholar]
- 14.Packer M, Medina N, Yushak M. Hemodynamic and clinical limitations of long-term inotropic therapy with amrinone in patients with severe chronic heart failure. Circulation 1984;70:1038–47. [DOI] [PubMed] [Google Scholar]
- 15.Mika D, Leroy J, Vandecasteele G, Fischmeister R. PDEs create local domains of cAMP signaling. J Mol Cell Cardiol 2012;52:323–9. [DOI] [PubMed] [Google Scholar]
- 16.Beca S, Ahmad F, Shen W, et al. Phosphodiesterase type 3A regulates basal myocardial contractility through interacting with sarcoplasmic reticulum calcium ATPase type 2a signaling complexes in mouse heart. Circ Res 2013;112:289–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wechsler J, Choi YH, Krall J, Ahmad F, Manganiello VC, Movsesian MA. Isoforms of cyclic nucleotide phosphodiesterase PDE3A in cardiac myocytes. J Biol Chem 2002;277:38072–78. [DOI] [PubMed] [Google Scholar]
- 18.Gao MH, Tang T, Guo T, Sun SQ, Feramisco JR, Hammond HK. Adenylyl cyclase type VI gene transfer reduces phospholamban expression in cardiac myocytes via activating transcription factor 3. J Biol Chem 2004;279:38797–802. [DOI] [PubMed] [Google Scholar]
- 19.Hai T, Wolfgang CD, Marsee DK, Allen AE, Ivaprasad U. ATF3 and stress responses. Gene Expression 1999;7:321–35. [PMC free article] [PubMed] [Google Scholar]
- 20.Yang CJ, Yang Y, Fan ZX. Activating transcription factor 3--an endogenous inhibitor of myocardial ischemia-reperfusion injury (Review). Mol Med Rep 2016;13:9–12. [DOI] [PubMed] [Google Scholar]
- 21.Luo W, Grupp IL, Harrer J, et al. Targeted ablation of the phospholamban gene is associated with markedly enhanced myocardial contractility and loss of beta-agonist stimulation. Circ Res 1994;75:401–9. [DOI] [PubMed] [Google Scholar]
- 22.Schwinger RH, Münch G, Bölck B, Karczewski P, Krause EG, Erdmann E. Reduced Ca(2+)-sensitivity of SERCA 2a in failing human myocardium due to reduced serine-16 phospholamban phosphorylation. J Mol Cell Cardiol 1999;31:479–91. [DOI] [PubMed] [Google Scholar]
- 23.Hambleton R, Krall J, Tikishvili E, et al. Isoforms of cyclic nucleotide phosphodiesterase PDE3 and their contribution to cAMP hydrolytic activity in subcellular fractions of human myocardium. J Biol Chem 2005;280:39168–74. [DOI] [PubMed] [Google Scholar]
- 24.Ponikowski P, Voors AA, Anker SD, et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Eur Heart J 2016;37:2129–2200. [DOI] [PubMed] [Google Scholar]
- 25.Assad-Kottner C, Chen D, Jahanyar J, et al. The use of continuous milrinone therapy as bridge to transplant is safe in patients with short waiting times. J Card Fail 2008;14:839–43. [DOI] [PubMed] [Google Scholar]
- 26.Price JF, Towbin JA, Dreyer WJ, et al. Outpatient continuous parenteral inotropic therapy as bridge to transplantation in children with advanced heart failure. J Card Fail 2006;12:139–43. [DOI] [PubMed] [Google Scholar]
- 27.Nakano SJ, Miyamoto SD, Movsesian M, Nelson P, Stauffer BL, Sucharov CC. Age-related differences in phosphodiesterase activity and effects of chronic phosphodiesterase inhibition in idiopathic dilated cardiomyopathy. Circ Heart Fail 2015;8:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miyamoto SD, Stauffer BL, Nakano S, et al. Beta-adrenergic adaptation in paediatric idiopathic dilated cardiomyopathy. Eur Heart J 2014;35:33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holmberg SR, Williams AJ. Phosphodiesterase inhibitors and the cardiac sarcoplasmic reticulum calcium release channel: differential effects of milrinone and enoximone. Cardiovasc Res 1991;25:537–45. [DOI] [PubMed] [Google Scholar]
- 30.Campos-Toimil M, Keravis T, Orallo F, Takeda K, Lugnier C. Short-term or long-term treatments with a phosphodiesterase-4 (PDE4) inhibitor result in opposing agonist-induced Ca(2+) responses in endothelial cells. Br J Pharmacol 2008;154:82–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin G, Xin ZC, Lue TF, Lin CS. Up and down-regulation of phosphodiesterase-5 as related to tachyphylaxis and priapism. J Urol 2003;170:S15–8. [DOI] [PubMed] [Google Scholar]
- 32.Sasseville M, Côté N, Vigneault C, Guillemette C, Richard FJ. 3’5’-cyclic adenosine monophosphate-dependent up-regulation of phosphodiesterase type 3A in porcine cumulus cells. Endocrinology 2007;48:1858–67. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.