

Abstract
We report a total synthesis of the pyridine-containing limonoid alkaloid (–)-xylogranatopyridine B in 11 steps from commercially available dihydrocarvone. The central pyridine ring was assembled by a late-stage fragment coupling approach employing a modified Liebeskind pyridine synthesis. One fragment was prepared by an allyl-palladium catalyzed oxidative enone β-stannylation, in which the key bimetallic β-stannyl palladium enolate intermediate undergoes a β-hydride elimination. This methodology also allowed introduction of alkyl and silyl groups to the β-position of enones.
Limonoid natural products are characterized by structural diversity generated through oxidative ring fragmentations and skeletal rearrangements.1–3 These structural reorganizations lead to unique scaffolds such as the mexicanolide 8α-hydroxycarapin (1)4 and the limonoid alkaloid xylogranatopyridine B (2).5 The range of limonoid architectures is responsible for the diverse biological activities of this family,6 including kinase inhibitors and compounds that reduce reactive oxygen species involved in neurodegenerative7 and other effects.8
Xylogranatopyridine B (2), a trace isolate from the leaves of a Chinese mangrove (Xylocarpus granatum), is a member of a recently discovered group of limonoid alkaloids typified by phosphatase-inhibitory activity and a pyridine ring embedded in the core structure.5,9 The putative biosynthesis of the pyridine-containing limonoid involves cleavage of the C9–C10 bond of a mexicanolide-type limonoid, such as 1, followed by condensation with an ammonia equivalent to form the pyridine in 2 (Figure 1A).9,10 Although a biomimetic approach could be pursued to generate the highly substituted pyridine, topological analysis suggested that a concise route would be possible by deconstruction of the pyridine substructure into two fragments that could be joined at a late stage.
Figure 1.
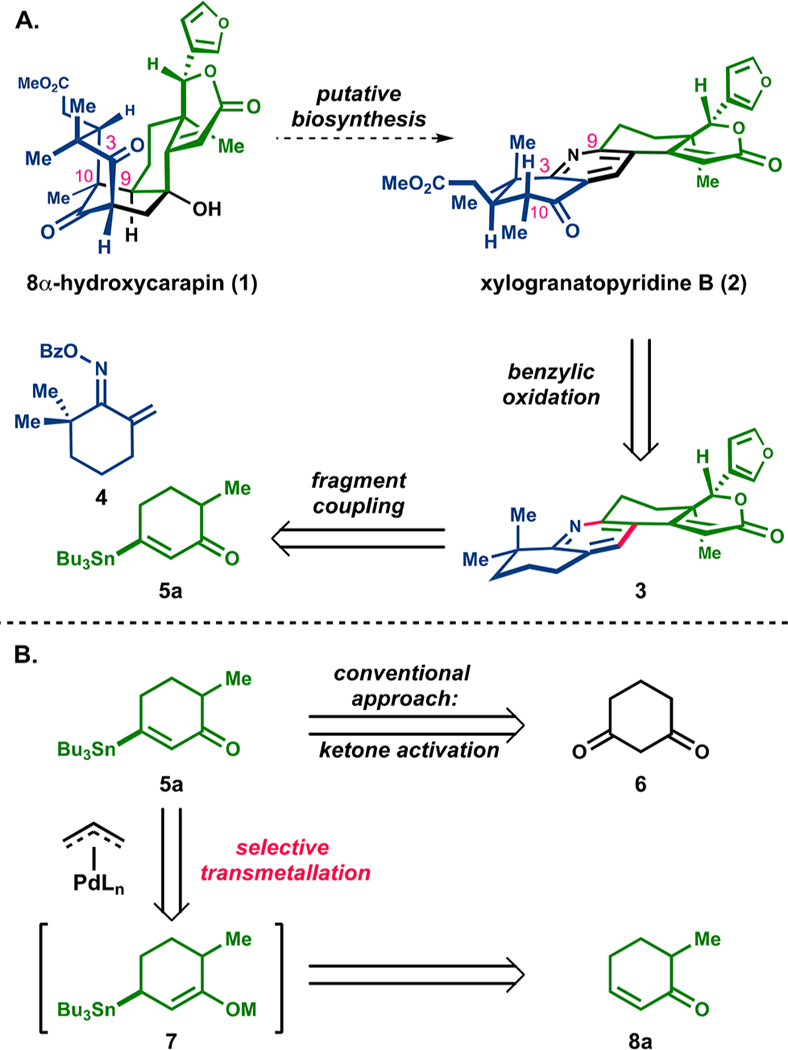
(A) Putative biosynthesis and abiotic retrosynthetic analysis. (B) Pd-mediated oxidative β-stannylation of enones to form 5a.
