

Graphical Abstract
Procedure
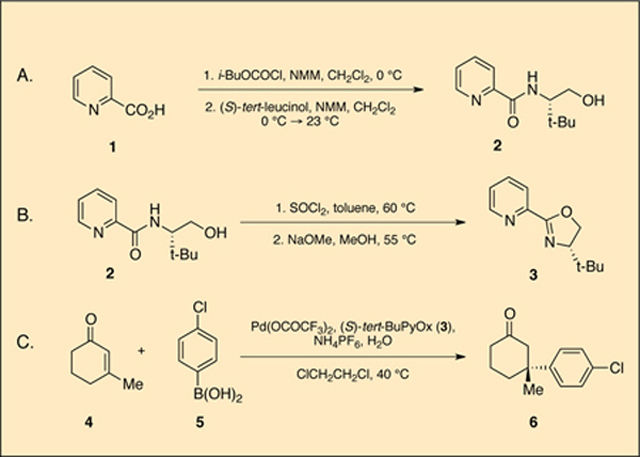
A. (S)-N-(l-Hydroxy-3,3-dimethylbutan-2-yl)picolinamide (2).
A 1-L onenecked round-bottomed flask equipped with a 3.0 cm × 1.4 cm, egg-shaped, Teflon-coated magnetic stirring bar is sealed with a septum and connected via needle adapter to a two-tap Schlenk adapter attached to an oil bubbler and a nitrogen/vacuum manifold (Note 1). The flask is dried with a heat gun under vacuum and cooled under a stream of nitrogen. The flask is charged with 2-picolinic acid (1) (6.15 g, 50.0 mmol, 1.00 equiv) (Note 2), evacuated and back-filled with nitrogen three times, then charged with dichloromethane (300 mL, 0.17 M) (Note 3) and N-methylmorpholine (7.59 g, 8.25 mL, 75.0 mmol, 1.50 equiv). The flask is cooled in an ice/water bath and iso-butylchloroformate (6.86 mL, 7.17 g, 52.5 mmol, 1.05 equiv) is added dropwise over 30 min by syringe pump. The reaction mixture is stirred for an additional 30 min while remaining submerged in the ice/water bath. A separate 100 mL flask is sealed with a septum and connected via needle adapter to the two-tap Schlenk adapter and manifold, dried with a heat gun under vacuum, and allowed to cool under a stream of nitrogen. This flask is charged with (S)-tert-leucinol (6.45 g, 55.0 mmol, 1.10 equiv), dichloromethane (40 mL), and N-methylmorpholine (6.07 mL, 5.56 g, 55.0 mmol, 1.10 equiv). The resulting clear solution is taken up in a syringe and transferred dropwise using a syringe pump over the course of 1 h to the stirring reaction mixture in the ice/water bath. The cooling bath is removed, and the pale gold colored reaction mixture is stirred for an additional 6 h at 23 °C. Upon consumption of starting material (Note 4), the mixture is quenched at ambient temperature with a single addition of an aqueous solution of saturated NH.Cl (50 mL), diluted with additional H20 (25 mL), and transferred into a 1 L separatory funnel. The phases are separated, and the aqueous phase is extracted with CH2Cl2 (3 × 100 mL). The combined organic phases are washed with an aqueous solution of saturated NaHCO3 (1 × 50 mL) and brine (1 × 50 mL). The combined organic phases are dried over Na2SO4 (10 g, 15 min while agitating), filtered through a M pore glass frit, and concentrated by rotary evaporation (28 °C, 15 mmHg). Excess N-methylmorpholine is further removed by placing the crude residue under high vacuum (< 12 mmHg, 12 h) to provide a pale red solid (Note 5). The crude residue is dissolved in 10 mL of acetone and purified via silica gel flash chromatography (Note 6). The combined product- containing fractions are concentrated by rotary evaporation (40 °C, 15 mmHg) to yield a solid, which is dried under high vacuum (<12 mmHg, 12 h) to afford (S)-N-(l-hydroxy-3,3-dimethylbutan-2-yl)picolinamide (2) as a white amorphous solid (9.88–9.95 g, 44.4–44.8 mmol, 89–90% yield) (Note 7).
B. (S)-4-(tert-Butyl)-2-(pyridin-2-yl)-4:,5-dihydrooxazole (3).
A 500 mL three-necked round-bottomed flask is equipped with a 3.0 cm × 1.4 cm, egg-shaped, Teflon-coated magnetic stirring bar. All necks are sealed with rubber septa and the center neck is attached via needle adapter to a two-tap Schlenk adapter connected to a bubbler and a nitrogen/vacuum manifold (Note 1). One side neck is fitted with a thermometer pierced through a rubber septum. The flask is dried with a heat gun under vacuum, cooled under a stream of nitrogen and charged with (S)-N-(l-hydroxy-3,3-dimethylbutan-2-yl)picolinamide (2) (8.89 g, 40.0 mmol, 1.00 equiv) and toluene (140 mL, 0.285 M) (Note 3). The resulting clear solution is heated at 60 °C in a preheated bath of armor beads (Note 8). In a separate 50 mL flask, SOCl2 (5.64 mL, 80.0 mmol, 2.00 equiv, Note 2) is dissolved in toluene (20 mL) and taken up with a syringe. The SOCL2 solution is added dr op wise over 70 min to the vigorously stirring reaction mixture using a syringe pump (Note 9). Upon complete addition, the mixture is stirred for an additional 4 h. The resulting slurry is allowed to cool to ambient temperature, concentrated by rotary evaporation (55 °C, 15 mmHg), crushed with a spatula, and further dried under high vacuum (<12 mmHg, 12 h) to afford (S)-N-(l-chloro-3,3-dimethylbutan-2-yl)picolinamide hydrochloride salt as a tan powder (10.68–10.86 g, 38.5–39.2 mmol, 96–98% yield) (Note 10). This material is used without purification.

