

Abstract
NsaS is one of four intramembrane histidine kinases (HKs) in Staphylococcus aureus that mediate the pathogen’s response to membrane active antimicrobials and human innate immunity. We describe the first integrative structural study of NsaS using a combination of solution state NMR spectroscopy, chemical-cross-linking, molecular modeling and dynamics. Three key structural features emerge: First, NsaS has a short N-terminal amphiphilic helix that anchors its transmembrane (TM) bundle into the inner leaflet of the membrane such that it might sense neighboring proteins or membrane deformations. Second, the transmembrane domain of NsaS is a 4-helix bundle with significant dynamics and structural deformations at the membrane interface. Third, the intracellular linker connecting the TM domain to the cytoplasmic catalytic domains of NsaS is a marginally stable helical dimer, with one state likely to be a coiled-coil. Data from chemical shifts, heteronuclear NOE, H/D exchange measurements and molecular modeling suggest that this linker might adopt different conformations during antibiotic induced signaling.
Graphical Abstract
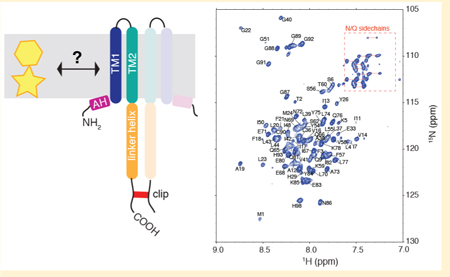
1. INTRODUCTION
Histidine kinases (HKs) are highly conserved intramembrane signaling proteins that help bacteria sense and adapt to a variety of environmental stimuli. In the context of antibiotic resistant pathogenic bacteria, these proteins play two major roles: (1) they orchestrate a large part of the transcriptional response to antibiotics, which can involve overexpression of proteases, transporters and biosynthetic enzymes to degrade and discard the antibiotic.1–3 (2) In commensal species like Staphylococcus aureus, HKs help the bacteria sense mammalian antimicrobial peptides so as to recognize and adapt to the innate immune response in different host environments. The mechanisms by which HKs sense and detect antibiotics and antimicrobials remain debated and there is significant complexity associated with physically defining the activating stimulus. In many cases, antibiotic sensing does not proceed via a simple ligand-binding event, and more complex phenomena such as membrane deformation, unfolded protein response and protein—protein interactions are invoked.4–7 Understanding how HKs have evolved to sense such complex stimuli requires characterizing the structures of HKs within the membrane and correlating structural changes to biological stimuli. This remains an outstanding challenge.
There are three major types of HKs, classified by the site of stimulus perception and protein domain architecture (Figure 1A): Extracytoplasmic-sensing HKs such as the nitrate sensor NarX8 and the carboxylate sensor DcuS9 have a extracellular ligand-binding domains. The transmembrane domain in these proteins acts as a signal transducer.10 Cytoplasmic HKs, such as NtrB,11 can be either soluble or membrane anchored; the sensor domains of these proteins bind to intracellular ligands and metabolites. In both these families, signaling is triggered by a discrete ligand-binding event. In contrast, intramembrane- sensing HKs such as the temperature sensitive DesK,12 and the autoinducer peptide sensitive AgrC,13 have transmembrane sensors that are made up of two to six TM helices connected by short extracellular loops. A large number of antibiotic and antimicrobial sensing HKs are intramembrane HKs. Sensory perception in this family is not well understood. It is expected to occur within the membrane either via direct interaction between the sensor and lipids, or indirectly via protein—protein interactions between the HK and other proteins that mediate the environmental response.14
Figure 1.
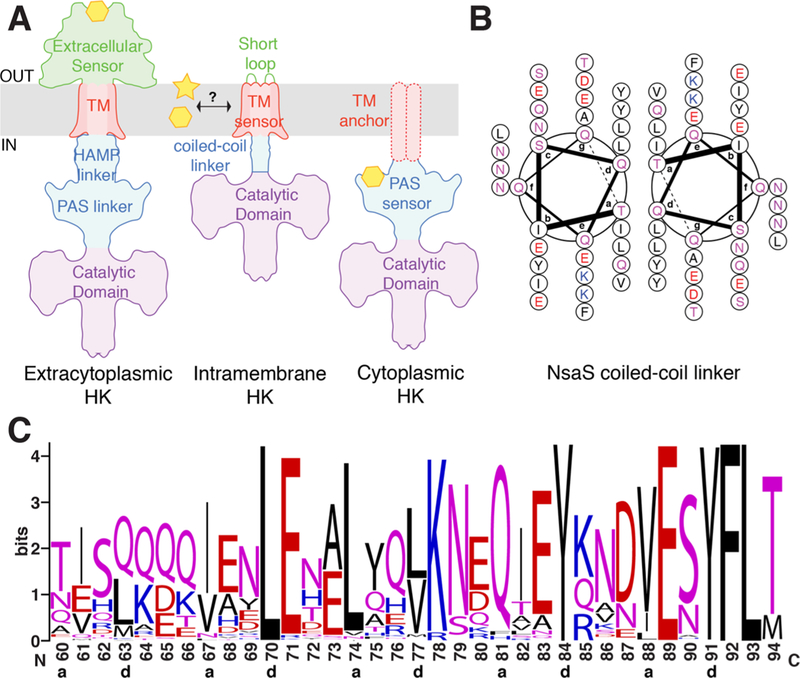
(a) Protein architecture of different classes of HKs. NsaS is an intramembrane HK and one of the smallest and simplest in the family. (b) Helical projection wheel for the linker domain of NsaS show that hydrophobic residues (black) cluster at the dimer interface with a heptad repeat. Coiled-coil predictors suggest that the linker domain of NsaS is a coiled-coil. (c) Sequence logo for the linker region of NsaS shows highly conserved heptad repeat sequence in the linker as the region. The logo contains data from 419 sequences obtained by Blasting the NsaS protein sequence from S. aureus Newman, against all annotated proteins from all Staphylocci, and using hits with >40% sequence identity.
NsaS (sometimes also called BraS4 and BceS) is one of four intramembrane HKs in the human commensal pathogen S. aureus, all of which have two transmembrane helices with a short extracellular loop. Previously, transcriptional profiling showed that the NsaS regulon is highly upregulated in the first few minutes after S. aureus is treated with sublethal doses of brilacidin, daptomycin (Cubicin) and the human antimicrobial peptide LL37.15,16 Genes controlled by NsaS include a variety of cell-wall biosynthesis and lipid modifying enzymes, proteases and notably several membrane transporters.15,17 These proteins presumably help S. aureus to sense the in vivo environment and to tolerate the effects of the antibiotics. Other studies have shown the NsaS is stimulated by the antibiotics nisin and bacitracin. Given the chemical diversity of the antibiotic agents sensed by NsaS—brilacidin is a ~900 Da rigid, fluorinated small-molecule; daptomycin is a ~1600 Da cyclic lipopeptide; nisin is a ~3300 Da lantibiotic with several non-natural amino acids and thioether linkages, and the human AMP LL-37 is a 4.5 kDa amphiphilic helix—we sought to characterize the structure of the transmembrane domain of NsaS, in an effort to understand how its structure might lead to this broad sensitivity to various antibiotics.
NsaS is also represents a relatively small class of HK with a particularly simple protein architecture. Though most HKs have a complex multidomain structure, the TM domain of NsaS is connected to its catalytic domain via a short 23 residue linker. We became especially interested in NsaS because bioinformatics analysis suggests that the linker has a very high propensity to be a left-handed coiled-coil (Figure 1B). Indeed, homologues of NsaS have different linker-lengths, but the coiled-coil character is always conserved (Figure 1C). There is much literature18–21 implicating coiled-coil structural transitions in HK signaling, so we sought to investigate the biophysical behavior of the NsaS coiled-coil.
Membrane domains of HKs remain under-studied. Structural studies of HKs have overwhelmingly focused on the cytoplasmic and catalytic domains of the protein. Indeed, there are over 25 structures of the DHp and catalytic domains and many more of the adaptor HAMP and PAS domains crystallized in various different functional states, but only 2 very recent structures that include the transmembrane domain.10 HKs are modular multidomain structures, in which individual domains can fold and function in a relatively independent manner, allowing signals to traverse via coupled conformational changes of the individual domains.22 Structural analysis has also suggested that asymmetry across the dimer interface in the cytoplasmic domains plays an important role in HK catalysis.22 Low-resolution structural models of the TM domains of PhoQ suggest that the TM helices might undergo a scissor-like diagonal displacement during signaling.23 Biochemical and computational studies of the intramembrane HK, DesK suggest that temperature induced changes in membrane fluidity remodel the TM domain leading to an unwinding of the coiled-coil linker.19,24 Recent structures of the nitrate/nitrite sensor histidine kinase NarQshow a symmetric ligand-free apo state and both symmetric and asymmetric ligand-bound states.10
Here we use a combination of high-resolution solution NMR, disulfide cross-linking and molecular modeling to construct a structural model of the dimeric NsaS transmembrane domain and its intracellular linker. Our studies reveal that the sensor domain of NsaS is a symmetric 4-helix bundle with three distinct features that were previously unknown: First, an N- terminal amphiphilic helix anchors the NsaS transmembrane bundle into the cytoplasmic leaflet of the lipid bilayer, where it is well positioned to read out membrane stress, antimicrobials that have gained access to the inner leaflet of the bilayer, or to interact with partner proteins that directly bind antibiotics. Second, the TM helix does not extend smoothly into the cytoplasmic linker: there is break in the helical structure and increased dynamics at the membrane interface. Finally, the cytoplasmic linker of NsaS is a marginally stable alpha helical coiled-coil, that can accommodate at least two different hydrophobic packing registries, with left-handed and right handed helical crossings, within its sequence. We propose that antibiotic induced signaling in NsaS proceeds by coiled-coil conformational switching in the linker. A similar model has been proposed for the S-helix of NarQ, leading from a HAMP to the DHp domain of this histidine kinase.
2. RESULTS
2.1. Biological Phenotype of the NsaS Knockout in S. aureus Newman.
In order to confirm the role of NsaS in S. aureus physiology, we made knockouts of the genes encoding both NsaS (nsaS) and its downstream response regulator NsaR (nsaR) in the S. aureus Newman strain background via allelic replacement (see Methods). The ΔnsaS strain grew at a rate similar to WT S. aureus Newman in rich media, but showed a mild growth defect in minimal media (Figure 2A), consistent with the concept that HKs, though not essential, provide important environmental cues that help increase bacterial fitness.
Figure 2.
