

SUMMARY
PD-L1 on the surface of tumor cells binds its receptor PD-1 on effector T cells, thereby suppressing their activity. Antibody blockade of PD-L1 can activate an anti-tumor immune response leading to durable remissions in a subset of cancer patients. Here, we describe an alternative mechanism of PD-L1 activity involving its secretion in tumor-derived exosomes. Removal of exosomal PD-L1 inhibits tumor growth, even in models resistant to anti-PD-L1 antibodies. Exosomal PD-L1 from the tumor suppresses T cell activation in the draining lymph node. Systemically introduced exosomal PD-L1 rescues growth of tumors unable to secrete their own. Exposure to exosomal PD-L1-deficient tumor cells suppresses growth of wild-type tumor cells injected at a distant site, simultaneously or months later. Anti-PD-L1 antibodies work additively, not redundantly, with exosomal PD-L1 blockade to suppress tumor growth. Together, these findings show that exosomal PD-L1 represents an unexplored therapeutic target, which could overcome resistance to current antibody approaches.
Graphical Abstract
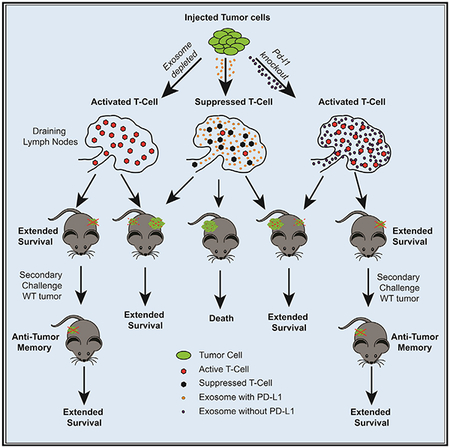
In Brief
Exosomal PD-L1 systemically acts to suppress the anti-tumor immune response, and its genetic blockage promotes T cell activity in the draining lymph node to induce systemic anti-tumor immunity and memory.
INTRODUCTION
Immunotherapy has revolutionized cancer therapy (Chen and Mellman, 2017). Immune checkpoint protein inhibitors, such antibodies against PD-L1 (aka CD274) and PD-1 (aka PDCD1), have shown effectiveness against a large number of cancer types, including melanoma, non-small-cell lung cancer, and renal cancer. This response includes durable remissions many patients who had previously failed multiple other therapeutic strategies. However, even in these cancers, only 10%–30% patients respond to anti-PD-L1/PD-1 therapy (Page et al., 2014). In other cancers, such as prostate cancer, responses are rare (Goswami et al., 2016; Sharma et al., 2017). The basis differential therapeutic success between patients and between cancers remains largely unknown.
PD-L1 is a membrane bound ligand found on the cell surface of many cell types that is upregulated in the setting of inflammation and/or a number of oncogenic lesions (Topalian et al., 2015). It binds the PD-1 receptor on immune T cells, leading to Sh2p-driven dephosphorylation of the T cell receptor and its co-receptor CD28, thereby suppressing antigen-driven activation of T cells (Hui et al., 2017; Yokosuka et al., 2012). This mechanism normally keeps inflammatory responses in check, and Pd-l1 knockout mice develop autoimmune-like diseases (Francisco et al., 2010). However, tumor cells can co-opt this mechanism to evade immune destruction. Therapeutic antibodies to PD-L1 and PD-1 block this interaction, which can then reactivate the anti-tumor immune response (Chen and Mellman, 2017).
It is generally thought that PD-L1 functions within the tumor bed, where cell-surface PD-L1 is directly interacting with PD-1 on the surface of tumor-infiltrating lymphocytes (TILs) (Mellman et al., 2011). However, PD-L1 also can be found on surface of extracellular vesicles (EVs). Furthermore, EV PD-L1 levels have been associated with tumor progression (Chen et al., 2018; Ricklefs et al., 2018; Theodoraki et al., 2018; Yang et al., 2018). Whether extracellular PD-L1 can promote tumor progression by inducing a local and/or systemic immunosuppression is unknown.
EVs are heterogeneous (Tkach et al., 2018). A particular form of EVs is exosomes, which derive from the endocytic pathway (van Niel et al., 2018). As endosomes mature, vesicles bud inward and are released in the lumen forming intravesicular bodies within the late endosomes. These late endosomes are also called multivesicular bodies (MVB). MVBs can either fuse with lysosomes for degradation and recycling of contents or fuse with the plasma membrane releasing the intravesicular bodies extra-cellularly, which are then called exosomes. Exosomes can be differentiated from other EVs based on their size, morphology, density, marker expression, and dependency for specific enzymes for their biogenesis. Key enzymes in their biogenesis include NSMASE2 (aka SMPD3), which promotes budding of intravesicular vesicles, and RAB27A, which is involved in the fusion of the MVB to the plasma membrane (Kosaka et al., 2010; Ostrowski et al., 2010). Genetic manipulation of these enzymes provides an opportunity to dissect the role of exosomes in vivo.
While evaluating mechanisms regulating levels of PD-L1 in different tumors, we discovered that cancer cells can secrete a vast majority of their PD-L1 on exosomes rather than present PD-L1 on their cell surface. Using genetic knockouts for Rab27a and nSMase2 and exogenously introduced exosomes, we show that exosomal PD-L1 from tumor cells promote tumor growth in an immune-dependent fashion. Exosomal PD-L1 suppresses T cell function in vitro and in vivo at the site of the draining lymph node. Exosomal PD-L1 appears to be resistant to anti-PD-L1 as a prostate cancer syngeneic model that is unresponsive to such therapy, is dependent on both PD-L1 and exosomes for their growth. Remarkably, even the transient presence of cancer cells deficient in exosomal PD-L1 results in long-term, systemic immunity against the cancer. A role for exosomal PD-L1 is also seen in a syngeneic colorectal model. In this model, anti-PD-L1 acts additively, not redundantly, with the suppression of PD-L1 secretion. These findings have significant implications for immunotherapeutic approaches to cancer therapy.
RESULTS
Differential Secretion of PD-L1 between CancerCell Lines
It has been reported that surface PD-L1 levels are low in prostate cancer cell lines and primary prostate tumor tissue, potentially explaining the general lack of therapeutic response to anti-PDL1 blockade (Martin et al., 2015; Zang et al., 2007). We sought to determine whether a transcriptional, post-transcriptional, translational, or post-translational mechanism underlies the reduced levels by comparing prostate cancer cell lines (PC3, DU145, LNCaP) to a melanoma cell line (SK-MEL-28). Measurements of mRNA and protein levels showed discordance between mRNA and protein levels across the different cancer cell lines. qRT-PCR showed a 15-fold increase of Pd-l1 mRNA levels in PC3 and DU145 relative to SK-MEL-28; LNCaP showed a near absence of transcripts (Figure 1A). In contrast to mRNA levels, western analysis showed similar cellular PD-L1 protein levels in PC3 and DU145 cells as SK-MEL-28 (Figure 1B); protein was undetectable in LNCaP cells. We asked whether the discordance in mRNA and protein levels between PC3 and SK-MEL-28 could be explained by differences in protein translation. Translation rates were determined by polysome profiling, a method by which transcripts bound by many ribosomes reflective of a high translation rate are separated from transcripts bound by one or a few ribosomes reflective of a low translation rate (Chassé et al., 2017). The two populations were separated on a sucrose gradient, and then the bound mRNA was measured by qRT-PCR (Figure S1A). This analysis showed an equal distribution of the Pd-l1 RNA across the fractions in PC3 and SK-MEL-28 (Figure S1B). Thus, differences in translation rates cannot explain the discordance between mRNA and protein levels between the lines.
Figure 1. Differential Secretion of PD-L1 in PC3 versus SK-MEL-28 Cells.

(A) qRT-PCR of Pd-l1 mRNA levels in one melanoma cell line (SK-MEL-28) and three prostate cancer cell lines (LNCAP, PC3, and DU145). n = 3.
(B) Western analysis of cellular levels of PD-L1 in the same cells as (A). Top: Representative western blot. Bottom: Quantification of three independent western blots.
(C) Top left: Schematic for crude isolation of secreted vesicles by sequential centrifugation of media through increasing centrifugal forces and finally pelleted at 100k g. Bottom left: Representative western blot of PD-L1 in cells versus secreted vesicles of either PC3 or SK-MEL-28 cells. Right: Quantification. n = 6.
(D) Nanoparticle tracking of size and quantity of secreted vesicles produced by PC3 and SK-MEL-28 cells. Left: Density plot for size. Right: Total vesicle number based on integration under the curve. n = 9.
(E) Schematic of steps involved in exosome purification including sequential centrifugations of supernatant at increasing gravitation force and then 100k g pellet run on sucrose gradient. Location of exosomes is followed by a western blot for CD63.
(F) Western blot for PD-L1 and exosome markers CD63 and HRS in different sucrose gradient fractions. n = 3.
(A and B) Error bars represent SD. (C and D) Error bars represent SEM. *p < 0.05; **p < 0.01; ***p < 0.001 (Student’s t test).
See also Figures S1 and S2.
Next, we evaluated potential differences in protein degradation. The two main pathways for protein degradation are the lysosome and the proteasome (Ciechanover, 2005). The small molecule Bafilomycin A1 (BafA1) inhibits lysosomal activity by blocking the V-ATPase hydrogen pump and thus acidification of the lysosome (Mauvezin and Neufeld, 2015). An increase in the protein LC3B, a known target of the lysosome, confirmed effectiveness of the small molecule on PC3 and SK-MEL-28 cells. However, levels of PD-L1 in both lines were unaltered, implying little turnover of PD-L1 by the lyso-some in these cells (Figures S1C and S1D). The small molecule MG132 suppresses proteasome activity by blocking the 26S proteasome complex and, consequentially, proteolysis (Lee and Goldberg, 1998). The proteasome recognizes ubiquitin side chains on its targets (Lee and Goldberg, 1998). An increase in the presence of ubiquitinated proteins confirmed the effectiveness of MG132 on the two cells lines. Again though, PD-L1 protein levels were minimally affected, and, if anything, the difference of the two lines was opposite from expected as PD-L1 levels were slightly up in SK-MEL-28, but not in PC3, cells (Figures S2E and S2F). These results show that differences in protein stability do not explain the discordance in mRNA and protein levels.