A 500 mL one-necked round-bottomed flask equipped with a 3.0 cm × 1.4 cm, egg-shaped, Teflon-coated magnetic stirring bar is charged with (S)-N-(l-chloro-3,3-dimethylbutan-2-yl)picolinamide hydrochloride salt (10.26 g, 37.0 mmol, 1.00 equiv) and methanol (100 mL, 0.37 M) (Note 3). To the clear, stirring solution is added powdered sodium methoxide (9.99 g, 185.0 mmol, 5.00 equiv) (Notes 2 and 11) in one portion, and the resulting mixture is lowered into an armor bead bath preheated at 55 °C. The slurry is stirred for 18 h (Note 12). Afterwards, the armor bead bath is removed, the reaction is allowed to cool to ambient temperature, and toluene (100 mL) is added (Note 13). The reaction mixture is partially concentrated by rotary evaporation to remove the methanol (40 °C, 60 mmHg), at which time the resulting slurry is transferred to a 250 mL separatory funnel and washed with water (100 mL) (Note 14). The layers are separated, and the aqueous layer is back extracted with toluene (3 × 40 mL). The combined organic extracts are dried over Na2S04 (20 g) with stirring for 20 min, filtered through a M pore glass frit, and concentrated by rotary evaporation (40 °C, 15 mmHg). The crude residue is purified by silica gel flash chromatography (Note 15). The combined product-containing fractions are concentrated by rotary evaporation (28 °C, 15 mmHg) to yield a solid, which is further dried under high vacuum (< 12 mmHg, 12 h) to afford (S)-tert-ButylPyOx (3) as a white solid (5.64–5.91 g, 27.6–28.9 mmol, 75–78% yield; 72–76% yield from 2) (Note 16).

C. (R)-3-(4-Chlorophenyl)-3-methylcyclohexanone (6).
A 500 mL, onenecked round-bottomed flask is equipped with a 4.2 × 2.0 cm egg-shaped, Teflon-coated magnetic stirring bar and charged sequentially with 1,2- dichloroethane (150 mL, 0.23 M), (S)-tert-ButylPyOx (3) (0.429 g, 2.10 mmol, 0.06 equiv), palladium(II) trifluoroacetate (0.582 g, 1.75 mmol, 0.05 equiv), ammonium hexafluorophosphate (1.71 g, 10.5 mmol, 0.30 equiv), and 4-chlorophenylboronic acid (5) (6.57 g, 42 mmol, 1.2 equiv) (Note 2). The suspension is stirred at ambient temperature for 5 min, at which time a pale yellow color is observed. Not all solids are dissolved at this time. 3-Methyl-2-cyclohexenone (4) (3.97 mL, 35.0 mmol, 1 equiv) is transferred via syringe to the stirring suspension at ambient temperature. Water (3.15 mL, 175 mmol, 5 equiv) (Note 3) is added in one portion via syringe to the stirring mixture and the flask is placed in an armor bead bath preheated at 40 °C. The reaction is vigorously stirred for 12–24 h (Note 17). Upon consumption of the starting material (Note 18), the reaction is cooled to ambient temperature and stirring is halted, allowing the solids to settle. The reaction mixture, including the solid matter, is filtered over a pad of silica gel (Note 19). The filter cake is washed with additional 1,2-dichloroethane (150 mL). The filtrate is concentrated by rotary evaporation (28 °C, 15 mmHg) and the crude residue is purified by silica gel flash chromatography (Note 20). The combined product-containing fractions are concentrated by rotary evaporation (28 °C, 15 mmHg) to yield a solid, which is further dried under high vacuum (< 12 mmHg, 12 h) to afford (R)-3-(4-chlorophenyl)-3-methylcyclohexanone (6) (6.80–7.07 g, 87–91% yield, 93% ee) as a colorless crystalline solid (Note 21).

Notes
A two-tap Schlenk adapter connected to a bubbler and an argon/vacuum manifold is illustrated in Yu, J.; True, V.; Riebel, P.; Hierl, E.; Mudryk, B. Org. Synth. 2008, 85, 64–71.
Iso-butylchloroformate (98%) and 2-picolinic acid (99%) were purchased from Acros Organics and used without further purification. N-Methylmorpholine (redistilled, >99.5%), thionyl chloride (>99%), sodium methoxide (95%), ammonium hexafluorophosphate (99.9%), and (S)-tert-leucinol (98%) were purchased from Sigma Aldrich and used as received. The submitters prepared (S)-tert-leucinol as described by Krout, M. R.; Mohr, J. T.; Stoltz, B. M. Org. Synth. 2009, 86, 181–193. 4-Chlorophenylboronic acid (98%) and 3-methylcyclohexen-l-one (98%) were purchased from Combi-Blocks and used as received. The submitters noted a variation in ee and yield with different commercial sources and batches of 4-chlorophenylboronic acid, presumably due to the presence of variable amounts of boroxine, which should be determined. Palladium(II) trifluoroacetate (97%) was purchased from Strem Chemicals and 3-methylcyclohexen-l-one (98%) was purchased from Combi Blocks, and each was used as received.
Methylene chloride (> 99.8%, anhydrous) toluene (99.8%, anhydrous), methanol (99.8%, anhydrous) and reagent grade 1,2-dichloroethane (99.8%, anhydrous) were purchased from Sigma Aldrich and used as received. In house deionized water was used without alteration.
The reaction can be monitored by TLC analysis using 3:2 hexanes/acetone as the eluent (E. Merck Silica gel 60 F254 precoated plates, 250 nm), visualizing with UV fluorescence quenching and p-anisaldehyde staining or iodine staining. Product amide (2) Rf= 0.42, impurity Rf = 0.38 (consistent with 3,3-dimethyl-2-(picolinamido)butyl picolinate), N-methylmorpholine Rf = 0.06 (iodine stain). Picolinic acid (1) and tert-leucinol remain at the baseline, as determined by TLC.
If N-methylmorpholine is not removed prior to chromatography, it will overload the silica gel column and will contaminate product fractions. Excess N-methylmorpholine complicates product solidification post chromatography.