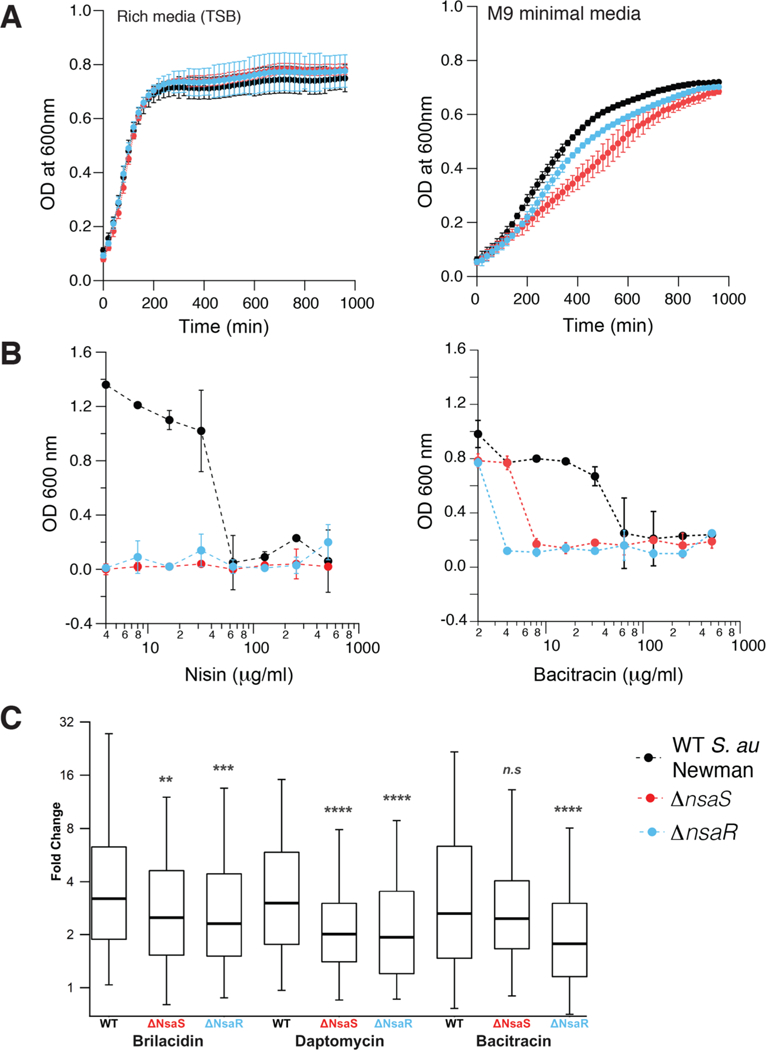
Phenotype of allelic knockouts of nsaS (histidine kinase) and nsaR (downstream response regulator) in S. aureus Newman (a) The ΔnsaS and ΔnsaR strains grow identically to the WT S. aureus Newman strain in rich media (Tryptic soy broth) but show a mild growth phenotype in M9 minimal media. Growth is measured by monitoring optical density at 600 nm, in a 96-well plate shaking at 250 rpm and 37 C. (b) The ΔNsaS and ΔNsaR knockout strains are much more susceptible to membrane active antibiotics Nisin and Bacitracin, when compared with WT S. aureus Newman indicating that the NsaS/R two-component system likely mediates a transcriptional response to these antibiotics. (c) Transcriptional profiling of the WT S. aureus and the nsaS and nsaR knockout strains shows reduced overall transcription of several genes in the knockout strains in response to sublethal amounts of different antibiotics.
We next tested the role of NsaS in bacterial susceptibility to various membrane active antibiotics. Previous studies suggest that nsaS transcription is stimulated by a variety of antibiotics including nisin, bacitracin brilacidin and daptomycin.4,15 We measured the minimum inhibitory concentration (MIC) of these antibiotics against WT S. aureus Newman, and the ΔnsaS and ΔnsaR knockout strains. The knockout strains are significantly more susceptible to nisin and bacitracin (Figure 2B), but do not show an MIC-phenotype against brilacidin and daptomycin (Figure S1). Comparative transcriptional profiling shows reduced gene activation in the NsaS regulon in the knockout strains vs WT S. aureus when cells are subject to sublethal doses of brilacidin, daptomycin and bacitracin (Figure 2C). Interestingly, the response was muted but not abolished in the ΔNsaS and ΔNsaR strains, suggesting that the NsaS/R regulon is activated by additional pathways. Furthermore, the similar transcriptional response between several different membrane-active antibiotics, suggests that the gene-products provide a productive selective advantage to the organism for only a subset of the antibiotics (namely, bacitracin and nisin).
2.2. Architecture of the NsaS Transmembrane Domain.
We cloned and expressed the transmembrane domain of NsaS from genomic DNA of S. aureus Newman. Several different constructs and truncations were tested. Neither the full-length NsaS, nor the isolated transmembrane domain of NsaS expressed well in Escherichia coli; however, a construct that includes both the TM domain and the intracellular coiled-coil linker (NsaS Δ1–86) was highly expressed in the membranes of E. coli C43 cells, so we used this construct for structural studies (Figure 3A). NsaS Δ1–86 also allowed us to investigate both the structure of the TM domain and its connection to the intracellular coiled-coil linker.
Figure 3.
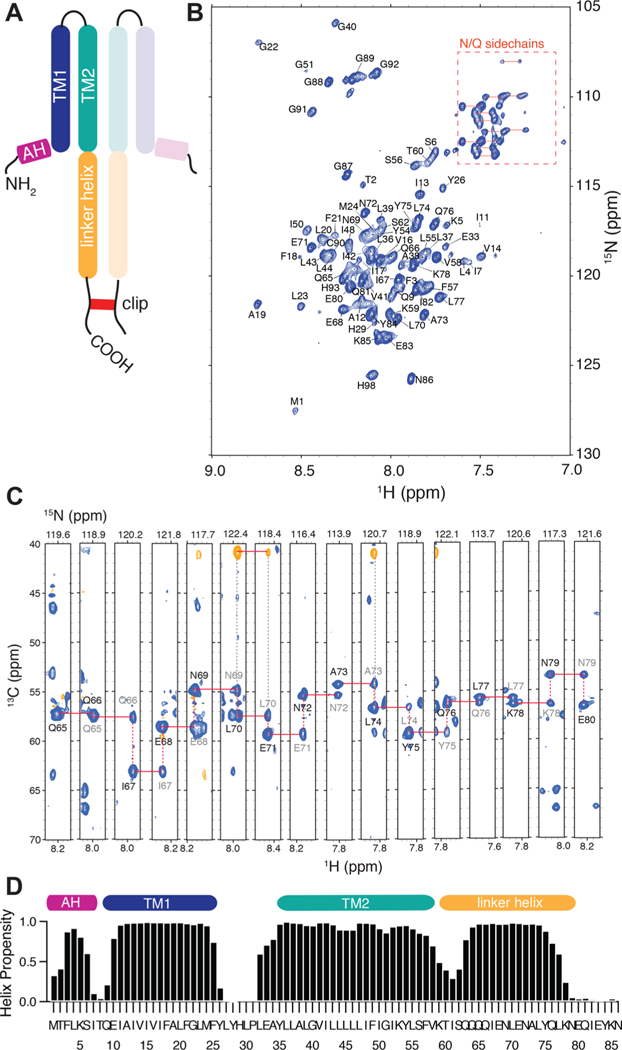
NMR characterization of the NsaS transmembrane bundle. (a) A schematic of the protein construct used for NMR studies. The construct includes the transmembrane domain and the intracellular linker domain of NsaS. A cysteine tag is attached to the C-terminal to ensure detergentindependent dimerization. (b) Fully assigned 2D 1H−15N TROSY spectrum and (c) 3D strip plot showing sequential backbone assignments of 2H−13C-15N labeled NsaS in 2H C12 betaine micelles at pH 6 (see Methods). Spectra were collected at 900 MHz using a cryo-probe set to 42 C. (d) Chemical shift based secondary structure predictions for NsaS show a helical break at residue 8, an extracellular loop between residues 25–33 and a second helical break around residue 60.
CD spectroscopy confirms that NsaS Δ1–86 highly α-helical when solubilized in the detergents DDM as well as C12-betaine (Figure S2). Analytical ultracentrifugation in density-matched C14-betaine suggests that NsaS Δ1–86 is predominantly dimeric (Figure S4), which is the physiologically relevant state of most HKs. Nevertheless, we added a C-terminal cysteine clip to NsaS Δ1–86 because of variations in the detergent: protein ratio is known to shift the monomer— oligomer equilibrium of membrane proteins25 (according to Le Chatelier’s principle of mass action), and we expected that it would be difficult for us to precisely control protein/detergent ratios across the purification procedure. Subsequent spectro-scopic studies were always done with this linker, which alleviated our concerns of minor monomeric species. The cysteine is attached via a 3-glycyl linker, is easily oxidized in air and ensures that regardless of changes in the protein: detergent ratio, the construct is completely dimeric (see Methods). NMR and CD spectra of NsaS Δ1—86 with and without this cysteine clip looked remarkably similar, suggesting that the clip does not perturb the structure of NsaS.
Solution NMR studies of the dimeric NsaS transmembrane domain in C12-betaine micelles show a well-folded α-helical bundle. We screened several different detergents and bicelle conditions to arrive at sample conditions that yield well- dispersed 1H—15N TROSY spectra (Figure S3). We were able to detect 78/86 amide resonances in the 2D TROSY. The peaks that are not assigned on the 2D include 3 residues at an N-terminal hinge (I7, T8, Q9), a proline and a phenylalanine from the loop (P31, F25), 2 leucines within a 5 leucine stretch in TM2 that are likely overlapped (L45, L46) and 1 glutamine in a 4-Gln stretch within the coiled-coil (Q63). Several of the resonances overlapped in the 2D were clearly assigned using 3D spectroscopy. Nearly complete 1H, 13C and 15N backbone assignments were obtained using a combination of 2D and 3D TROSY based NMR spectroscopy (Figure 3B,C, Table S1). We also were able to detect and assign signals from the C-terminal glycines, the cysteine clip and the histidine tag, all of which appeared disordered suggesting that they did not perturb the overall structure of the protein. Although our construct is dimeric, only one set of resonances was observed, indicating that our sample conditions preferentially stabilize a symmetric conformation of the homodimer, or one in which small deviations from symmetry average rapidly on the submsec time scale. Overall, the spectral quality and dispersion was good, considering the highly helical structure of NsaS.
Secondary structure analysis via TALOS26 and RCI27 show that the two transmembrane helices are connected by a loop, which is approximately 7–10 residues and contains two charged residues: H29 and E33. Proline 31 likely induces the start of the second transmembrane helix, and our chemical shifts indicate that L32-Y35 are also helical. There is a break in the TALOS-predicted helicity at around T8 (Figure 3D), which we interpret as a kink in TM1, that allows the N-terminal residues to reside on the surface of the micelle. The NMR intensities I7, T8 and Q9 are very weak, likely due to enhanced dynamics in this region due to the helical break. A second break in helicity is observed between residues 50–60 in the region where the second transmembrane helix connects to the signaling coiled-coil.