Our retrosynthetic analysis begins with structural simplification of the A-ring to intermediate 3. In a forward sense, a selective benzylic oxidation and vicinal difunctionalization would allow for the conversion of 3 to 2. The tetrasubstituted pyridine 3 could be fashioned by the Liebeskind pyridine synthesis,11 which entails a convergent Chan-Lam-type cross-coupling of achiral oxime 4 and a stannane fragment, such as 5a, followed by oxidative 6π-electrocyclization.
Existing methods for the synthesis of β-stannyl enones similar to 5a require the activation and cross-coupling of 1,3-diketones starting materials like 6,12,13 which themselves are often challenging substrates to prepare (Figure 1B). Alternatively, based on our previously reported Pd-catalyzed dehydrogenation,14 we anticipated that stannane 5a could be accessed by the direct conversion of an abundantly available enone precursor (8a) via bimetallic intermediate 7. This would require a selective transmetalation of Pd with the metal enolate, rather than with the allyl stannane, to form a Pd enolate that could regenerate the enone functionality via a β-hydride elimination. If feasible, this approach would allow for the introduction of a diverse array of nucleophiles directly to the β-position of enones, obviating the activation of 1,3-diketones.
Various stannane nucleophiles were employed to perturb the identity of the transient metal enolate 7 (Scheme 1). In contrast to our previous reports of zinc enolate-mediated carbonyl α,β-dehydrogenation, we found that a lithium enolate performed well in this context: conjugate addition of tributylstannyllithium to enone 8a provided 5a in 50% isolated yield using the Pd and allyl oxidant system developed for ketone dehydrogenation (entry l).14d In contrast, when a distannylzinc nucleophile15 was employed, 5a was formed with decreased efficiency (25%, entry 2). The use of a Et2Zn-derived stannylzincate16 provided a somewhat lower yield (42%) due to incomplete dehydrogenation (entry 3).
Scheme 1. Optimization of the Synthesis of Stannane 5a.
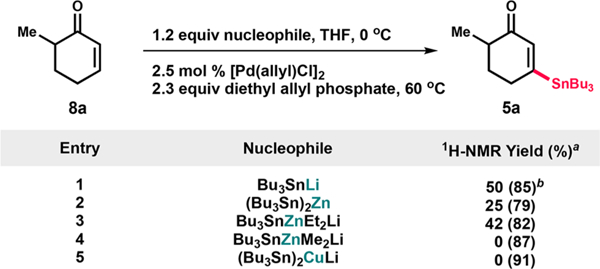
a1H-NMR yield was determined using 1,3,5-trimethoxybenzene as an internal standard. The conversion of starting material is indicated in parentheses. bIsolated yield using 1.0 equiv of diethyl allyl phosphate.
Interestingly, the zincate nucleophile derived from Me2Zn was completely ineffective for dehydrogenation and instead yielded a complex mixture of byproducts (entry 4). The Pd-catalyzed dehydrogenation of the enolate accessed from a stannylcuprate nucleophile17 led to complete decomposition of 7 (entry 5). Due to the increased equivalents of nucleophilic species in entries 2–5, a proportionate increase in the amount of oxidant was necessary to observe any conversion of 7. Large quantities of stannyl enone 5a necessary for the synthesis of xylogranatopyridine B could be prepared in 43–50% isolated yield by employing this methodology on up to 35-g scale. Though efficient dehydrogenation occurred at room temperature, obtaining 5a reproducibly and preventing product decomposition required heating to 60 °C, which led to precipitation of Pd.