Silica gel column dimensions: 5 cm diameter × 20 cm height, ca. 220 g silica gel (RediSep RF column from Teledyne Isco, catalog 69–2203-422), eluting with 4:1 hexanes/acetone. Pre-run of 200 mL is collected before fractions are taken. Fractions are collected in 18 mm × 150 mm test tubes. Fraction purity can be assayed by TLC analysis using 3:2 hexanes/acetone with UV visualization. Product (2) Rf = 0.42, impurity Rf = 0.38 (consistent with 3,3-dimethyl-2-(picolinamido)butyl picolinate), N-methylmorpholine Rf = 0.06 (iodine stain). Both starting materials remain at the baseline (Rf= 0).
(S)-N-(l-Hydroxy-3,3-dimethylbutan-2-yl)picolinamide (2) exhibited the following characterization data: Rf = 0.42 with 3:2 hexanes / acetone; mp 78.7–79.8 °C; H NMR (400 MHz, CDCl3) δ: 1.05 (s, 9 H), 2.60 (br s, 1 H), 3.70 (dd, J = 8.4, 11.2 Hz, 1 H), 3.96–4.03 (m, 2 H), 7.44 (dd, J = 5.2, 6.8 Hz, 1 H), 7.85 (dt, J = 1.2, 8.0 Hz, 1 H), 8.19 (d, J = 8.0 Hz, 1 H), 8.32 (br d, J = 8.0 Hz, 1 H), 8.56 (d, J = 4.8 Hz, 1 H); 13C NMR (100 MHz, CDCl3) δ: 27.0, 33.7, 60.5, 63.6, 122.4, 126.2, 137.4, 148.0, 149.6, 165.5; IR (film): 3390, 3241, 2966, 1646, 1535, 1464, 1435, 1368, 1293, 1241, 1083, 1054, 1022, 999, 863, 750, 697 cm-1; HRMS (Multimode ESI/FTMS) m/z calcd for C12H19N2O2 [M + H]+: 223.1441, found: 223.1439; [α]D24 = –9.09 (c 1.728, CHCl3); Anal calcd for C12H18N2O2: C, 64.84; H, 8.17; N, 12.61. Found: C, 64.65; H, 8.19; N, 12.60.
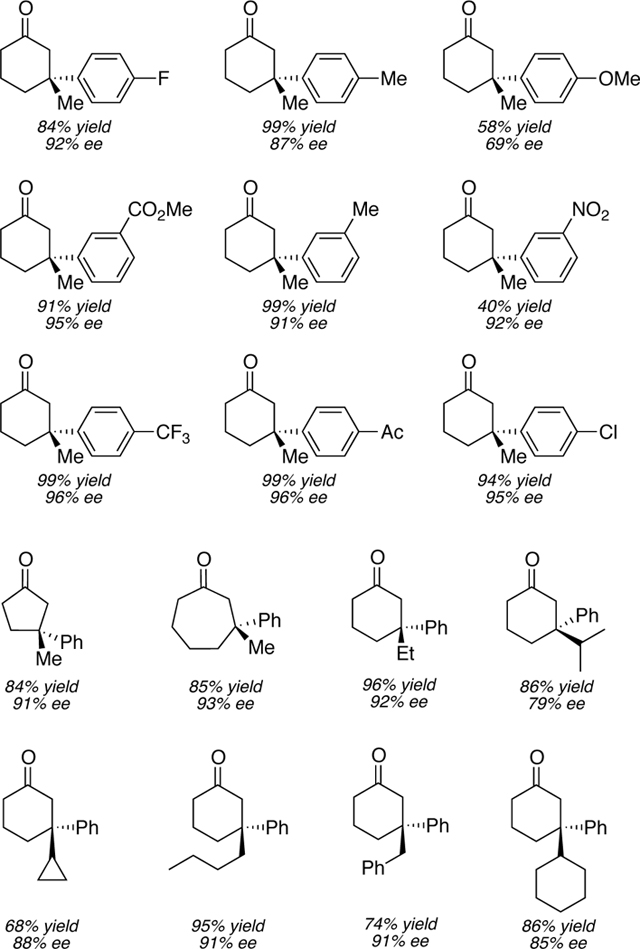
The submitters used an oil bath heated to 60 °C.
Care should be taken to ensure the addition rate is initially very slow to avoid formation of a brown, tar-like byproduct. By addition of the first 3.5 mL over 30 min and the remaining solution (~ 23 mL) over 40 min by syringe pump, no byproduct was seen.
(S)-N-(l-Chloro-3,3-dimethylbutan-2-yl)picolinamide hydrochloride salt exhibited the following characterization data: 1H NMR (400 MHz, DMSO-d6) δ: 0.93 (s, 9 H), 3.88–3.97 (m, 2 H), 4.08 (dt, J = 3.6, 9.6 Hz, 1 H), 7.68 (ddd, J = 1.2, 4.8, 7.2 Hz, 1 H), 8.09 (dt, J = 1.2, 7.6 Hz, 1 H), 8.15 (d, J = 7.6 Hz, 1 H), 8.68–8.71 (m, 2 H), 13.24 (br s, 1 H); 13C NMR (100 MHz, DMSO-d6) δ: 26.6, 35.3, 45.1, 59.4, 122.5, 126.9, 138.8, 147.9, 149.0, 163.7; IR (Neat): 3192, 3023, 2953, 2922, 1680, 1601, 1561, 1518, 1472, 1345, 1291, 1211, 1181, 1036, 1002, 971 cm-1; HRMS (MultiMode ESI/FTMS) m/z calcd for C12H18C1N2O [M+H]: 241.1102, found 241.1103; [α]22D = +25.09 (c 1.132, MeOH). Anal calcd for C12H18Cl2N2O: C, 52.00; H, 6.55; N, 10.11; Cl 25.58. Found: C, 52.29; H, 6.59; N, 10.10; Cl, 25.57. A broad resonance at ~ 5 ppm in the ‘H NMR spectrum is found with incomplete drying of the product; by D20 quenching we determined this resonance to be the hydrate.