Next, we tried to obtain long-range NOE based distance restraints to help fold the tertiary structure. We ran 15N filtered 1H-1H NOESY experiments on a perdeuterated sample of NsaS Δ1–86. We detected several sequential NOEs between amides protons and i–i+4 contacts defining helical regions. However, given the perdeuteration, the NOE intensities between amide and Hα or side chains were not easily quantifiable. Similar experiments on a fully protonated sample yielded several side chain methyl-backbone NOE cross-peaks; however, given the sequence degeneracy and the presence of multiple valine, isoleucine and leucines, it was very difficult to unambiguously assign the methyl groups to yield distance restraints. Given the complexity of the spectra, we decided to utilize a biochemical approach via cysteine cross-linking to obtain tertiary distance restraints within bilayers. These restraints were then combined with MD simulations to build a structural model of NsaS (discussed in detail later).
2.3. Helical Break and Enhanced Dynamics at the TM- Linker Interface.
In many proteins, regions involved in signal transduction often show enhanced structural plasticity. We therefore measured the site-specific structural plasticity of NsaS using two orthogonal measurements: 1H–15N heteronuclear NOE and H/D exchange.
The 1H-15N heteronuclear NOE is sensitive to backbone motions on the picosecond–nanosecond time scale (Figure 4A, Methods). Site-specific 1H–15N NOE measurements show a high degree of backbone order in transmembrane helical regions, indicated by heteronuclear NOE values between 0.8 and 1. Significantly reduced NOE values are seen for the termini and the extracellular loop, which connects the two TM helices, indicating that these regions have enhanced backbone flexibility. The NOEs indicate that the extracellular loop is ~10 residues long and bracketed by two tyrosine’s (Y26 and Y35). It is likely that these tyrosines interact with the lipid headgroups. A second region of enhanced dynamics is observed between residues 55 and 65. This is the region that connects the TM domain to the intracellular coiled-coil linker. Lower hetero- nuclear NOEs for this region suggest a break in the helix or a frustrated structure, i.e., the second TM helix does not seamlessly connect to the catalytic domains.
Figure 4.
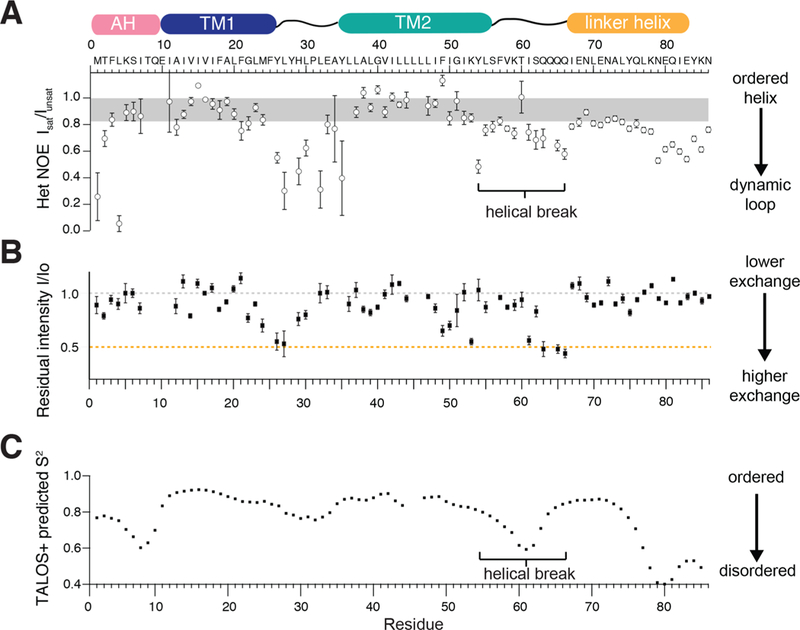
Site-specific measurements of protein dynamics at a variety of time scales indicate a helical break between the transmembrane helix and the coiled-coil linker. (a) 1H–15N Heteronuclear NOE experiments show lowered values between residue 55 and 64 (b) Backbone 15N–1H H/D exchange measurements conducted by measuring the loss of intensity after 6 h in 50% D2O, at pH 6 and 42C show two regions of heightened exchange between residues 49–53 and residues 61–66. (c) Predicted order parameter (S2) from chemical shift based secondary structure calculations using TALOS+ suggest reduced helicity that peaks around residue 61. These data together suggest that there is a discontinuity between the second transmembrane helix and the linker helix.
We also conducted H/D exchange measurements by NMR (Figure 4B, Methods) to monitor site-specific signal-loss upon exchanging the buffer with 50% D2O. The rate of H/D exchange for the transmembrane residues was slow28 and it took several hours to detect any signal loss (Figure S5). The observed protection extended into the cytoplasmic helical linker, indicating that the linker has a defined secondary structure. In contrast, signals corresponding to the C-terminal glycyl-cysteine clip exchanged rapidly.
Residues near and within the loop also showed increased backbone H/D exchange. We also observed increased H/D exchange for residues I50, K53 and I61-L66. These residues fall at the interface of the transmembrane domain and the intracellular linker, and further support a break in idealized helical structure. Finally, TALOS based helical propensity predictions suggest that TM2 does not extend into the linker via a continuous idealized helix (Figure 4C). MD simulations of the full-length NsaS in membranes (details below) also show that the region between the TM and the linker has a distorted a-helical conformation.
2.4. Transmembrane Domain Is Anchored by a Short N-Terminal Amphiphilic Helix.
The first ~10 residues of NsaS form a short helix whose sequence has amphiphilic properties (Figure 5A). This amphiphilic nature is conserved across many annotated homologues of NsaS (Figure 5B). We therefore used two complementary paramagnetic agents to map the water and lipid accessibility of this helix.
Figure 5.
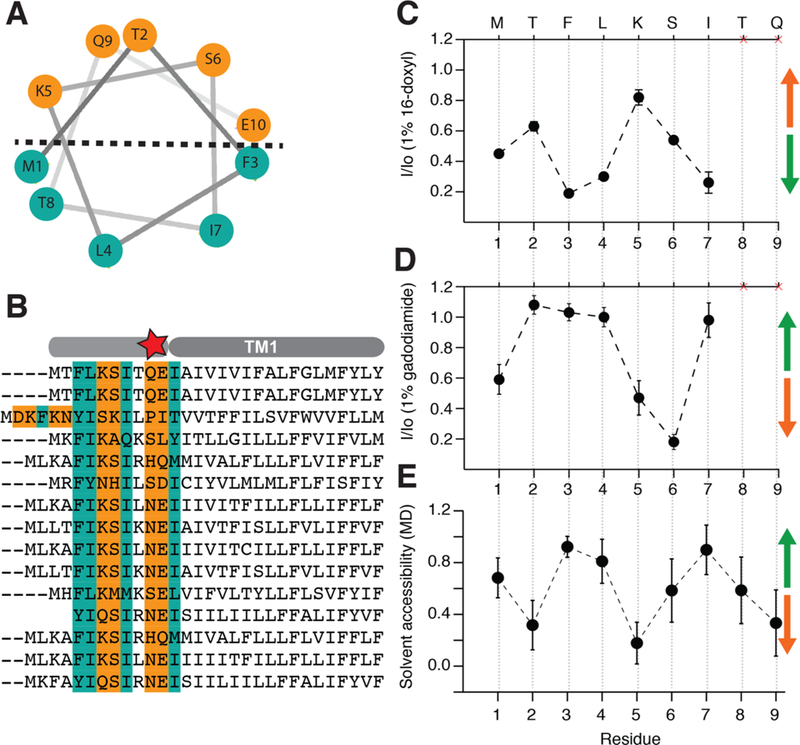
Short amphiphilic helix exists at the N-terminus of NsaS. (a) Helical wheel projection of the first 10 residues at the N-terminus of NsaS shows a clear division between hydrophobic and hydrophilic residues. (b) Sequence alignment of NsaS homologues annotated in Uniprot show that the amphiphilic character of the N-terminus is conserved across the family. (c) Paramagnetic quenching with 16-DOXYL shows a clear periodicity suggesting that the N-terminus residues on the surface of the micelle. (d) Complementary solvent quenching experiments with gadodiamide confirm amphiphilic character of the N-terminus. (e) Water accessibility by MD simulations also suggests that the first helix lies at the membrane interface
We used the lipophilic paramagnetic agent 16-DOXYL (see Methods) to map detergent facing residues. 16-DOXYL partitions into the micelle and enhances the relaxation of residues that contact the hydrophobic regions.29 Thus, the NMR intensity of peaks in the presence of 16-DOXYL is correlated with their burial in the micelle. We observed periodicity in the lipid accessibility of the N-terminal helix that was significantly different from the flatter pattern observed for membrane spanning helices (Figure 5C, S6). This result suggested that the first few residues of NsaS likely form a short helix that lies on the surface of the micelle. To further test this idea, we used a polar paramagnetic agent gadodiamide30 to selectively quench signals from residues that are solvent accessible. Again, we observed a similar periodic pattern for the amphiphilic helix, albeit with an inverted phase (Figure 5D). The results strongly support the presence of a short N- terminal amphiphilic helix that likely anchors the transmembrane bundle within the membrane. MD simulations of the full-length NsaS anchored in POPE membrane also showed that as NsaS equilibrated in the membrane, the first 10 amino acids of NsaS preferred an orientation on the surface of the membrane and retained a surficial conformation throughout the simulation (Figure 5E).
2.5. Handedness of the Coiled-Coil Linker.
Unlike most canonical HKs, the transmembrane domain of NsaS connects directly to the DHp via a ~23 residue linker. Sequence analysis shows that the linker has a sequence of conserved hydrophobic residues (63–77, Figure S7, panel c) that follow a 7/2 hydrophobic periodicity that is consistent with a left-handed coiled-coil. Given the well-known thermodynamic stability of left-handed coiled-coils, we expected this conformation to be stabilized in our NMR sample of NsaS. Instead, our data suggest that the linker adopts a different, possibly more righthanded conformation under our sample conditions.