Next, we considered the possibility that PD-L1 may be differentially secreted from cells in the form of membrane vesicles. Extra-cellular vesicles can be enriched using sequential centrifugation transferring supernatant through increasing gravitational forces to remove cellular debris and apoptotic bodies, before finally pelleting at 100,000 gravitational forces (100k g) (Xu et al., 2016). Western analysis showed 2- to 3-fold more PD-L1 in the 100k g fraction of PC3 cells relative to SK-MEL-28 cells (Figure 1C). This difference could have been due to more PD-L1 being loaded per vesicle or release of more vesicles. A nanoparticle tracking instrument can track the size and number of vesicles by using light diffraction off particles moving under Brownian motion (Carr and Warren, 2012). Analysis of conditioned media from the two cell types showed a slightly elevated total vesicle count in SK-MEL-28 (Figure 1D). Thus, even though PC3 and SKMEL-28 cells had similar levels of cellular PD-L1 protein, PC3 cells packaged greater amounts of PD-L1 into extracellular vesicles. This difference appears to underlie the discordance between mRNA and protein levels between the two cell lines.
In vivo, tumor cells can mediate adaptive resistance by upregulating PD-L1 in response to interferon-gamma (IFNγ) released by the cytotoxic T lymphocytes within the tumor bed (Garcia-Diaz et al., 2017). Therefore, we asked whether, in addition to up-regulating cellular PD-L1, IFNγ may increase the secretion of PD-L1. Treatment of cancer cells with IFNγ led to an increase in PD-L1 in both the cellular and 100k g fractions (Figure S2A). The increase was proportionally similar between the two fractions, suggesting no direct impact of IFNγ on PD-L1 secretion (Figure S2B). In addition, IFNγ did not increase the number of vesicles secreted (Figure S2C). Thus, similar to the cell-surface membrane PD-L1, extracellular vesicular PD-L1 increases in response to IFNγ.
PD-L1 Is Specifically Secreted within Exosomes
Extracellular vesicles come in multiple forms differing in size, density, protein markers, and biogenesis (Xu et al., 2016). Given that PD-L1 is endocytosed from the surface of cells (Burr et al., 2017; Mezzadra et al., 2017), we hypothesized that PD-L1 is being specifically secreted in the form of exosomes. Exosomes can be enriched relative to other vesicles based on their density by spinning the crude 100k g pellet on a sucrose gradient (Shurtleff et al., 2016). The exosomal marker CD63 traveled in the 20%–40% sucrose fractions (Figure 1E). PDL1 and the additional exosomal marker HRS colocalized with CD63 (Figure 1F). These data support that PD-L1 is packaged in exosomes.
Next, to determine if PD-L1 is specifically found in exosomes, we took a genetic approach to remove exosomes. We focused on the Rab27a and nSMase2 genes using CRISPR/Cas9-mediated mutagenesis to knock them out in PC3 cells (Figures S3A–S3C). Deletion of these two genes did not affect the proliferation of PC3 cells (Figures S3D and S3E). To follow exosomes, we ectopically expressed CD63-GFP in the three genetic backgrounds. Measurement of the conditioned media showed an almost complete absence of CD63-GFP+ particles arising from the two mutant lines versus wild-type (WT) (Figure 2A). To further evaluate the effect of the Rab27a and nSMase2 deletion on exosomes, we performed electron microscopy on sucrose gradient concentrated particles. The resultant images showed very few exo-some-like particles in the Rab27a and nSMase2 null samples, with a greater loss in the Rab27a null cells (Figures 2B–2dD). Consistent with these images, western analysis showed an absence of endogenous CD63 in the 100k g preps from Rab27a null cells and a small amount remaining in the nSMase2 null cells (Figure 2E). In contrast to CD63 levels, nSMase2 showed a complete absence, while Rab27a null cells showed a dramatic reduction in PD-L1 in the 100k g fraction (Figure 2E). This difference appears to be due to an additional role for NSMASE2 on Pd-l1 transcription (Figures S3F and S3G). Together, these data show critical roles for both Rab27a and nSMase2 in exosome biogenesis and PD-L1 secretion.
Figure 2. PD-L1 Is Secreted in the Form of Rab27a- and nSMase2-Dependent Exosomes.

(A) Nanoparticle tracking of size and quantity of GFP+ vesicles from WT and Rab27a null PC3 cells expressing CD63-GFP. Left: Density trace. Right: Integration under curve for total particles. n = 3.
(B–D) Electron microscopy images of WT (B), Rab27a null (C), and nSMase2 (D) null PC3 exosomes, respectively, purified on sucrose gradient.
(E) Representative western for PD-L1 and CD63 in WT PC3, Rab27a null, and nSMase2 null PC3 cells.
(F) Quantification of PD-L1 in WT PC3, Rab27a null, and nSMase2 null PC3 cells. n = 6.
(G) Schematic of in vitro functional assay of exosomal PDL1.
(H) Quantification of IL-2 released by Jurkat cells following Raji presentation of super-antigen. Jurkat (PD1), Jurkat T cells overexpressing PD1; Raji (Parental), Raji B cells expressing low levels of PD-L1; Raji (PD-L1), Raji B cells overexpressing PD-L1; PC3 vesicles, 100k g pellet from conditioned media of WT PC3 cells; PC3 Pd-l1−/− Vesc, 100k g pellet from conditioned media of Pd-l1 null PC3 cells. n = 4.
(A, F, and H) Error bars represent SD. *p < 0.05; **p < 0.01; ***p < 0.001 (Student’s t test).
See also Figures S3 and S4.
Given that PD-L1 is a trans-membrane protein, exosomal PDL1 must arise from the limiting membrane of the late endosome. The limiting membrane of the late endosome initially arises from the endocytosis of the plasma membrane. However, material provided directly from the ER and Golgi as well as cytoplasm is added as the resultant endosome matures (van Niel et al., 2018). Flow cytometry and immunofluorescence showed the presence of PD-L1 on both the surface and within vesicle-like structures inside PC3 cells (Figures S4A and S4B). Using standard cell fractionation techniques, PD-L1 and CD63 co-localized in the sucrose light, or endolysosomal fraction (Shurtleff et al., 2016) (Figure S4C). To determine whether exosomal PD-L1 directly arises from the ER or early Golgi, we took advantage of the fact that PD-L1 is glycosylated (Li et al., 2016). This glycosylation was easily removed with the amidase PNGaseF, consistent with it being a N-linked oligosaccharide chain (Figure S4D). In contrast, the majority of cellular and all the exosomal PD-L1 was resistant to EndoH cleavage, consistent with maturation of the oligosaccharide chain in the distal Golgi (Stanley, 2011). Therefore, exosomal PD-L1 does not appear to come directly from the ER or early Golgi. To measure the plasma membrane as a source, we performed a cell-surface biotinylation assay (Figure S4E). Biotin-labeled PD-L1 was found in the cells and exosomes of treated cells, showing that exosomal PD-L1 originates from the surface of the PC3 cells.
Exosomal PD-L1 Suppresses T Cell Activation In Vitro
Next, we asked whether exosomal PD-L1 could function similarly to cell-surface PD-L1 in the suppression of T cell activation. PD-L1 function can be measured in vitro in the setting of Raji B cell presentation of antigen to Jurkat T cells (Hui et al., 2017). Normally, this presentation would activate T cells, which can be measured by secretion of interleukin-2 (IL-2). However, if PD-L1 is exogenously expressed in the Raji B cells and PD-1 is exogenously expressed in the Jurkat T cells, T cell activation is suppressed (Hui et al., 2017). In this setting, we asked whether exosomal PD-L1 from PC3 cells could replace exogenous expression of PD-L1 on the Raji B cells (Figure 2G). As expected, in the absence of exogenous expression of PD-L1 in Raji B cells, IL-2 secretion was high (Figure 2H, compare first two bars). However, the introduction of the PC3 100k g extracellular fraction re-repressed IL-2 secretion showing that PC3 vesicles can replace Raji cell PD-L1 expression (Figure 2H, third bar). To determine whether exosomal PD-L1 was responsible for this effect, we used CRISPR/Cas9 editing to delete the Pd-l1 gene in the PC3 cells (Figure S4F). Deletion of the Pd-l1 gene did not affect the proliferation of the PC3 cells or alter the number of vesicles released (Figures S4G and S4H). However, introduction of the Pd-l1 null PC3 100k g extracellular fraction failed to repress IL2 secretion (Figure 2H, fourth bar). These data show that exosomal PD-L1 does function to suppress T cell activation in vitro.
Exosomal PD-L1 Promotes Tumor Progression
Given its ability to suppress T cell activation in vitro, we wanted to know whether exosomal PD-L1 can function in vivo to promote tumor progression. To do so, we turned to a syngeneic model of prostate cancer, the TRAMP-C2 model (Foster et al., 1997). This preclinical model, like human prostate cancer, is resistant to anti-PD-L1 blockade (Yu et al., 2012). CRISPR/Cas9-mediated deletion of Rab27a (Figure S5A) and Pd-l1 (Figure S5B) resulted in a loss of PD-L1 in the EV fraction (Figure 3A). Loss of Rab27a did not influence cell-surface PD-L1 levels, nor did loss of Pd-l1 influence exosome production (Figures S5C and S5D). Like with the PC3 cells, deletion of Rab27a or Pd-l1 did not affect the proliferation of the cells (Figures 3B and 3C). The WT, Rab27a null, and Pd-l1 null TRAMP-C2 cells were injected into the flanks of C57BL6/J syngeneic mice and were followed for over 4 months. All mice injected with the WT TRAMP-C2 cells had visible tumors by around 35 days and had to be euthanized between 40 and 71 days (Figures 3D and 3E). In contrast, all mice injected with Rab27a null or Pd-l1 null TRAMP-C2 cells showed no tumor growth during the same time period (Figure 3D). Similarly, the mice injected with Rab27a and Pd-l1 null TRAMP-C2 cells showed a dramatically extended lifespan relative to their WT counterparts (Figure 3E). Indeed, a majority of the mice remained alive after 90 days, at which point they were used for the memory experiments described below.
Figure 3. Exosomal PD-L1 Suppresses Tumor Progression in Syngeneic Prostate Cancer Model.

(A) Western blot for PD-L1 in WT, Rab27a null, and Pd-l1 null TRAMP-C2 100k g extracellular fraction.
(B) Cell counts over time for Pd-l1 null versus WT TRAMP-C2 cells. n = 3. Error bars represent SD.
(C) Cell counts over time for Rab27a null versus WT TRAMP-C2 cells. n = 3. Error bars represent SD.
(D) Tumor growth volume over time following subcutaneous injection of 1 × 106 WT, Rab27a null, or Pd-l1 null TRAMP-C2 cells into immuno-competent B6 mice. n = 5 for each genotype. Error bars represent SEM. Tramp WT versus TRAMP-C2 Rab27a null, p < 0.001. Tramp WT versus TRAMP-C2 Pd-l1 null, p < 0.0001. Tramp WT versus TRAMP-C2 nSMase2 null, p < 0.001 (twoway ANOVA test).
(E) Mouse survival curve following injection of cells like in (D). n = 10 for each genotype. Tramp WT versus TRAMP-C2 Rab27a null, p < 0.001. Tramp WT versus TRAMP-C2 Pd-l1 null, p < 0.001. Tramp WT versus TRAMP-C2 nSMase2 null, p < 0.001 (log rank test).