Sodium methoxide is used from a freshly opened bottle or retrieved from storage in a nitrogen-atmosphere glovebox, free of adventitious moisture. The addition to the reaction mixture is exothermic.
The reaction can be monitored by TLC analysis using 3:2/hexanes:acetone as the eluent, visualizing with UV fluorescence quenching and p-anisaldehyde staining. Product Rf = 0.44, starting material Rf = 0.32.
Toluene is added to prevent the crude ligand from exposure to concentrated sodium methoxide. The ligand is unstable to concentrated acids or bases.
After toluene was added, approx 140–160 mL of solvent were removed on the rotovap. During the work-up and phase separation, a persistent rag and emulsions formed. The rag is to be kept with the top organic layer.
Silica gel column dimensions: 5 cm diameter × 20 cm height, ca. 200 g silica gel (Silica Gel ZEOprep® 60 ECO 40–63 Micron from American International Chemical, Inc. It is necessary to use this specific silica gel to suppress reversion to (S)-N-(l-hydroxy-3,3-dimethylbutan-2-yl)picolinamide during chromatography, which was observed when employing silica gel from other commercial sources) (Note: The checker used this brand of Silica Gel as received from Prof. Stoltz’s lab. No other silica brands were examined) eluting with 4:1 hexanes / acetone. Fractions are collected in 18 mm × 150 mm test tubes. Fraction purity can be assayed by TLC analysis using 3:2 hexanes / acetone with UV visualization. Product (3) Rf = 0.44. This slow eluent is used for chromatography to remove a yellow impurity of similar polarity that contaminates the white solid product if the column is eluted with higher polarity eluent. This impurity appears to be an indiscrete decomposition product not readily identified. This impurity is not easily removed by recrystallization, and contaminated material should be resubmitted to flash chromatography.
(S)-tert-ButylPyOX (3) exhibited the following characterization data Rf = 0.44 with 3:2 hexanes / acetone; mp 70.4–71.0 °C; 1H NMR (400 MHz, CDCL3) δ: 0.99 (s, 9 H), 4.13 (dd, J = 8.8, 10.4 Hz, 1 H), 4.32 (t, J = 8.4 Hz, 1 H), 4.46 (t, J = 8.8 Hz, 1 H), 7.39 (ddd, J = 1.2, 4.4, 7.2 Hz, 1 H), 7.77 (dt, J = 1.2, 8.0 Hz, 1 H), 8.10 (d, J = 8.0 Hz, 1 H), 8.72 (d, J = 4.0 Hz, 1 H); 13C NMR (100 MHz, CDCl3) δ: 26.0, 34.0, 69.3, 76.5, 124.0, 125.4, 136.5, 147.0, 149.7, 162.5; IR (film): 2954, 2903, 2867, 1640, 1565, 1466, 1358, 1345, 1272, 1244, 1211, 1095, 1037, 967 cm-1; HRMS (MultiMode ESI/APCI) m/z calcd for C12H17N2O [M+H]+: 205.1335, found 205.1336; [α]22D = −91.85 (c 5.09, CHCl3); Anal calcd for C12H17N2O, C 70.56, H 7.90, N 13.71, found C 71.09, H 8.12, N 13.29. Purityof 3 was assessed at 99.3 wt% by quantitative 1H NMR in CDCl3 using methyl phenyl sulfone as a standard. If desired, the white solid can be recrystallized from minimal hot n-heptane, with crystals collected at –20 °C (after cooling to room temperature) as white needles. However, the ligand is more difficult to weigh in this state due to its tendency to cling with static electricity. Enantiomeric excess can be determined via analytical chiral SFC (Jasco SFC utilizing a Chiralcel OB-H column (4.6 mm × 25 cm) obtained from Daicel Chemical Industries, Ltd) with visualization at 210 nm and flow rate of 5 mL/min eluting with 10% MeOH/CO2. Major enantiomer retention time: 2.51 min, minor enantiomer retention time: 2.20 min. (In addition to the high optical purity as seen by polarimetry, the material was analysed by SFC using the method above and the ligand was determined to be 99.7% ee.) Attempts were made to separate enantiomers via analytical chiral HPLC using various columns (Chiralcel OD-H, Chiralcel OJ-H, Chiralpak AD, Chiralpak AS, Chiralcel OB-H) and solvent systems, however adequate separation of peaks was not achieved. In the absence of an analytical chiral SFC, optical rotation measurements can be utilized to give the enantiomeric excess by way of optical purity calculations . The optical rotation listed above may be used as αmaximal and a sufficiently large concentration (e.g. c 5.00) as well as multiple trials should be used in order to minimize error. It is imperative to store the ligand in a desiccator to minimize hydrolysis, which results in diminished enantiocontrol in the conjugate addition reaction. For long-term storage, the ligand should be stored frozen under an inert atmosphere; we recommend a nitrogen atmosphere glovebox freezer.
Vigorous stirring that results in a visible vortex is essential for reaction conversion, as dispersion of water throughout the 1,2-dichloroethane is necessary. After water is added to the reaction, a viscous gel is formed which prevented the magnetic stir bar from spinning. The reaction was manually agitated until the stir bar became free.
The reaction progress can be monitored by TLC analysis with 4:1 hexanes/ethyl acetate, using a p-anisaldehyde stain; product (6) Rf =0.38 (stains yellow /orange with fresh p-anisaldehyde stain), 3-methyl-2- cyclohexenone (4) Rf = 0.19 (stains tan/brown), 4,4’-dichloro-l,l’- biphenyl Rf = 0.65 (does not stain). Reaction times typically are 12–24 h, with conversion slowing considerably after the first several hours.
Silica gel plug dimensions: 3 cm diameter × 4 cm height, ca. 12 g silicagel.