Right-handed coiled-coils have a helical periodicity of >3.6 residues/turn. The periodicity of the amide secondary chemical shift can be used to detect the handedness of a coiled-coil. Chemical shifts of amide protons on the solvent accessible side of helices tend to be shifted upfield compared to the more strongly hydrogen bonded amides in solvent inaccessible or buried parts of the helix. Previous work on coiled-coil proteins such as α3D and GCN4 has shown an excellent correlation between the periodicity of the proton secondary shift and the phase of a coiled-coil.31,32 For a classically left-handed coiled- coil such as GCN4, there are 7 residues in 2 turns leading to a periodicity of 3.5 residues/turn (Figure 6A). An ideal straight helix has a periodicity of 3.6 residues/turn. A canonical right handed coiled-coil could have an 11/3 repeat or a 15/4 repeat, both of which yield frequencies of greater than 3.6 residues/ turn. Secondary shift analysis on the NsaS linker also shows a periodic pattern for ~10 residues between 65 and 75. Given the heptad like repeat present in the sequence, we expected a periodicity of ~3.5; however, the data fit best to a repeat of nearly 3.7 residues/turn (Figure 6B, Methods) suggesting that the linker is stabilized conformation that is significantly less left-handed compared to a canonical coiled-coil and might even reach a right-handed conformation.
Figure 6.
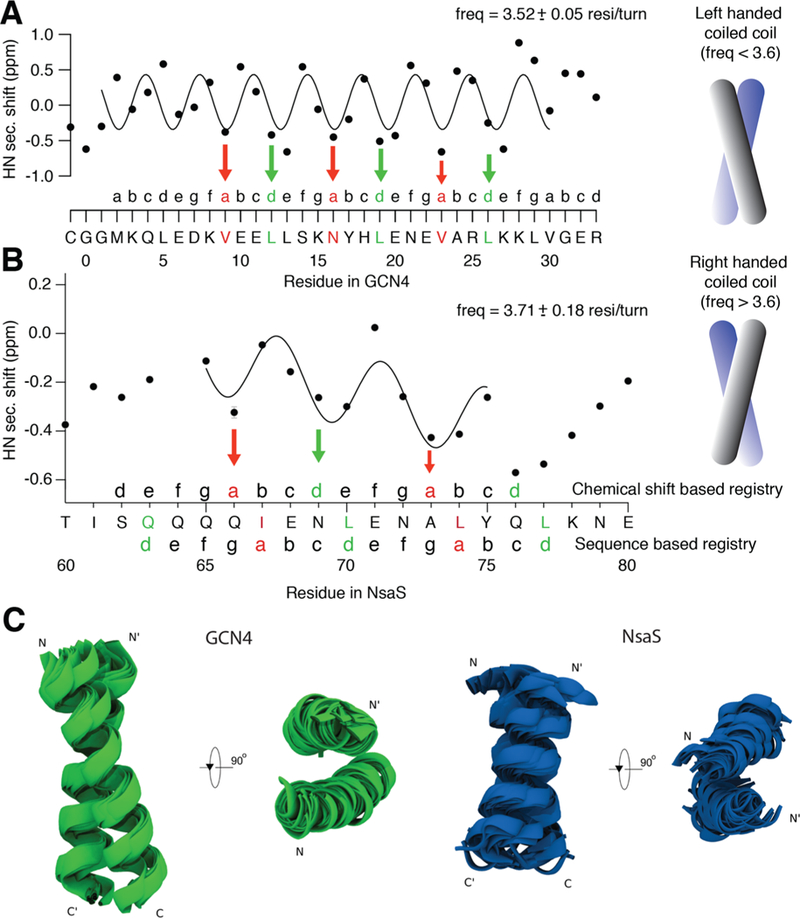
Amide secondary shift is sensitive to the phase of a coiled coil. (a) Secondary shift analysis on the model coiled-coil GCN4 shows a sinusoidal pattern, in which the A and D positions of the heptad, indicated in red and green respectively, are at the troughs of the sinusoid. These positions prefer hydrophobic residues that mediate the zipper like packing which forms the basis of a coiled-coil. (b) Similar analysis on the putative coiled-coil linker domain of NsaS shows that there is a 1-residue gap between the heptad registry obtained by secondary shift analysis and the registry based on the sequence analysis. Quantitative analysis suggests that the periodicity fits to a frequency of ∼3.7 residues/turn indicating a right handed conformation. This suggests that the NsaS linker might be able to adopt two different types of structures: one with canonical coiled-coil packing that follows the sequence propensity, and a second noncanonical packing that we have serendipitously stabilized under our NMR conditions. (c) De novo folding of the model coiled-coil GCN4 and the NsaS linker helix show that GCN4 has a clear preference adopt a left-handed structure whereas the NsaS linker folds into a right-handed conformation. A structural ensemble of 50 lower energy states are shown in cartoon.
Given the small number of data points and the large error in the fit, we conducted a ChiSq analysis to gain more confidence in the results (see methods). χ2 is a statistical metric used to assess the goodness of fit; a lower χ2 indicates a better fit. We fit our data to a range of different helical frequencies ranging from 3.3 to 3.9 residues/turn and a minimum χ2 corresponds to a frequency of 3.7 residues/turn (Figure S9). Thus, although noisy, we speculate that the data represent a more right-handed conformation for the linker helix. Analysis of the peaks and troughs reveal that the hydrophobic registry stabilized in the right-handed conformations is one residue off from the canonical heptad repeat found in the sequence (Figure 6B). Details of the data fitting are discussed in the Methods.
To further corroborate this surprising result, we conducted ROSETTA based de novo folding to map the conformational preference of the NsaS linker. We first benchmarked the de novo folding algorithm to ensure that it can properly fold known coiled-coil structures such as GCN4 and predict the correct handedness (backbone RMSD 1.9 Å ± 0.49; Figure 6C). ROSETTA de novo folding of the NsaS linker yielded several conformations of right handed dimeric structures with different helical crossing angles, but no left-handed structures.
Because the NMR and de novo folding data support more right-handed helical crossings for the linker, though the sequence homology favored a left-handed, we hypothesize that the NsaS linker is a marginally stable coiled-coil that can adopt different conformations during signaling.
2.6. Marginal Stability of the Coiled-Coil Linker.
Given the apparent marginal stability of the intracellular coiled-coil linker, we wanted to investigate its biophysical characteristics independent of the TM domain and the DHp. We therefore synthesized a short peptide corresponding to the linker residues (Figure 7), with an appended N-terminal cysteine to control its dimerization as in previous work on GCN432 CD measurements showed that the linker peptide is largely unstructured and only becomes helical when the cysteine is oxidized, i.e., the monomers are forced into proximity. CD melting curves show that the helical character increases with lowered temperature (Figure 7B,C), suggesting that at ambient temperatures the hydrophobic packing from residues 70–80 is not sufficient limit the structure of the linker into a unique coiled-coil conformation.
Figure 7.
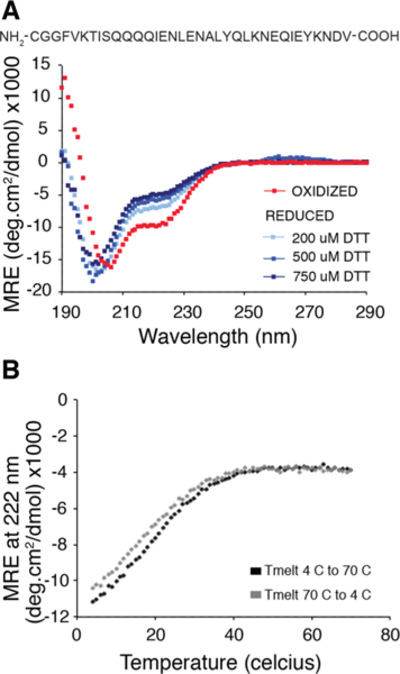
Marginal stability of the coiled-coil linker is shown using CD spectroscopy. (a) CD spectrum of the linker peptide (40 μM peptide in 10 mM NaPi buffer, pH 7) under air oxidized (red) vs reduced conditions (shades of blue). Helical character at 222 nm is only seen when the cysteine is oxidized indicating that the coiled-coil sequence is not sufficient to fold the linker at micromolar concentrations in the absence of tethering. (b) CD melting curve (at 222 nm) for the oxidized form of the peptide shows marginal helicity at room temperature and significant stabilization of the helix at lower temperatures. The melting curves are reversible (black, 4C to 70C and gray, 70C to 4C).
Our data show that the linker in NsaS is a marginally stable coiled-coil whose helical structure is likely dictated by its structural connections to both the transmembrane bundle and the DHp bundle. The marginal stability is likely due to an inherent tension between the linker’s sequence-based propensity to adopt a left-handed helical structure and its conformational propensity to adopt a less left-handed structure.
2.7. Structural Modeling of NsaSΔ1–86.
2.7.1. Restraints from Disulfide Cross-Linking.
We obtained key tertiary distance restraints, in the form of intermonomer restraints using disulfide cross-linking experiments on a series of cysteine mutants of NsaS. This method allowed us to probe the structure in native membranes.
Native chemical cross-linking has previously been used to determine the helical phase of TM bundles.23 The underlying principle is that residues at the dimer interface cross-link better than residues on the outer surface, so by quantifying the differential cross-linking of sequential cysteine mutations, one can infer helical phase. We used Cu(II) phenanthroline as a membrane soluble oxidizing agent to stimulate cysteine oxidation, and the reactions were conducted within membrane preparations so that the structure was sampled within a nativelike environment (see Methods).
First, we generated a library of 48 single cysteine mutations within the NsaS Δ1–86 construct. The construct was modified to remove the C-terminal cysteine clip, but the 6-His tag was retained so that we could use an Anti-His Western blot to detect the protein. The mutations covered TM1 (residues 5–24), TM2 (residues 34–50) and the first few residues (residues 51–64) of the intracellular coiled-coil. Protein expression and membrane insertion for each of these mutations was assessed by conducting anti-His Western blots on membrane preparations of each mutation, without any oxidizing agent. Not surprisingly, several mutants showed significantly lower expression and/or membrane insertion. Some mutants also showed altered expression when grown in rich vs minimal media. A detection limit was established based on the quantitative reproducibility of the analyzed data across three experimental replicates (Figure S8). A standard sample made with equimolar amounts of the monomer and the C-terminally clipped dimer was used as an internal standard to calibrate monomer and dimer intensities on each gel. Cysteine mutants that crossed the detection limit were used for disulfide cross-linking. These included stretches of 7–10 residues in TM1, TM2 and the coiled-coil.
An example of a blot showing relative dimer vs monomer populations is shown in Figure 8A. The results for a series of residues on TM1 and TM2 are shown in Figure 8B. The fits are approximate because there is uncertainty in quantification of monomer and dimer populations based on blots, but the data can be grossly fit to a periodic function with a frequency of 3.6 residues/turn to yield approximate helical phase of TM1 and TM2. There was no periodicity observable in the region between TM2 and the coiled-coil linker (residues 48–55). The cross-link distance restraints in TM1 and TM2 were subsequently used to guide structural modeling of the TM domain (see below) and by combining this information with secondary structure information from NMR, we were able to build a a structural model for the dimeric transmembrane and linker domains of NsaS.