(F) Mouse survival curve following NOD-SCID IL2rg−/− (NSG) mice injected with 1 × 106 WT, Rab27a null, and Pd-l1 null TRAMP-C2 cells. n = 10 for Pd-l1 null and WT genotypes, n = 8 for Rab27a null genotype, n = 5 for nSMase2 null genotype.
See also Figure S5.
To further confirm that the loss of Rab27a was blocking tumor growth through its role in exosome biogenesis, we repeated the experiments using nSMase2 null cells. nSMase2 was deleted again using CRISPR/Cas9-mediated mutagenesis (Figure S5E). Similar to PC3, nSMase2 null TRAMP-C2 cells showed a decrease in both cellular and extracellular levels of PD-L1 (Figures S5F–S5H). These cells were injected in syngeneic mice and compared to their WT counterparts. Similar to Rab27a null TRAMP-C2 cells, the nSMase2 null TRAMP-C2 cells failed to form palpable tumors during the time period when mice receiving WT cells had to be euthanized (Figure 3D). Again, a majority of the mice (8 of 10) were still alive after 90 days (Figure S3E). Together, these data show that blocking exosome biogenesis or removing PD-L1 result in a very similar tumor growth suppression phenotype.
It remained plausible that the exosome biogenesis factors were acting cell autonomously to suppress tumor growth, rather than non-autonomously via exosomal PD-L1 to suppress tumor growth via the anti-tumor immune response. To differentiate these alternative possibilities, we next asked whether the effect of Rab27a and/or nSMase2 loss was dependent on an active immune system. WT, Rab27a, nSMase2, and Pd-l1 null TRAMP-C2 cells were injected into NOD-scid IL2rγnull (NSG)-immunodeficient mice. In striking contrast to the results seen in immunocompetent mice, the four lines led to end-stage tumors at a similar rate in the immunodeficient background (Figure 3E). Therefore, RAB27A and NSMASE2, like PD-L1, promote tumor growth through the suppression of the immune system.
Exosomal PD-L1 Suppresses T Cell Activity in the Draining Lymph Node
Next, we asked whether loss of exosomes and PD-L1 have a similar effect on the immune response to the tumor. To address this question, we measured the response in the lymphoid tissues following the injection of WT, Rab27a null, and Pd-l1 null TRAMP-C2 cells (Figure 4A). At 14 days, the spleens of mice injected with either mutant cell line were significantly larger than those injected with WT cells (Figure 4B). Immunophenotyping of the spleen showed equal percentages of CD8, CD4, and regulatory T cells across the three genotypes (Figure S6). These data are consistent with an enhanced generalized systemic immune response in the absence of exosomes or PD-L1. In contrast, immunophenotyping of the draining lymph nodes showed striking differences between the WT and mutant animals. CD8 positive cells made up a much greater fraction of the T cells following injection of the two mutant tumor cell lines relative to the WT line (Figure 4C). The fraction of CD4 was relatively down in the mutants, whereas FoxP3+ T regulatory cells composed a constant fraction of the T cells (Figures 4D and 4E). Given the relative increase in CD8 cells in the absence of exosomes or PD-L1, we evaluated markers of T cell exhaustion and activation within the CD8 and CD4 T cell populations. The fraction of CD8 and CD4 cells that were PD-1 high were trending down in mice receiving the mutant cells (Figures 4F and 4G). Much more significant was a decrease in the percentage of cells expressing the exhaustion marker Tim3 and increase in the percentage of cells expressing the activation marker Granzyme B (Figures 4H–4K). Furthermore, the fraction of cells positive for the proliferation marker Ki67 was significantly up among the CD8 T cells and trending up among the CD4 T cells (Figure 4L and 4M). These data are consistent with tumor-derived exosomal PD-L1 traveling to the draining lymph node and suppressing T cell activation.
Figure 4. Immunophenotypes of Draining Lymph Node T Cells Predicts Immune Memory and Tumor Suppression.

(A) Schematic of experimental design for (B)–(M).
(B) Spleen weight in grams 14 days post-injection with 1 × 106 WT, Pd-l1 null, and Rab27a null TRAMP-C2 cells.
(C–E) Flow cytometric quantification of the percentage of CD8+ (C), CD4+ (D), and regulatory T cells (T-reg) (E), respectively, among CD45+, CD3+ cells in the draining lymph node (DLN) (n = 5 mice/genotype) 14 days post-tumor cell injection (1 × 106 WT, Pd-l1 null, and Rab27a null TRAMP-C2 cells).
(F and G) Quantification of PD-1+ cells among CD8 (F) and CD4 (G) T cells (n = 5 mice/genotype).
(H and I) Quantification of Tim3+ in CD8 (H) and CD4 (I) cells (n = 5 mice/genotype).
(J–M) Quantification of granzyme B (GzmB) (J and K) or Ki67 (L and M) CD8 (J and L) and CD4 (K and M) T cells (n = 5 mice/genotype). *p < 0.5; **p < 0.01 (Student’s t test).
(N) Schematic of experimental design for (O).
(O) Tumor growth volume over time following secondary subcutaneous injection of 1 × 106 WT TRAMP-C2 cells into immunocompetent B6 mice previously sham treated or injected with Pd-l1, Rab27a, or nSMase2 null TRAMP-C2 cells. n = 5 for each condition. Tramp WT versus TRAMP-C2 WT pre-injected with TRAMP-C2 Rab27a null, p < 0.001. Tramp WT versus TRAMP-C2 WT pre-injected with TRAMP-C2 Pd-l1 null, p < 0.001. Tramp WT versus TRAMP-C2 WT pre-injected with TRAMP-C2 nSMase2 null, p < 0.001 (two-way ANOVA test). Error bars represent SEM.
See also Figure S6.
We hypothesized that if exosomes are acting through PD-L1 to suppress the T cell activation in the lymph node, then in the absence of exosomal PD-L1, immune-competent mice will not only suppress immediate growth of the mutant tumor cells but will also develop memory toward the tumor cells. To test this hypothesis, mice injected with either Rab27a or Pd-l1 null TRAMP-C2 cells on one flank were rechallenged 90 days later with WT TRAMP-C2 cells on the other flank (Figure 4N). Age matched mice that had not been previously injected with any tumor cells were used as control. Remarkably, WT TRAMP-C2 tumors failed to grow in any of the mice previously injected with either mutant cell line, but showed normal growth in the age matched controls (Figure 4O). Thus, exposure to tumor cells lacking exosomes or PD-L1 results in a robust memory response even against cells that secrete exosomal PD-L1. We interpret this to mean that once the T cells have been exposed to tumor antigens in the absence of exosomal PD-L1, they become resistant to the suppressive effects of exosomal PD-L1.
Exosomal PD-L1 Promotes Growth across Different Tumor Types
Next, we asked whether the effect of exosomal PD-L1 on tumor progression is unique to the TRAMP-C2 model. To address this question, we evaluated a colorectal cancer model, MC38. Unlike the TRAMP-C2 model, this model shows a partial response to anti-PD-L1 and therapy (Deng et al., 2014). Western analysis showed that MC38 cells, like TRAMP-C2 cells, secrete PD-L1 (Figure 5A). They also express low levels on the cell surface (Figure 5B). Using identical guide RNAs as in the TRAMP-C2 model, Pd-l1 and Rab27a were knocked out in MC38 cells via CRISPR/Cas9-mediated mutagenesis (Figures 5B and S7A). The Rab27a knockout cells showed a loss of PD-L1 in the secreted fraction, confirming the packaging of PD-L1 in exosomes (Figure 5A). Also, similar to TRAMP-C2 cells, the Rab27a and Pd-l1 knockout MC38 cells showed no change in proliferation (Figures 5C and 5D). These cells were injected in syngeneic WT mice (Figures 5E and 5F). WT MC38 tumors grew rapidly, and mice had to be euthanized starting at 9 days. Loss of either Rab27a or Pd-l1 slowed tumor growth and extended lifespan. However, unlike the TRAMP-C2 model, Pd-l1 loss had a greater effect than Rab27a loss (Figure 5F). Therefore, exosomal PD-L1 appears to play an important, but partial role, in this model.
Figure 5. Exosomal PD-L1 Suppresses Tumor Progression in Syngeneic Colorectal Cancer Model.

(A) Western for PD-L1 in WT and Rab27a null MC38 100k g extracellular fraction.
(B) Flow cytometry for surface PD-L1 on MC38 cells.
(C) Cell counts over time for Rab27a null versus WT MC38 cells. n = 3. Error bars represent SD.
(D) Cell counts over time for Pd-l1 null versus WT MC38 cells. n = 3. Error bars represent SD.
(E) Tumor growth over time following subcutaneous injection of 1 × 106 WT, Rab27a null, or Pd-l1 null MC38 cells into immunocompetent B6 mice. n = 5 for each genotype. Error bars represent SEM. MC38 WT versus MC38 Rab27a null, p < 0.05. MC38 WT versus MC38 Pd-l1 null, p < 0.05. MC38 WT versus MC38 Pd-l1 null; Rab27a null, p < 0.05 (two-way ANOVA test).
(F) Mouse survival curve following injection of cells like in (D). n = 10 for each genotype. MC38 WT versus MC38 Rab27a null, p < 0.001. MC38 WT versus MC38 Pd-l1 null, p < 0.001. MC38 WT versus MC38 Pd-l1 null; Rab27a null, p < 0.001 (log rank test).
(G) Survival curve for mice injected with WT, Rab27a null, or Pd-l1 null MC38 cells followed by treatment with either anti-PD-L1 or isotype control antibody. n = 5 for each condition. MC38 WT isotype versus MC38 WT anti-PD-L1, p < 0.01. MC38 Rab27a isotype versus MC38 Rab27a anti-PD-L1, p < 0.05. MC38 Pd-l1 isotype versus MC38 Rab27a anti-PD-L1, ns. (log rank test).
See also Figure S7.
The difference in the effect of Pd-l1 versus Rab27a loss allowed for an epistasis analysis to address the question of whether exosomes are acting specifically through PD-L1 to influence tumor growth. If exosomes have a PD-L1 independent role, their removal should further reduce tumor growth and extend lifespan in the Pd-l1 null background. Therefore, we made Rab27a; Pd-l1 double knockout MC38 cells (Figure S7). These cells grew at a rate similar to that of the Pd-l1 single mutant cells (Figures 5E and 5F). Thus, we conclude that exosomes are functioning predominantly, if not entirely, through their presentation of PD-L1.