The product is very crystalline. When loading onto the column, the product may crystallize. Silica gel column dimensions: 5 cm diameter × 20 cm height, ca. 220 g silica gel (RediSep RF column from Teledyne Isco, catalog 69–2203-422). The column is eluted with 92:8 hexanes /ethyl acetate until the product is collected. Fractions are collected in 18 mm × 150 mm test tubes. Fraction purity can be assayed by TLC analysis using 4:1 hexanes/ethyl acetate with UV fluorescence quenching visualization and heating with p-anisaldehyde. Product (6) Rf = 0.38, stains yellow/orange with fresh p-anisaldehyde stain. 3-Methyl-2-cyclohexenone (4) Rf = 0.19, stains tan/brown. 4,4’-Dichloro-l,l’- biphenyl Rf = 0.65, does not stain.
(R)-3-(4-Chlorophenyl)-3-methylcyclohexanone (6) exhibited the following characterization data Rf = 0.38 (hexanes /ethyl acetate 4:1), 1H NMR (400 MHz, CDCl3) δ: 1.30 (s, 3 H), 1.60–1.69 (m, 1 H), 1.85–1.94 (m, 2 H), 2.12–2.19 (m, 1 H), 2.31 (t, J = 6.8 Hz, 2 H), 2.42 (d, J = 14.4 Hz, 1 H), 2.83 (d, J = 14.4 Hz, 1 H), 7.23–7.25 (m, 2 H), 7.27–7.30 (m, 2 H); 13C NMR (100 MHz, CDCl3) δ: 21.9, 29.9, 37.9, 40.7, 42.6, 52.9, 127.1, 128.6, 132.0, 145.8, 210.9; IR (thin film): 2944, 2865, 1693, 1589, 1492, 1450, 1432, 1352, 1265, 1229, 1092, 1010, 952, 824 759, 737, 721 cm-1; HRMS (MultiMode ESI/APCI) m/z calcd for C13H14C1O [M+H]: 223.0884, found 223.0885; [α]D22 = −65.8 (c 1.912, CDCl3); Anal calcd C13H15C10: C 70.11, H 6.79, O 7.18, Cl 15.92 found C 69.73, H 7.02, O 7.49, Cl 15.88. Enantiomeric excess of 93% is determined via analytical chiral HPLC (Agilent 1100 Series HPLC utilizing a Chiralcel OB-H column (4.6 mm × 25 cm) obtained from Daicel Chemical Industries, Ltd) with visualization at 254 nm and flow rate of 1 mL/min eluting with 1% iso-propanol/hexanes. The sample was prepared in a 1:1 v/v solution of iso-propanol/ hexanes. Major enantiomer retention time 16.47 min, minor enantiomer retention time 14.55 min.
Working with Hazardous Chemicals
The procedures in Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as “Prudent Practices in the Laboratory” (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red “Caution Notes” within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one’s own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
Discussion
Pyridinooxazoline (PyOx) ligands represent a growing class of bidentate dinitrogen ligands used in asymmetric catalysis.2 Our laboratory has reported the palladium-catalyzed asymmetric conjugate addition of arylboronic acids to cyclic, β,β-disubstituted enones utilizing (S)-tert-ButylPyOx as the chiral ligand.3 This robust reaction does not require an inert atmosphere, is highly tolerant of water,4 and provides cyclic ketones bearing [β-benzylic quaternary stereocenters in high yields and enantioselectivities. While the reaction itself proved to be amenable to multi-gram scale, the ligand is not commercially available, and no reliable method for the large-scale synthesis of (S)-tert-ButylPyOx was known.5 We sought to address this shortcoming by developing an efficient route starting from a cheap, commercially available precursor to pyridinooxazoline ligands. In our initial experiments, (S)-tert-ButylPyOx was synthesized by methanolysis of 2-cyanopyridine, and subsequent acid-catalyzed cyclization with (S)-tert-leucinol to afford the (S)-tert-ButylPyOx ligand.6 We found the yields of this reaction sequence to be highly variable, and the purification by silica gel chromatography to be tedious due to the presence of numerous impurities.
The above reported procedure begins with activation of 2-picolinic acid by treatment with iso-butylchloroformate and N-methylmorpholine, facilitating the desired amidation with (S)-tert-leucinol in high isolated yield, albeit requiring column chromatography.7 Many alternative coupling methods were screened, but mixed anhydride activation gave the highest yield, and the chromatographic purification proved simple even on multigram scale. Furthermore, unlike the methoxyimidate intermediate from the initial synthesis, (S)-N-(l-hydroxy-3,3-dimethylbutan-2-yl)picolinamide is bench stable at room temperature, allowing long-term storage of material. The cyclization of the amide alcohol to (S)-tert-ButylPyOx proved more challenging than anticipated. Activation as mesylate and tosylate followed by in situ cyclization gave the desired product in low yield and incomplete conversion. This could be a result of ligand hydrolysis under the reaction conditions.’ As an alternative to in situ cyclization of an activated intermediate, the amide alcohol was treated with thionyl chloride to yield the hydrochloride salt of (S)-N-(l-chloro-3,3-dimethylbutan-2-yl)picolinamide. After drying under vacuum, this compound proved to be bench stable and was spectroscopically unchanged after exposure to ambient atmosphere and adventitious moisture for more than one week. Furthermore, this chloride salt proved to be a competent cyclization substrate. A series of bases were screened, and sodium methoxide was found to be optimal, as slower rates of hydrolysis to (S)-N-(l-hydroxy-3,3-dimethylbutan-2-yl)picolinamide were observed when compared to the use of other bases.