Figure 8.
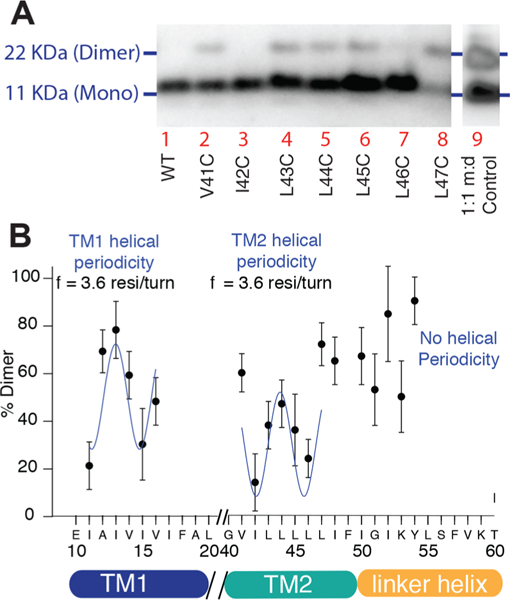
Native chemical cross-linking experiments on single-point cysteine mutants across the TM domain of NsaS yields longer range intermonomer restraints. (a) Anti-His Western blot showing data used to quantify the extent of cross-linking by measuring the intensity of monomer vs dimer bands. Lane 9 shows control that contains a 1:1 mixture of monomer and dimer, used to calibrate intensities. (b)Analysis of cross-linking measurements. The periodicity is indicative of the phase of the helix in the bundle, i.e., positions such as I13, V41 and L47 that are highly cross-linked, likely point into the helical bundle, whereas positions such as I11, I15 on TM1 and I42, L46 on TM2 likely point toward the lipids.
2.7.2. NsaS Integrative Model Building and MD Simulations.
Structural models were built using a combination of three computational methods and integrating available experimental and structural information: (1) the BCL::MP- Fold method was used to model the TM domain restrained by cross-linking data, (2) Rosetta de novo structure prediction generated models of the linker and DHp domains, and (3) Rosetta homology modeling based on a recent CpxA structure (PDB id: 4biu) was applied to the cytoplasmic DHp and kinase domains of NsaS (see Methods and Figure 10A). This multistep approach was used to best leverage the available experimental and structural data for model building.
Figure 10.
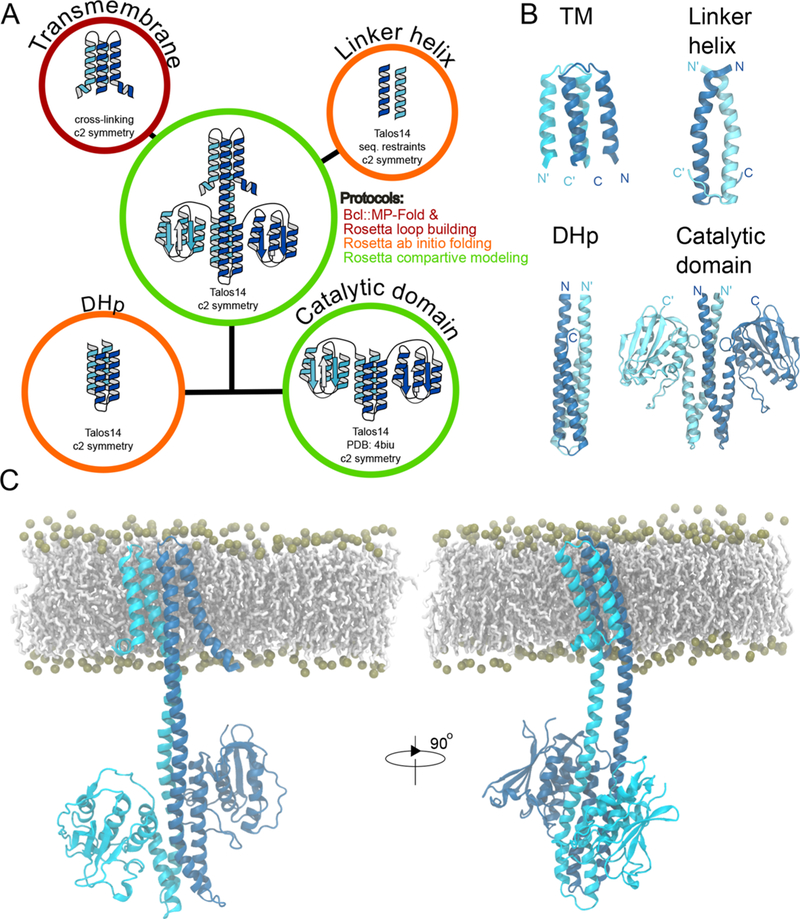
Integrative molecular model of NsaS based on NMR secondary structure, cross-linking, homology modeling and Rosetta ab initio folding. (a) Summary of the different strategies used for the modeling of NsaS. (b) Structural model of each domain. (c) Full length model of NsaS after a 250 ns of molecular dynamics simulation. The carbon atoms from POPC membrane are shown with white sticks and the phosphate group with gold spheres.
BCL::MP-Fold builds TM helix topologies through the assembly of entire helices allowing for efficient conformational sampling. Our experimental cross-linking data were converted into intersubunit contact restraints to guide this TM helix assembly (see Methods for details). The final model of the NsaS TM domain is a 4-helix bundle characterized by a left-handed crossing of the helices. The bundle is rhombohedral- shaped and TM2 forms the tighter interface. P31 is at the first helical position of TM2. The model shows good agreement between the experimentally observed disulfide cross-linking data and corresponding Cβ—Cβ distances in the dimer, which display a nearly anticyclic phase pattern (Figure 9).
Figure 9.
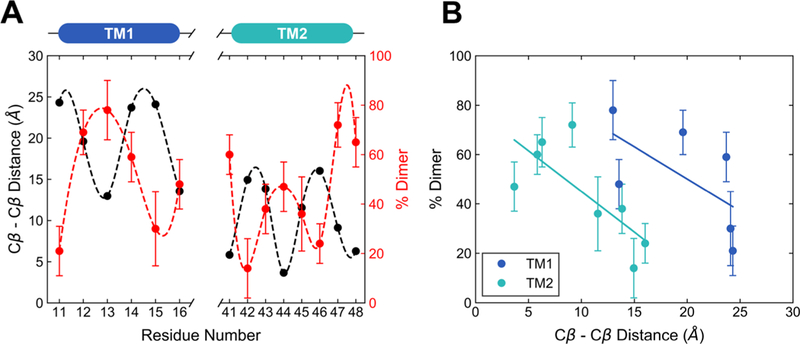
Comparison between the disulfide cross-linking data and Cβ−Cβ distance predicted from models of the NsaS–TM domain built with BCL::MPFold method. (a) The periodicity obtained from cross-linking experiments of the NsaS−TM is compared with distance measured from the final BCL model. Strong anticorrelation between the observed cross-linking efficiency and modeled Cβ−Cβ distance is seen for the best model. (b) The relative BCL model fit to the experimental data is shown for each of the two TM helices in NsaS.
In order to understand its conformational character, we modeled the linker in two different ways: (l) as an independent domain and (2) tethered to the TM and the DHp domains on the N- and C-termini, respectively. The linker was modeled as an independent coiled-coil using Rosetta de novo structure prediction protocols. The structure of the linker converged to a well-defined right-handed coiled coil with a hydrophobic packing interface similar to the one predicted by the NMR secondary amide shift data (Q66xxN69xxxA73xxQ76). This suggests that in the absence of the DHp and the TM domain, the linker preferentially populates a right-handed conformation.
To understand the effect of the TM and the DHp, we produced a full-length model of NsaS, in which the lowest- energy model of the TM and linker domains was connected with the homology model of the DHp and kinase domains using the Rosetta comparative modeling procedure. In this model, the transmembrane and DHp domain are both left handed. Including the TM and the DHp on each end of the linker did not change the handedness of the linker but forced it into an alternative hydrophobic packing conformation (l67xxL70xxxL74xxL77) (Figure 10B), with a hydrophobic registry predicted from sequence analysis. To further refine this model of NsaS, it was embedded into a POPE bilayer and subjected to an all atom MD simulation with a length of up to 250 ns. The model remained stable with only marginal fluctuations of the secondary structure. A representation of the refined structural model of NsaS is shown in Figure 10C.
Thus, it appears that the linker is an unstable coiled-coil whose conformational preference is dependent on the structure of the TM and the DHp, making it an excellent structural transducer. One might even speculate that signal-induced changes in the packing of the TM domain would alter the structure of the linker, and perhaps even change its handedness.
3. DISCUSSION
Our initial studies on NsaS indicate that it mediates the S. aureus response to the membrane active antibiotics nisin and bacitracin. The nsaS knockout is not lethal in S. aureus, but it confers a mild growth phenotype in minimal media. The TM domain of NsaS is a small dimeric 4-helix bundle that is anchored into the membrane of S. aureus by a short N-terminal amphiphilic helix. The bundle extends into the cytoplasm and connects with the catalytic domain via a structurally dynamic coiled-coil. Enhanced plasticity is observed in the region connecting the TM domain to the coiled-coil domain. Our data also imply that signal transduction in this family likely involves helical bundle rearrangements that manifest as changes between left-handed and right-handed helical crossings in the coiled-coil linker.
Marginally stable coiled-coils have long been implicated in HK signaling. Structural and mutational studies on several HKs such as YF1 18,33 and NarX20 have shown that linker domains of these proteins are imperfect coiled coils, i.e., instead of the characteristic 7-residue repeating sequence motif, they have l, 3 and 4 residue insertions that disrupt an idealized coiled-coil geometry. Perturbing the linkers with amino acid insertions and deletions often completely alter the signaling phenotype of the kinase. Recent analysis by Schmidt et al. suggested that the structural feature that differentiates the kinase and phosphatase signaling states is the “accommodation length” of the linker.34 They propose that the structural frustrations in the linkers are delocalized over a large region in the resting state, thereby stabilizing the structure in a more symmetric conformation. In the presence of an activating stimulus, structural frustrations are concentrated into a very short helical region which manifests as helical kinks, bulges, bends and significant local asymmetric structures. On the basis of analysis of signaling-helix structures of CpxA and Af1503, Schmidt et al. suggest signaling occurs with a change in the helical crossing angle, which can switch from left- to right-handed in some situations, depending on the length of the accommodation length and the type of the insertion or deletion.34
NsaS is different from canonical HKs like CpxA in that it does not have an intracellular HAMP or PAS domain. The coiled-coil linker of NsaS is a primitive S-helix and it directly connects the TM domain to functional catalytic domains. Our data show that even in this simple architecture, the linker is a marginally stable coiled-coil that can accommodate both a lefthanded and a right-handed helical crossing. The left-handed conformation is accommodating by using a built-in hydrophobic heptad repeat that is highly conserved in its sequence. The right-handed conformation was surprisingly stabilized in our NMR sample, in which the linker is only tethered to the TM domain and not to the DHp.