The difference in the effect of Pd-l1 versus Rab27a loss on tumor growth also implied that there is an exosome independent pool of PD-L1 that is also functioning to suppress the immune response in this model. We hypothesized that unlike exosomal PD-L1, this pool may be sensitive to anti-PD-L1 antibody. This pool could explain why unlike the TRAMP-C2 model, the MC38 model is partially responsive to anti-PD-L1 antibodies (Deng et al., 2014). To test this hypothesis, we evaluated the effect on survival of anti-PD-L1 alone and in combination with exosome depletion. Similar to the loss of Rab27a, the treatment of the mice with anti-PD-L1 antibody extended survival, but not to the same degree as loss of Pd-l1 (Figure 5G). Remarkably, the combination of Rab27a deletion and treatment with anti-PD-L1 antibodies lead to similar survival curve as the Pd-l1 deletion. This finding strongly supports the conclusion that while exosomal PD-L1 is resistant to anti-PD-L1, there is another pool in this model, likely cell-surface PD-L1, which is both a significant suppressor of the anti-tumor immune response and sensitive to the antibodies.
Exosomal PD-L1-Deficient Tumor Cells Can Suppress WT Tumor Growth at a Distance Site
Exosomes can enter the blood and lymphatic systems and travel throughout the body, potentially influencing tumor growth at distant sites (Ruivo et al., 2017). Similarly, immune cells educated at one tumor site can travel throughout the body potentially influencing growth of tumors at distant sites (Chen and Mellman, 2013). Therefore, we next asked whether the simultaneous injection of WT and mutant cells at different sites would affect growth of either tumor. In particular, could exosomal PD-L1 released from WT tumor cells promote growth of mutant cells at a distant site, and/or could the T cells activated by the mutant cells suppress growth of WT cells at a distant site? To address this question, WT TRAMP-C2 cells were injected in one flank of each mouse simultaneously with Pd-l1, Rab27a, or nSMase2 null TRAMP-C2 cells on the other flank (Figure 6A). Tumor growth on each flank was then followed over time. Remarkably, growth of the WT tumor in these double injections was dramatically reduced relative to mice where WT cells alone were injected (Figure 6B). The effects of Pd-l1, Rab27a, or nSMase2 null cells on the distant WT tumor were similar. In contrast, the WT cells did not have any effect on the growth of Rab27a or nSMase2 null cells (Figure 6C). However, the WT cells did promote the growth of Pd-l1 null cells. This effect was small as tumor growth was much slower than the WT counterparts (Figure 6C, compare y axis to 6B). The reason for the difference between the Pd-l1 null cells and the exosome-deficient cells is unclear and requires further study. Overall, the double-injected mice had a greatly extended survival relative to those that received WT TRAMP-C2 cells alone (Figure 6D). These findings strongly supported the notion that immune cells activated in the draining lymph node of the mutant side were able to travel and attack the WT tumor cells on the opposite flank. Indeed, histological analysis of the WT tumors showed a dramatic increase in the number of infiltrating lymphocytes when co-injected with the mutant cells on the opposite flank (Figures 6E and S7E). Together, these data show communication between the tumors, with the effect of the mutant tumor being dominant over that of the WT tumor.
Figure 6. Cross-Communication between WT and Mutant TRAMP-C2 Cells at Distant Sites.

(A) Schematic of the experiment: briefly, immuno-competent B6 mice were co-injected with 1 × 106 mutant TRAMP-C2 cells in one flank and with 1 × 106 WT TRAMP-C2 cells in the other flank.
(B) Tumor growth of WT TRAMP-C2 cells in mice singly injected or co-injected with either Pd-l1 null or Rab27a null cells. n = 5 for each condition. Tramp WT versus TRAMP-C2 WT co-injected with TRAMP-C2 Rab27a null, p < 0.001. Tramp WT versus TRAMP-C2 WT co-injected with TRAMP-C2 Pd-l1 null, p < 0.001. Tramp WT versus TRAMP-C2 WT co-injected with TRAMP-C2 nSMase2 null, p < 0.01 (two-way ANOVA test). Error bars represent SEM.
(C) Tumor growth of Pd-l1 null or Rab27a null TRAMP-C2 cells in mice singly injected or co-injected with WT TRAMP-C2 cells. n = 5 for each condition. Error bars represent SEM.
(D) Mouse survival curve mice singly injected with WT TRAMP-C2 cells or doubly injected with WT and Pd-l1 null or Rab27a null cells. n = 5 for each genotype.
(E) Histological analysis of lymphocyte infiltration of tumors under the noted conditions. Each symbol represents an individual mouse. Representative images for each state (severe, moderate, mild, and none) can be found in Figure S7E.
Exogenously Introduced Exosomal PD-L1 Can Rescue Immune Suppression and Tumor Growth
Next, we asked whether exogenously introduced exosomal PDL1 could suppress the anti-tumor immune response and promote tumor growth. To address the effect on the immune response, we transplanted Rab27a null TRAMP-C2 cells in the flank of syngeneic mice followed by tail vein injections of in vitro collected exosomes from either their WT or Pd-l1 null counterparts (Figure 7A). Exosomes from the WT, but not Pd-l1 null, cells were able to induce a systemic immunosuppression as evidenced by nearly a 50% reduction in spleen size (Figure 7B). Furthermore, the WT exosomes were able to suppress the immune response in the draining lymph node of the Rab27a-deficient cells. Compared to the Pd-l1-deficient exosomes, the WT exosomes led to a reduced CD8/CD4 ratio, while having little effect on the T-reg cells (Figures 7C–7E). More importantly, they led to an increase in the fraction of cells expressing high levels of the exhaustion markers PD-1 and TIM3 and low levels of the activation marker Gramzyme B (Figures 7F–7K). These findings paralleled the findings seen in mice transplanted with WT versus Rab27a null cells (cf. Figures 4 and 7). To address the effect on the tumor growth, we transitioned to the MC38 model, given its more rapid growth characteristics. As with the TRAMP-C2 model, Rab27a-deficient MC38 cells were injected in the flank of syngeneic mice followed by exosome injection in the tail vein of the same mice. Consistent with the immune suppression seen in the TRAMP-C2 model, injection of exosomes collected from WT, but not Pd-l1-deficient MC38, cells promoted tumor growth and reduced survival (Figures 7L and 7M). These results confirm that exosomes are functioning through PD-L1 to suppress the anti-tumor immune response and thus promote tumor growth.
Figure 7. Exogenously Introduced Exosomal PD-L1 Rescues Immune Suppression and Tumor Growth of Rab27a Null Tumors.

(A) Schematic of experimental design (B)–(K); briefly, mice were transplanted with 1 × 106 Rab27a null TRAMP-C2 cells, followed by 3 times per week tail-vein injections of exosomes that were collected from either WT or Pd-l1 null TRAMP-C2 cells grown in vitro. Immune analysis was performed at 14 days.
(B) Spleen weight in grams.
(C–E) Flow cytometric quantification of the percentage of CD45+ CD3+ cells that are CD8 (C), CD4 (D), and regulatory T cells (T-reg) (E), respectively, in the draining lymph node (DLN).
(F and G) Quantification of the percentage PD-1+ cells among the CD8 (F) and CD4 (G) T cell populations.
(H and I) Quantification of the percentage of Tim3+ cells among the CD8 (H) and CD4 (I) T cell populations.
(J and K) Quantification of the percentage of granzyme B (GzmB+) cells among the CD8 (J) and CD4 (K) T cell populations.
(B–K) Each dot represents an individual mouse. Error bars represent mean and SD. *p < 0.05; **p < 0.01; ***p < 0.001 (Student’s t test).
(L) Tumor growth over time following subcutaneous injection of 1 × 106 MC38 Rab27a null cells followed by tail vein exosomes derived from MC38 WT and MC38 Pd-l1 null cells grown in vitro. MC38 Rab27a null treated with WT versus Pd-l1 null exosomes, p < 0.01 (two-way ANOVA test).(M) Survival curve following injection of cells as in (L). MC38 Rab27a null tumors treated with WT versus Pd-l1 null exosomes, p < 0.01 (log rank test).
(L and M) Error bars represent SEM.
DISCUSSION
All together, these data uncover a key role for exosomal PD-L1 in enabling cancer cells to evade anti-tumor immunity. Indeed, in the presence of exosomal PD-L1, T cells in the tumor’s draining lymph node express markers of exhaustion and the spleens are smaller. Genetically blocking exosome biogenesis or deleting Pd-l1 reverses the phenotype by strongly promoting T cells activation, proliferation, and killing potential. This effect is reversed again with the introduction of exogenous exosomal PD-L1. Therefore, tumor exosomes have the ability to travel to the draining lymph node, where they present PD-L1 inhibiting T cell activation. Remarkably, blocking the release of exosomal PD-L1 not only suppresses growth of the local tumor cells but also blocks WT tumor cells injected at a distant site either simultaneously or months later. Therefore, enabling T cell activation at the local lymph node leads to a durable systemic immune response that is no longer affected by the secretion of exosomal PD-L1. The end result is extended survival of the afflicted mice.
A role for EVs, including exosomes, in tumor progression has been proposed in a number of settings (Ruivo et al., 2017). EVs can carry tumor antigens. When taken up by dendritic cells, these antigens can be presented, thereby inducing an immune response (Wolfers et al., 2001). As such, EVs were originally thought to have an anti-tumor effect. However, more recent studies have suggested a number of immunosuppressive effects. For example, EVs can inhibit dendritic cell maturation, natural killer (NK) cell function, and directly kill CD8 T cells (Abusamra et al., 2005; Liu et al., 2006; Yu et al., 2007). EVs also have been shown to promote directional migration of tumor cells (Sung et al., 2015), home tumor cells to lymph nodes (Hood et al., 2011), induce neovascularization and leakiness of tumor vessels (Peinado et al., 2012; Skog et al., 2008; Zhou et al., 2014), and, even, establish pre-metastatic niches (Fong et al., 2015; Hoshino et al., 2015). The mechanisms behind these various functions remain largely speculative.
Here, we present extensive evidence that EVs, specifically exosomes, can function to promote tumor progression by presenting PD-L1. In vitro, exosomes suppressed T cells in a PD-L1-dependent fashion. In vivo, the removal of tumor exosomes from TRAMP-C2 cells using two independent genetic mutations recapitulated the effects of deleting Pd-l1. These recapitulated effects included suppression of tumor growth, increased cellularity of the spleen, and the activation of a T cell response in lymph nodes with similar effects on the various activation, exhaustion, and proliferation markers. All these outcomes were reversed with the injection of in vitro collected exosomes carrying PD-L1. In the absence of PD-L1, the exogenously introduced exosomes had little effect. Therefore, at least in the TRAMP-C2 model, presentation of PD-L1 appears to be the major mechanism by which exosomes promote cancer progression.