We examined the scope of the oxidative stannylation (Scheme 2A). α-Santonin was oxidatively stannylated to provide 5b in 44% yield. The oxidative stannylation of (–)-carvone required a zincate nucleophile for modest product formation (30%) as the use of the stannyllithium nucleophile was less effective. Silyllithium18 nucleophiles were also used to provide the oxidatively silylated products 5d–5f. The higher yield of the silylation to form 5d (79%) as compared to the stannylation to form 5a reflects the more efficient dehydrogenation in the presence of a silane. The synthesis of 5f demonstrates that the reaction system is amenable to a vinylogous dehydrogenation of lactones. Synthetic modification of the C–Si bond may allow for further elaboration of these products.
Scheme 2. Scope of Oxidative Enone Functionalization.
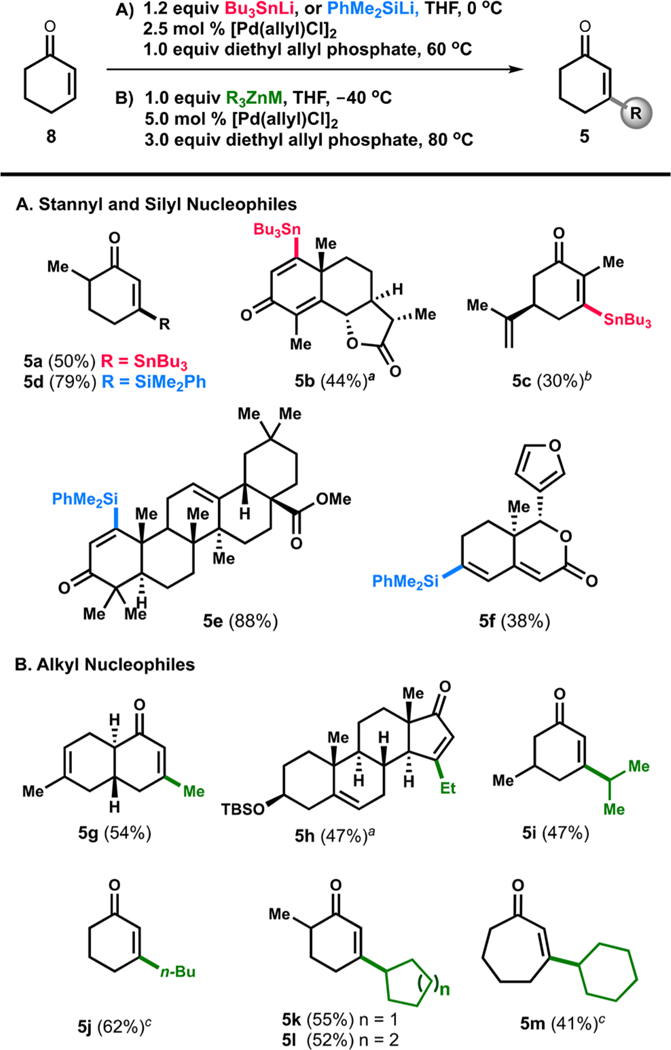
aConjugate addition was conducted at 23 °C. b1.0 equiv of Bu3SnZnEt2Li and 2.3 equiv of diethyl allyl phosphate was used. cConjugate addition was conducted at −78 °C.
Under a set of modified conditions, alkyl groups were also incorporated to the β-position of enones (Scheme 2B).19 Mixed trialkylzincate20 nucleophiles derived from commercial dialkylzinc (5g–5i, 5k), organolithium (5j), and Grignard reagents (5l, 5m) readily underwent conjugate addition to cycloalkenones to give zincate enolates that could be dehydrogenated. The functional group tolerance of organozincate conjugate additions partially restricts the substrate scope of this oxidative alkylation.
With a scalable protocol to form 5a we undertook the synthesis of xylogranatopyridine B (Scheme 3). The preparation of oxime 4 involved methylcuprate addition to 3-methyl-2-cyclohexenone (9) and trapping with TMSCl to provide enoxysilane 10. Using Yamamoto’s conditions for oxime formation with in situ generated TIPS-NO2,21 10 was oxidized to the α-keto oxime (11) in 63% yield over the two-step vicinal difunctionalization of enone 9. Methylenation and benzoylation of the α-keto oxime provided fragment 4.
Scheme 3. Total Synthesis of Xylogranatopyridine B (2).