Rosetta de novo structure prediction showed that in isolation the linker domain always adopted right-handed conformations with a packing pattern that did not match the regular left-handed heptad. However, for the full length model, in which this coiled coil was connected to both the TM domain and the DHp domain, the hydrophobic registry switched to the more canonical sequence heptad, inducing significant frustration in the right-handed crossing. A 250 ns MD simulation on the full length structure showed that this conformation is stable but frustrated: the linker often bends significantly leading to highly asymmetric structures, much like the conformations observed in several recent crystallographic studies.10,35 We therefore propose a signaling mechanism in which the TM and the DHp act as tethers that impose left-handed structural frustration on an inherently right-handed linker. When the structure of the TM is perturbed in response to an antibiotic stimulus, the signal is transmitted to the linker as a structural frustration that changes its handed-ness and induces asymmetry.
Our studies on NsaS also provide some of the first structural insights into the architecture and dynamics of an intramembrane HK. One defining feature of NsaS is an N-terminal amphiphilic helix. Amphiphilic helices are present in a number of different membrane-sensing proteins such as the various BAR proteins that sense membrane curvature36 and the M2 channel that localizes at the neck of the budding virus and induces negative Gaussian curvature.37 Because NsaS transcription is triggered by several different membrane-active antibiotics, one hypothesis is that its amphiphilic helix might interact with the lipid bilayer sensing the physical state of the bilayer. An alternate hypothesis is that this helix anchors and orients the entire TM domain into the membrane so that it does not bend and tilt within the membrane, and the appropriate intramembrane surfaces are stabilized for protein—protein interactions. Indeed, the NsaS might engage in intramembrane protein—protein interactions with the ABC transporter BraD/E4. Finally, the positively charged residues on the N-terminal end might also serve as a signal peptide and help NsaS fold and insert into the membrane correctly.
The second structural feature we observed is a helical break and increased plasticity in the regions between the TM and the coiled-coil linker. One could argue that our measurements (heteronuclear NOE, HD exchange etc.) are made in a detergent environment at a relatively high temperature so the micelle environment might be perturbing protein structure; however, chemical cross-linking experiments in lipid bilayers also suggest a lack of clear helical periodicity between the TM2 helix and the linker, suggesting that this region is disordered even under milder conditions of a membrane environment and room temperature. Thus, the original piston-like signaling mechanism10,38 (which postulated small vertical displacements of transmembrane helices, which propagate in a rigid manner into the cytoplasmic domains) appears unlikely for NsaS, although lateral helical displacements might be one component of the conformational change that changes in structure and dynamics in NsaS. We speculate that this plasticity allows the TM domain of NsaS to be softly coupled to the signaling coiled-coil,22 so that when the TM domain senses the antibiotic, it perturbs the structural equilibrium between the left- and right-handed states of the coiled-coil linker, which in turn alters the propensity of the protein to be a kinase vs phosphatase.
The outstanding question is how does NsaS sense chemically diverse membrane active antibiotics? Nisin’s antimicrobial activity stems from binding to and sequestering the pyrophosphate moiety of lipid-II, an important intermediate for cell-wall biosynthesis. The nisin-lipid II complex also leads to the formation of pores in the membrane, causing cell death.39 Bacitracin has many different effects on cells. Most notably, it interferes with the dephosphorylation of C55- isoprenyl pyrophosphate, a membrane carrier molecule that transports cell-wall precursors between the membrane and the cell-wall.40 Both these antibiotics interfere with cell-wall biosynthesis and there is good evidence from genetic knockouts, and detailed biochemical studies of the homologous BceS system in Bacillus subtilis6,7 suggesting that Nisin and Bacitracin are sensed by an ABC transporter, which then interacts with and activates the enzymatic activity of NsaS. The NsaS regulon is also triggered by several other membrane active compounds such as brilacidin, daptomycin and the antimicrobial peptide LL16.15 Further studies need to be done to understand how the architecture of NsaS leads to its ability to sense these various antibiotics. Our structural approach provides a first physical model for the structure of the dimeric NsaS sensor and paves the way for future studies of antibiotic resistance mechanisms in S. aureus.
4. METHODS
4.1. Molecular Biology.
Allelic knockouts of NsaS and the cognate response regulator NsaR were made in the S. aureus Newman background using the knockout strategy via the pIMAY plasmid as described previously. Briefly, plasmid integration is achieved using a temperature sensitive origin of replication, and temporarily confined cultivation in the presence of antibiotic selection under nonpermissive conditions for pIMAY (37 °C). Enrichment of desired doublecrossover candidates was achieved by using an ATc-induced antisense secY RNA based counter-selection that removes single-crossover events. The genomic identity of resulting strains was checked by PCR and sequencing to confirm deletion of the genes. To prevent polar effects, the knockout was designed so that the first 3 and last 3 amino acids of NsaS remained intact. All plasmid transformations into S. aureus Newman were conducted by electroporation (BioRad electro- porator, 1 mm cuvette, 2100 V, 1.1 ms pulse) using ~1 ug of plasmid in 150 μL of competent cells prepared using the sucrose method (reference) with 3 h of recovery in BHI broth. Typical transformation efficiency for the subcloned NsaS-pIMAY plasmids was 10–20 colonies/ug of DNA. Primers and strains are listed in Table S1. For the structural biology, NsaS was cloned out of S. aureus Newman genomic DNA and inserted into the pEXP5 T7-promoter based expression plasmid using a Gibson cloning strategy. The expression of several different constructs and tags was tested. Best results were obtained for a construct that contained the first 86 residues of NsaS followed by a GGCGG and a 6-His tag for purification. The plasmid and protein sequence for this is in the SI. Single cysteine mutations were generated for at 52 positions with the construct of NsaS using a standard Quikchange mutation strategy.
4.2. Transcriptional Profiling.
Daptomycin (Cubicin) was used without further purification. Brilacidin was purified as an HCl salt by reverse-phase high-performance liquid chromatography (HPLC). Bacitracin was purchased from Sigma. Three cultures of strains S. aureus Newman, S. aureus Newman DNsaS and S. aureus Newman DNsaR were grown to an OD600 of 0.5 and split 2-fold into prewarmed LB medium with drug, and aliquots were collected by centrifugation every 20 min for 2 h. Pellets were flash-frozen in liquid N2 to halt transcription. Total RNA was purified from pellets using TRIzol reagent per manufacturer’s instructions. mRNA was enriched by rRNA removal using a Microb-express bacterial mRNA purification kit (Ambion).
4.2.1. Illumina Sequencing Library Generation.
Barcoded Illumina sequencing libraries were constructed using a modified protocol obtained from the DeRisi lab at UCSF.41 Briefly, random hexamer primers with a 5’ adapter sequence (“3Sol_N”‘, primer 1) were used to synthesize first strand cDNA from enriched mRNA using a cDNA synthesis kit (Invitrogen). The second strand was synthesized using the same primer and Thermo Sequenase DNA polymerase (GE healthcare). cDNA library was PCR amplified using KAPA polymerase (10 cycles) using primer 2 (“3Sol”). After PCR-clean up, concentrations were determined by nanodrop and samples were normalized to 2 ng/μL concentration. This library was barcoded using custom built 7 bp barcode Illumina sequencing primers (DeRisi lab, primer 3, “SolM2”; primer 4, “5SolM2”) using Kapa Hi-fi polymerase (Kapa Biosystems) for 2 amplification cycles, and barcoded library amplified in the same reaction using hot-start primers (primer 5, “5SolM2_18”; primer 6, “5SolM2_19”) for an additional 6 cycles. Hot-start primers were activated by heating reaction at 94 °C for 10 min. Resulting barcoded libraries were multiplexed (3 samples of 18 conditions each), size selected on a DNA agarose gel (300–400 bp) and quantified by nanodrop. These 3 samples were then single-read sequenced (50 bp reads) using a HiSeq Illumina sequencer with a 7 bp barcode sequencing per manufacturer’s instruction at the Center for Advanced Technology at UCSF.
4.2.2. Deep-Sequencing Data Analysis.
Demultiplexed sequencing data were first checked for sequencing quality and then mapped on to the Newman strain genome using BowTie (galaxy project).42–44 Mapped reads were then binned into gene ORFs and analyzed for differential expression. Fold enrichment was calculated using an expectation weighted normalization to minimize over-estimation of fold-change in genes with low read counts as shown below.
where
4.3. Protein Expression, Purification and NMR Sample Preparation.
pEXP5-NsaS was transformed into E. coli C43 cells. For NMR samples, the cells were grown in LB media and then transferred isotopically labeled M9 media (95% D2O for 2H labeling, 13C-glucose for 13C labeling and 15NH4Cl for 15N labeling). Cells were grown to an OD600 ~ 0.8 at 37C, and protein expression was induced with 1 mM IPTG. The cells were harvested after 8–10 h of growth at 37 °C. Cell pellets were treated with 0.1% lysozyme and the lysed using sonication. The membranes in the lysate was solubilized by incubating with 5% Empigen detergent and gentle shaking at 4C for ~30 min. Cell debris was removed by centrifugation at 15,000 rpm for 45 min. The solubilized membrane component was incubated with Ni-NTA agarose beads (Qiagen) for 2 h at 25 °C with gentle agitation and loaded on a gravity-flow column. The nickel beads were then washed with 10 column volumes of purification buffer (50 mM Tris, 300 mM NaCl, 25 mM imidazole, 1% OG detergent, pH = 8) and NsaS was eluted using the same buffer with 250 mM imidazole. Protein purity was analyzed by SDS-PAGE (Figure S11). The sample typically ran as a mixture of monomer and dimer when the sample was boiled in the SDS loading buffer. The eluted protein was concentrated to 5 mL in a 3000 Da—MWCO Amicon Ultra centrifugal filter and further purified by gradient separation on a reverse-phase HPLC on a C4 column. The peptide eluted at 95% B’ (70% isopropyl alcohol, 20% acetonitrile, 10% water, 0.1% TFA), and was recovered by lyophilization. The lyophilized NsaS peptide was redissolved in a suitable buffer (40 mM MES, 100 mM 2H—C12 betaine, 10% D2O, pH = 6) for NMR experiments.