This role for exosomal PD-L1 is not limited to the TRAMP-C2 model. Removal of exosomes in the colorectal MC38 model also suppressed tumor growth and extended survival. Once again, the effect was dependent on PD-L1 as the removal of exosomes had no additional effect in the Pd-l1 null background. Interestingly though, unlike the TRAMP-C2 model, the loss of exosomes alone did not have as much of an impact as Pd-l1 loss, suggesting a combined role of exosomal and cellular PD-L1 in the MC38 model. Remarkably, combining exosome loss with anti-PD-L1 treatment extended survival of these mice to a similar degree as removing PD-L1 altogether. These data show that in the MC38 model, both exosomal and cellular PD-L1 play an important role in promoting tumor progression in the latter, but not in the former, being sensitive to anti-PD-L1 therapy. Understanding what regulates the relative surface versus exosomal presentation of PD-L1 will be an important avenue of research going forward.
Several recent papers have identified exosomal PD-L1 in the blood of patients with a variety of cancers including head and neck cancer and melanoma (Chen et al., 2018; Theodoraki et al., 2018). In melanoma, it has been further shown that patients resistant anti-PD-1 therapy have elevated levels of exosomal PD-L1 in their blood prior to treatment (Chen et al., 2018). Therefore, our findings on the immunosuppressive and tumor-promoting roles of exosomal PD-L1 are likely to be relevant across many cancer types. However, the degree to which exosomal versus surface PD-L1 is driving immunosuppression will vary between patients and cancer types. Identifying patients who are more or less dependent on exosomal PD-L1 will be critical going forward in deciding those who are more likely to respond to therapy. Measuring levels in the blood as shown in these recent studies is likely to be one such strategy.
Going forward, targeting both cell-surface and exosome presentation of PD-L1 should be considered in any therapeutic strategy. The TRAMP-C2 model is resistant to current anti-PDL1 antibody blockade (Yu et al., 2012). However, the deletion of Pd-l1 in the tumor cells had a striking effect. Similarly, although the MC38 model shows partial responsiveness to anti-PD-L1 therapy (Deng et al., 2014), deletion of the Pd-l1 gene has a greater effect (Figure 5). These data are all consistent with exosomal PD-L1 being resistant to current anti-PD-L1 therapy. The reason exosomal PD-L1 is resistant is unclear. It is possible that how PD-L1 is presented on the exosome makes it less responsive to the current antibodies. Alternatively, exosomal PD-L1 may be produced at high enough levels that it can compete with the delivered antibody. It is also possible that exosomes can reach targets that are sequestered from the effects of the antibody. It will be important to tease apart the interactions of exosomal PD-L1 with current therapeutics.
Potentially the most promising therapeutic implication of our findings is that inhibition of exosome secretion at one tumor site can lead to a systemic and durable immune response against distant tumor sites or secondary tumor challenges. This observation is reminiscent of the abscopal effect, originally seen in patients treated with irradiation (Ngwa et al., 2018). In particular, irradiation of the primary tumor can lead to secondary regression of metastases. It is thought that this phenomenon is driven by activation of anti-tumor immune response and preclinical studies combining irradiation with immunosuppression are underway (Ngwa et al., 2018). It will be interesting to determine whether localized anti-exosomal therapy combined with systemic anti-PD-L1 blockade could synergize to induce a systemic immune response against multiple tumor sites simultaneously.
Here, we used genetics to dissect the role of exosomal PD-L1 in tumor progression by deleting two important exosomal biogenesis genes: Rab27a and nSMNase2. The deletion of Rab27a led to loss of all exosomes as measured by markers (CD63, HRS), particle tracking, and electron microscopy. The deletion of nSMNase2 led to a loss of a majority, but not all, exosomes, as measured by the same assays. Therefore, both enzymes represent potential therapeutic targets. Small molecule inhibitors toward NSMNASE2 and RAB27A already exist, GW4869 and Nexinhib-20, respectively (Johnson et al., 2016; Luberto et al., 2002). However, these small molecules are unable to inhibit exosome release in multiple cancer cell lines, including PC3, or show an in vivo effect in the MC38 model (Phuyal et al., 2014) (Figures S7D–S7H), even though their respective knockouts had a profound effect (this paper). Therefore, there is a need for more effective small molecules toward these targets before any hope of developing them into drugs. Based on our findings, such drugs could have the potential to act alone or in combination with current immune-checkpoint therapies, reaching a large population of cancer patients.
In summary, we have discovered that exosomal PD-L1 is a major regulator of tumor progression through its ability to suppress T cell activation in draining lymph nodes and that its inhibition can lead to a long-lasting, systemic anti-tumor immunity.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for all original resources and reagents presented in this manuscript should be directed and will be fulfilled by the Lead Contact, Robert Blelloch (robert.blelloch@UCSF.edu). MTAs may be required for some reagents per UCSF policy.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human tumor cell lines
PC3, DU145, LNCaP, SK-MEL-28 and 293T cells were obtained from ATCC, where they had been authenticated. PC3, DU145 and LNCaP are prostate cancer cell lines derived from male patients. SK-MEL-28 is a male melanoma cell line. PC3 were cultured in F-12K Medium (Kaighn’s Modification of Ham’s F-12 Medium) (GIBCO, ref 21127–022), 10% Fetal Bovine Serum (Corning, ref. 35-010-CV) and Penicillin/Streptomycin (Sigma, cat. P4333). DU145 were cultured in Eagle’s Minimum Essential Medium (EMEM) (Lonza, cat. 12–125Q), 2 mM L-glutamine (Sigma, cat. G7513), 10% Fetal Bovine Serum (Corning, ref. 35-010-CV) and Penicillin/Streptomycin (Sigma, cat. P4333). LNCaP and SK-MEL-28 were cultured in RPMI-1640 Medium (Corning, 15-040-CM), 10% Fetal Bovine Serum (Corning, ref. 35-010-CV), 2 mM L-glutamine (Sigma, cat. G7513), and Penicillin/Streptomycin (Sigma, cat. P4333). SK-MEL-28. Raji B cells and Jurkat T cells were cultured as previously reported (Hui et al., 2017). All cells were cultured at 37°C in a humidified atmosphere containing 5% CO2.
Mouse tumor cell lines
TRAMP-C2 cells were obtained from ATCC, where they had been authenticated, TRAMP-C2 cells were derived from a primary large T antigen expressing tumor that developed in a C57BL/6 male mouse (Foster et al., 1997). TRAMP-C2 cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (UCSF cell culture facility), 5% Nu-Serum IV (Fisher, cat. CB-55004, 500 ml, to be diluted to 5%), 5% Fetal Bovine Serum (Corning, ref. 35-010-CV), Bovine Insulin (Sigma, cat. I0516), DHEA (Sigma, D-063), and Penicillin/Streptomycin (Sigma, cat. P4333). MC38 cells were kindly provided by Jeffrey Schlom’s Lab and cultured in DME H-21 (Dulbecco’s Modified Eagle Medium) High Glucose (UCSF cell culture facility), 10% Fetal Bovine Serum (Corning, ref. 35-010-CV), 1 mM sodium pyruvate (UCSF cell culture facility). MC38 cells are a colon cancer cell line derived from a female C57BL/6. All cells were cultured at 37°C in a humidified atmosphere containing 5% CO2.
Mice
C57BL/6J (stock # 000664) mice were purchased from “The Jackson Laboratory” and/or bred in the UCSF animal house. Immunodeficient mice NOD-Prkdcem26Cd52Il2rgem26Cd22/NjuCrl (catalog # 24110022) were obtained from Charles River. Only male mice (approximately 8–10 weeks old unless otherwise specified in text) were used for all experiments. Mice were kept under the pre-approved guidelines of the Institutional Animal Care and Use Committee at UCSF. Mice were housed in pathogen free conditions. Mice dying for nonrelated cancer causes were excluded from the studies (i.e., fights or infections). Additional information (i.e., sample size, replicates) is described in the figure legends. Each experimental group was created with mice derived from different brood.
METHOD DETAILS
Cell lines treatments
To inhibit lysosome function, cells were cultured with 250 nM BafA1 (Cell Signaling, cat. 54645S) for six hours. To inhibit proteasome function, cells were cultured in 10 uM of MG132 (SIGMA, cat, M7449) for six hours.
To investigate PD-L1 glycosylation, PC3 cell lysate were treated with PNGase F (Sigma-Aldrich, cat. P7367). To investigate further glycosyl modification in the Golgi apparatus, PC3 cell lysate were treated with Endo H (NEB, cat. P0702S).
For IFN-γ treatments, 10ng/mL IFN-γ (PeproTech – human cells, cat. 300–02) (Abcam – mouse cells, cat. Ab9922) to media for 48 hours prior to exosome and cell collections.
For GW4869 treatments, 10μM GW4869 (Sigma-Aldrich, cat. D1692) to media for 48 hours prior media collections.
Exosome/Secreted Vesicle isolation
PC3 and SK-Mel-28 cells were plated at a density of 5 million per 15 cm plate (Corning 430599), cultured for 48 hours and media from six plates were pooled.
For TRAMP-C2 and MC38 secreted vesicle extractions, cells were plated at a density of 3 million per 15 cm plate for 48 hours and media from 6 plates were pooled.
For vesicle enrichment, media was centrifuged at 300 g for 10 minutes at room temperature, followed by 2k g for 20 min at 4°C then 12k g for 40 minutes at 4°C. Supernatant was then centrifuged twice at 100k g, for 1 hour then 10 minutes at 4°C. For exosome purification, the 100k g pellet was further spun on a sucrose gradient (20%–60% sucrose) for 16 hours at 4°C at 100k g. Fractions were collected and sucrose concentration measured using a refractometer.
It was noted that components of FBS lacked any reactivity to anti-human or anti-mouse PD-L1 and thus in experiments where only proteins were measured, vesicles were collected directly from culture media described above. In experiments where vesicles or exosomes would be counted or visualized, FBS was replaced by Knock-out Serum Replacement (KSR, GIBCO 10828028) to exclude particles present in the FBS.