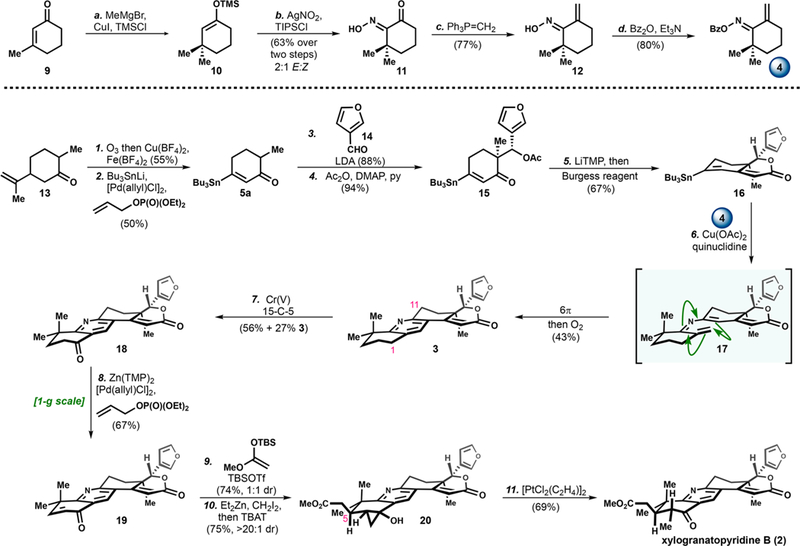
aReagents and conditions: (a) LiCl (0.1 equiv), CuI (5 mol %), TMSCl (1.1 equiv), MeMgBr (1.2 equiv), −40 °C, THF, 10 min; (b) AgNO2 (1.2 equiv), TIPSCl (1.4 equiv), −40 °C, MeCN, 2 h; 10 (1.0 equiv), −40 to −20 °C, 2 h, 63% over 2 steps; (c) Ph3P=CH2 (3.0 equiv), PhMe, 23 to 60 °C, 1.5 h, 77%; (d) Bz2O (1.1 equiv), Et3N (2.0 equiv), DMAP (0.1 equiv), CH2Cl2 0 to 23 °C, 1 h, 80%; (1) O3, MeOH, −40 °C, 4 h; Cu(BF4)2 (1.5 equiv), Fe(BF4)2 (1.2 equiv), −40 to 23 °C, 1 h, 55%; (2) Bu3SnLi (1.2 equiv), [Pd(allyl)Cl]2 (2.5 mol %), diethyl allyl phosphate (1.0 equiv), THF, 60 °C, 1 min, 50%; (3) LDA (1.1 equiv), 14 (1.2 equiv), −78 °C, THF, 0.5 h, 88%; (4) Ac2O (2.0 equiv), py (3.0 equiv), DMAP (0.1 equiv), 0 °C, CH2Cl2, 0.5 h, 94%; (5) LiTMP (2.5 equiv), −78 to 23 °C, THF; Burgess reagent (3.0 equiv), 60 °C, 2 h, 67%; (6) Cu(OAc)2 (1.0 equiv), quinuclidine (2.0 equiv), 4 (1.5 equiv), 60 °C, DMF, 12 h; O2, 100 °C, 1 h, 43%; (7) Na[OCr(O2COC(CH3)C2H5)2] (10.0 equiv), 15-C-5 (0.5 equiv), 75 °C, MeCN, 14 h, 56% + 27% 3; (8) Zn(TMP)2 (1.4 equiv), [Pd(allyl)Cl]2 (5 mol %), diethyl allyl phosphate (1.2 equiv), 85 °C, 1 h, 67%; (9) TBSOTf (5 mol %), 1-(TBS)-1-methoxyethene (1.5 equiv), CH2Cl2, 0 to 23 °C, 0.5 h, 75%, 1:1 dr at C5; (10) Et2Zn (2.0 equiv), CH2I2 (4.0 equiv), 0 to 23 °C, PhMe, 6 h; TBAT (10.0 equiv) in THF, 12 h, 75%; (11) [PtCl2(C2H4)]2 (10 mol %), CH2Cl2, 23 °C, 5 h, 69%.
The synthesis of stannane fragment 16 began with inexpensive dihydrocarvone (13), which was converted to 6-methylcyclohexenone (8a) in 55% yield via ozonolytic fragmentation22 followed by the aforementioned Pd-catalyzed oxidative stannylation. A diastereoselective aldol between the lithium enolate of 5a and 3-furaldehyde (14) provided 15 after acylation. Treatment of acetate 15 with LiTMP initiated an intramolecular acetate aldol, in which the intermediate adduct was trapped with Burgess reagent to provide fragment 16.