4.4. NMR Spectroscopy.
All NMR experiments were conducted at 42 °C on a Bruker 800 or 900 MHz spectrometers equipped with cryogenic probes. A variety of different detergent and sample conditions were evaluated based on chemical shift dispersion in a 2D 1H—15N TROSY. The final sample used for assignment contained 350 μL of 750 μM 2H—13C—15N NsaS solubilized in 40 mM MES and 100 mM 2H—C12 betaine at pH = 6 in a Shigemi tube.
4.4.1. Assignments.
Sequence-specific assignment of backbone 1Hn, 15N, 13Ca, 13Cb and 13C’ chemical shifts was achieved with the 2H decoupled TROSY versions of HNCA, HNCACB, HNCO and HN(CO)CACB using standard out-and-back experiments. A recycle delay of 1.2 s was used to balance efficient signal averaging with the longer T1s due to extensive deuteration. RF carriers were at 4.75 ppm for 1H, at 119 ppm for 15N, and at 176 ppm for 13C’ and 55 ppm for 13Ca. The backbone connectivity was validated using 3D 15N-edited TROSY-NOESY experiments (Tmix = 200 ms). Partial side chain assignments were obtained by measuring 3D 15N-filtered TROSY- NOESY experiments (Tmix = 60 ms) and CHHC-TOCSY with 80 ms of mixing on a 1H—13C—15N NsaS. All chemical shifts were referenced to DSS at 25 °C. NMR spectra were processed with NMRPipe and analyzed with SPARKY. Chemical shift based secondary structure was obtained using TALOS+. Secondary chemical shifts were calculated by subtracting the residue specific random coil chemical shift from the assignment.
4.4.2. Heteronuclear NOE.
The 15N—1H heteronuclear NOE and reference spectra were recorded in an interleaved manner, with either an 15N—1H saturation time for the NOE experiment or the equivalent recovery time for the reference experiment. The heteronuclear NOE is reported as the residue specific ratio of peak intensity between the saturated and unsaturated experiments.
4.4.3. PRE Measurements.
Solvent accessibility of NsaS was probed by titration with the water-soluble paramagnetic agent gadodiamide. Briefly, 1H—15N TROSY spectra were recorded at 800 MHz for 300 μM 1H—15N labeled NsaS in Cu betaine. Gadodiamide was titrated in at concentrations of 100 μM, 1 mM, 10 and 25 mM. The effective volume of methanol added to the NMR sample was 0—6 μL, which is very small relative to the sample volume (~600 μL) and unlikely to perturb the membrane structure. The reported PRE effect was calculated as the ratio of assigned resonance intensities to the reference intensities at 25 mM Gadodiamide. The lipid accessibility of NsaS was probed using the lipophilic paramagnetic agent 16-DOXYL, which partitions into detergent micelles. Similar to the solvent PRE, 16- DOXYL was titrated in at 1% and 2%. The reported PRE is the ratio of assigned resonance intensities to the reference intensities at 1% 16- DOXYL.
4.4.4. HDX Measurements.
In order to measure H/D exchange, a concentrated sample of NsaS (400 μL at 800 μM) was prepared. The sample was divided into two aliquots of 200 μL each: the first aliquot diluted 2× with 100% H2O buffer (40 mM MES and 100 mM 2H—C12 betaine at pH = 6). This served as a reference sample. The second aliquot was diluted with 100% D2O buffer (40 mM MES and 100 mM 2H—C12 betaine at pH = 6) immediately before measurement. Serial 1H—15N TROSY spectra were recorded for the D2O exchanged sample at various time intervals (SI). The data presented in the main text represent relative intensities of assigned resonances at 6 h. Remaining data are in the supplement.
4.4.5. Amide Chemical Shift Analysis.
The secondary 1Hn amide shift for residues in GCN4 and NsaS were calculated using δ(ss) = δ(1HN) — δ(1HN)random coil. The chemical shifts for GCN4 were obtained from Kim et al.32 and the Cβ—Cβ distances were calculated using coordinates in the PDB file 2ZTA.
Data Fitting.
Secondary shift data were fit to the following equation:
where A is the amplitude, r0 is the starting residue of the fit, f is the frequency, i.e., residues/turn, p is the phase at residue r0 in degrees and B and C are linear and constant baseline offsets. The data were first fit to a linear equation to estimate B, whose value was then held constant in the periodic fit (Figure S9 panel A). The amplitude (A) was held at half of the difference between the minimum and maximum values (0.22) and allowed to vary by the standard deviation (0.14). R0 was set to be the first residue in the series (residue 65). Thus, the only two parameters that were varied in the fit were the frequency and the phase. A multiparameter fit was conducted in Igor with the following constraints: A = 0.22 ± 0.14; r0 = 65; 3.4 < f < 3.8 to capture the range of helical frequencies; 0 < p < 360. The best fit (judged by lowest χ2) yielded a helical frequency of 3.71. Because of the large error in the fit parameters and the small number of data points, we also conducted a ChiSq (χ2) analysis. For this analysis we forced the frequency to be held at different values in the range of 3.3 and 3.9 and assessed the quality of the fit using χ2. Note that the degrees of freedom were identical across these fits, so we did not need to use the reduced chisq metric. A plot of χ2 vs frequency is shown in Figure S8 and shows that the minimum χ2 occurs at a frequency of 3.7 (Figure S9, panel C).
4.5. Peptide Synthesis and CD Spectroscopy.
The linker domain of NsaS was chemical synthesized and purified by reverse phase HPLC. A cysteine separated two glycines were added to the N-terminus of the linker. NsaS solutions were prepared at ~50 μM in 50 mM phosphate buffer (pH 7.0) and 10 mM C12 betaine and oxidized in air overnight. CD studies were conducted using a 0.1 mm cuvette at 25 °C on a JASCO J-810 CD spectrophotometer equipped with a Peltier temperature control unit. DTT was titrated into the samples at 200, 500 and 750 μM initiate reduction of the cysteine. The mean residue ellipticity was calculated using a molecular weight of 4086.5 g/mol and an extinction coefficient of 254.8 M–1 cm–1.
4.6. Disulfide Cross-Linking.
Single cysteine mutations of NsaS were transformed into E. coli C43 cells. Freshly plated colonies were picked and used to inoculate 25 mL LB + 100 mg/mL ampicillin. Cultures were grown at 37 C for 3 h shaking at 220 rpm. Cultures were induced with IPTG at OD600–0.4 because we found that the level of leaky expression was not sufficient to accurately quantify crosslinking. Cells pelleted by centrifugation at 3,700G for 10 min at 4C, treated with 10 mg/mL lysozyme and lysed via sonication. Cell debris was removed by spinning at 3700G for 10 min, and the supernatant was transferred to an ultracentrifuge and spun at 105,000 rpm for 15 min at 4C to pellet the membranes. The membranes were then resuspended in 50 mM Tris, 300 mM NaCl, 10% glycerol, pH = 8. Twenty-five ul of membranes was combined with 25ul of the oxidative catalyst Cu(II)(1,10-phenanthroline) so that the final concentration of Cu(II) was 0.5 mM. The cross-linking reaction was allowed to proceed for 30 min at 25C and quenched with 25ul of Invitrogen LDS, 8 M Urea, 20 mM NEM, 20 mM EDTA buffer that allowed the sample to be loaded onto gels. Ten ul of this sample was loaded onto a 5–20% Bis-Tris gel(NuPage). Proteins were separated by electrophoresis and transferred to nitrocellulose membrane (iBlot2). For cross-linking reactions in the TM region, membranes were washed with Tris- buffered saline with Tween (TBST) buffer (10 mM Tris, pH 7.5, 2.5 mM EDTA, 50 mM NaCl, 0.1% Tween 20) and blocked with 2% BSA in TBST. NsaS was probed using a penta-His-HRP conjugated antibody (QIAGEN) and the chemiluminescent signal was detected using an ECL reagent (Amersham, GE Health Sciences) for 1 min and exposure of 30s. The relative intensities of monomer vs the dimer were calculated using ImageJ software and the relative % of dimer is reported as dimer/(monomer + dimer). Error bars represent the standard deviation from two experimental replicates.
4.7. Molecular Modeling.
The TM domain of NsaS was modeled using the BCL::MP-Fold method,45 which creates membrane protein topologies through the assembly of entire transmembrane helices. Helix regions were predicted using Jufo9D46 and Octopus.47 During TM-helix assembly, a C2-symmetry of the NsaS homodimer was enforced. In addition, experimental cross-linking data were converted into intersubunit contact restraints and applied during folding. To this end, the observed frequency of NsaS dimer formation was treated as contact probability and multiplied with the BCL contact score:
Here, d is the Cβ—Cβ model distance between the restrained amino acid pair and values d0 and d1 were set to 8 and 25 Å, respectively. Because BCL::MP-Fold yields models consisting only of unconnected secondary structure elements, interhelical loop regions were built using the Rosetta (version 3.7) loop modeling protocol, employing both the CCD48 and KIC49 algorithm.
The linker helix (G51– N86) and the catalytic domain (L70–E295) were modeled using Rosetta version XXX 3.7 and de novo50 and comparative modeling51 protocols. C2 symmetry was assumed for the dimer. Fragments libraries were constructed using the Robetta Web server (http://robetta.bakerlab.org/). The Talaris2014 energy function was used for the modeling of the linker helix. The generated models were filtered for extended helices. A homology model of NsaS catalytic domain was built based on the X-ray structure of CpxA (PDB id: 4biu) as a template. In order to compensate for the lower sequence identity of the first helix of the Dhp, de novo folding was carried out for the Dhp helical bundle alone (L70–T145). The model with the lowest score and RMSD compared to the Dhp domain of 4biu was selected and connected to the homology model of the catalytic domain. Finally, to construct a full-length model of NsaS, models of the four individual domains (TM, linker, Dhp and catalytic domain) were assembled using Rosetta comparative modeling. Regions connecting neighboring domains were relaxed by energy minimization to allow for a smooth transition of the protein backbone.