CRISPR–Cas9-mediated gene disruption
sgRNA oligonucleotides (ELIMBIO), were cloned into pSpCas9(BB)-2A-GFP (PX458, ADDGENE) according to the published protocol (Ran et al., 2013). For each gene disrupted, two different guides were simultaneously transfected. 1ug of each vector was transfected using FUGENE HD (Promega, cat. E2311). Pd-l1 null, Rab27a null and nSMase2 null clones were obtained by GFP+ single cell cloning, 48 hours post transfection. Knockout clones were identified either by western (RAB27a) or by flow cytometry analysis for cell surface PD-L1.
| sgRNA oligonucleotides: | Sequences: |
|---|---|
| human Pd-l1 guide 1: | GGTTCCCAAGGACCTATATG |
| human Pd-l1 guide 2: | ACAGAGGGCCCGGCTGTTGA |
| human Rab27a guide 1: | CCAAAGCTAAAAACTTGATG |
| human RAB27a guide 2: | CAACAGTGGGCATTGATTTC |
| Human nSMase2 guide 1: | GAGAAACGCAAAGGGCAGCG |
| Human nSMase2 guide 2: | CGGTCCACCAGCCAGTAGCA |
| mouse Pd-l1 guide 1: | GTTTACTATCACGGCTCCAA |
| mouse Pd-l1 guide 2: | GGGGAGAGCCTCGCTGCCAA |
| mouse RAB27a guide 1: | CCAAGGCCAAGAACTTGATG |
| mouse RAB27a guide 2: | CACAGTGGGCATTGATTTCA |
| mouse nSMase2 guide 1: | CGTTAATGGCCGACTGGCTC |
| mouse nSMase2 guide 2: | AATGCCAAGTGGTTAAAGGA |
Electron Microscopy
Exosome sucrose float fractions where prepared as described above and resuspended in PBS. Sample aliquots of 4 μL were pipetted onto 200 mesh formvar/carbon grids (EMS) which had been glow-discharged for 15 s. Samples were incubated on grids for 30 s and subsequently negatively stained with a 2% uranyl acetate solution. Data were acquired using a Philips CM200F electron microscope operating at 200 keV equipped with an UltraScan 1000 CCD Camera (Gatan).
Nanoparticle Tracking Analysis
200K cells were seeded per well (6 wells plate, Corning, cat. 3516) in KSR media 24 hours before collection for PC3 DU145 SK-MEL-28. 100K cells cells were seeded in KSR media 24 hours before collection for TRAMP-C2 and MC38. Media was pre-processed at 300 g for 10 minutes at room temperature, followed by 2k g for 20 min at 4°C then 12k g for 40 minutes at 4°C. The processed media was analyzed on a NanoSight LM10.
Antibodies
Immunoblotting
Anti-human PD-L1 (E1L3N) (Cell Signaling technology, cat. 13684T), anti-mouse PD-L1 (mouse) (EPR20529) (Abcam, cat. AB213480), anti-human CD63 (MEM-259) (ABCAM, cat. ab8219) GAPDH (FL-335) (Santa Cruz Biotechnology, cat. Sc25778), anti-human HRS (c-7) (Santa Cruz biotechnology, cat. SC-271455). Anti-human/mouse RAB27A (D7Z9Q) (Cell Signaling Technology, cat. 69295S). Anti-human nSMase2 (Santa Cruz Biotechnology, sc-166637)
Flow Cytometry
Anti-human CD274 (B7-H1) PE (MIH1) (eBioscience, cat. 125983-42), PE mouse IgG2b, k Isotype control (MG2b-57) (Biolegend, cat. 401209).
Immunofluorescence
Primary antibody: Anti-human PD-L1 (E1L3N) (cell signaling technology, cat. 13684T). Secondary Antibody: Anti-Rabbit Alexa Fluor 680 (Thermo Fisher Scientific, cat. A10043).
Western Blot Analysis
Samples were lysed in RIPA buffer (Thermo Scientific, cat. 89900) with the addition of PhosSTOP (Sigma-Aldrich, 4906837001) and Complete Mini proteasome inhibitors (Sigma-Aldrich, 05892791001). Western Blot was performed as described in the commercially available antibody protocols listed above. Western blot band intensity quantification was calculated using ImageJ.
Raji - Jurkat T cell stimulation assay
Raji B cells were pre-loaded by adding 30 ng/ml SEE superantigen in the media and incubating for 45 min at 37°C. Raji B cells were then washed and irradiated 10,000 rads prior to coculture. In a 96 wells plate (flat bottom) 17,000 Raji B cells were co-cultured with 70,000 Jurkat T cells. Vesicles were derived from 32 pates (15 cm plate, each plate seeded with 5 millions of cells 48 hours prior media collection) and added to the co-cultured cells as illustrated in Figure 2H. Human IL-2 was quantified from the media by ELISA (R&D Systems,#DY202–05).
Immunofluorescence
Cells were cultured in 8-wells chambered coverglass (Nunc, cat. 154534). Cells were fixed with 4% PFA in PBS (Fisher Scientific cat. NC9245948) and incubated for 10 minutes at room temperature. After, PBS (Sigma, cat. RNBG9030) + 0.2% of Triton-X (VWR, cat. VW8609–0) was added and the samples permeabilized in solution for 10 minutes at room temperature. Following permeabilization, samples were blocked in 5% Goat Serum (Invitrogen, cat. 16210064) PBS for 30 minutes at room temperature, and incubated at 4C overnight with primary antibodies (1:20). The following day, secondary was added (1:500) to the samples after washing, then incubated at room temperature for 3 hours. Prior imaging, sample were mounted with VECTASHIELD Antifade Mounting Medium with DAPI (Fisher Scientific, cat. NC9524612). Samples were imaged using NIKON spinning disk confocal.
Polysome Fractionation and RNA isolation
PC3 and SK-Mel-28 cells were pre-treated for 10 min with 100 μg/ml cycloheximide (Sigma) added in media for 10 min. Cells were washed with cold PBS (Sigma, cat. RNBG9030) with 100 μg/ml cycloheximide (Sigma, cat. C4859) and scraped. Cell pellets were lysed in 10mM Tris-HCl pH8 (Fisher Scientific, cat. BP1531), 140mM NaCl (VWR, cat. JT4058–7), 1.5mM MgCl2 (Thermo Fisher Scientific, cat. R0971), 0.25% NP-40 (Fisher Scientific, cat. PI85124), 0.1% Triton X-100 (VWR, cat. VW8609–0), 50mM DTT (Thermo Fisher Scientific, cat. R0861), 150 μg/ml cyclohexamide, (Sigma, cat. C4859) 640U/ml RNasin (Life Technology, cat. AM2696). After 30 min on ice, supernatant was separated by centrifugation at 9300 g and the supernatant transferred to a 10%–50% sucrose gradient. Samples were centrifuged at 35k g for 3 hours at 4C. After centrifugation the sample was fractionated (BIOCOMP). Samples were DNase treated.
Reverse transcriptase-quantitative PCR
DU145, PC3, LNCaP and SK-Mel-28 RNA was isolated with QIAZOL Reagent (QIAGEN, cat. 79306), and purified with Pure Link RNA mini kit (QIAGEN, 12183018A). RNA was eluted in H2O and reverse-transcribed to cDNA following the kit protocol (Thermo Scientific, cat. FERK1672). Eluted (1:50) cDNA samples were run on 7900HT fast real-time PCR system using SYBR green (Roche, cat. 04913914001).
Pd-l1 qPCR forward primer
GGCATTTGCTGAACGCAT, Pd-l1 qPCR reverse primer: CAATTAGTGCAGCCAGGT (Casey et al., 2016).
Rpl7 qPCR forward primer
GAACCATGGAGGGTGTAGAAGAG, Rpl7 qPCR reverse primer: GGCAAACTTCTTTCTCAGGCG.
Cell Fractionation
Five million PC3 cells were collected (trypsin) from a 15 cm plate. Cells were washed with 10 mL of media. Cells were then re-suspended in 5 mL of lysosome lysis buffer (1M Sucrose, 1 M Tricine pH 7.2, 0.5M EDTA pH 8, and water). Cells were spun at 900 g for 5 min at 4C. Cells were suspended up to 1.5 mL or working buffer (lysis buffer plus Protease Inhibitor, phosTOP). 1.4 ml of cell suspension were passed through cell homogenizer (Isobiotec) using a 12um ball bearing (#7,9880) for 15 times. Cell lysate were spun at 700 g for 10 minutes and the supernatant collected. Pellet was re-suspended with working buffer equivalent to the previous collected supernatant and re-spun at 700 g. Pool of supernatants was spun at 3,000 g for 15 min. Pellet was collected for western blot. The supernatant from 3kg was further spun at 25,000 g for 25 minutes in a TLA 100.3 tube (Beckman Coulter). The pellet was processed into a sucrose gradient and the supernatant was further spun at 100,000 g for 30 minutes. The pellet and the supernatant were collected for western. The pellet from the 25k g spin was spun in a sucrose gradient made of layers of 1.25M, 1.10M, and 0.25M sucrose in a TLS-55 tube (Beckman Coulter) at 120000 g for 1.5 hours. The interphase between 1.25M and 1.1M (Sucrose Light) and the pellet were collected. Both collections were run in the Western Blot.
Biotin-Streptavidin Assay
Cell were biotin-labeled (Sigma Aldrich, cat. B4639). Cells were washed and incubated for 24 hours in fresh media. Exosomes were then isolated from cell-conditioned media, lysed, and then biotin-labeled proteins were pulled down by streptavidin-conjugated beads (Thermo Scientific, cat. 65001).
Tumor cells injections
Mice were injected (S.C.) with a million TRAMP-c2 WT or TRAMP-c2 Pd-l1 null or TRAMP-c2 Rab27a null or TRAMP-c2 nSMase2 null cells. Mice were injected (S.C. with a million MC38 WT or MC38 Pd-l1 null or MC38 Rab27a null cells. Mice were considered “end stage” when the tumor reached 2 cm in at least one dimension. Tumor growth was monitored three times a week by measuring tumor length and width. Tumor volume was calculated according to the following equation: length × width × 0.5 × width (Faustino-Rocha et al., 2013).
Mice Treatments
Where indicated, mice were treated with 200 mg antibody I.P. at days 7, 10, and 13 post tumor injection: anti–PD-L1 (10F.9G2) (BioXCell, cat. BE0101), isotype control (10F.9G2) (BioXCell, cat. LTF-2). Were indicated, mice were treated with 2.5 mg/kg GW4869 (Sigma-Aldrich, cat. D1692) from day 0 to day 14 once a day. Where indicated mice were treated with exosomes injected IV in the tail vein. 15 million cells were seeded in five 15 cm dishes (Corning CLS430599), and cultured for 48 hours. Vesicles were isolated as a 100k g pellet as described above. MC38 vesicles were resuspended in 1 mL PBS and TRAMP-C2 vesicles in 600 μl. Each mouse was injected with 100 μl of PBS containing vesicles.
Immune-Profiling
Mice were implanted subcutaneously (S.C.) with a million of TRAMP-C2 wt, Pd-l1 null, or Rab27a null cells. Alternately, mice were injected with TRAMP-C2 Rab27a and treated with exosomes derived from TRAMP-C2 WT or TRAMP-C2 Pd-l1 cells. Mice were euthanized and spleens and lymph nodes collected 14 days after tumor cell injection.