With access to both fragments in 4–5 steps on decagram-scale, we forged the central pyridine.11 The ring synthesis employed oxime benzoate 4 and stannane 16 rather than the previously reported boronic acid due to the stannane’s relative ease of preparation and tolerance to subsequent reactions. Typical Chan-Lam coupling conditions11,23 predominantly resulted in decomposition of oxime 4 and destannylation of 16. We determined that the use of quinuclidine was more optimal than previously reported amine additives.24 The tetracyclic core of xylogranatopyridine B (3) was obtained in 43% yield by heating the same reaction vessel to initiate a 6π-electrocyclization and aromatization. In practice, an isomeric mixture of 4 was employed, but independent subjection of each of the oxime isomers demonstrated both are competent substrates for the Chan-Lam coupling, suggesting that either 4 or 17 may be undergoing thermal isomerization. This mechanistic pathway is supported by the control experiment wherein substitution of des-stannyl-16 for 16 in the reaction conditions does not lead to the product 3.
With the completed carbon framework in hand, we turned our attention to the selective oxidation of the benzylic position adjacent to the more electron-rich pyridine meta-position (Cl) over the more electron-deficient ortho-position (C11).25 A broad examination of oxidants identified the Cr(V) complex26 as the optimal reagent to provide ketone 18 in 56% yield along with 27% recovered 3. It is remarkable that selective oxidation occurred in the presence of numerous oxidizable functionalities (e.g. furan, pyridine nitrogen, etc.).
Ketone 18 underwent smooth α,β-dehydrogenation with the conditions previously developed in our laboratory14d to provide enone intermediate 19 in 67% yield on 1-g scale. It is interesting to note that additional optimization of the reported conditions was not required even in the presence of the basic pyridine functionality in 19. A Mukaiyama–Michael reaction with 19 and the enoxysilane derived from methyl acetate, in the presence of catalytic TBSOTf, provided an intermediate enoxysilane as a 1:1 mixture of diastereomers at C5. Although direct methylation of the corresponding enolate was feasible, the alkylations were inefficient owing to the base sensitivity of 2. To circumvent the unselective alkylation, Simmons–Smith cyclopropanation proceeded with high diastereoselectivity (>20:1 dr) and deprotection of the siloxycyclopropane with TBAT occurred smoothly to give a combined yield of 75% for the mixture of C5 epimers and a 31% isolated yield of the single diastereomer 20. The use of Zeise’s dimer, [PtCl2(C2H4)]2,27 was optimal for opening cyclopropanol 20 to form xylogranatopyridine B (2) in 69% yield.
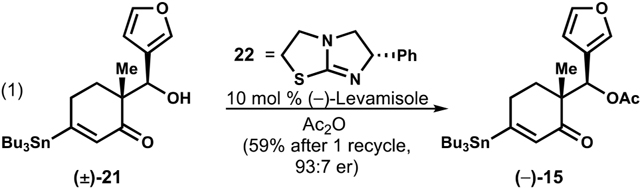
The racemic synthesis of 2 was rendered asymmetric through Birman’s acylative kinetic resolution of alcohol (±)-21 using (–)-Levamisole (22),28 providing acetate (–)-15 in 93:7 er and 43% yield on decagram scale (eq 1). The unreacted alcohol could be converted back to 5a by a retro-aldol elicited by K2CO3/MeOH in 89% yield, and a total yield of 59% of (–)-15 could be obtained after one round of recycling. The enantioenriched material provided (–)-xylogranatopyridine B.
The total synthesis of the limonoid alkaloid, (–)-xylogranatopyridine B, required a longest linear sequence of 11 steps from commercially available materials. This synthesis was enabled by the development of an oxidative enone β-functionalization, which provided rapid access to stannane 16 and demonstrated the use of allyl-Pd catalysis for enabling C–C and C–X bond constructions. Furthermore, the strategic combination of a modified Liebeskind pyridine ring synthesis and a late-stage benzylic oxidation allowed for a convergent assembly of the limonoid alkaloid skeleton. With a scalable route established to the core scaffold, current efforts are underway to further explore the biological activities of the limonoid alkaloids and their synthetic derivatives.