Our asymmetric conjugate addition method is noteworthy for its simple procedure, owing to its tolerance of both water and atmospheric oxygen. As the catalytic, enantioselective construction of all-carbon quaternary stereocenters remains a challenging problem in synthetic chemistry,9 asymmetric conjugate addition of carbon-based nucleophiles to suitable α,β-unsaturated carbonyl acceptors has garnered much attention as a reliable method for the formation of quaternary stereocenters.10 However, the majority of well-established asymmetric conjugate additions involve the use of highly reactive and water-sensitive organometallic reagents (e.g., diorganozinc,11 triorganoaluminum,12 and organomagnesium reagents13) and, therefore, require rigorously anhydrous reaction conditions. The notable exception is the Hayashi and Shintani rhodium /diene system,14 one of few quaternary-center forging reactions that avoid use of organozinc or organoaluminum reagents.15 Furthermore, our system advantageously uses commercially available arylboronic acids, as opposed to sodium tetraarylborates or boroxins, and palladium as the transition metal catalyst, as opposed to relatively rare and expensive rhodium.16 We believe these advantages uniquely dispose our system toward use in large-scale asymmetric conjugate addition reactions. Furthermore, a large number of 1,4-addition products have been prepared utilizing this methodology featuring a wide array of functional groups (Table 1).
Table 1.
β-Quaternary Ketones Prepared via Asymmetric Conjugate Addition17
 |
Biography

Jeffrey C. Holder graduated from Harvard University in 2009 where he conducted research with Professors Daniel Kahne and E. J. Corey. In 2014, he completed his Ph.D. in the laboratory of Professor Brian M. Stoltz at the California Institute of Technology, where he studied palladium-catalyzed asymmetric conjugate addition reactions and their application in total synthesis. Currently, he is a postdoctoral scholar in the laboratory of Professor John F. Hartwig at the University of California, Berkeley. His research interests include the development and study of transition-metal catalyzed reactions, and their applications in natural product synthesis.

Samantha E. Shockley was born in Birmingham, AL in 1990. She received her B.S. degree in Chemistry from the University of Chicago in 2012 where she conducted research for Professor Richard F. Jordan. After graduating from the College, she worked with Professor Martin Banwell at the Australian National University as a U.S. Fulbright scholar. She is now pursuing her graduate studies at the California Institute of Technology under the guidance of Professor Brian M. Stoltz. Her graduate research focuses on the total synthesis of novel natural products.

Mario P. Wiesenfeldt was born in 1989 in Ludwigshafen am Rhein, Germany. He received his B.S. degree in Chemistry from Ruprecht- Karls-Universität Heidelberg in 2012 where he conducted research with Professor Lutz H. Gade. He started his Masters studies in Heidelberg in the same year with an exchange semester at the University of York, United Kingdom. He is currently a visiting student in the laboratory of Professor Brian M. Stoltz where he is conducting research on palladium-catalyzed enantioselective conjugate addition reactions towards his Master’s Thesis. His research interests include organometallic chemistry and the development of new synthetic methodology.

Hideki Shimizu was born in 1976 in Osaka, Japan. He received his M. S. with Professor Munehiro Nakatani from Kagoshima University in 2001 and Ph. D. in Organic Chemistry from Kyushu University in 2004 under the supervision of Professors Tsutomu Katsuki. He joined the Department of Process Chemistry at Shionogi & Co., Ltd. in 2004. During his time at Shionogi, he worked as a visiting postdoctoral fellow under the direction of Professor Brian M. Stoltz at the California Institute of Technology from 2010 to 2011. He is currently an Associate Director at Shionogi, working on production planning management in the Global Supply Chain Management Division.

Brian M. Stoltz was born in Philadelphia, PA in 1970 and obtained his B.S. degree from the Indiana University of Pennsylvania in Indiana, PA. After graduate work at Yale University in the labs of John L. Wood and an NIH postdoctoral fellowship at Harvard in the Corey labs he took a position at the California Institute of Technology. A member of the Caltech faculty since 2000, he currently is a Professor of Chemistry. His research interests lie in the development of new methodology for general applications in synthetic chemistry.

Brian Cochran obtained his Bachelor’s degree in chemistry in 1999 at Grinnell College. His organic chemistry career started as a medicinal chemist while working for Albany Molecular Research, Inc. followed by Genzyme, Inc. Brian returned to academia and received his Ph. D. in 2009 developing and studying new amination methods at the University of Washington under Professor Forrest Michael. He moved to Colorado to pursue his postdoctoral studies with Professor Tomislav Rovis developing new cyclic anhydride desymmetrization methods with application towards the total synthesis of Ionomycin. In 2012, Brian joined the process chemistry group at Amgen in Thousand Oaks, California.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
2-Picolinic Acid: Pyridine-2-carboxylic acid; (98–98-6)
iso-Butylchloroformate; (543–27-1 )
N-Methylmorpholine: NMM; (109–02-4)
(S)-tert-Leucinol: (S)-2-Amino-3,3-dimethyl-l-butanol; (112245–13-3)
(S)-N-(l-Hydroxy-3,3-dimethylbutan-2-yl)picolinamide; (1476785–62-2)
thionyl chloride; (7719–09-7)
Sodium methoxide: sodium methylate; (124–41-4)
(S)-tert-ButylPyOx: (S)-4-(tert-Butyl)-2-(pyridin-2-yl)-4,5-dihydrooxazole
(117408–98-7)
Palladium(II) trifluoroacetate (42196–31-6)
Ammonium hexafluorophosphate (16941–11-0)
3-Methyl-2-cyclohexenone (1193–18-6)
4-Chlorophenylboronic acid (1679–18-1)
(R)-3-(4-Chlorophenyl )-3-methylcyclohexanone (1235989–03-3)
References
- 1.The Warren and Katharine Schlinger Laboratory for Chemistry and Chemical Engineering, Division of Chemistry and Chemical Engineering, California Institute of Technology, 1200 East California Boulevard, MC 101–20, Pasadena, California, 91125, United States. E-mail: stoltz@caltech.edu. This publication is based on work supported by NIH-NIGMS (R01GM080269–01), and the authors additionally thank Amgen, Abbott, Boehringer Ingelheim and Caltech for financial support. J.C.H. wishes to thank the American Chemical Society Division of Organic Chemistry for a graduate fellowship. We also thank Shionogi & Co., Ltd. For a research grant and a fellowship to H.S.