Spleens were surgically removed with sterilized surgical equipment, weighed, and crushed with the blunt end of a 10 mL syringe on Petri dishes containing 5 mL of PBS. The spleen mixtures were separately filtered through a 70 mM filter into a 50 mL conical tube, centrifuged at 1500 rpm for 5 minutes at 4°C. After wash, cell pellets were resuspended in 5mL of red blood cell lysis solution (Santa Cruz Biotechnology; Cat sc-296258) on ice for 5 minutes and stopped with the addition of 30mL of PBS. After wash, cells were reconstituted for counting by Vi-Cell (Beckman Coulter, U.S.A.). Draining lymph nodes (DLNs) were extracted with sterilized surgical equipment and crushed between the frosted surfaces of super-frosted microscope slides into wells containing PBS. Cell mixtures were then filtered through a 70 mm filter into 15 mL conical tubes. Cells were then washed and counted.
Single cell suspensions (1 million cells) were first incubated with Fc Block (Tombo Biosciences, cat. 70–0161) for 10 minutes, then co-incubated with antibodies for 20 minutes at 4°C followed by washing with staining buffer (PBS + 1% FBS). FoxP3 and intracellular staining were performed using an eBioscience intracellular kit (Cat#00-5523-00) according to the manufacturer’s protocol. Flow cytometry was performed on Fortessa X20 Dual, and data analyzed by FlowJo software (TreeStar). Details on flow cytometry antibodies used in this study can be found in Table S1.
Haematoxylin and eosin staining
Mice were sacrificed and the subcutaneous tumors were dissected. Tumors were fixed in 10% formalin (Fisher, cat. SF98–4) at room temperature overnight and consequentially dehydrated in 70% ethanol. Samples were mounted in paraffin blocks and sectioned at 5 μm of thickness. Haematoxylin and eosin staining was performed in Lecia Autostainer XL. Briefly, slides were baked at 60°C for 15 minutes, then treated with Xylene Substitute (Citrisolv) (Fisher chemical cat. x3p-1gal) then rehydrated and stained with Hematoxylin, followed with washes with running water. An incubation with Acid Alcohol (Sigma, cat. A3429) was performed, followed with washes with running water. The samples were then stained with eosin (Thermo Scientific) then dehydrated with ethanol followed by Xylene Substitute (Citrisolv). Samples were mounted with Permount (Fisher Scientific, cat. SP15–500). Slides were submitted for pathologic evaluation. Pathologist was blinded to sample origin (i.e., experimental versus control)
QUANTIFICATION AND STATISTICAL ANALYSIS
Differences between groups were calculated using Student t test. Logrank test was performed for survival analysis. 2-way ANOVA was performed for tumor growth analysis. Further statistical methods are detailed in the figure legends. Data was processed with GraphPad Prism, Version 7 (GraphPad Software). In all cases a P value of 0.05 and below was considered significant: p < 0.05 (*), p < 0.01(**) and p < 0.001 (***). Details regarding number of replicates and the definition of center/error bars can be found in legends.
DATA AVAILABILITY
Mendeley dataset: https://doi.org/10.17632/4d8h97z4c8.1
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Immunoblotting: anti-human PD-L1 (E1L3N) | Cell Signaling technology | cat. 13684; RRID:AB_2687655 |
| Immunoblotting: anti-mouse PD-L1 (mouse) (EPR20529) | Abcam | cat. AB213480 |
| Immunoblotting: anti-human CD63 (MEM-259) | Abcam | cat. AB8219; RRID:AB_306364 |
| Immunoblotting: anti-human/mouse GAPDH (FL-335) | Santa Cruz Biotechnology | cat. SC-25778; RRID:AB_10167668 |
| Immunoblotting: anti-human HRS (c-7) | Santa Cruz biotechnology | cat. SC-271455; RRID:AB_10648901 |
| Immunoblotting: Anti-human/mouse RAB27A (D7Z9Q) | Cell Signaling Technology | cat. 69295 |
| Immunoblotting: Anti-human nSMase2 (G-6) | Santa Cruz Biotechnology | cat. SC-166637; RRID:AB_2270817 |
| Flow Cytometry: Anti-human CD274 (B7-H1) PE (MIH1) | eBioscience | cat. 255983–42; RRID:AB_1907368 |
| Flow Cytometry: PE mouse IgG2b, k Isotype control (MG2b-57) | Biolegend | cat. 401209 |
| immunofluorescence: Primary antibody: Anti-human PD-L1 (E1L3N) | Cell Signaling Technology | cat. 13684; RRID:AB_2687655 |
| immunofluorescence: Secondary Antibody: Anti-Rabbit Alexa Fluor 680 | Thermo Fisher Scientific | cat. A10043; RRID:AB_2534018 |
| Antibodies for Immunophenotyping see Table S1 | This paper | N/A |
| anti-PD-L1 (10F.9G2) | BioXCell | cat. BE0101; RRID:AB_10949073 |
| isotype control 971 (10F.9G2) | BioXCell | cat. LTF-2; RRID:AB_1107780 |
| ELISA: IL-2 | R&D System | cat. DY202–05 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| BafA1 | Cell Signaling | cat. 54645S |
| MG132 | SIGMA | cat. M7449 |
| PNGase F | Sigma-Aldrich | cat. P7367 |
| Endo H | NEB | cat. P0702S |
| Human IFN-γ | PeproT ech | cat. 300–02 |
| Mouse IFN-γ | Abcam | cat. AB9922 |
| GW4869 | Sigma-Aldrich | cat. D1692 |
| RIPA buffer | Thermo Scientific | cat. 89900 |
| PhosSTOP | Sigma-Aldrich | cat. 4906837001 |
| Complete Mini proteasome inhibitors | Sigma-Aldrich | cat. 05892791001 |
| Triton-X | VWR | cat. VW8609–0 |
| VECTASHIELD Antifade Mounting Medium with DAPI | Fisher Scientific | cat. NC9524612 |
| cycloheximide | Sigma | cat. C4859 |
| Tris-HCl pH8 | Fisher Scientific | cat. BP1531 |
| NaCl | VWR | cat. JT4058–7 |
| MgCl2 | Thermo Fisher Scientific | cat. R0971 |
| NP-40 | Fisher Scientific | cat.PI85124 |
| DTT | Thermo Fisher Scientific | cat. R0861 |
| RNasin | Life Technology | cat. AM2696 |
| SYBR green | Roche | cat. 04913914001 |
| Biotin | Sigma Aldrich | cat. B4639 |
| streptavidin-conjugated beads | Thermo Scientific | cat. 65001 |
| Formalin | Fisher | cat. SF98–4 |
| Xylene Substitute (Citrisolv) | Fisher chemical | cat. x3p-1gal |
| Acid Alcohol | Sigma | cat. A3429 |
| Permount | Fisher Scientific | cat. SP15–500 |
| Critical Commercial Assays | ||
| FUGENE HD | Promega | cat. E2311 |
| QIAZOL Reagent | QIAGEN | cat. 79306 |
| Pure Link RNA mini kit | QIAGEN | cat. 12183018A |
| Thermo Scientific Maxima First Strand cDNA Synthesis Kit for RT-qPCR | Thermo Scientific | cat. FERK1672 |
| Red blood cell lysis solution | Santa Cruz Biotechnology | cat. SC-296258 |
| Zombie UV Fixable Viability Kit | Biolegend | cat. 423107 |
| Intracellular kit | eBioscience | cat. 00-5523-100100 |
| Deposited Data | ||
| Uncropped Western Blots | Mendeley dataset | https://doi.org/10.17632/4d8h97z4c8.1 |
| Experimental Models: Cell Lines | ||
| PC3 | ATCC | CRL-1435; RRID:CVCL_0035 |
| DU145 | ATCC | HTB-81; RRID:CVCL_0105 |
| LnCap | ATCC | CRL-1740; RRID:CVCL_1379 |
| SK-MEL-28 | Robert Judson Lab (UCSF) | RRID:CVCL_0526 |
| 293 T | ATCC | CRL-3216; RRID:CVCL_0063 |
| RAJI B CELLS | Ron Vale Lab (UCSF) | RRID:CVCL_0511 |
| JURKAT T-CELLS Overexpressing PD-1 | Ron Vale Lab (UCSF) | https://doi.org/10.1126/science.aaf1292 |
| RAJI B CELLS Overexpressing PD-L1 | Ron Vale Lab (UCSF) | https://doi.org/10.1126/science.aaf1292 |
| TRAMP-C2 | ATCC | CRL-2731; RRID:CVCL_3615 |
| MC38 | Jeffrey Schlom lab (NCI) | RRID:CVCL_B288 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J | The Jackson Laboratory | RRID:IMSR_JAX:000664 |
| NCG (NOD-Prkdcem26Cd52Il2rgem26Cd22/NjuCrl) | Charles River Laboratories | RRID:IMSR_CRL:572 |
| Oligonucleotides | ||
| Sequences for sgRNAs, see table in STAR Methods | This paper | N/A |
| Pd-l1 qPCR forward primer: GGCATTTGCTGAACGCAT | Casey et al., 2016 | https://doi.org/10.1126/science.aac9935 |
| Pd-l1 qPCR reverse primer: CAATTAGTGCAGCCAGGT | Casey et al., 2016 | https://doi.org/10.1126/science.aac9935 |
| Rpl7 qPCR forward primer: GAACCATGGAGGGTGTAGAAGAG | This paper | N/A |
| Rpl7 qPCR reverse primer: GGCAAACTTC lllCTCAGGCG | This paper | N/A |
| Recombinant DNA | ||
| pSpCas9(BB)-2A-GFP | ADDGENE | PX458 |
| pMH243-CD63-GFP | Hebrok Lab (UCSF) | N/A |
| Software | ||
| FlowJo | 10.5.2 | https://www.flowjo.com/ |
| Prism | 7.0b | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ | 1.49V | https://imagej.nih.gov/ij |
Highlights.