Supplementary Material
ACKNOWLEDGMENTS
This work is dedicated to Prof. E. J. Corey on the occasion of his 90th birthday. We are grateful for financial support from Yale University, the Sloan Foundation, the NSF (CAREER, 1653793 and GRF to A.W.S.), Bristol-Myers Squibb (Graduate Fellowship to A.W.S.), Nalas Engineering, and the NIH-funded Chemistry/Biology Interface Training Program (D.H., T32GM067543), and a Rudolph J. Anderson postdoctoral fellowship (Y.C.).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b13189.
Experimental procedures and spectroscopic data for all new compounds including 1H- and 13C NMR spectra (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Tan Q Luo Chem. Rev. 2011, 111, 7437. [DOI] [PubMed] [Google Scholar]
- (2).Heasley B Eur. J. Org. Chem. 2011, 2011, 19. [Google Scholar]
- (3).For recent limonoid total syntheses, see: [Google Scholar]; (a) Behenna DC; Corey EJ J. Am. Chem. Soc. 2008, 130, 6720. [DOI] [PubMed] [Google Scholar]; (b) Faber JM; Eger WA; Williams CM J. Org. Chem. 2012, 77, 8913. [DOI] [PubMed] [Google Scholar]; (c) Yamashita S; Naruko A; Nakazawa Y; Zhao L; Hayashi Y; Hirama M Angew. Chem., Int. Ed. 2015, 54, 8538. [DOI] [PubMed] [Google Scholar]; (d) Lv C; Yan X; Tu Q; Di Y; Yuan C; Fang X; Ben-David Y; Xia L; Gong J; Shen Y; Yang Z; Hao X Angew. Chem., Int. Ed. 2016, 55, 7539. [DOI] [PubMed] [Google Scholar]
- (4).Schuppe AW; Newhouse TR J. Am. Chem. Soc. 2017, 139, 631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).For the isolation of xylogranatopyridine B, see: Zhou Z-F; Liu HL; Zhang W; Kurtán T; Mán di A; Bényei A; Li J; Taglialatela-Scafati O; Guo Y-W Tetrahedron 2014, 70, 6444. [Google Scholar]
- (6).For reviews on limonoid biological activity, see: [Google Scholar]; (a) Champagne DE; Koul O; Isman MB; Scudder GGE; Towers GHN Phytochemistry 1992, 31, 377. [Google Scholar]; (b) Saraf S; Roy A Biol. Pharm. Bull. 2006, 29, 191. [DOI] [PubMed] [Google Scholar]
- (7).(a) Hett EC; Slater LH; Mark KG; Kawate T; Monks BG; Stutz A; Latz E; Hung DT Nat. Chem. Biol. 2013, 9, 398. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) English AW; Liu K; Nicolini JF; Mulligan AM; Ye K Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 16217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) Miller EG; Porter JL; Binnie WH; Guo YI; Hasegawa SJ Agric. Food Chem. 2004, 52, 4908. [DOI] [PubMed] [Google Scholar]; (b) Ejaz S; Ejaz A; Matsuda K; Lim CW J. Sci. Food Agric. 2006, 86, 339. [Google Scholar]
- (9). For recent isolation of related limonoid alkaloids: [Google Scholar]; (a) Cui J; Ouyang J; Deng Z; Lin W Magn. Reson. Chem. 2008, 46, 894. [DOI] [PubMed] [Google Scholar]; (b) Wu J; Zhang S; Bruhn T; Xiao Q; Ding H; Bringmann G Chem. - Eur. J. 2008, 14, 1129. [DOI] [PubMed] [Google Scholar]; (c) Pan J-Y; Chen S-L; Li M-Y; Li J; Yang M-H; Wu JJ Nat. Prod. 2010, 73, 1672. [DOI] [PubMed] [Google Scholar]
- (10).See Supporting Information for more details.
- (11).Liu S; Liebeskind LS J. Am. Chem. Soc. 2008, 130, 6918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).For seminal studies on β-substituted enone synthesis via 1,3-diones, see: Stork G; Danheiser RL J. Org. Chem. 1973, 38, 1775. [Google Scholar]
- (13).A conventional activation approach to access 5a, as outlined in Figure 1B, resulted in a 26% overall yield; see ref 10.