- 2.(a) Podhajsky SM; Iwai Y; Cook-Sneathen A; Sigman MS Tetrahedron 2011, 67, 4435–4441. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Aranda C; Cornejo A; Fraile JM; García-Verdugo E; Gil MJ; Luis SV; Mayoral JA; Martínez- Merino V; Ochoa Z Green Chem. 2011, 13, 983–990. [Google Scholar]; (c) Pathak TP; Gligorich KM; Welm BE; Sigman MS J. Am. Chem. Soc. 2010, 132, 7870–7871. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Jiang F; Wu Z; Zhang W Tetrahedron Lett. 2010, 51, 5124–5126. [Google Scholar]; (e) Jensen KH; Pathak TP; Zhang Y; Sigman MS J. Am. Chem. Soc. 2009, 131, 17074–17075. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) He W; Yip K-T; Zhu N-Y; Yang D Org. Lett. 2009, 11, 5626–5628. [DOI] [PubMed] [Google Scholar]; (g) Dai H; Lu X Tetrahedron Lett. 2009, 50, 3478–3481. [Google Scholar]; (h) Linder D; Buron F; Constant S; Lacour J Eur. J. Org. Chem. 2008, 5778–5785. [Google Scholar]; (i) Schiffner JA; Machotta AB; Oestreich M Synlett 2008, 2271–2274. [Google Scholar]; (j) Koskinen AMP; Oila MJ; Tois JE Lett. Org. Chem. 2008, 5, 11–16. [Google Scholar]; (k) Zhang Y; Sigman MS J. Am. Chem. Soc. 2007, 129, 3076–3077. [DOI] [PubMed] [Google Scholar]; (l) Yoo KS; Park CP; Yoon CH; Sakaguchi S; O’Neill J; Jung KW Org. Lett. 2007, 9, 3933–3935. [DOI] [PubMed] [Google Scholar]; (m) Dhawan R; Dghaym RD; St. Cyr DJ; Arndtsen BA Org. Lett 2006, 8, 3927–3930. [DOI] [PubMed] [Google Scholar]; (n) Xu W; Kong A; Lu XJ Org. Chem. 2006, 71, 3854–3858. [DOI] [PubMed] [Google Scholar]; (o) Malkov AV; Stewart Liddon AJP; Ramírez-López P; Bendová L; Haigh D; Kocovsky P Angezw. Chem, Int. Ed. 2006, 45, 1432–1435. [DOI] [PubMed] [Google Scholar]; (p) Abrunhosa I; Delain-Bioton L; Gaumont A-C; Gulea M; Masson S Tetrahedron 2004, 60, 9263–9272. [Google Scholar]; (q) Brunner H; Kagan HB; Kreutzer G Tetrahedron: Asymmetry 2003, 14, 2177–2187. [Google Scholar]; (r) Cornejo A; Fraile JM; García JI; Gil MJ; Herrerías CI; Legarreta G; Martínez-Merino V; Mayoral JAJ Mol. Catal. A: Chem. 2003, 196, 101–108. [Google Scholar]; (s) Zhang Q; Lu X; Han XJ Org. Chem. 2001, 66, 7676–7684. [DOI] [PubMed] [Google Scholar]; (t) Zhang Q; Lu XJ Am. Chem. Soc. 2000, 222, 7604–7605. [Google Scholar]; (u) Perch NS; Pei T; Widenhoefer RA J. Org. Chem. 2000, 65, 3836–3845. [DOI] [PubMed] [Google Scholar]; (v) Bremberg U; Rahm F; Moberg C Tetrahedron: Asymmetry 1998, 9, 3437–3443. [Google Scholar]; (w) Brunner H; Obermann U; Wimmer P Organometallics 1989, 8, 821–826. [Google Scholar]
- 3.(a) Kikushima K; Holder JC; Gatti M; Stoltz BM J. Am. Chem. Soc. 2011, 233, 6902–6905. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Holder JC; Zou L; Marziale AN; Liu P; Lan Y; Gatti M; Kikushima K; Houk KN; Stoltz BM J. Am. Chem. Soc. 2013, 135, 14996–15007; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Holder JC; Marziale AN; Gatti M; Mao B; Stoltz BM Chem. Eur. J. 2013,19, 74–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.The reaction requires a small amount of water to run efficiently. Typically 5 equiv of water are added to small-scale reactions.
- 5.A number of syntheses are known, including: Brunner H; Obermann. U. Chem. Ber. 1989,122, 499–507, and ref 3b. [Google Scholar]
- 6.This route is adapted from the synthesis reported in ref 6a.
- 7.Jensen KH; Webb JD; Sigman MSJ Am. Chem. Soc. 2010, 132, 17471–17482. While Sigman and coworkers utilize these conditions to make derivatives of PyOx ligands, such conditions are not reported for the synthesis of tert-ButylPyOx. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Degradation experiments demonstrate that (S)-tert-ButylPyOx is susceptible to hydrolysis. Exposure of tert-ButylPyOx to 3N HC1 results in complete hydrolysis to (S)-N-(l-hydroxy-3,3-dimethylbutan-2- yl)picolinamide as observed by 1H NMR.
- 9.For reviews on the synthesis of quaternary stereocenters, see: (a) Denissova I; Barriault L Tetrahedron 2003, 59, 10105–10146.(b) Douglas CJ; Overman LE Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 5363–5367.(c) Christoffers J; Baro A Adv. Synth. Catal. 2005, 347, 1473–1482.(d) Trost BM; Jiang C Synthesis 2006, 369–396.(e) Mohr JT; Stoltz BM Chem.-Asian J 2007, 2, 1476–1491.(f) Cozzi PG; Hilgraf R; Zimmermann N Eur. J. Org. Chem. 2007, 36, 5969–5994.