Genetic blockade of exosomal PD-L1 extends survival by promoting anti-tumor immunity
Exosomal PD-L1 suppresses T cell activity in the draining lymph node
Exosome-deficient tumor cells induce systemic anti-tumor immunity and memory
Exosomal PD-L1 appears to be resistant to anti-PD-L1 antibody blockade
ACKNOWLEDGMENTS
This publication is part of the NIH Extracellular RNA Communication Consortium paper package and was supported by the NIH Common Fund’s exRNA Communication Program. We thank Mark Ansel, Andrei Goga, Bo Huang, Noelle L’Etoile, and Robert Raffai for helpful discussions throughout development of this project. We thank Jason Cyster for helpful feedback on the manuscript. We thank Adam Olshen for helpful advice on the statistical tests. We thank Atsushi Urano and Matthias Hebrok for the CD63-GFP construct. This work was funded by the NIH Common Fund Extracellular RNA Consortium (U19 CA179512), the George and Judy Marcus Innovation Fund, and NIH training grant (F31CA200163) (to T.H.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2019.02.016.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abusamra AJ, Zhong Z, Zheng X, Li M, Ichim TE, Chin JL, and Min WP (2005). Tumor exosomes expressing Fas ligand mediate CD8+ T-cell apoptosis. Blood Cells Mol. Dis 35, 169–173. [DOI] [PubMed] [Google Scholar]
- Burr ML, Sparbier CE, Chan YC, Williamson JC, Woods K, Beavis PA, Lam EYN, Henderson MA, Bell CC, Stolzenburg S, et al. (2017). CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature 549, 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr B, and Warren J (2012). Company profile: NanoSight: delivering practical solutions for biological nanotechnology. Nanomedicine (Lond.) 7, 1129–1132. [DOI] [PubMed] [Google Scholar]
- Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, Gouw AM, Baylot V, Gütgemann I, Eilers M, and Felsher DW (2016). MYC regulates the antitumor immune response through CD47 and PD-L1. Science 352, 227–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chassé H, Boulben S, Costache V, Cormier P, and Morales J (2017). Analysis of translation using polysome profiling. Nucleic Acids Res. 45, e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DS, and Mellman I (2013). Oncology meets immunology: the cancer-immunity cycle. Immunity 39, 1–10. [DOI] [PubMed] [Google Scholar]
- Chen DS, and Mellman I (2017). Elements of cancer immunity and the cancer-immune set point. Nature 541, 321–330. [DOI] [PubMed] [Google Scholar]
- Chen G, Huang AC, Zhang W, Zhang G, Wu M, Xu W, Yu Z, Yang J, Wang B, Sun H, et al. (2018). Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 560, 382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A (2005). Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat. Rev. Mol. Cell Biol. 6, 79–87. [DOI] [PubMed] [Google Scholar]
- Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RR, and Fu YX (2014). Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J. Clin. Invest 124, 687–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faustino-Rocha A, Oliveira PA, Pinho-Oliveira J, Teixeira-Guedes C, Soares-Maia R, da Costa RG, Colaço B, Pires MJ, Colaço J, Ferreira R, and Ginja M (2013). Estimation of rat mammary tumor volume using caliper and ultrasonography measurements. Lab Anim. (NY) 42, 217–224. [DOI] [PubMed] [Google Scholar]
- Fong MY, Zhou W, Liu L, Alontaga AY, Chandra M, Ashby J, Chow A, O’Connor ST, Li S, Chin AR, et al. (2015). Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat. Cell Biol. 17, 183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster BA, Gingrich JR, Kwon ED, Madias C, and Greenberg NM (1997). Characterization of prostatic epithelial cell lines derived from transgenic adenocarcinoma of the mouse prostate (TRAMP) model. Cancer Res. 57, 3325–3330. [PubMed] [Google Scholar]
- Francisco LM, Sage PT, and Sharpe AH (2010). The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev 236, 219–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, Zaretsky JM, Sun L, Hugo W, Wang X, et al. (2017). Interferon receptor signaling pathways regulating PD-L1 and PD-L2 Expression. Cell Rep. 19, 1189–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami S, Aparicio A, and Subudhi SK (2016). Immune checkpoint therapies in prostate cancer. Cancer J. 22, 117–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood JL, San RS, and Wickline SA (2011). Exosomes released by melanoma cells prepare sentinel lymph nodes for tumor metastasis. Cancer Res. 71, 3792–3801. [DOI] [PubMed] [Google Scholar]
- Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic Mark M, Molina H, Kohsaka S, Di Giannatale A, Ceder S, et al. (2015). Tumour exosome integrins determine organotropic metastasis. Nature 527, 329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, Sasmal DK, Huang J, Kim JM, Mellman I, and Vale RD (2017). T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 355, 1428–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JL, Ramadass M, He J, Brown SJ, Zhang J, Abgaryan L, Biris N, Gavathiotis E, Rosen H, and Catz SD (2016). Identification of neutrophil exocytosis inhibitors (Nexinhibs), small molecule inhibitors of neutrophil exocytosis and inflammation: druggability of the small GTPase Rab27a. J. Biol. Chem 291, 25965–25982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosaka N, Iguchi H, Yoshioka Y, Takeshita F, Matsuki Y, and Ochiya T (2010). Secretory mechanisms and intercellular transfer of microRNAs in living cells. J. Biol. Chem 285, 17442–17452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DH, and Goldberg AL (1998). Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol. 8, 397–403. [DOI] [PubMed] [Google Scholar]
- Li CW, Lim SO, Xia W, Lee HH, Chan LC, Kuo CW, Khoo KH, Chang SS, Cha JH, Kim T, et al. (2016). Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat. Commun 7, 12632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Yu S, Zinn K, Wang J, Zhang L, Jia Y, Kappes JC, Barnes S, Kimberly RP, Grizzle WE, and Zhang HG (2006). Murine mammary carcinoma exosomes promote tumor growth by suppression of NK cell function.J. Immunol 176, 1375–1385. [DOI] [PubMed] [Google Scholar]
- Luberto C, Hassler DF, Signorelli P, Okamoto Y, Sawai H, Boros E, Hazen-Martin DJ, Obeid LM, Hannun YA, and Smith GK (2002). Inhibition of tumor necrosis factor-induced cell death in MCF7 by a novel inhibitor of neutral sphingomyelinase. J. Biol. Chem 277, 41128–41139. [DOI] [PubMed] [Google Scholar]
- Martin AM, Nirschl TR, Nirschl CJ, Francica BJ, Kochel CM, van Bokhoven A, Meeker AK, Lucia MS, Anders RA, DeMarzo AM, and Drake CG (2015). Paucity of PD-L1 expression in prostate cancer: innate and adaptive immune resistance. Prostate Cancer Prostatic Dis. 18, 325–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauvezin C, and Neufeld TP (2015). Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy 11, 1437–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellman I, Coukos G, and Dranoff G (2011). Cancer immunotherapy comes of age. Nature 480, 480–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezzadra R, Sun C, Jae LT, Gomez-Eerland R, de Vries E, Wu W, Logtenberg MEW, Slagter M, Rozeman EA, Hofland I, et al. (2017). Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature 549, 106–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngwa W, Irabor OC, Schoenfeld JD, Hesser J, Demaria S, and Formenti SC (2018). Using immunotherapy to boost the abscopal effect. Nat. Rev. Cancer 18, 313–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrowski M, Carmo NB, Krumeich S, Fanget I, Raposo G, Savina A, Moita CF, Schauer K, Hume AN, Freitas RP, et al. (2010). Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat Cell Biol 12, 19–30, sup pp 11–13. [DOI] [PubMed] [Google Scholar]
- Page DB, Postow MA, Callahan MK, Allison JP, and Wolchok JD (2014). Immune modulation in cancer with antibodies. Annu. Rev. Med 65, 185–202. [DOI] [PubMed] [Google Scholar]
- Peinado H, Alečković M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, Hergueta-Redondo M, Williams C, García-Santos G, Ghajar C, et al. (2012). Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med 18, 883–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phuyal S, Hessvik NP, Skotland T, Sandvig K, and Llorente A (2014). Regulation of exosome release by glycosphingolipids and flotillins. FEBS J. 281, 2214–2227. [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricklefs FL, Alayo Q, Krenzlin H, Mahmoud AB, Speranza MC, Nakashima H, Hayes JL, Lee K, Balaj L, Passaro C, et al. (2018). Immune evasion mediated by PD-L1 on glioblastoma-derived extracellular vesicles. Sci Adv 4, eaar2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruivo CF, Adem B, Silva M, and Melo SA (2017). The biology of cancer exosomes: insights and new perspectives. Cancer Res. 77, 6480–6488. [DOI] [PubMed] [Google Scholar]
- Sharma P, Hu-Lieskovan S, Wargo JA, and Ribas A (2017). Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168, 707–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shurtleff MJ, Temoche-Diaz MM, Karfilis KV, Ri S, and Schekman R (2016). Y-box protein 1 is required to sort microRNAs into exosomes in cells and in a cell-free reaction. eLife 5, e19276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skog J, Würdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, Curry WT Jr., Carter BS, Krichevsky AM, and Breakefield XO (2008). Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 10, 1470–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley P (2011). Golgi glycosylation. Cold Spring Harb. Perspect. Biol 3, a005199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung BH, Ketova T, Hoshino D, Zijlstra A, and Weaver AM (2015). Directional cell movement through tissues is controlled by exosome secretion. Nat. Commun 6, 7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodoraki MN, Yerneni SS, Hoffmann TK, Gooding WE, and White-side TL (2018). Clinical significance of PD-L1+ exosomes in plasma of head and neck cancer patients. Clin. Cancer Res. 24, 896–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkach M, Kowal J, and Théry C (2018). Why the need and how to approach the functional diversity of extracellular vesicles. Philos. Trans. R. Soc. Lond. B Biol. Sci 373, 20160479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian SL, Drake CG, and Pardoll DM (2015). Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 27, 450–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Niel G, D’Angelo G, and Raposo G (2018). Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 19, 213–228. [DOI] [PubMed] [Google Scholar]
- Wolfers J, Lozier A, Raposo G, Regnault A, Théry C, Masurier C, Flament C, Pouzieux S, Faure F, Tursz T, et al. (2001). Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat. Med 7, 297–303. [DOI] [PubMed] [Google Scholar]
- Xu R, Greening DW, Zhu HJ, Takahashi N, and Simpson RJ (2016). Extracellular vesicle isolation and characterization: toward clinical application.J. Clin. Invest 126, 1152–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Li CW, Chan LC, Wei Y, Hsu JM, Xia W, Cha JH, Hou J, Hsu JL, Sun L, and Hung MC (2018). Exosomal PD-L1 harbors active defense function to suppress T cell killing of breast cancer cells and promote tumor growth. Cell Res. 28, 862–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, and Saito T (2012). Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J. Exp. Med 209, 1201–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Liu C, Su K, Wang J, Liu Y, Zhang L, Li C, Cong Y, Kimberly R, Grizzle WE, et al. (2007). Tumor exosomes inhibit differentiation of bone marrow dendritic cells. J. Immunol 178, 6867–6875. [DOI] [PubMed] [Google Scholar]
- Yu P, Steel JC, Zhang M, Morris JC, Waitz R, Fasso M, Allison JP, and Waldmann TA (2012). Simultaneous inhibition of two regulatory T-cell subsets enhanced interleukin-15 efficacy in a prostate tumor model. Proc. Natl. Acad. Sci. USA 109, 6187–6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang X, Thompson RH, Al-Ahmadie HA, Serio AM, Reuter VE, East-ham JA, Scardino PT, Sharma P, and Allison JP (2007). B7-H3 and B7x are highly expressed in human prostate cancer and associated with disease spread and poor outcome. Proc. Natl. Acad. Sci. USA 104, 19458–19463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Fong MY, Min Y, Somlo G, Liu L, Palomares MR, Yu Y, Chow A, O’Connor ST, Chin AR, et al. (2014). Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell 25, 501–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Mendeley dataset: https://doi.org/10.17632/4d8h97z4c8.1