- (14).(a) Chen Y; Romaire JP; Newhouse TR J. Am. Chem. Soc. 2015, 137, 5875. [DOI] [PubMed] [Google Scholar]; (b) Chen Y; Turlik A; Newhouse TR J. Am. Chem. Soc. 2016, 138, 1166. [DOI] [PubMed] [Google Scholar]; (c) Turlik A; Chen Y; Newhouse TR Synlett 2016, 27, 331. [Google Scholar]; (d) Chen Y; Huang D; Zhao Y; Newhouse TR Angew. Chem., Int. Ed. 2017, 56, 8258. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Zhao Y; Chen Y; Newhouse TR Angew. Chem., Int. Ed. 2017, 56, 13122. [DOI] [PubMed] [Google Scholar]
- (15).Matsubara S; Hibino J-I; Morizawa Y; Oshima K; Nozaki HJ Organomet. Chem. 1985, 285, 163. [Google Scholar]
- (16).Morita Y; Suzuki M; Noyori RJ Org. Chem. 1989, 54, 1785. [Google Scholar]
- (17).Piers E; Morton HE; Chong JM Can. J. Chem. 1987, 65, 78. [Google Scholar]
- (18).Lee TW; Corey EJ Org. Lett. 2001, 3, 3337. [DOI] [PubMed] [Google Scholar]
- (19).For alternative methods to synthesize β-alkyl enones from enones, see: [Google Scholar]; (a) Matsuo J.-i.; Aizawa Y Chem. Commun. 2005, 2399. [DOI] [PubMed] [Google Scholar]; (b) Kerr WJ; Pearson CM; Thurston GJ Org. Biomol. Chem. 2006, 4, 47. [DOI] [PubMed] [Google Scholar]; (c) McMahon CM; Alexanian EJ Angew. Chem., Int. Ed. 2014, 53, 5974. [DOI] [PubMed] [Google Scholar]; (d) Huber T; Kaiser D; Rickmeier J; Magauer TJ Org. Chem 2015, 80, 2281 See also ref 12. [DOI] [PubMed] [Google Scholar]
- (20).(a) Isobe M; Kondo S; Nagasawa N; Goto T Chem. Lett. 1977, 6, 679. [Google Scholar]; (b) Watson RA; Kjonaas RA Tetrahedron Lett. 1986, 27, 1437. [Google Scholar]
- (21).Baidya M; Yamamoto HJ Am. Chem. Soc. 2011, 133, 13880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Huang D; Schuppe AW; Liang MZ; Newhouse TR Org. Biomol. Chem. 2016, 14, 6197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).(a) Liu S; Yu Y; Liebeskind LS Org. Lett. 2007, 9, 1947. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhang Z; Yu Y; Liebeskind LS Org. Lett. 2008, 10, 3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).For a review on the classical Chan-Lam coupling, see: Qiao JX; Lam PYS Synthesis 2011, 2011, 829. [Google Scholar]; For a recent mechanistic study, see: Vantourout JC; Miras HN; Isidro-Llobet A; Sproules S; Watson AJB J. Am. Chem. Soc. 2017, 139, 4769. [DOI] [PubMed] [Google Scholar]
- (25).Newhouse TR; Baran PS Angew. Chem., Int. Ed. 2011, 50, 3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).(a) Krumpolc M; Roček JJ Am. Chem. Soc. 1979, 101, 3206. [Google Scholar]; (b) Wilde NC; Isomura M; Mendoza A; Baran PS J. Am. Chem. Soc. 2014, 136, 4909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).(a) Ikura K; Ryu I; Ogawa A; Sonoda N; Harada S; Kasai N Organometallics 1991, 10, 528. [Google Scholar]; (b) Hoberg JO; Jennings PW Organometallics 1996, 15, 3902. [Google Scholar]
- (28).(a) Birman VB; Li X Org. Lett. 2006, 8, 1351. [DOI] [PubMed] [Google Scholar]; (b) Ortiz A; Benkovics T; Beutner GL; Shi Z; Bultman M; Nye J; Sfouggatakis C; Kronenthal DR Angew. Chem., Int. Ed. 2015, 54, 7185. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.