- 10.For an excellent comprehensive review, see: Hawner C; Alexakis A Chem. Commun. 2010, 46, 7295–7306. [DOI] [PubMed] [Google Scholar]
- 11.(a) Wu J; Mampreian DM; Hoveyda AH J. Am. Chem. Soc. 2005, 127, 4584–4585. [DOI] [PubMed] [Google Scholar]; (b) Hird AW; Hoveyda AH j. Am. Chem. Soc. 2005, 127, 14988–14989. [DOI] [PubMed] [Google Scholar]; (c) Lee K; Brown MK; Hird AW; Hoveyda AH J. Am. Chem. Soc. 2006, 128, 7182–7184. [DOI] [PubMed] [Google Scholar]; (d) Brown MK; May TL; Baxter CA; Hoveyda AH Angezv. Chem., Int. Ed. 2007, 46, 1097–1100. [DOI] [PubMed] [Google Scholar]; (e) Wilsily A; Fillion EJ Am. Chem. Soc. 2006, 128, 2774–2775. [DOI] [PubMed] [Google Scholar]; (f) Wilsily A; Fillion EJ Org. Chem. 2009, 74, 8583–8594. [DOI] [PubMed] [Google Scholar]; (g) Dumas AM; Fillion E Acc. Chem. Res. 2010, 43, 440–454. [DOI] [PubMed] [Google Scholar]; (h) Feringa BL Acc. Chem. Res. 2000, 33, 346–353. [DOI] [PubMed] [Google Scholar]; (i) Wilsily A; Fillion E Org. Lett. 2008,10, 2801–2804. [DOI] [PubMed] [Google Scholar]
- 12.(a) d’Augustin M; Palais L; Alexakis A Angezv. Chem, Int. Ed. 2005, 44, 1376–1378. [DOI] [PubMed] [Google Scholar]; (b) Vuagnoux-d’Augustin M; Alexakis A Chem. Eur. J. 2007,13, 9647–9662. [DOI] [PubMed] [Google Scholar]; (c) Palais L; Alexakis A Chem. Eur. J. 2009, 15, 10473–10485. [DOI] [PubMed] [Google Scholar]; (d) Fuchs N; d’Augustin M; Humam M; Alexakis A; Taras R; Gladiali S Tetrahedron: Asymmetry, 2005, 16, 3143–3146. [Google Scholar]; (e) Vuagnoux-d’Augustin M; Kehrli S; Alexakis A Synlett, 2007, 2057– 2060. [Google Scholar]; (f) May TL; Brown MK; Hoveyda AH Angew. Chem., Int. Ed. 2008, 47, 7358–7362. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Ladjel C; Fuchs N; Zhao J; Bernardinelli G Alexakis A J. Org. Chem. 2009, 4949–955.19480413 [Google Scholar]; (h) Palais L; Mikhel IS; Bournaud C; Micouin L; Falciola CA; Vuagnoux-d’Augustin M; Rosset S; Bernardinelli G; Alexakis A Angew. Chem., Int. Ed. 2007, 46, 7462–7465. [DOI] [PubMed] [Google Scholar]; (i) Hawner C; Li K; Cirriez V; Alexakis A Angew. Chem., Int. Ed. 2008, 47, 8211–8214. [DOI] [PubMed] [Google Scholar]; (j) Takaya Y; Ogasawara M; Hayashi TJ Am. Chem. Soc. 1998, 120, 5579–5580. [Google Scholar]; (k) Müller D; Hawner C; Tissot M; Palais L; Alexakis A Synlett, 2010, 1694–1698. [Google Scholar]; (l) Hawner C; Müller D; Gremaud L; Felouat A; Woodward S; Alexakis A Angew. Chem., Int. Ed. 2010, 49, 7769–7772. [DOI] [PubMed] [Google Scholar]
- 13.(a) Martin D; Kehrli S; d’Augustin M; Clavier H; Mauduit M; Alexakis AJ Am. Chem. Soc. 2006, 128, 8416–8417. [DOI] [PubMed] [Google Scholar]; (b) Kehrli S; Martin D; Rix D; Mauduit M; Alexakis A Chem. Eur. J. 2010, 16, 9890–9904. [DOI] [PubMed] [Google Scholar]; (c) Hénon H; Mauduit M; Alexakis A Angew. Chem., Int. Ed. 2008, 47, 9122–9124. [DOI] [PubMed] [Google Scholar]; (d) Matsumoto Y; Yamada K.-i.; Tomioka K J. Org. Chem. 2008, 73, 4578–4581. [DOI] [PubMed] [Google Scholar]
- 14.(a) Shintani R; Tsutsumi Y; Nagaosa M; Nishimura T; Hayashi TJ Am. Chem. Soc. 2009, 131, 13588–13589. [DOI] [PubMed] [Google Scholar]; (b) Shintani R; Takeda M; Nishimura T; Hayashi T Angew. Chem., Int. Ed. 2010, 49, 3969–3971. [DOI] [PubMed] [Google Scholar]
- 15.(a) Mauleón P; Carretero JC Chem. Commun. 2005, 4961–4963. [DOI] [PubMed] [Google Scholar]; (b) Shintani R Duan W-L; Hayashi T J. Am. Chem. Soc. 2006, 128, 5628– 5629. [DOI] [PubMed] [Google Scholar]
- 16.A paper describing the use of a Rh•OlefOx (olefin-oxazoline) complex provided a single example of a phenyl boronic acid addition to 3-methylcyclohexenone. Unfortunately, the product was isolated in only 36% yield and 85% ee, see: Hahn BT; Tewes F; Fröhlich R; Glorius F Angew Chem., Int. Ed. 2010, 49, 1143–1146. For more recent examples of palladium-catalyzed conjugate addition of arylboronic acids, see: Gottumukkala, A. L.; [DOI] [PubMed] [Google Scholar]; (a) Matcha K; Lutz M; de Vries JG; Minnaard AJ Eur. J. Chem. 2012, 18, 6907–6914. [DOI] [PubMed] [Google Scholar]; (b) Buter J; Moezelaar R; Minnaard AJ Org. Biomol. Chem. 2014,12, 5883–5890. [DOI] [PubMed] [Google Scholar]
- 17.Yields in Table 1 are without ammonium hexafluorophosphate as an additive, which can greatly improve isolated yield of many arylboronic acid additions.