4.8. Molecular Dynamics.
We used MD simulations to further refine the model of the full length NsaS structure in a phospholipid bilayer. The atomistic model was inserted in a 120 × 120 Å palmitoyl oleoylphosphatidylethanolamine (POPE) bilayer patch. The system was then solvated in a 120 × 120 × 150 Å water box, neutralized through the addition of NaCl at a concentration of 150 mM. The MD simulation was performed using NAMD 2.10 engine,52 with the CHARMM36 force field for the protein and POPE membrane53,54 TIP3P water parametrization was used to describe the water molecules.55 The periodic electrostatic interactions were computed using the particle-mesh Ewald (PME) summation with a grid spacing <1 Å. Constant temperature (310 K) was imposed by using Langevin dynamics, with damping coefficient of 1.0 ps. Constant pressure of 1 atm was maintained with a Langevin piston dynamics, 200 fs decay period and 50 fs time constant. During equilibration, NsaS backbone atoms were restrained with harmonic potential (force constant: 1 kCal/mol/Å2). The system was first minimized by 5000 conjugate gradient steps and then equilibrated by using a linear temperature gradient, which heated up the system from 0 to 310 K in 5 ns. An additional 5 ns was carried out before removing all restraints. An unrestrained molecular dynamics was performed up to 250 ns with a 2 fs integration time step using the RATTLE algorithm applied to all bonds.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Tim Forster (Trinity College) for promptly sharing the pIMAY plasmid and the associated DC10B and SA08B strains, which were critical to producing the nsaS knockouts. We also thank Lindsey Shaw from University of Southern Florida for generously sharing several NsaS strains and the pMK4 plasmid. We thank Mark Kelly (NMR Facility at UC San Francisco) for assistance with NMR instrumentation and Hyunil Jo for assistance with synthesis and purification of the NsaS linker peptide fragment. M.P.B. was supported by a fellowship from the Jane Coffin Childs Foundation. Funds for the 900 MHz NMR spectrometer were provided by the NIH through grant GM68933.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b09670. Bio Mag Res Bank (BMRB) accession code 27502.
Chemical shift assignments (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Blair JM; Webber MA; Baylay AJ; Ogbolu DO; Piddock LJ Nat. Rev. Microbiol. 2015, 13 (l), 42–51. [DOI] [PubMed] [Google Scholar]
- (2).Qin Z; Lee B; Yang L; Zhang J; Yang X; Qu D; Jiang H; Molin S FEMS Microbiol. Lett 2007, 273 (2), 149–56. [DOI] [PubMed] [Google Scholar]
- (3).Matsuo M; Kato F; Oogai Y; Kawai T; Sugai M; Komatsuzawa H J.Antimicrob. Chemother. 2010, 65 (7), 1536–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Hiron A; Falord M; Valle J; Debarbouille M; Msadek T Mol. Microbiol. 2011, 81 (3), 602–22. [DOI] [PubMed] [Google Scholar]
- (5).Yang SJ; Bayer AS; Mishra NN; Meehl M; Ledala N; Yeaman MR; Xiong YQ; Cheung AL Infect. Immun. 2012, 80,(1), 74–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Fritz G; Dintner S; Treichel NS; Radeck J; Gerland U; Mascher T; Gebhard S mBio 2015, 6 (4), e00975–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Dintner S; Heermann R; Fang C; Jung K; Gebhard SJ Biol. Chem. 2014, 289 (40), 27899–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Cheung J; Hendrickson W.a. Structure (Oxford, U. K.) 2009, 17,(2), 190–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Kramer J; Fischer JD; Zientz E; Vijayan V; Griesinger C; Lupas A; Unden GJ Bacteriol. 2007, 189 (11), 4290–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Gushchin I; Melnikov I; Polovinkin V; Ishchenko A; Yuzhakova A; Buslaev P; Bourenkov G; Grudinin S; Round E; Balandin T; Borshchevskiy V; Willbold D; Leonard G; Buldt G; Popov A; Gordeliy V Science 2017, 356 (6342), No. eaah6345. [DOI] [PubMed] [Google Scholar]
- (11).Song Y; Peisach D; Pioszak AA; Xu Z; Ninfa AJ Biochemistry 2004, 43 (21), 6670–8. [DOI] [PubMed] [Google Scholar]
- (12).Aguilar PS; Hernandez-Arriaga AM; Cybulski LE; Erazo AC; de Mendoza D EMBO J. 2001, 20 (7), 1681–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Wang B; Zhao A; Novick RP; Muir TW Mol. Cell 2014, 53 (6), 929–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Mascher T; Helmann JD; Unden G Microbiology and molecular biology reviews: MMBR 2006, 70 (4), 910–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Mensa B; Howell GL; Scott R; DeGrado WF Antimicrob. Agents Chemother. 2014, 58 (9), 5136–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Thennarasu S; Tan A; Penumatchu R; Shelburne CE; Heyl DL; Ramamoorthy A Biophys. J. 2010, 98 (2), 248–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Kolar SL; Nagarajan V; Oszmiana A; Rivera FE; Miller HK; Davenport JE; Riordan JT; Potempa J; Barber DS; Koziel J; Elasri MO; Shaw LN Microbiology 2011, 157, 2206–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Diensthuber RP; Bommer M; Gleichmann T; Moglich A Structure (Oxford, U. K.) 2013, 21 (7), 1127–36. [DOI] [PubMed] [Google Scholar]
- (19).Saita E; Abriata LA; Tsai YT; Trajtenberg F; Lemmin T; Buschiazzo A; Dal Peraro M; de Mendoza D; Albanesi D Mol. Microbiol. 2015, 98 (2), 258–71. [DOI] [PubMed] [Google Scholar]
- (20).Stewart V; Chen L-L Journal of bacteriology 2010, 192 (3), 734–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Ferris HU; Zeth K; Hulko M; Dunin-Horkawicz S; Lupas AN J. Struct. Biol. 2014, 186 (3), 349–356. [DOI] [PubMed] [Google Scholar]
- (22).Bhate MP; Molnar KS; Goulian M; DeGrado WF Structure 2015, 23 (6), 981–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Molnar KS; Bonomi M; Pellarin R; Clinthorne GD; Gonzalez G; Goldberg SD; Goulian M; Sali A; DeGrado WF Structure (Oxford, U. K.) 2014, 22 (9), 1239–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Inda ME; Vandenbranden M; Fernandez A; de Mendoza D; Ruysschaert JM; Cybulski LE Proc. Natl. Acad. Sci. U. S. A. 2014, 111 (9), 3579–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Seddon AM; Curnow P; Booth PJ Biochim. Biophys. Acta, Biomembr. 2004, 1666 (1–2), 105–17. [DOI] [PubMed] [Google Scholar]
- (26).Shen Y; Delaglio F; Cornilescu G; Bax AJ Biomol NMR 2009, 44 (4), 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Berjanskii MV; Wishart DS J. Am. Chem. Soc. 2005, 127 (43), 14970–1. [DOI] [PubMed] [Google Scholar]
- (28).Bai Y; Milne JS; Mayne L; Englander SW Proteins: Struct., Funct., Genet 1994, 20 (1), 4–14. [DOI] [PubMed] [Google Scholar]
- (29).Bhunia A; Domadia PN; Mohanram H; Bhattacharjya S Proteins: Struct., Funct., Genet. 2009, 74 (2), 328–43. [DOI] [PubMed] [Google Scholar]
- (30).Respondek M; Madl T; Gobl C; Golser R; Zangger K J. Am. Chem. Soc. 2007, 129 (16), 5228–34 [DOI] [PubMed] [Google Scholar]
- (31).Walsh ST; Cheng RP; Wright WW; Alonso DO; Daggett V; Vanderkooi JM; DeGrado WF Protein Sci. 2003, 12, 520–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Goodman EM; Kim PS Biochemistry 1991, 30 (50), 11615–20. [DOI] [PubMed] [Google Scholar]
- (33).Möglich A; Ayers R a.; Moffat K J. Mol. Biol. 2009, 385 (5), 1433–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Schmidt NW; Grigoryan G; DeGrado WF Protein Sci. 2017, 26 (3), 414–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Mechaly AE; Sassoon N; Betton J-M; Alzari PM PLoS Biol. 2014, 12 (1), e1001776–e1001776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Peter BJ; Kent HM; Mills IG; Vallis Y; Butler PJ; Evans PR; McMahon HT Science 2004, 303 (5657), 495–499. [DOI] [PubMed] [Google Scholar]
- (37).Schmidt NW; Mishra A; Wang J; DeGrado WF; Wong GC J. Am. Chem. Soc. 2013, 135 (37), 13710–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Chervitz S. a.; Falke JJ Proc. Natl. Acad. Sci. U. S. A. 1996, 93(68), 2545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Garg N; Oman TJ; Andrew Wang T-S; De Gonzalo CVG; Walker S; van der Donk WA J. Antibiot. 2014, 67 (1), 133–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Joo HS; Otto M Biochim. Biophys. Acta, Biomembr. 2015, 1848 (11), 3055–3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Stenglein MD; Sanders C; Kistler AL; Ruby JG; Franco JY; Reavill DR; Dunker F; Derisi JL mBio 2012, 3 (4), e00180–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Goecks J; Nekrutenko A; Taylor J Genome Biol. 2010, 11 (8), R86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Blankenberg D; Von Kuster G; Coraor N; Ananda G; Lazarus R; Mangan M; Nekrutenko A; Taylor J Galaxy: a web- based genome analysis tool for experimentalists In Current Protocols in Molecular Biology; Wiley, 2010; Chapter 19, Unit 19.10, pp 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Giardine B; Riemer C; Hardison RC; Burhans R; Elnitski L; Shah P; Zhang Y; Blankenberg D; Albert I; Taylor J; Miller W; Kent WJ; Nekrutenko A Genome Res. 2005, 15 (10), 1451–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Weiner BE; Woetzel N; Karakas M; Alexander N; Meiler J Structure 2013, 21 (7), 1107–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Leman JK; Mueller R; Karakas M; Woetzel N; Meiler J Proteins: Struct., Funct., Genet. 2013, 81 (7), 1127–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Viklund H; Elofsson A Bioinformatics 2008, 24 (15), 1662–8. [DOI] [PubMed] [Google Scholar]
- (48).Canutescu AA; Dunbrack RL Jr. Protein Sci. 2003, 12 (5), 963–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Mandell DJ; Coutsias EA; Kortemme T Nat. Methods 2009, 6 (8), 551–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Bradley P; Misura KM; Baker D Science 2005, 309 (5742), 1868–1871. [DOI] [PubMed] [Google Scholar]
- (51).Song Y; DiMaio F; Wang RY; Kim D; Miles C; Brunette T; Thompson J; Baker D Structure 2013, 21 (10), 1735–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Phillips JC; Braun R; Wang W; Gumbart J; Tajkhorshid E; Villa E; Chipot C; Skeel RD; Kale L; Schulten KJ Comput. Chem. 2005, 26 (16), 1781–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Best RB; Zhu X; Shim J; Lopes PE; Mittal J; Feig M; Mackerell AD Jr. J. Chem. Theory Comput. 2012, 8 (9), 3257–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Guvench O; Mallajosyula SS; Raman EP; Hatcher E; Vanommeslaeghe K; Foster TJ; Jamison FW 2nd; Mackerell AD Jr. J. Chem. Theory Comput. 2011, 7 (10), 3162–3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Jorgensen WL; Jayaraman C; Madura JD J. Chem. Phys. 1983, 79 (2), 926. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.